

Abstract
Progressive alteration of the extracellular matrix (ECM) is the characteristic of hypertensive nephropathy (HN). Both mesangial and endothelial cells have the ability to synthesize and degrade ECM components, including collagens through the activation of matrix metalloproteinases (MMPs) in stress conditions, such as in hypertension. On the other hand, hydrogen sulfide (H2S) has been shown to mitigate hypertensive renal matrix remodeling. Surprisingly, whether H2S ameliorates receptor-mediated (urokinase plasminogen activator receptor-associated protein, uPARAP/Endo180) collagen dysregulation in Ang-II hypertension is not clear. The purpose of this study was to determine whether Ang-II alters the expression of Endo180, tPA (tissue plasminogen activator), MMPs and their tissue inhibitors (TIMPs) leading to the dysregulation of cellular collagen homeostasis and whether H2S mitigates collagen turnover. Mouse mesangial cells (MCs) and glomerular endothelial cells (MGECs) were treated without or with Ang-II and H2S donor, GYY (GYY4137) for 48 hrs. Cell lysates were analyzed by Western blot and RT-PCR, and cells by immunocytochemistry. The results indicated that, while Ang-II differentially expressed MMP-13 and TIMP-1 in MCs and in MGECs, it predominantly decreased tPA, Endo 180, and increased PAI-1 (plasminogen activator inhibitor-1), MMP-14, and collagen IIIA and IV in both the cell types. Interestingly, H2S donor GYY treatment normalized the above changes in both the cell types. We conclude that Ang-II treatment causes ECM remodeling in MCs and MGECs through PAI-1/tPA /Endo180 and MMP/TIMP-dependent collagen remodeling, and H2S treatment mitigates remodeling, in part, by modulating these pathways.
Keywords: H2S, PAI-1, tPA, MMP, TIMP, Endo180, Collagen
Introduction
Hypertensive nephropathy (HN) is a common cause of chronic kidney disease (CKD), which is due to high blood pressure in the kidney vasculature, causing glomerulosclerosis and tubular atrophy [1, 2]. Hydrogen sulfide (H2S) has emerged in the recent past as an endogenous physiological blood pressure regulator along with two other established gaseous molecules, nitric oxide (NO) and carbon monoxide (CO) [3, 4]. The kidney is a major organ, which expresses several H2S producing enzymes, such as CSE (cystathionine β-synthase), CBS (cystathionine γ-lyase), and 3MST (3-mercaptopyruvate sulfurtransferase), and thus likely to contribute to local blood pressure regulation in the kidney vasculature [5–7]. It is not well known, however, whether H2S potentially modulate kidney cellular matrix in different cell types that are often associated with hypertensive conditions.
CKD, including HN, is characterized by an imbalance in matrix degradation and synthesis, leading to the accumulation of extracellular matrix (ECM) proteins [8]. The complex cross-linked network of macromolecules that constitute the ECM serves to maintain tissue architecture and regulate cellular functions such as migration, proliferation, and apoptosis [9, 10]. The remodeling of ECM is centrally engaged in numerous physiological and pathological conditions [11]. In fibrosis, aberrant ECM remodeling is the hallmark and the cause and symptoms of the disease progression[12].
A subset of matrix metalloproteinases (MMPs) is capable of cleaving collagen structures and hence plays a regulatory role in the progression of renal fibrosis by regulating the extracellular collagen degradation pathway [13–15]. On the other hand, endogenous tissue inhibitor of MMPs (TIMPs) regulates MMPs activity. Thus, an imbalance to MMPs and TIMPs plays a crucial role in matrix synthesis and degradation. In addition to this, another mechanism for intracellular endocytic degradation of collagen exists, which regulates and maintains the ECM homeostasis through the interaction of tissue plasminogen activator (tPA), plasminogen activator inhibitor- 1 (PAI-1) and endocytic collagen receptor, uPARAP/Endo 180 [16–18].
In kidney disease, such as in HN glomerular mesangial cells (MCs) and glomerular endothelial cells (GECs) undergo excessive ECM alteration, cell proliferation, and cell apoptosis, leading to glomerular dysfunction and kidney failure. However, it is unclear in terms of matrix remodeling, whether glomerular mesangial and endothelial cells behave differently in response to hypertensive stress. We therefore aimed at investigating how mesangial and endothelial cells respond to hypertension condition by treating these cells with or without angiotensin II (Ang-II). We also tested whether H2S has any positive impact on these cells against Ang-II stress. Our results showed that these two cells respond differently in hypertensive conditions by altering tPA, PAI-1, Endo180, MMP-13 /-14, TIMP-1, and Col IIIA / IV mRNAs and protein expression. We also showed that H2S donor, GYY (GYY4137), ameliorated Ang-II-mediated ECM remodeling in these cells.
Material and method
Antibodies and reagents
MMP-14 primary antibody and fluorescently conjugated secondary antibodies were from Abcam (Cambridge, CA). Collagen IV antibody was purchased from Novus Biologicals LLC (Centennial, CO); MMP-13, PAI-1, and uPARAP/Endo180 antibodies were purchased from Thermo Fisher Scientific (Waltham, MA). Antibodies against TIMP-1, Collagen-IIIA, tPA, GAPDH (Glyceraldehyde 3-phosphate dehydrogenase), small interfering RNAs (siRNAs) against Endo 180 (sc-62277), scrambled siRNA (as a negative control, sc-37007), and all HRP conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). DAPI, Angiotensin II, GYY4137, and all other analytical reagents were from Sigma-Aldrich, St. Louis, MO.
Cell culture
Mouse kidney mesangial cells (MCs) and glomerular endothelial cells (MGECs) were purchased from ATCC (Manassas, VA). MCs were maintained in DMEM/F-12 (50:50) medium containing 5% fetal bovine serum (ATCC, Manassas, VA), antibiotics, and L-glutamine (Mediatech, Inc, Manassas, VA). The complete MGECs medium was from Cell Biologics (Chicago, IL). Cells were grown in culture flasks at the 37°C humid chamber with an atmosphere of 5% CO2 and 95% air. Semi-confluent cells were trypsinized for a subculture or plated onto cell culture plates and grown to 60–70% confluency before the treatment started. At the end of the experiments, cells were processed and analyzed accordingly.
Cell treatment and transfection of siRNA
Cells were divided into four groups: Group 1, no treatment (control); Group 2, treated with 200 nM Angiotensin II (Ang-II); Group 3, treated with GYY4137 (GYY; 250 μM, an H2S donor); and Group 4, treated with Ang-II in combination with GYY. At the end of 48 h experimentation, the cells were washed with PBS and processed accordingly for mRNA, protein, and immunostaining. Transfection of siRNAs into both cell types was performed by Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific, Waltham, MA), using a standard transfection protocol at a final siRNA concentration of 10 nM for 48 h. The protocol was adopted following the manufacturer’s instruction, and as we have previously described [19].
RNA extraction
Cells were washed with ice-cold PBS, and RNA was extracted using the TRIzol reagent isolation kit following the manufacturer’s protocol (Life Technologies, Grand Island, NY). The quality of total RNA was measured by NanoDrop ND-1000, and only high-quality RNA (260/280–2.00 and 260/230–1.80) was used for reverse transcriptase PCR (RT-PCR).
Semi-quantitative RT-PCR
The total RNA (2 μg) was reverse transcribed by using the Easyscript cDNA synthesis kit (MidSci G0234) according to the manufacturer’s instructions. PCR program for amplification of cDNA was done using GoTaq Hot start green Master Mix (Promega) according to the manufacturer’s guidelines. The primer sequences are presented in Table 1.
Table 1.
Primers and primer sequences
| Primer | Forward primer | Reverse primer |
|---|---|---|
| MMP-14 | GGATGGACACAGAGAACTTCGT | GTGACCCTGACTTGCTTCCATA |
| MMP-13 | CAGTTGACAGGCTCCGAGAA | TTCACCCACATCAGGCACTC |
| TIMP-1 | AGTGATTTCCCCGCCAACTC | GGGGCCATCATGGTATCTGC |
| Endo180 | CATCACCAAACAGATCAAGC | TCGGACCACTCAAAATTCAT |
| Col IV | GACCACTATGCTTGAAGTGA | ACAGAAGGCCTTAGTAGTCT |
| Col IIIA | AATTCGGACTAGACATTGGC | GGGAAATTGAGTTTGGGTTG |
Abbreviations: MMP, matrix metalloproteinase; TIMP, tissue inhibitor of matrix metalloproteinase; Col, collagen; Endo180, an endocytic recycling glycoprotein and collagen receptor
Western blot
Protein was extracted from cells by RIPA buffer (Boston BioProducts, Worcester, MA) with 1 mM phenylmethylsulfonyl fluoride (PMSF), and 1% protease inhibitor cocktail (Sigma, St Louis, MO). After sonication, protein lysate was centrifuged at 12,000 × g for 10 min at 4°C. Protein concentration in the samples was measured by Bradford assay. An equal amount of protein extract (25 μg) was electrophoresed by SDS-PAGE and immunoblotted onto PVDF membrane. The membranes were blocked by 5% non-fat milk for 60 min at room temperature and subsequently incubated with respective primary antibodies overnight at 4°C. Membrane washed 3 times in PBS and then incubated with respective secondary antibodies for 2 hr at room temperature. The membranes were then washed, and protein bands were visualized using ECL Luminata Forte (Millipore, Temecula, CA) in a Bio-Rad ChemiDoc system. GAPDH was used as a loading control, and band intensity was quantified using ImageJ software.
Immunocytochemistry (ICC)
Cells were fixed in 4% paraformaldehyde for 10 min and washed thrice with ice-cold PBS. Cells were then permeabilized with 0.25% Tween 20 in PBS for 10 min followed by washing thrice with PBST (0.1% Tween 20). Cells were incubated for 1 h with blocking buffer (1% BSA in PBST), washed, and then incubated with primary antibody overnight at 4°C. Appropriate to the primary antibody species, secondary antibodies conjugated with fluorescence were applied to the cells for immune-detection. After washing with PBST, cells were immediately visualized and analyzed for fluorescence intensity under a laser scanning confocal microscope (Olympus FluoView1000, Pittsburgh, PA) using an appropriate filter.
Statistical Analysis
ImageJ software was used to calculate the mean values of mRNA or protein expression. Data were expressed as mean ± SEM of 9 independent experiments or as stated in each of the figure legends. The difference in treatment effects was measured within and across effectors or inhibitors using two-way ANOVA for repeated measures. The significant differences between groups were accepted at p<0.05.
Results
GYY normalized Ang-II-induced increased expression of tPA and PAI-1
The immunoblotting results indicated PAI-1 and tPA expression in control cells, i.e., without any treatment, were at the basal level both in MCs and in MGECs. These basal level expressions did not change significantly in cells treated with only GYY (Fig. 1a–b). Results from Ang-II treated MCs and MGECs indicated upregulated PAI-1 and downregulated tPA protein expressions in these two cell types compared to their respective control groups (Fig. 1a–b). Interestingly, GYY treatment reversed and normalized both PAI-1 and tPA expression in these two cell types in Ang-II treated condition (Fig. 1a–b). These results suggest that H2S plays a regulatory role in PAI-1 and tPA expression during hypertensive conditions in MCs and MGECs.
Fig. 1. GYY reversed Ang-II-induced altered PAI-1 and tPA expression in MCs and MGECs.
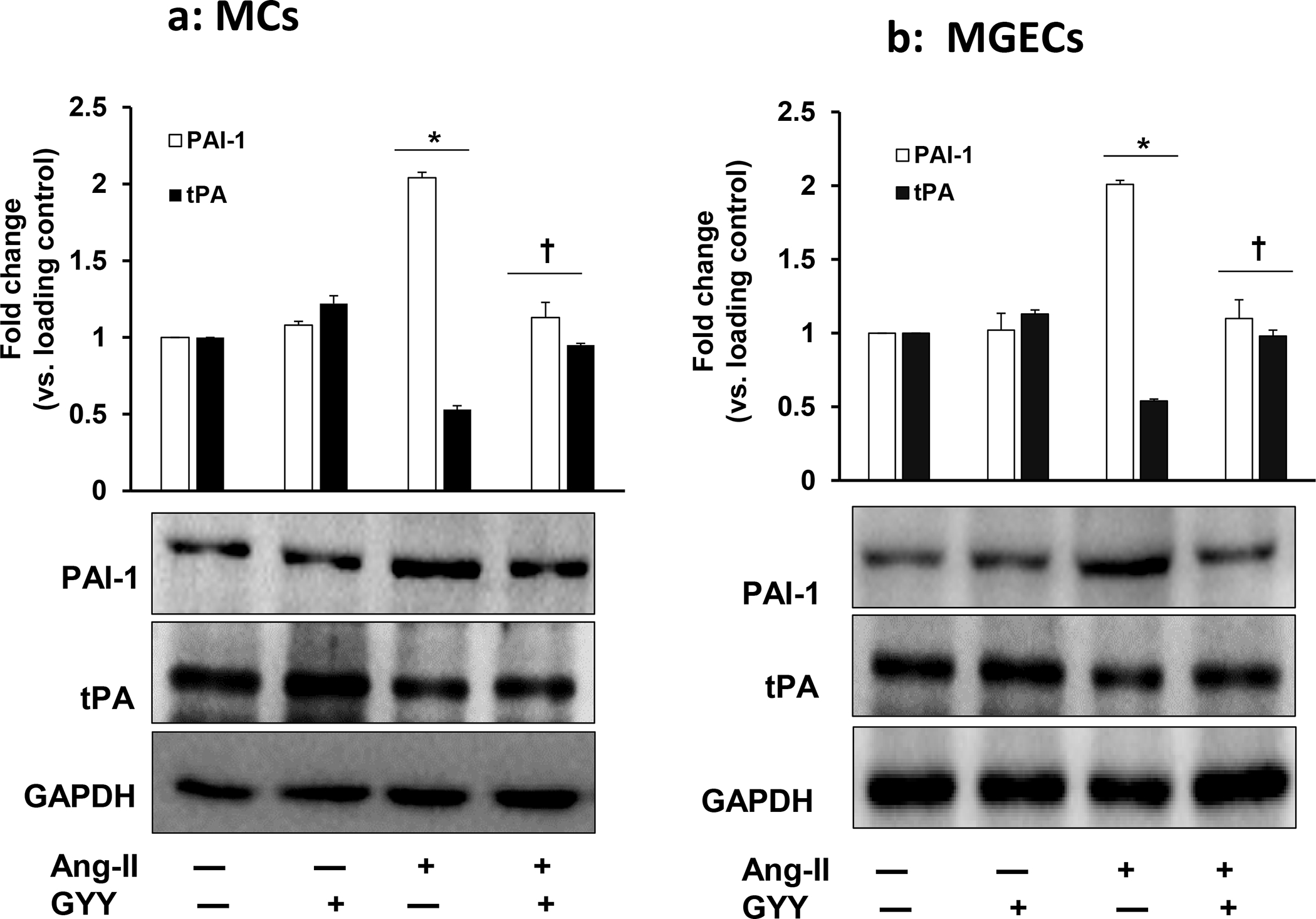
Mouse (a) mesangial cells (MCs), and (b) glomerular endothelial cells (MGECs) were treated with or without Ang-II for 48 h. GYY (GYY4137) was added 1 h prior to Ang-II treatment. The expressions of PAI-1 (~48 kDa) and tPA (~70 kDa) protein were measured by Western blotting using respective primary and HRP-conjugated appropriate secondary antibodies. GAPDH was used as a loading control. Data are presented as mean ± SEM, n = 9 independent experiments / group; *p <0.05 vs control, †p <0.05 vs Ang-II.
Ang-II-induced increased MMP-14 expression was normalized by GYY
The expression of MMP-14 at mRNA and protein level in MCs and in MGECs was at the basal level in control cells without Ang-II or GYY (Fig. 2a–b). The expression of MMP-14 protein and mRNA in these two cell types were significantly increased in Ang-II treated cells compared to their respective controls (Fig. 2a–b). Interestingly, the protein and mRNA expression in these cells were normalized by GYY treatment, suggesting that GYY was able to mitigate MMP-14 expression Ang-II condition (Fig. 2a–b). The expression of MMP-14 in control and in Ang-II condition with or without GYY in MCs and MGECs was further confirmed by immunostaining (Fig. 3a–d), which indicated a similar pattern of expression as obtained by immunoblotting.
Fig 2. Increased MMP-14 expression in Ang-II condition was normalized by GYY.
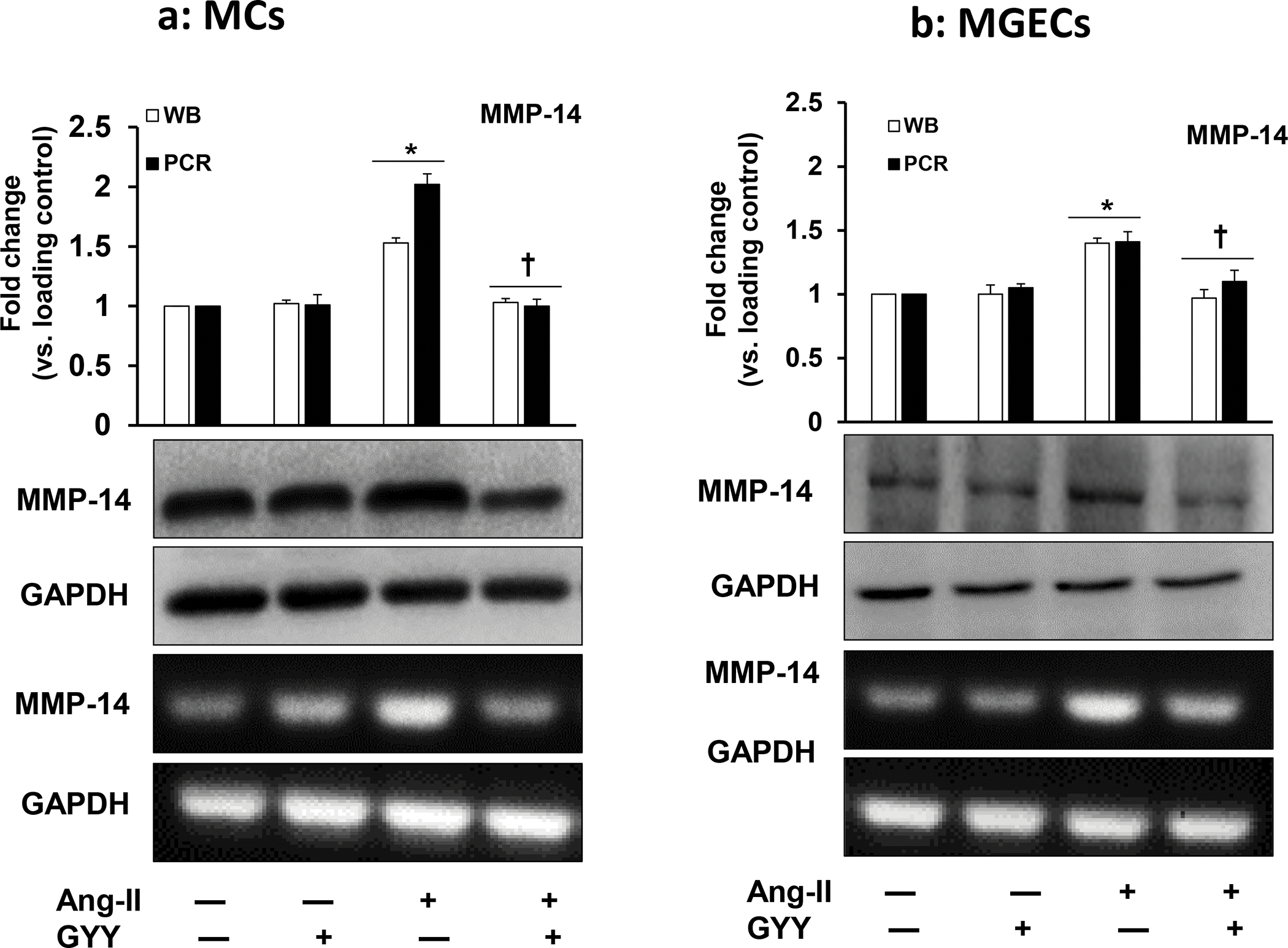
Mouse (a) mesangial cells (MCs) and (b) glomerular endothelial cells (MGECs (B) were treated with or without Ang-II for 48 h. GYY (GYY4137) was added 1 h prior to the Ang-II treatment. MMP-14 protein (~65 kDa) and gene expression were measured by Western blot and RT-PCR, respectively. GAPDH was used as a respective gene and protein loading control. Data are presented as mean ± SEM, n=9 independent experiments / group. *p <0.05 vs control, †p <0.05 vs Ang-II.
Fig. 3. Increased expression of MMP-14 in Ang-II was mitigated by GYY.
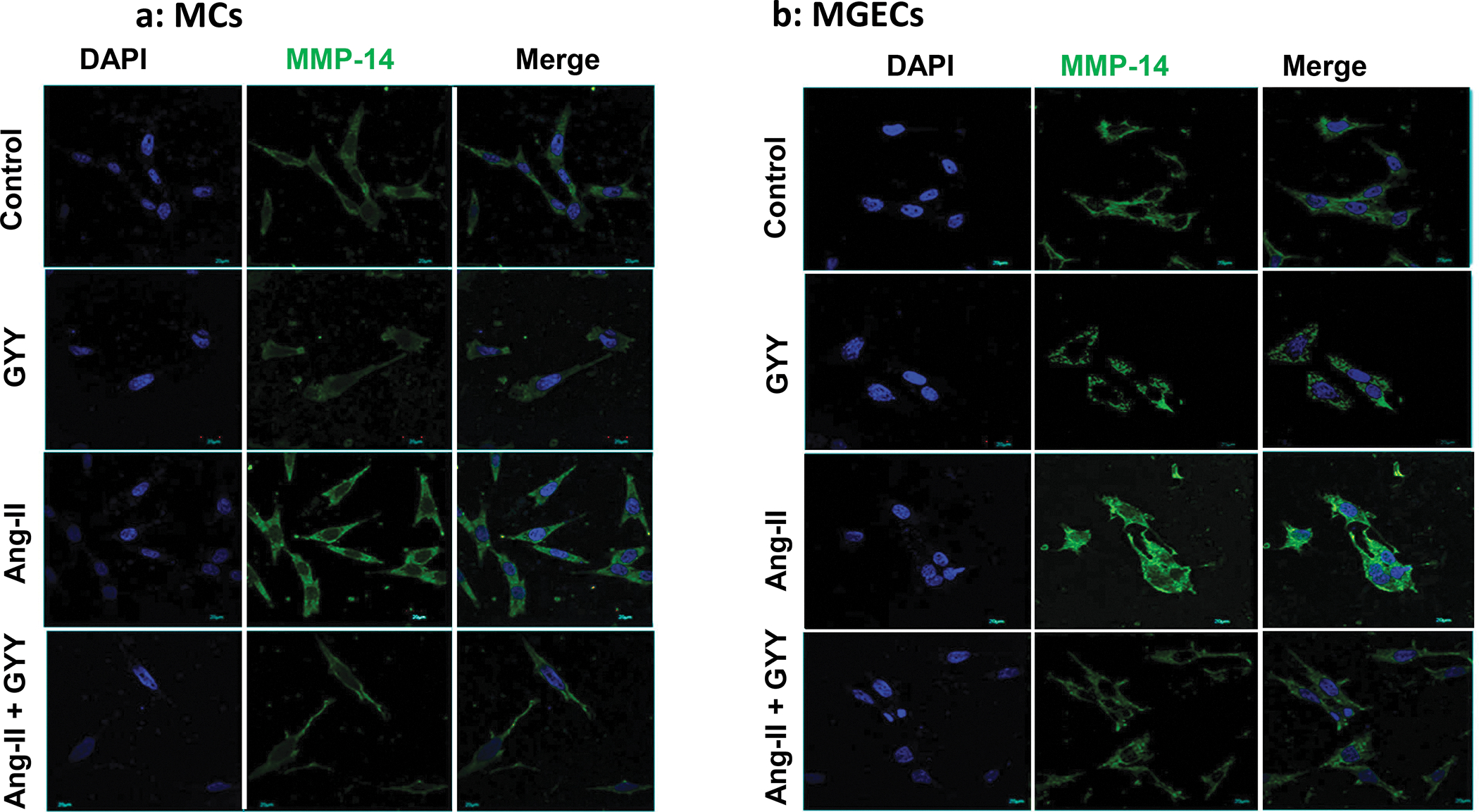
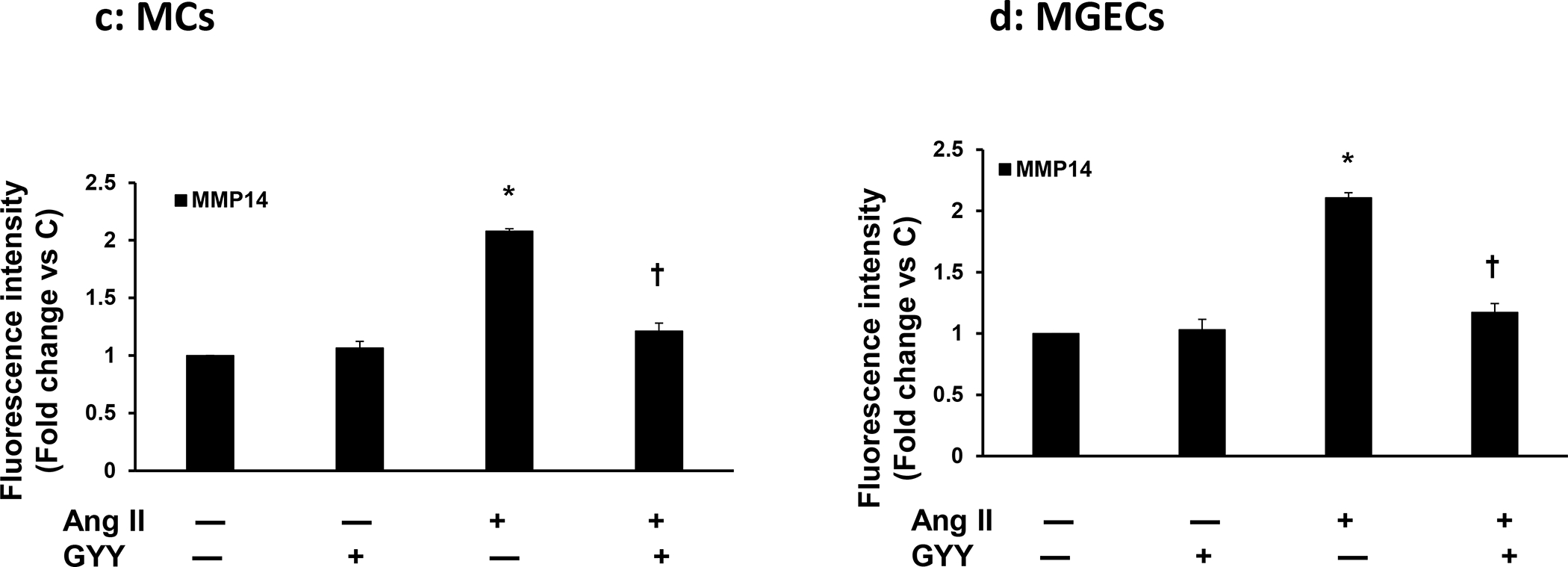
Both cells, MCs (a) and MGECs (b) were treated with or without Ang-II for 48 h. GYY (GYY4137) was added 1 h prior to the Ang-II treatment. Cells were immunostained against MMP-14 (green) and nuclei counterstained with DAPI (blue). (c) and (d) summarized data of MMP-14 expression in MCs and MGECs, respectively. Data are presented as mean ± SEM, n=9 independent experiments / group. *p <0.05 vs control, †p <0.05 vs Ang-II. Scale bar, 20 μm; magnification 60X.
Ang-II-induced altered MMP-13 expression was mitigated by GYY
The expression of MMP-13 was at the basal level in control cells, both in MCs and in MGECs, although the general expression was apparently higher in the MGECs compared to MCs (Fig. 4a–b). The GYY alone treatment did not change MMP-13 expression in these cell lines compared to their respective controls. On the other hand, while the protein expression increased in MCs, it was decreased in MGECs in Ang-II treated cells compared to their respective controls (Fig. 4a–b). Interestingly, GYY treatment normalized MMP-13 expression in these cells in Ang-II condition, which was comparable to their basal levels of expression. The mRNA expression and analysis also revealed a similar pattern as that of protein expression in these cells across their respective groups (Fig. 4a–b). Experiments on similar groups of cells treated with or without Ang-II and with or without GYY and immunostaining with MMP-13 antibody revealed a similar result as that of mRNA and protein expression by RT-PCR and immunoblotting, respectively (Fig. 4c–f). Together, these results suggest that Ang-II has a differential effect on MMP-13 expression in MCs and MGECs and that GYY reverses this effect in Ang-II hypertensive condition.
Fig. 4. Effect of GYY on Ang-II-induced MMP-13 gene and protein expression.
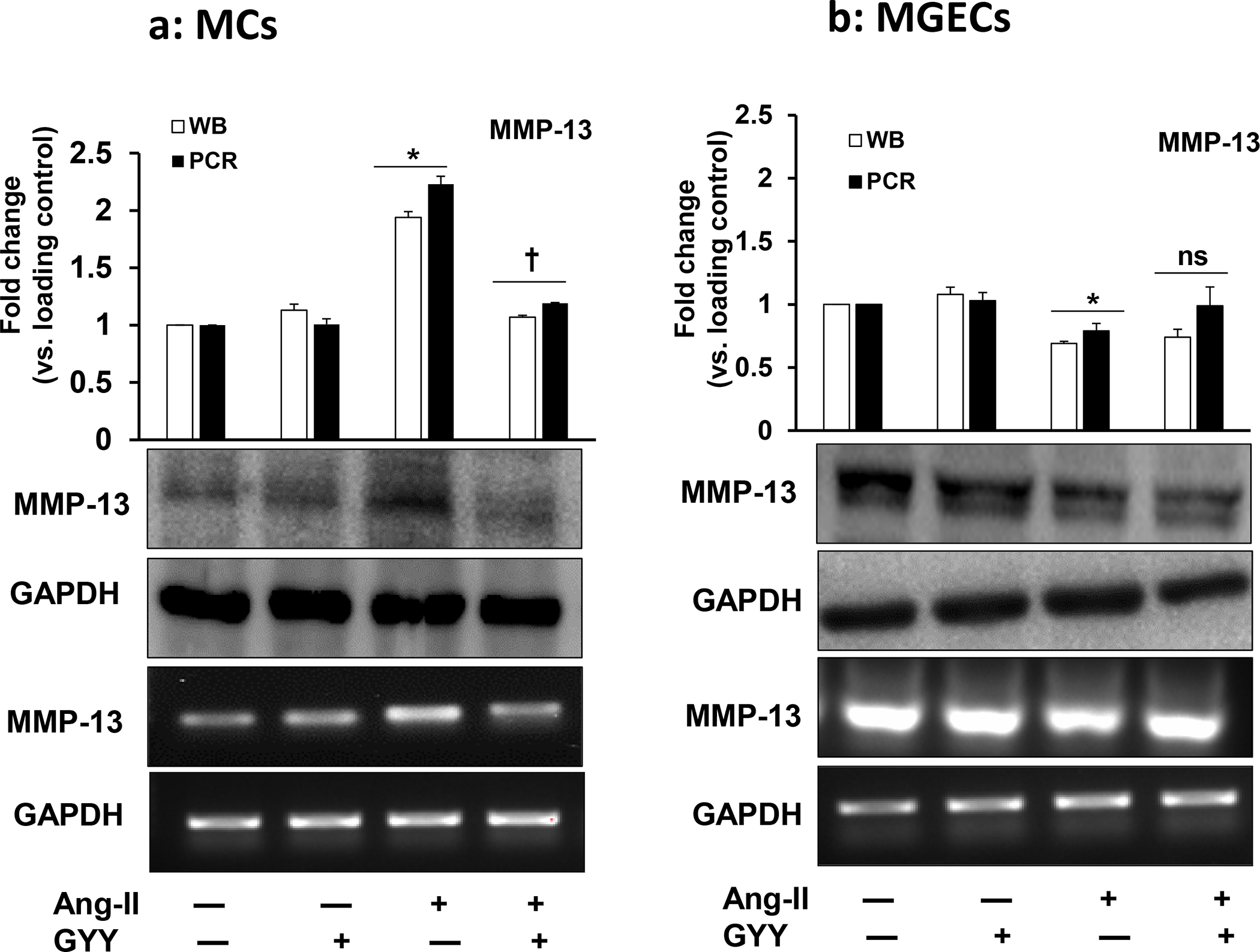
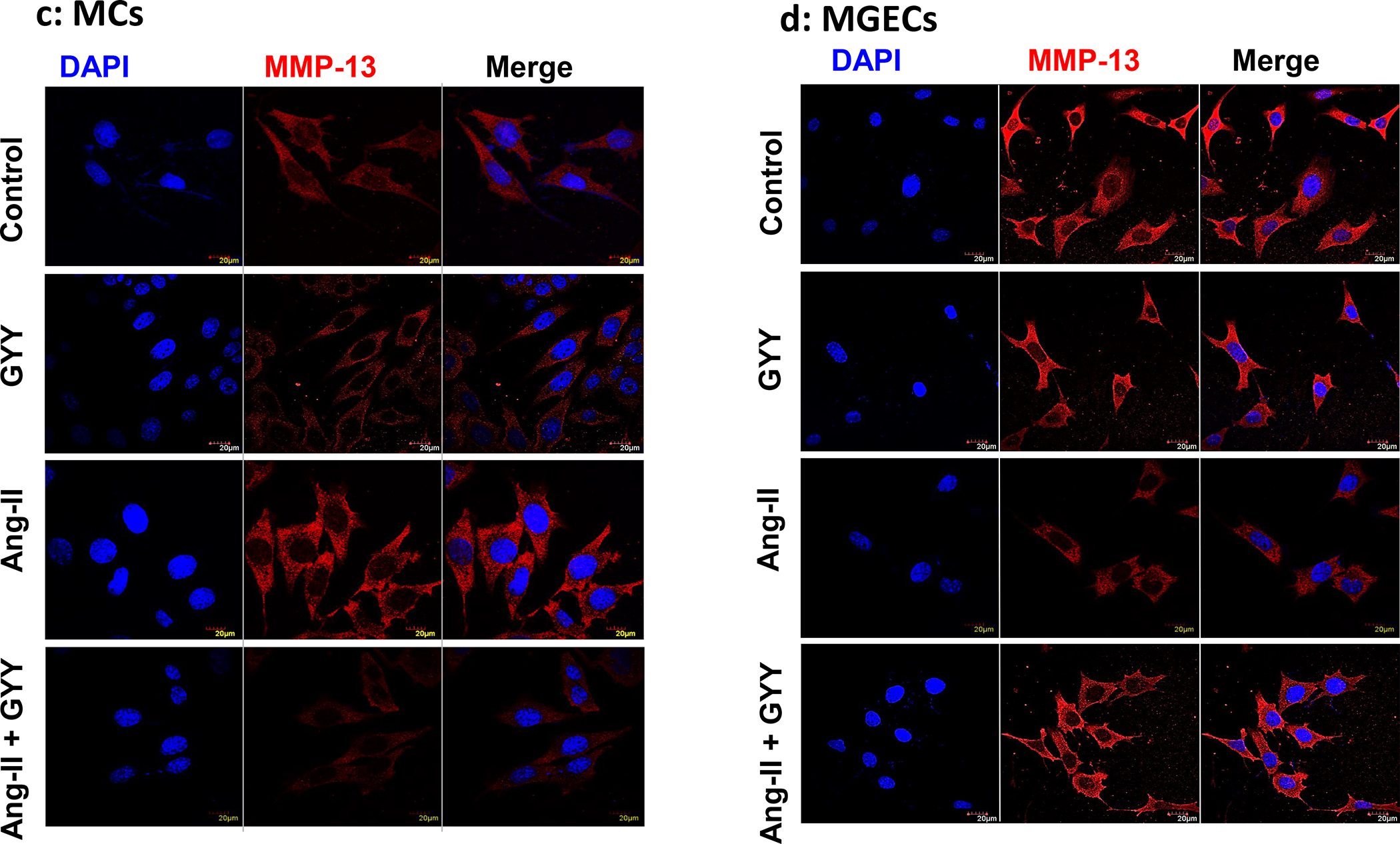
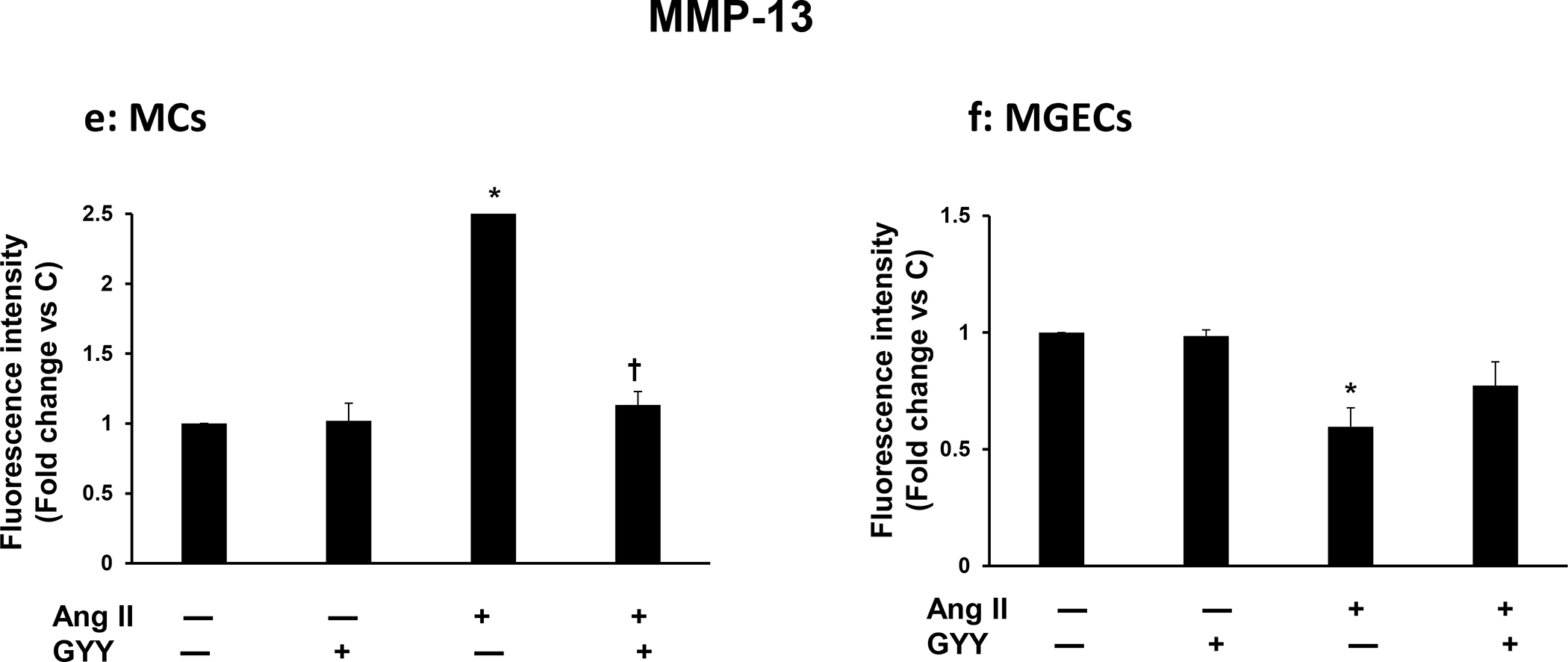
Both MCs (a, c) and MGECs (b, d) were treated with or without Ang-II for 48 h. GYY (GYY4137) was added 1 h prior to the Ang-II treatment. MMP-13 protein (~53 kDa) and gene expressions of were determined by Western blot and RT-PCR, respectively (a, b). GAPDH was used as a loading control. Data are presented as mean ±SEM, n = 9 independent experiments / group. *p <0.05 vs control, †p <0.05 vs Ang-II. Scale bar, 20 μm, magnification 60 X. (c, d) cells were immunostained against MMP-13 (red), and nuclei counterstained with DAPI (blue). (e) and (f) summarized data of MMP-13 expression as observed in immunostaining of MCs and MGECs, respectively. Data are presented as mean ± SEM, n=9 independent experiments / group. *p <0.05 vs control, †p <0.05 vs Ang-II.
GYY reversed Ang-II-induced altered TIMP-1 expression
Since TIMPs regulate MMPs, and TIMP-1 is the main regulator of most MMPs, we determined whether TIMP-1 expression changes in Ang-II condition and whether GYY has any role in regulating TIMP-1. Our results indicated that a basal level of TIMP-1 protein and mRNA expression was observed in both the control cell types, i.e., in MCs and MGECs (Fig. 5a–b). The expression of TIMP-1 protein and mRNA did not change in cells treated with only GYY compared to their respective controls. In Ang-II treated MCs, the expression of both protein and mRNA was mitigated compared to the control, which was normalized with GYY treatment (Fig. 5a).
Fig. 5. Effect of GYY on Ang-II-induced TIMP-1 gene and protein expression MCs.
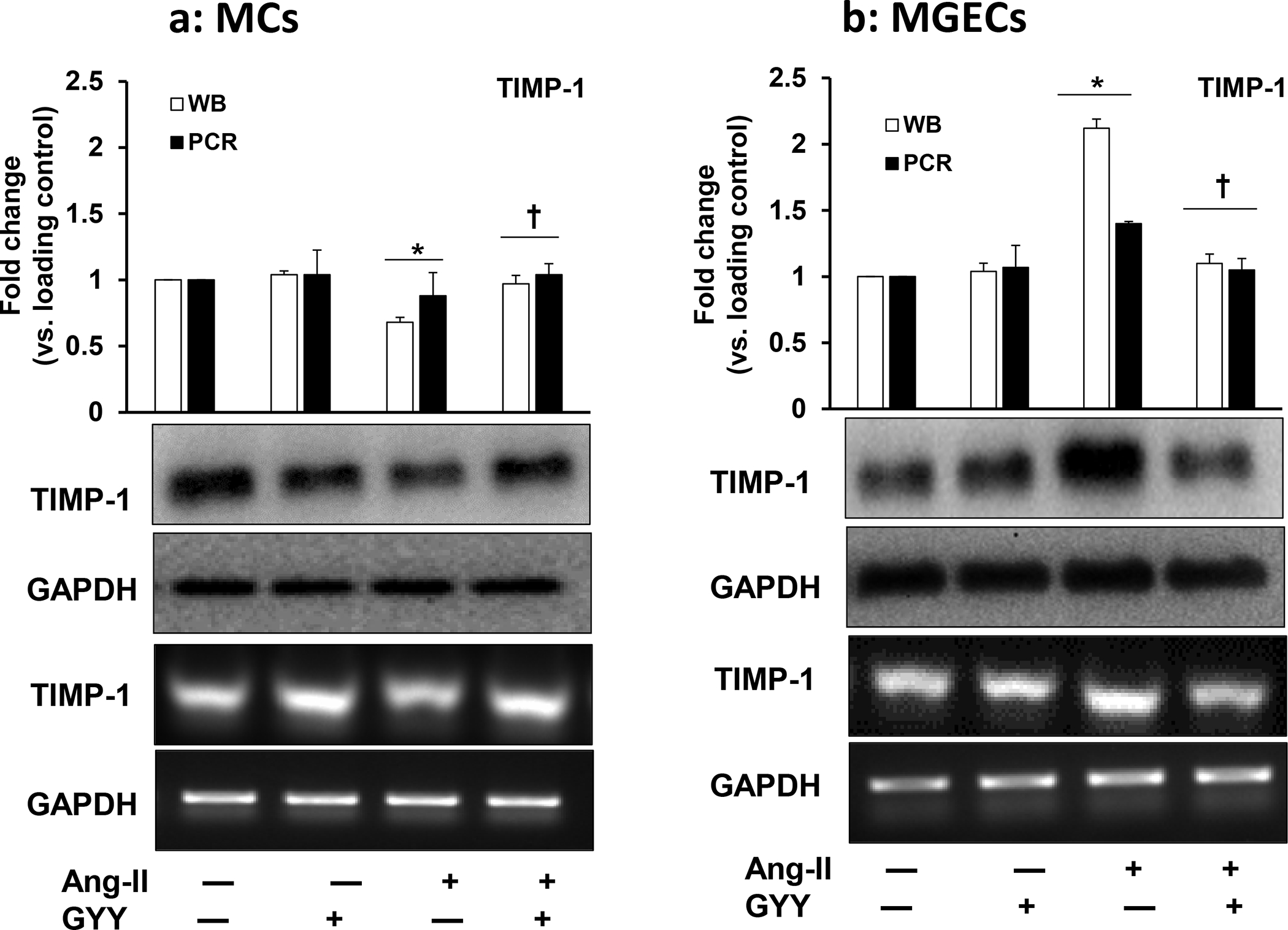
(a) and MGECs (b) were treated with Ang-II for 48 h. GYY (GYY4137) was added 1 h prior to the Ang-II treatment. TIMP-1 protein (~23 kDa) and gene expressions were determined by Western blot and RT-PCR, respectively. GAPDH was used as a loading control. Data are presented as mean ±SEM, n = 9 independent experiments / group. *p <0.05 vs control, †p <0.05 vs Ang-II.
Contrary to MCs, the mRNA and protein expression of TIMP-1 in MGECs was highly upregulated in Ang-II treated cells, which was normalized by GYY treatment (Fig. 5b). Further to the differential expression patterns in MCs vs. MGECs, the TIMP-1 expression was opposite to that of MMP-13 in both these cell lines (Fig. 4a–f). These results suggest that TIMP-1 expression in Ang-II condition is cells specific and that GYY regulates TIMP-1 expression in both of these cell lines (Fig. 5a–b).
GYY normalized increased Col IIIA and Col IV expression
Ang-II plays an active role in inducing renal fibrosis by accumulating excess ECM proteins, including collagen [20, 21]. We determined whether Ang-II induced collagen both in MCs and in MGECs cells and whether GYY has any role in modulating collagen expression in hypertensive condition. Our results indicated that both the cells had expressed a basal level of collagen IIIA at mRNA and protein level, and these basal levels of expressions did not change in cells treated with only GYY (Fig. 6a–b). The expression of collagen IIIA mRNA and protein increased significantly in both the cell type treated with Ang-II compared to their respective controls (Fig. 6a–b). Treatment of cells with GYY mitigated the increased expression of collagen IIIA towards basal levels suggesting that GYY has the potential to inhibit the upregulation of collagen IIIA in hypertensive condition. Similar to the above results, we also observed collagen IV expression both at mRNA and protein levels in all experimental and control groups in these cell types, and GYY mitigated collagen IV expression in hypertensive condition (Fig. 7a–b). Together, these results suggest that GYY has the potential to mitigate increased collagen remodeling in hypertensive kidney cells.
Fig. 6. Effect of GYY on Ang-II-induced Col IIIA gene and protein expression MCs.
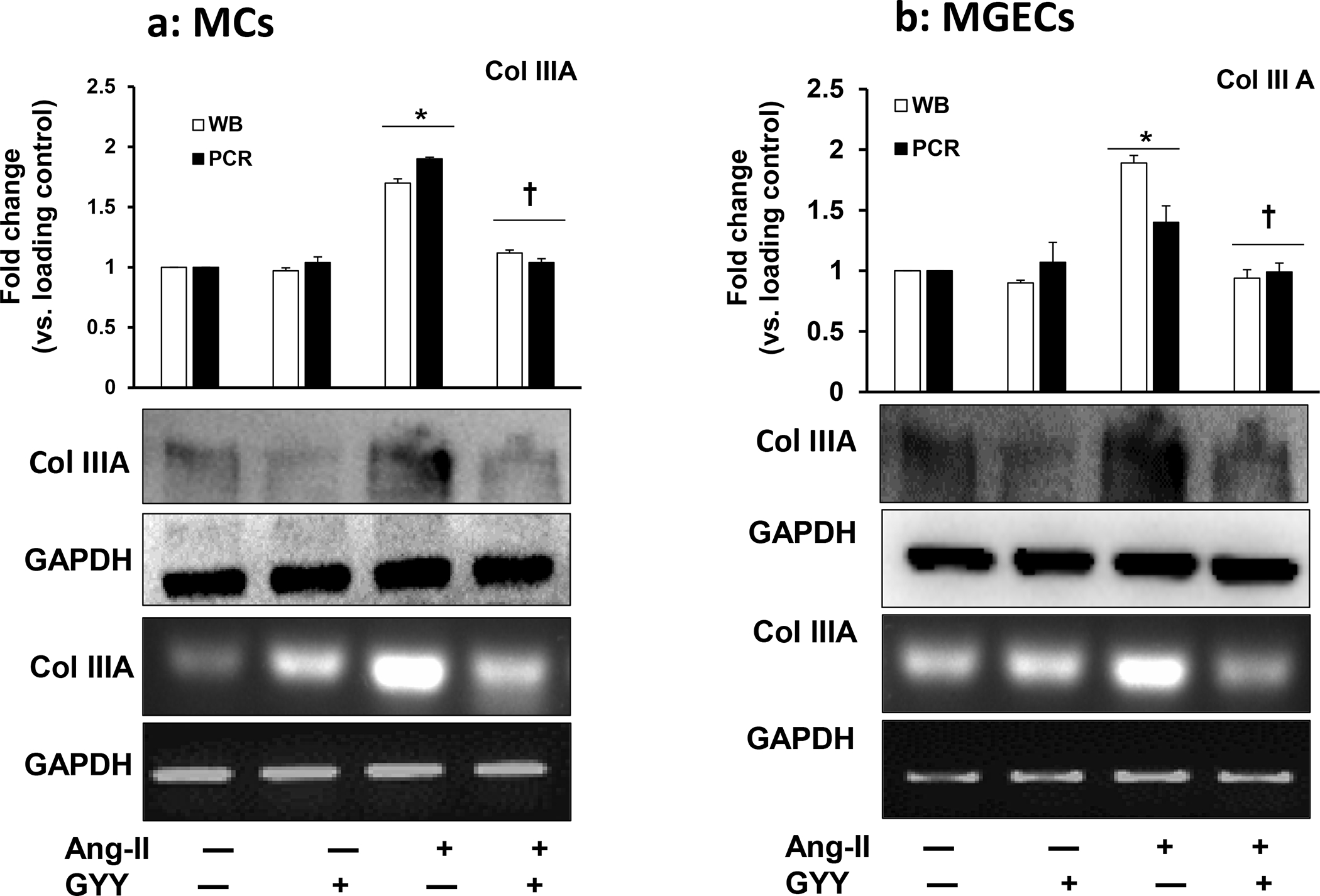
(a) and MGECs (b) were treated with Ang-II for 48 h. GYY (GYY4137) was added 1 h prior to the Ang-II treatment. Col IIIA protein (~138 kDa) and gene expressions were determined by Western blot and RT-PCR, respectively. GAPDH was used as a loading control. Data are presented as mean ±SEM, n = 9 independent experiments / group. *p <0.05 vs control, †p <0.05 vs Ang-II.
Fig. 7. Effect of GYY on Ang-II-induced Col IV gene and protein expression MCs.
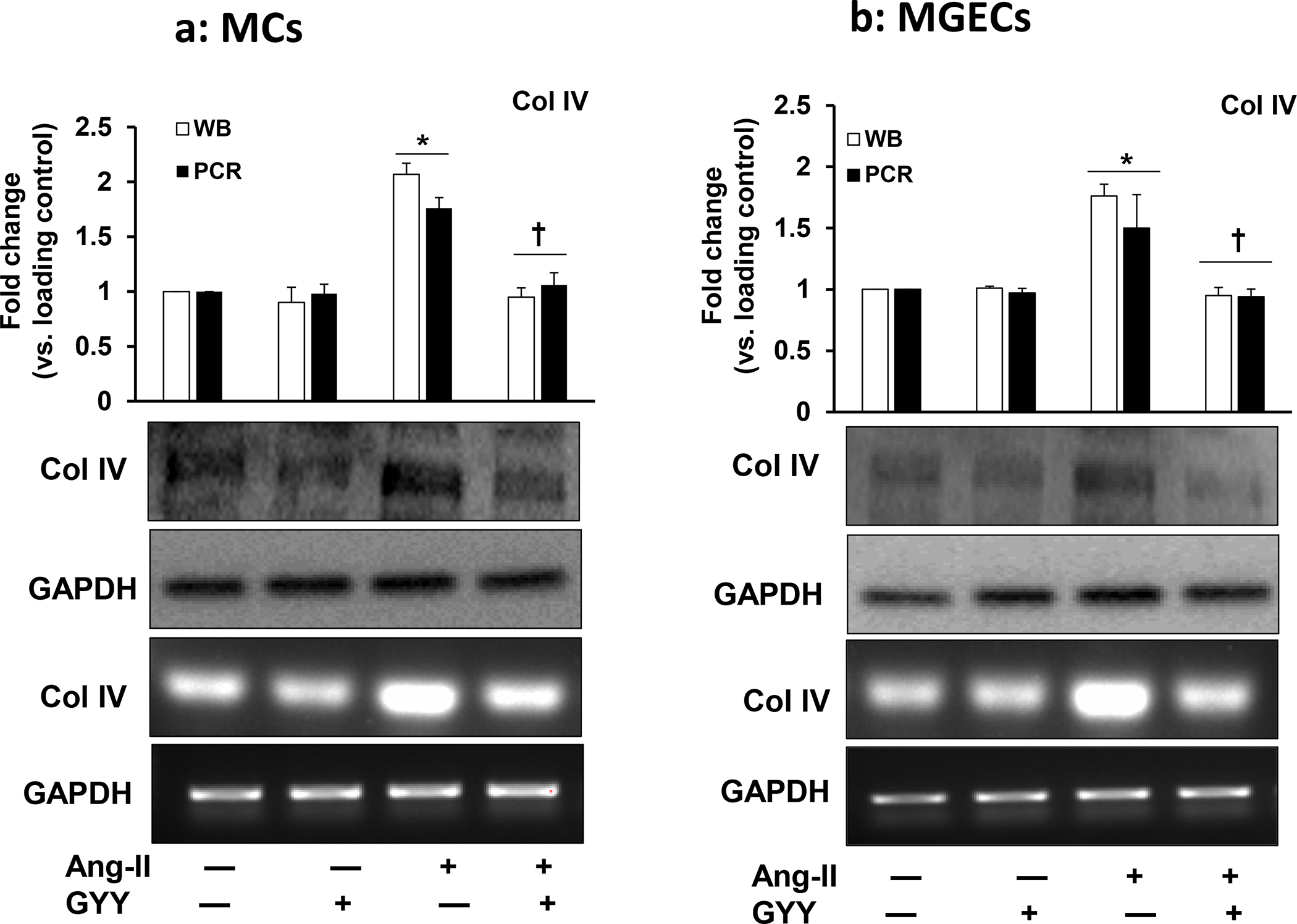
(a) and MGECs (b) were treated with Ang-II for 48 h. GYY (GYY4137) was added 1 h prior to the Ang-II treatment. Col IV protein (~160 kDa) and gene expressions were determined by Western blot and RT-PCR, respectively. GAPDH was used as a loading control. Data are presented as mean ±SEM, n = 9 independent experiments / group. *p <0.05 vs control, †p <0.05 vs Ang-II.
GYY ameliorated Ang-II-mediated decreased Endo180 expression
The uPARAP / Endo180 plays an important role in collagen degradation and ECM turnover [22]. We determined whether Ang-II-induced collagen upregulation was because of uPARAP / Endo180 downregulation and whether GYY could ameliorate this change. MCs and MGECs with no treatment showed a basal level of uPARAP / Endo180 mRNA and protein expression, and these expressions level did not change in GYY alone treated cells compared to their respective controls in both the cell types (Fig. 8a–b). Ang-II however, significantly decreased mRNA and protein expression in these cells compared to their respective controls. Interestingly, GYY mitigated and normalized both mRNA and protein expression in these cells (Fig. 8a–b), suggesting that GYY is a potential drug, which helps MCs and MGECs to maintain a basal level of uPARAP / Endo180 expression in hypertensive condition. Immunostaining of cells with uPARAP / Endo180 and collagen IV antibodies also demonstrated similar results as that of mRNA and Western blot data, and further support the hypothesis that GYY can normalize Ang-II-mediated altered uPARAP / Endo180 and collagen expression in both the renal cell lines (Fig. 9a–d).
Fig. 8. Effect of GYY on Ang-II induced uPARAP/Endo180 gene and protein expression MCs.
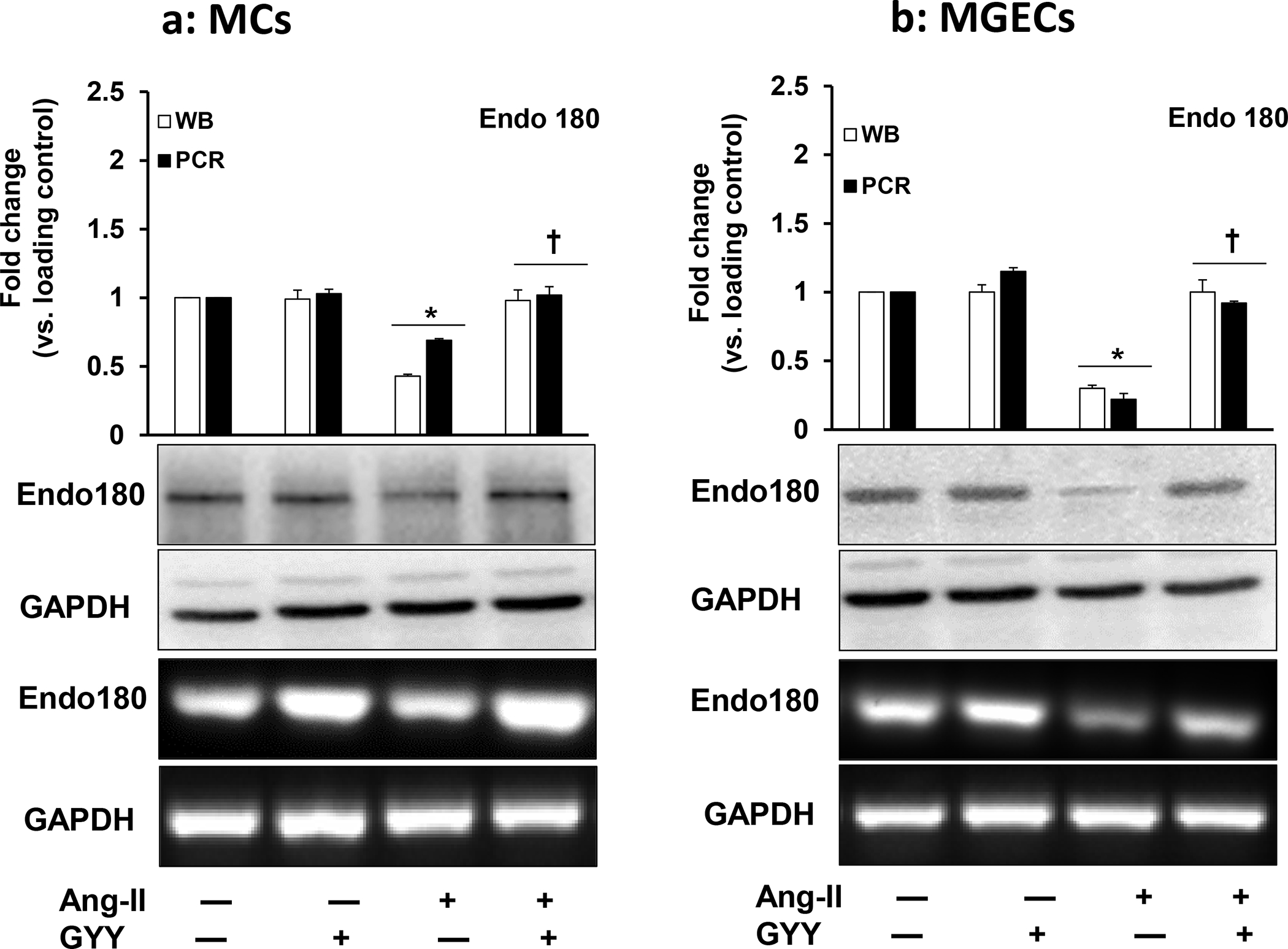
(a) and MGECs (b) were treated with Ang-II for 48 h. GYY (GYY4137) was added 1 h prior to the Ang-II treatment. The uPARAP/Endo180 (urokinase plasminogen activator receptor-associated protein / Endo180) protein (~167 KDa) and gene expressions were determined by Western blot and RT-PCR respectively. GAPDH was used as a loading control. Data are presented as mean ±SEM, n = 9 independent experiments / group. *p <0.05 vs control, †p <0.05 vs Ang-II.
Fig. 9. Decreased uPARAP/Endo180 and increased Col IV in Ang-II condition was normalized by GYY.
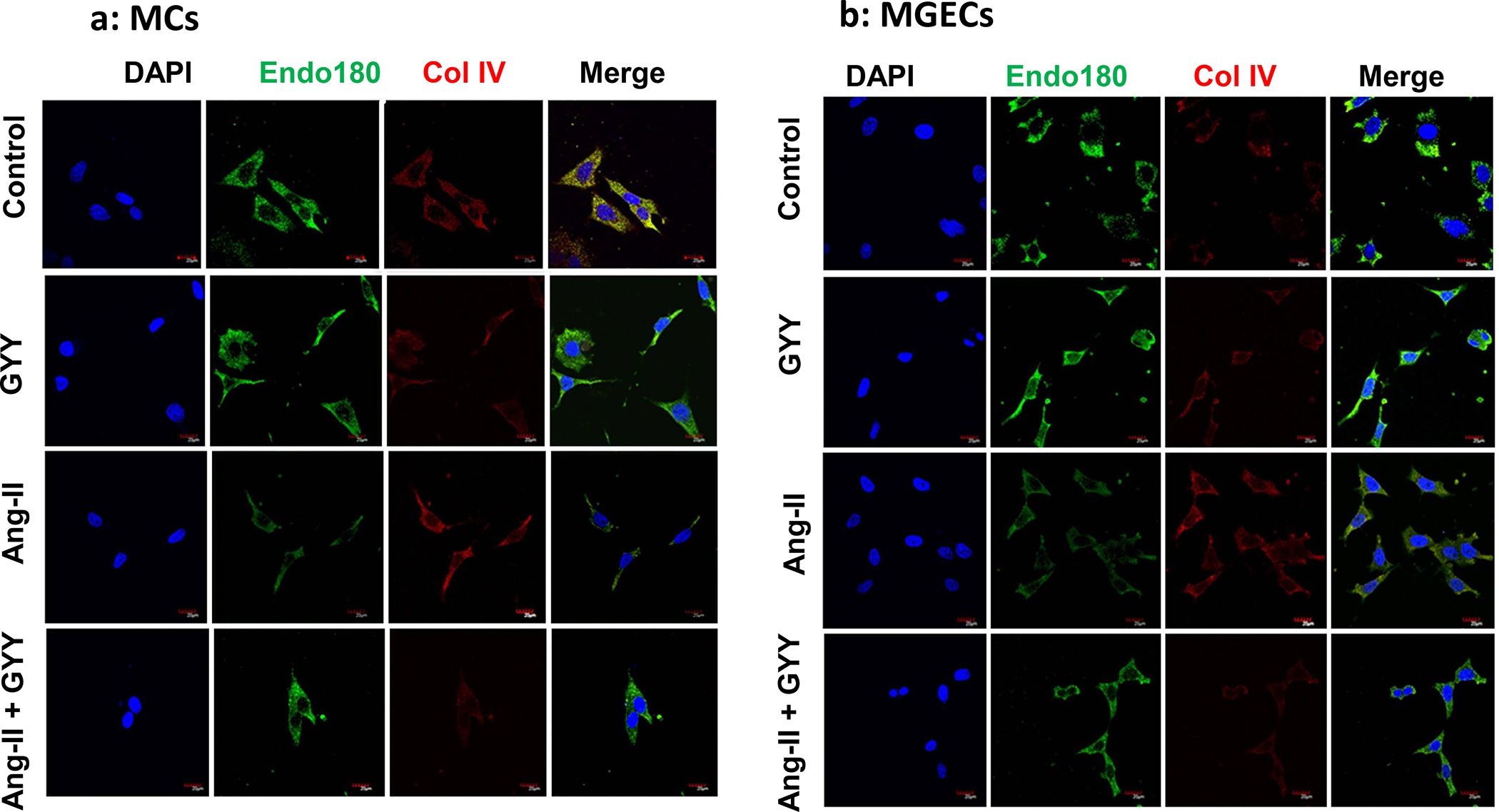
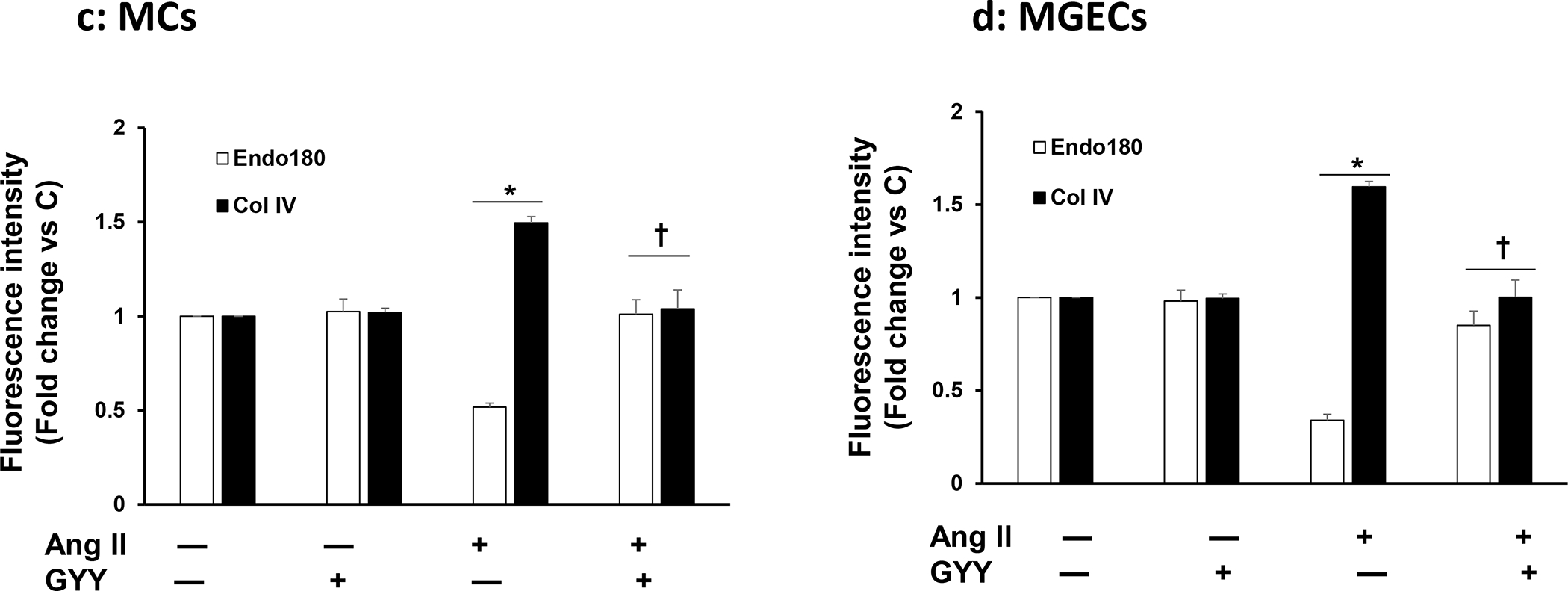
Plated MCs (a) and MGECs (b) on the chamber slides were treated with Ang-II for 48 h. GYY (GYY4137) was added 1 h prior to the Ang-II treatment. Following experiments, cells were immunostained against appropriate primary and fluorescence-tagged secondary antibodies against uPARAP/Endo180 (green) and Col IV (red), and nuclei counterstained with DAPI (blue). Co-localization of MMP-14 and Col IV are shown in the merged image. Scale bar, 20 μm, magnification 60 X. (c) and (d) summarized data of Endo 180 and Col IV expression in MCs and MGECs, respectively. Data are presented as mean ± SEM, n=9 independent experiments / group. *p <0.05 vs control, †p <0.05 vs Ang-II.
Ang-II-mediated collagen remodeling was Endo180-dependent
To determine whether Ang-II-mediated collagen remodeling was Endo180-dependent, we silenced Eno180 prior to Ang-II treatment and then measured collagens and MMPs expression. Our results indicated that siRNA suppressed Endo180 expression in both the cell types (Fig. 10a). Ang-II-induced Col IIIA and IV in both MCs and MGECs compared to their respective controls (Fig. 10b,c). Interestingly, when Endo180 was silenced with siRNA, the induction was even greater. Additionally, when cells were treated with GYY along with Ang-II in Endo180 silenced cells, the induction of collagens were diminished, compared to Ang-II alone or Ang-II plus GYY, which was similar to the basal level of expression in these two cell lines (Fig. 10b,c).
Fig. 10. Endo180 suppression further altered MMP-13, MMP-14, Col IIIA, and Col IV expression in Ang-II hypertension, which was restored by GYY.
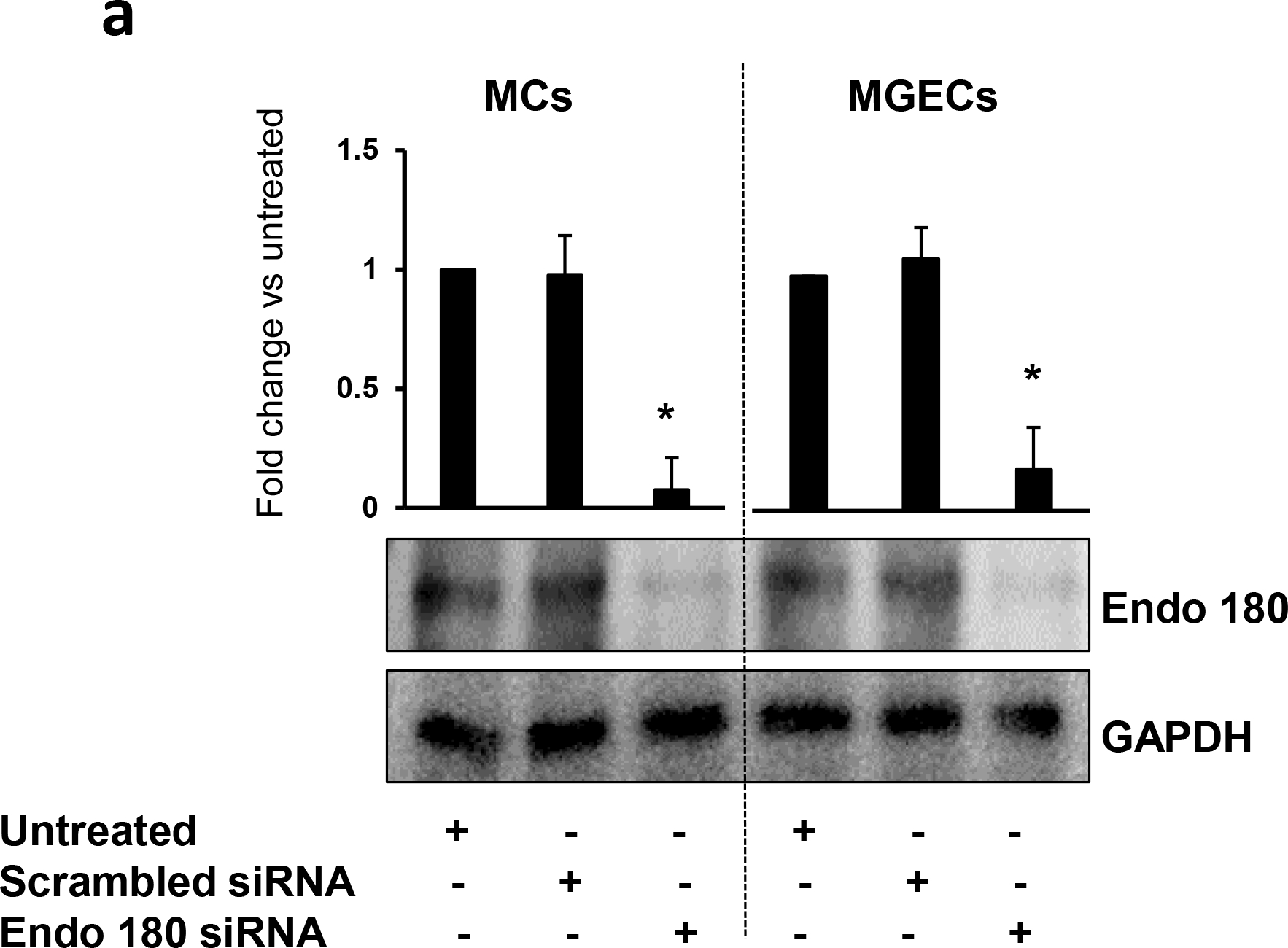
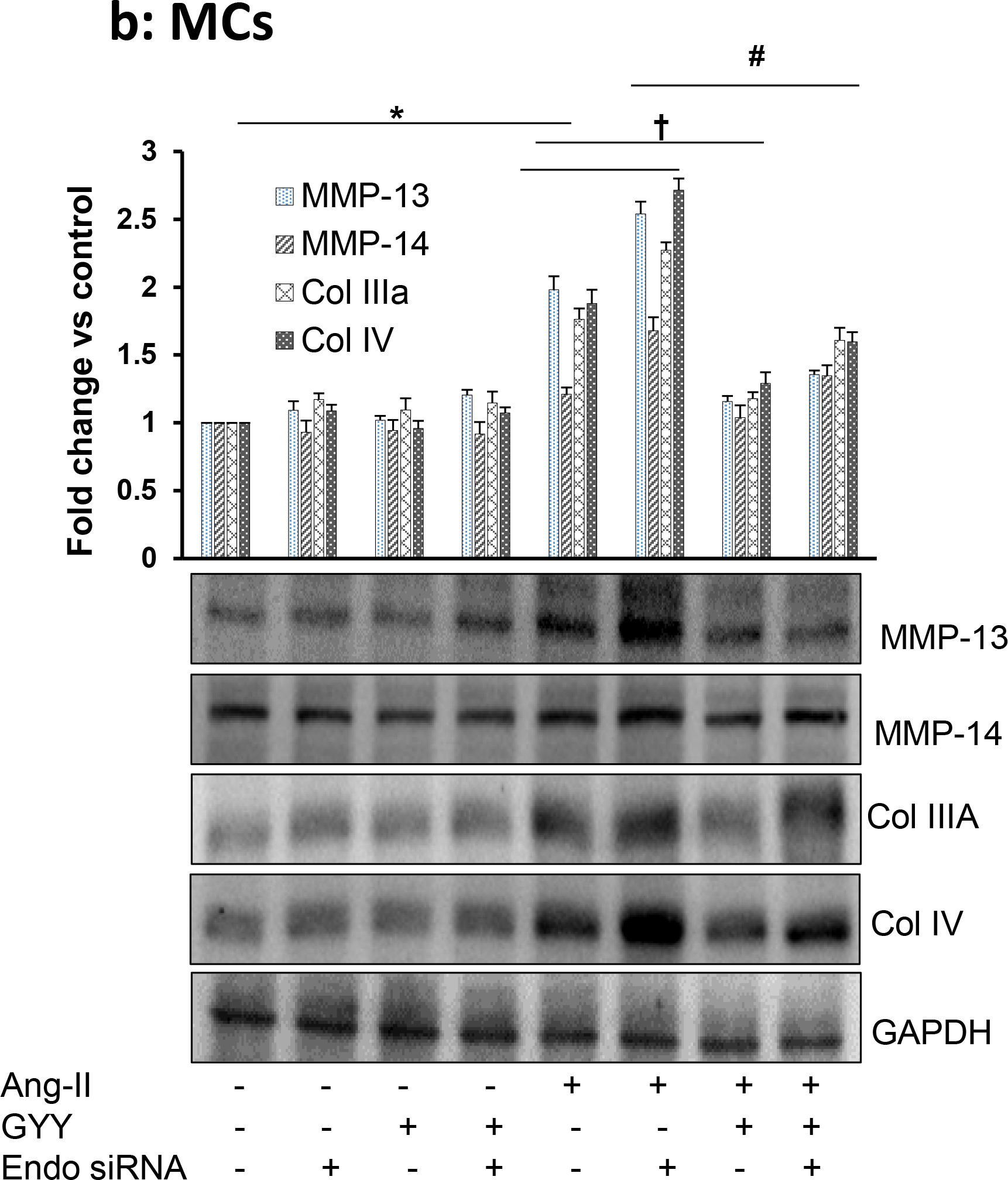
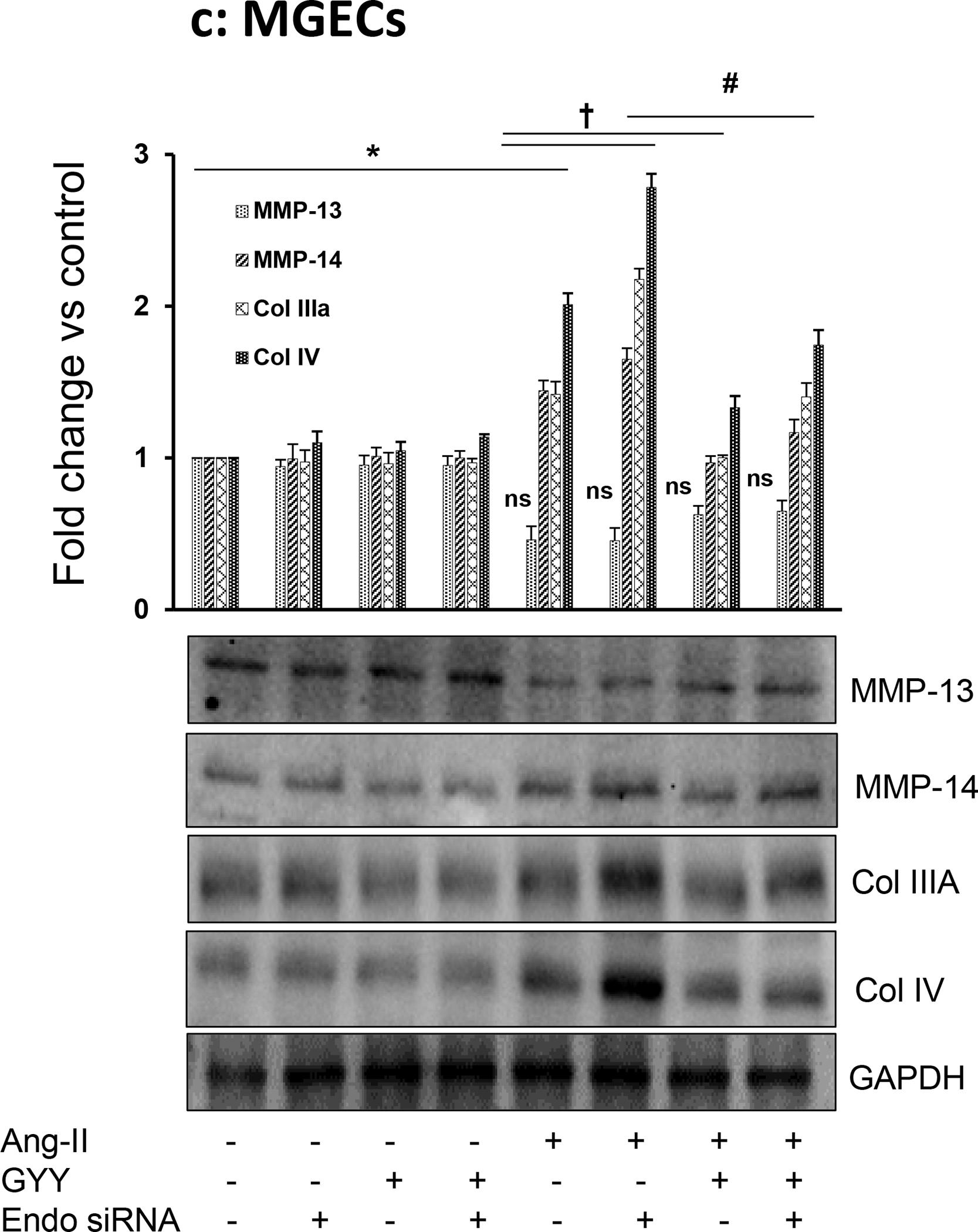
MCs and MGECs were transfected with scrambled siRNA (as negative control) and Endo 180 siRNA (10 nM, 48 hrs). Western blot analysis indicated suppression of Endo180 by siRNA in both the cell types (a). In siRNA transfected MCs (b) and MGECs (c), GYY (GYY4137) was added 1 h prior to the Ang-II treatment. Endo 180, MMP-13, -14, Col IIIA, and Col IV protein expressions were determined by Western blot. GAPDH was used as a loading control. Data are presented as mean ±SEM, n = 9 independent experiments / group. *,†, #p <0.05 vs respective controls; ns=non-significant difference of MMP-13 expression among these groups.
The expression of MMP-13 was similar to Col IIIA and Col IV expression patterns among the control and experiment groups in both the cell types (Fig. 10b,c). Interestingly, while Ang-II induced MMP-13 expression in MCs (Fig. 10b), it diminished MMP-13 expression in MGECs (Fig. 10c). A similar trend was also observed in MC and MGEC cells treated with Ang-II following Endo180 silencing (Fig. 10b,c). GYY treatment, either alone or in combination with Endo180-silencing normalized MMP-13 expression in Ang-II-treated both cell types (Fig. 10b,c).
On the other hand, Ang-II induced MMP-14 expression in both MCs and MGECs, and it was further augmented by Endo180 knockdown (Fig. 10b,c). Interestingly, Ang-II-mediated MMP-14 expression in both cell types, whether silenced with Endo180 siRNA or not, was normalized or mitigated with GYY treatment (Fig. 10b,c). The expression of MMP-14 in control, Endo180 siRNA transfected cells, GYY or GYY plus Endo180 siRNA transfected cells were similar and was at the basal level (Fig. 10b,c).
Discussion
In this present study, we demonstrate comparative stress response in two different mouse kidney cell types, mesangial (MCs) and glomerular endothelial cells (MGECs), exposed to in vitro Ang-II hypertensive condition. Results from our study indicate these two types of cells are highly responsive to Ang-II treatment and both the cells expressed similar pattern of mRNA and protein that include PAI-1, tPA, uPARAP /Endo 180, MMP-14, Collagen IIIA and IV with the exception of TIMP-1 and MMP-13, which were differentially expressed. It is also evident from our study that the MMP-13, -14 and TIMP-1, as well as intracellular endocytosis receptor (uPARAP / Endo 180), are involved in collagen dynamics in response to hypertensive stress. Our results further suggest that GYY (GYY4137), an H2S donor, plays a protective role in maintaining normal ECM homeostasis in these two cell types in Ang-II hypertensive condition. The overall findings of our study are depicted in fig. 11 for easy comprehension.
Fig. 11. Schematic of overall finding.
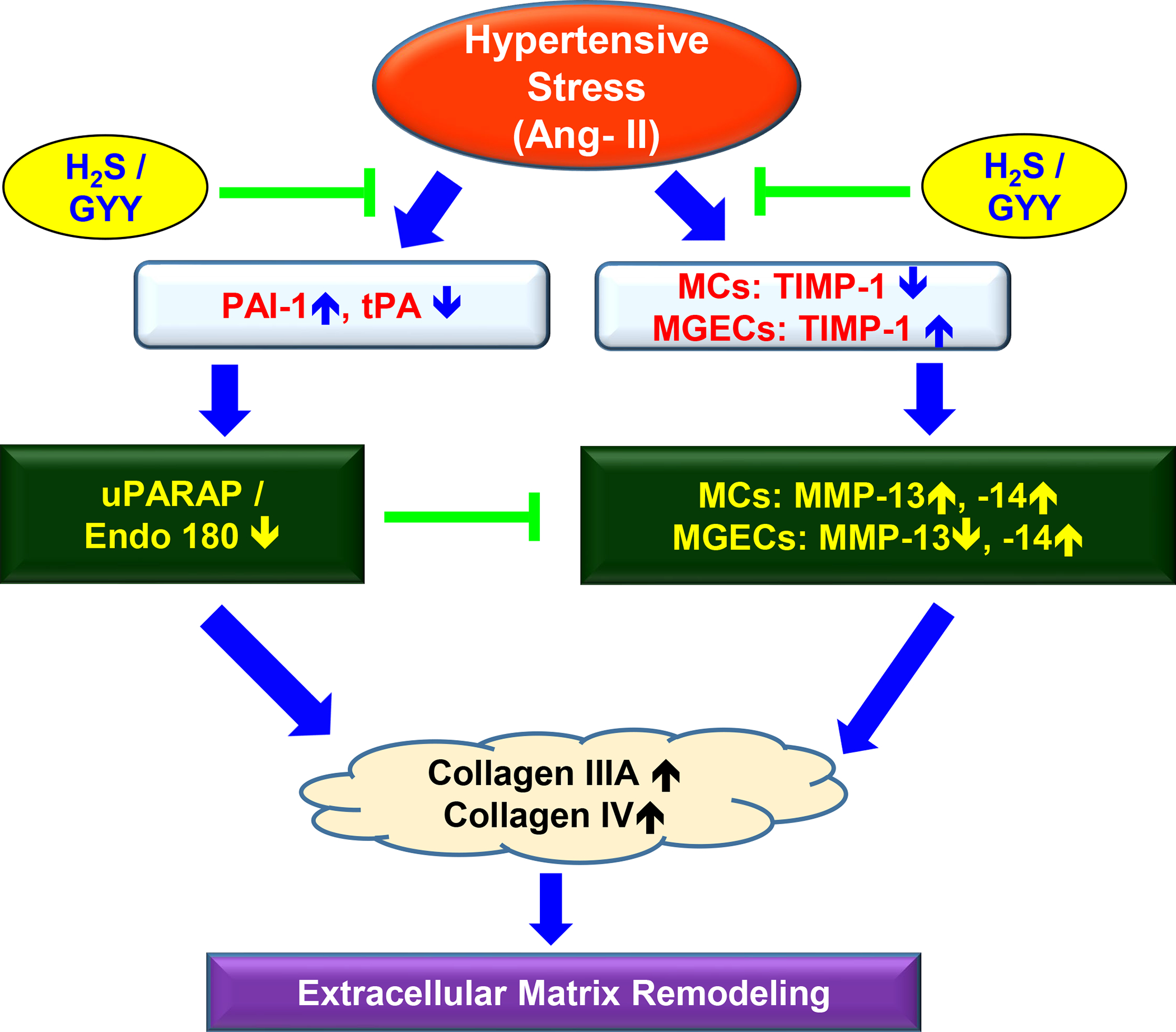
In response to hypertensive stress, upregulated PAI-1 inhibited tPA resulting in Endo 180 downregulation both in MCs and MGECs. On the other hand, while TIMP-1 was downregulated in MCs, it was upregulated in MGECs during hypertensive stress conditions. Because of this differential TIMP-1 deregulation, an opposing effect on MMP-13 expression was also observed in these two cell lines; whereas, the expression of MMP-14 was upregulated in both the cells. Silencing expression of Endo 180 by siRNA alters Ang-II-induced MMP/Collagen remodeling in both cell types. Together, Endo 180 and MMP/TIMP imbalance contributed collagen deposition and ECM remodeling in MCs and MGECs.
Hypertension is a recognized cause of chronic kidney disease (CKD) worldwide, which is also characterized by renal fibrosis due to excess accumulation of extracellular matrix (ECM) protein, cellular damage, and dysfunction [23]. Angiotensin II (Ang-II) is an active hormone of the renin-angiotensin system (RAS) known to play a pivotal role in the development of hypertension and contribute to the pathogenesis of reno-cardiovascular diseases [24–26]. Ang-II has also been implicated in pathological ECM accumulation, both in experimental models and in chronic human renal diseases [27–30]. Hence, Ang-II is considered as an active player to control renal function through cellular- and ECM remodeling [30–33]. In recent years, H2S has been shown to have renoprotective effects in many renal diseases, including HN [34, 35]. However, whether the renoprotective effects of H2S is consistent among different renal cell types in the hypertensive condition is unclear. To mimic Ang-II-mediated hypertensive stress, we treated MCs and MGECs with Ang-II and evaluated their cellular matrix turnover, and tested the role of H2S in protecting kidney cells in vitro. Our results indicated that Ang-II hypertensive condition in these two cell types induced cellular remodeling involving plasminogen system, metalloproteinases, and their tissue inhibitors, and H2S ameliorated remodeling in Ang-II hypertensive condition (Fig. 11).
The plasminogen, an inactive zymogen, is an important cellular mechanism of inflammation, tissue remodeling, and glomerulosclerosis in CKD [36]. The plasminogen activation system consists of the circulating zymogen plasminogen (PLG) and the tissue- and urokinase-type plasminogen activators, tPA and uPA, respectively, and plasminogen activator inhibitor type-1, PAI-1 [37]. Plasminogen is cleaved to generate the active protease plasmin by urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA), both of which are inhibited by PAI-1. Plasmin cleaves numerous substrates, but relevant to potential fibrotic functions; it can directly degrade matrix proteins by activating metalloproteinases (MMPs) [27, 38, 39]. Our study indicated that Ang II downregulated tPA and upregulated PAI-1 both in MCs and MGECs, suggesting that hypertensive condition inhibited PAI-1, which in turn upregulated tPA (Fig. 1a–b). Our study further supports previous findings, which reported the induction of plasminogen/plasmin system by Ang-II in cultured brain astrocytes, vascular endothelial, and rat mesangial cells [27, 40–42]. Although we have observed differential expression of MMP-13/-14 and TIMP-1 in MCs and MGECs, the commonality between these two cell types was dysregulation of MMP/TIMP in hypertensive condition. This finding is in accordance with our earlier report, which revealed that altered the MMP/TIMP ratio is an important contributor in renal remodeling in Ang-II induced hypertension conditions [28].
In this study, we also demonstrated the effect of GYY on altered PAI-1 and tPA protein expression in Ang-II exposed MCs and MGECs. We found that Ang-II-induced PAI-1 expression subsequently inhibited tPA protein expression in both the cell types, which was significantly reversed by GYY treatment (Fig. 1a–b). We then observed that Ang-II-induced increased MMP-14 mRNA and protein expression were attenuated by GYY in both the cell types (Fig. 2a–b). Immunocytochemistry study further confirmed mRNA results demonstrating increased MMP-14 expression in Ang-II induced hypertensive condition in both the cell types, which was normalized by GYY treatment (Fig. 3a–d). We also examined MMP-13 mRNA and protein expression in both the cell types treated with or without Ang-II and GYY. The MMP-13 mRNA and protein expression were increased in MCs; however, it was decreased in MGECs by Ang-II treatment. Interestingly, GYY treatment normalized MMP-13 mRNA and protein expression in both the cell types (Fig. 4a–b), suggesting the potential role of H2S as an MMP-13 modulator in hypertensive condition. Immunostaining results further confirmed the mRNA and Western blotting results, and the role of GYY in modulating this proteinase (Fig. 4c–f). As specific tissue inhibitors of MMPs, TIMPs control the activity of MMPs. We then examined the effect of Ang-II on TIMP-1 mRNA and protein expression. Results showed that Ang-II-induced TIMP-1 protein and mRNA expression was decreased in MCs, while it was increased in MGECs. GYY normalized the TIMP-1 protein and mRNA expression in both the cell types (Fig. 5a–b), suggesting the potential role of GYY to normalize TIMP-1 dysregulation in hypertensive conditions.
Previous studies have reported the involvement of Ang-II induced ECM remodeling in the hypertensive kidney [43–45], and H2S ameliorated excess ECM deposition in nephropathy [46]. In this study, we examined the effect of GYY on Ang-II induced collagen IIIA and collagen IV mRNA and protein expression in both MCs and MGECs. Ang-II treatment showed increased collagen IIIA mRNA and protein expression in both the cell types, which was mitigated by GYY treatment (Fig. 6a–b). Similarly, collagen IV mRNA and protein expression were significantly increased by Ang-II, and GYY attenuated the Collagen IV expression in both the cell types, which confirmed that GYY prevented Ang-II induced collagen deposition in these cells (Fig. 7a–b).
Intracellular collagen degradation mediated by uPARAP/Endo180 is an important pathway for ECM degradation, which involves a highly organized interplay between proteases and their cellular binding sites as well as specific substrates and internalization receptors [47]. The urokinase plasminogen activator receptor (uPAR) and the uPAR-associated protein (uPARAP; also named as Endo180), that are considered crucially engaged in matrix degradation. The uPAR is a glycosyl-phosphatidylinositol-anchored glycoprotein on the surface of various cell types that serves to bind the uPA and localize the activation reactions in the proteolytic cascade system of plasminogen activation. The uPARAP/Endo180 is a type-1 membrane protein belonging to the mannose receptor family, is an endocytic receptor for collagen having a pronounced role in the internalization of collagen for intracellular degradation [18, 48], which also involves in anomalous renal function in CKD [49]. To determine whether collagen deposition by Ang-II involves the intracellular collagen degradation pathway, we investigated the effect of Ang-II on uPARAP/Endo180 expression in MCs and MGECs. Interestingly, we detected significant inhibition of uPARAP/Endo180 mRNA and protein expression by Ang-II in both cell types. GYY treatment reversed the altered uPARAP/Endo180 mRNA and protein expression in both the cells (Fig. 8a–b). Immunolocalization study indicated that in both cell types, Ang-II induced altered uPARAP/Endo180 and Col IV protein expressions are inversely related to each other, and the effect of Ang-II on this protein expression was normalized by GYY treatment (Fig. 9a–d). These results suggest the involvement of the uPARAR/Endo 180 pathway in collagen remodeling and the potential role of GYY to modulate this pathway in hypertensive condition.
Experimental evidence indicated that membrane-type 1 matrix metalloproteinase (MT1-MMP, also known as MMP-14) is one of the key collagenolysins, which is regulated by Endo180 collagen receptor [50]. MMP-14 not only degrades ECM protein, including collagens I-IV, it also activates pro-MMP-2 and pro-MMP-9 [51], which are also gelatino- and collagenolysins [52]. Interestingly, Endo180 receptors co-express with MMP-13 [53], and Endo180 activates pro-MMP-13 [54]. In our previous publication, we have shown that Ang-II downregulates MMP-2 and upregulates MMP-9 expression as well as activity in mouse kidney, and H2S normalizes these two MMPs [55]. It is known that MMP-13 and -14 have significant renal, including glomerular expression [56, 57]. In addition, TIMP-1 has been shown to instigate renal fibrosis [58]. To our knowledge, it is unknown whether Endo180-mediates collagen remodeling in hypertensive kidney involving, TIMP-1, MMP-13, and -14. Since Endo180 regulates MMP-14 and activates MMP-13, in this present study, we measured TIMP-1, MMP-14, and MMP-13 expression in two distinctly important kidney cell lines, i.e., MGECs and MCs, in response to Ang-II. Additionally, we determined whether H2S can modulate Ang-II effects. Our results indicated that Ang-II mediated alteration of MMP-13,-14 and Collagen-IIIA and -IV expression was Endo180 dependent, and GYY was able to restore these changes during Ang-II conditions (Fig. 10a–c). It is worthy of mentioning that the current study was designed to determine in vitro cellular mechanisms of collagen remodeling involving the MMPs and TIMP-1 as mentioned above and whether GYY/ H2S can modulate remodeling in Ang-II hypertensive conditions. Nevertheless, the in vivo experiments could have provided more in-depth information regarding the role of MMP-13, -14, and TIMP-1 in Ang-II hypertensive kidney, and needs future attention.
In conclusion, our study demonstrates that Ang-II differentially instigates MMPs/TIMPs in MCs and MGECs, whereas PAI-1, tPA and uPARAP/Endo 180 followed a similar pattern of expression in both the cell types leading to collagen accumulation. This study also demonstrates a protective role of GYY in Ang-II-induced ECM remodeling in MCs and MGECs, by involving MMP/TIMP-mediated inhibition of collagen accumulation as well as PAI-1/tPA and uPARAP/Endo180-mediated endocytic pathway of collagen remodeling.
Acknowledgments
We thank Naira Metreveli for her technical assistance related to cell culture experiments and the acquisition of confocal images. This work was supported in part by National Institutes of Health Grants, DK104653 and DK116591 (to U.S.), and American Heart Association Scientist Development Grant, 15SDG25840013 (to S.P.)
Footnotes
Compliance with ethical standards
Conflict of interest The authors declare no conflicts of interest, financial or otherwise.
Data availability:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References:
- 1.Luke RG (1999) Hypertensive nephrosclerosis: pathogenesis and prevalence. Essential hypertension is an important cause of end-stage renal disease. Nephrol Dial Transplant 14:2271–8. [DOI] [PubMed] [Google Scholar]
- 2.Hill GS (2008) Hypertensive nephrosclerosis. Curr Opin Nephrol Hypertens 17:266–70. doi: 10.1097/MNH.0b013e3282f88a1f [DOI] [PubMed] [Google Scholar]
- 3.Szijarto IA, Marko L, Filipovic MR, Miljkovic JL, Tabeling C, Tsvetkov D, Wang N, Rabelo LA, Witzenrath M, Diedrich A, Tank J, Akahoshi N, Kamata S, Ishii I and Gollasch M (2018) Cystathionine gamma-Lyase Produced Hydrogen Sulfide Controls Endothelial NO Bioavailability and Blood Pressure. Hypertension 71:1210–1217. doi: 10.1161/Hypertensionaha.117.10562 [DOI] [PubMed] [Google Scholar]
- 4.Rose P, Moore PK and Zhu YZ (2017) H2S biosynthesis and catabolism: new insights from molecular studies. Cellular and Molecular Life Sciences 74:1391–1412. doi: 10.1007/s00018-016-2406-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishii I, Akahoshi N, Yu XN, Kobayashi Y, Namekata K, Komaki G and Kimura H (2004) Murine cystathionine gamma-lyase: complete cDNA and genomic sequences, promoter activity, tissue distribution and developmental expression. Biochemical Journal 381:113–123. doi: 10.1042/Bj20040243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kimura H (2011) Hydrogen sulfide: its production, release and functions. Amino Acids 41:113–121. doi: 10.1007/s00726-010-0510-x [DOI] [PubMed] [Google Scholar]
- 7.Sen U, Sathnur PB, Kundu S, Givvimani S, Coley DM, Mishra PK, Qipshidze N, Tyagi N, Metreveli N and Tyagi SC (2012) Increased endogenous H2S generation by CBS, CSE, and 3MST gene therapy improves ex vivo renovascular relaxation in hyperhomocysteinemia. American Journal of Physiology-Cell Physiology 303:C41–C51. doi: 10.1152/ajpcell.00398.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Genovese F, Manresa AA, Leeming DJ, Karsdal MA and Boor P (2014) The extracellular matrix in the kidney: a source of novel non-invasive biomarkers of kidney fibrosis? Fibrogenesis Tissue Repair 7:4. doi: 10.1186/1755-1536-7-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gelse K, Poschl E and Aigner T (2003) Collagens--structure, function, and biosynthesis. Adv Drug Deliv Rev 55:1531–46. [DOI] [PubMed] [Google Scholar]
- 10.Schuppan D, Ruehl M, Somasundaram R and Hahn EG (2001) Matrix as a modulator of hepatic fibrogenesis. Semin Liver Dis 21:351–72. doi: 10.1055/s-2001-17556 [DOI] [PubMed] [Google Scholar]
- 11.Rowe RG and Weiss SJ (2009) Navigating ECM barriers at the invasive front: the cancer cell-stroma interface. Annu Rev Cell Dev Biol 25:567–95. doi: 10.1146/annurev.cellbio.24.110707.175315 [DOI] [PubMed] [Google Scholar]
- 12.Bataller R and Brenner DA (2005) Liver fibrosis. J Clin Invest 115:209–18. doi: 10.1172/JCI24282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lauer-Fields JL, Juska D and Fields GB (2002) Matrix metalloproteinases and collagen catabolism. Biopolymers 66:19–32. doi: 10.1002/bip.10201 [DOI] [PubMed] [Google Scholar]
- 14.Mott JD and Werb Z (2004) Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol 16:558–64. doi: 10.1016/j.ceb.2004.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arpino V, Brock M and Gill SE (2015) The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol 44–46:247–54. doi: 10.1016/j.matbio.2015.03.005 [DOI] [PubMed] [Google Scholar]
- 16.Madsen DH, Jurgensen HJ, Ingvarsen S, Melander MC, Vainer B, Egerod KL, Hald A, Rono B, Madsen CA, Bugge TH, Engelholm LH and Behrendt N (2012) Endocytic collagen degradation: a novel mechanism involved in protection against liver fibrosis. Journal of Pathology 227:94–105. doi: 10.1002/path.3981 [DOI] [PubMed] [Google Scholar]
- 17.Ghosh AK and Vaughan DE (2012) PAI-1 in tissue fibrosis. J Cell Physiol 227:493–507. doi: 10.1002/jcp.22783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engelholm LH, Ingvarsen S, Jurgensen HJ, Hillig T, Madsen DH, Nielsen BS and Behrendt N (2009) The collagen receptor uPARAP/Endo180. Front Biosci (Landmark Ed) 14:2103–14. [DOI] [PubMed] [Google Scholar]
- 19.John A, Kundu S, Pushpakumar S, Fordham M, Weber G, Mukhopadhyay M and Sen U (2017) GYY4137, a Hydrogen Sulfide Donor Modulates miR194-Dependent Collagen Realignment in Diabetic Kidney. Sci Rep 7:10924. doi: 10.1038/s41598-017-11256-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Satoh M, Kashihara N, Yamasaki Y, Maruyama K, Okamoto K, Maeshima Y, Sugiyama H, Sugaya T, Murakami K and Makino H (2001) Renal interstitial fibrosis is reduced in angiotensin II type 1a receptor-deficient mice. J Am Soc Nephrol 12:317–25. [DOI] [PubMed] [Google Scholar]
- 21.Pushpakumar S, Ren L, Kundu S, Gamon A, Tyagi SC and Sen U (2017) Toll-like Receptor 4 Deficiency Reduces Oxidative Stress and Macrophage Mediated Inflammation in Hypertensive Kidney. Sci Rep 7:6349. doi: 10.1038/s41598-017-06484-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Curino AC, Engelholm LH, Yamada SS, Holmbeck K, Lund LR, Molinolo AA, Behrendt N, Nielsen BS and Bugge TH (2005) Intracellular collagen degradation mediated by uPARAP/Endo180 is a major pathway of extracellular matrix turnover during malignancy. J Cell Biol 169:977–85. doi: 10.1083/jcb.200411153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Humphreys BD (2018) Mechanisms of Renal Fibrosis. Annual Review of Physiology, Vol 80 80:309–326. doi: 10.1146/annurev-physiol-022516-034227 [DOI] [PubMed] [Google Scholar]
- 24.Wolf G (1998) Angiotensin II is involved in the progression of renal disease: importance of non-hemodynamic mechanisms. Nephrologie 19:451–6. [PubMed] [Google Scholar]
- 25.Kim S and Iwao H (2000) Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol Rev 52:11–34. [PubMed] [Google Scholar]
- 26.Weir MR and Dzau VJ (1999) The renin-angiotensin-aldosterone system: a specific target for hypertension management. Am J Hypertens 12:205S–213S. [DOI] [PubMed] [Google Scholar]
- 27.Kagami S, Kuhara T, Okada K, Kuroda Y, Border WA and Noble NA (1997) Dual effects of angiotensin II on the plasminogen/plasmin system in rat mesangial cells. Kidney Int 51:664–71. [DOI] [PubMed] [Google Scholar]
- 28.Pushpakumar S, Kundu S, Pryor T, Givvimani S, Lederer E, Tyagi SC and Sen U (2013) Angiotensin-II induced hypertension and renovascular remodelling in tissue inhibitor of metalloproteinase 2 knockout mice. J Hypertens 31:2270–81; discussion 2281. doi: 10.1097/HJH.0b013e3283649b33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ichikawi I and Harris RC (1991) Angiotensin actions in the kidney: renewed insight into the old hormone. Kidney Int 40:583–96. [DOI] [PubMed] [Google Scholar]
- 30.Wolf G and Neilson EG (1993) Angiotensin II as a renal growth factor. J Am Soc Nephrol 3:1531–40. [DOI] [PubMed] [Google Scholar]
- 31.Egido J (1996) Vasoactive hormones and renal sclerosis. Kidney Int 49:578–97. [DOI] [PubMed] [Google Scholar]
- 32.Ruiz-Ortega M and Egido J (1997) Angiotensin II modulates cell growth-related events and synthesis of matrix proteins in renal interstitial fibroblasts. Kidney Int 52:1497–510. [DOI] [PubMed] [Google Scholar]
- 33.Mezzano SA, Ruiz-Ortega M and Egido J (2001) Angiotensin II and renal fibrosis. Hypertension 38:635–8. [DOI] [PubMed] [Google Scholar]
- 34.Frenay ARS, Snijder PM, Koning AM, Bachtler M, Pasch A, Kwakernaak AJ, van den Berg E, Bos EM, Hillebrands JL, Navis G, Leuvenink HGD and van Goor H (2014) Hydrogen sulfide attenuates angiotensin II-induced hypertension, proteinuria and renal damage. Nitric Oxide-Biology and Chemistry 39:S23–S24. doi: 10.1016/j.niox.2014.03.075 [DOI] [PubMed] [Google Scholar]
- 35.Holwerda KM, Burke SD, Faas MM, Zsengeller Z, Stillman IE, Kang PM, van Goor H, McCurley A, Jaffe IZ, Karumanchi SA and Lely AT (2014) Hydrogen Sulfide Attenuates sFlt1-Induced Hypertension and Renal Damage by Upregulating Vascular Endothelial Growth Factor. Journal of the American Society of Nephrology 25:717–725. doi: 10.1681/Asn.2013030291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Madhusudhan T, Kerlin BA and Isermann B (2016) The emerging role of coagulation proteases in kidney disease. Nature Reviews Nephrology 12:94–109. doi: 10.1038/nrneph.2015.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Svenningsen P, Hinrichs GR, Zachar R, Ydegaard R and Jensen BL (2017) Physiology and pathophysiology of the plasminogen system in the kidney. Pflugers Arch 469:1415–1423. doi: 10.1007/s00424-017-2014-y [DOI] [PubMed] [Google Scholar]
- 38.Eddy AA (2002) Plasminogen activator inhibitor-1 and the kidney. Am J Physiol Renal Physiol 283:F209–20. doi: 10.1152/ajprenal.00032.2002 [DOI] [PubMed] [Google Scholar]
- 39.Zhang G, Kernan KA, Collins SJ, Cai X, Lopez-Guisa JM, Degen JL, Shvil Y and Eddy AA (2007) Plasmin(ogen) promotes renal interstitial fibrosis by promoting epithelial-to-mesenchymal transition: role of plasmin-activated signals. J Am Soc Nephrol 18:846–59. doi: 10.1681/ASN.2006080886 [DOI] [PubMed] [Google Scholar]
- 40.Olson JA Jr., Shiverick KT, Ogilvie, Buhi WC and Raizada MK(1991) Angiotensin II induces secretion of plasminogen activator inhibitor 1 and a tissue metalloprotease inhibitor-related protein from rat brain astrocytes. Proc Natl Acad Sci U S A 88:1928–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zelezna B, Rydzewski B, Lu D, Olson JA, Shiverick KT, Tang W, Sumners C and Raizada MK (1992) Angiotensin-II induction of plasminogen activator inhibitor-1 gene expression in astroglial cells of normotensive and spontaneously hypertensive rat brain. Mol Endocrinol 6:2009–17. doi: 10.1210/mend.6.12.1491687 [DOI] [PubMed] [Google Scholar]
- 42.Vaughan DE, Lazos SA and Tong K (1995) Angiotensin II regulates the expression of plasminogen activator inhibitor-1 in cultured endothelial cells. A potential link between the renin-angiotensin system and thrombosis. J Clin Invest 95:995–1001. doi: 10.1172/JCI117809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao W, Chen SS, Chen Y, Ahokas RA and Sun Y (2008) Kidney fibrosis in hypertensive rats: role of oxidative stress. Am J Nephrol 28:548–54. doi: 10.1159/000115289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tharaux PL, Chatziantoniou C, Fakhouri F and Dussaule JC (2000) Angiotensin II activates collagen I gene through a mechanism involving the MAP/ER kinase pathway. Hypertension 36:330–6. [DOI] [PubMed] [Google Scholar]
- 45.Boffa JJ, Lu Y, Placier S, Stefanski A, Dussaule JC and Chatziantoniou C (2003) Regression of renal vascular and glomerular fibrosis: role of angiotensin II receptor antagonism and matrix metalloproteinases. J Am Soc Nephrol 14:1132–44. [DOI] [PubMed] [Google Scholar]
- 46.Song K, Wang F, Li Q, Shi YB, Zheng HF, Peng HJ, Shen HY, Liu CF and Hu LF (2014) Hydrogen sulfide inhibits the renal fibrosis of obstructive nephropathy. Kidney Int 85:1318–1329. doi: 10.1038/ki.2013.449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Melander MC, Jurgensen HJ, Madsen DH, Engelholm LH and Behrendt N (2015) The collagen receptor uPARAP/Endo180 in tissue degradation and cancer. International Journal of Oncology 47:1177–1188. doi: 10.3892/ijo.2015.3120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Behrendt N (2004) The urokinase receptor (uPAR) and the uPAR-associated protein (uPARAP/Endo180): membrane proteins engaged in matrix turnover during tissue remodeling. Biol Chem 385:103–36. doi: 10.1515/BC.2004.031 [DOI] [PubMed] [Google Scholar]
- 49.Zhang G and Eddy AA (2008) Urokinase and its receptors in chronic kidney disease. Front Biosci 13:5462–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Messaritou G, East L, Roghi C, Isacke CM and Yarwood H (2009) Membrane type-1 matrix metalloproteinase activity is regulated by the endocytic collagen receptor Endo180. Journal of cell science 122:4042–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheng Z, Limbu MH, Wang Z, Liu J, Liu L, Zhang X, Chen P and Liu B (2017) MMP-2 and 9 in Chronic Kidney Disease. Int J Mol Sci 18. doi: 10.3390/ijms18040776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Itoh Y and Seiki M (2006) MT1-MMP: a potent modifier of pericellular microenvironment. J Cell Physiol 206:1–8. doi: 10.1002/jcp.20431 [DOI] [PubMed] [Google Scholar]
- 53.Engelholm LH, Nielsen BS, Netzel-Arnett S, Solberg H, Chen XD, Lopez Garcia JM, Lopez-Otin C, Young MF, Birkedal-Hansen H, Dano K, Lund LR, Behrendt N and Bugge TH (2001) The urokinase plasminogen activator receptor-associated protein/endo180 is coexpressed with its interaction partners urokinase plasminogen activator receptor and matrix metalloprotease-13 during osteogenesis. Lab Invest 81:1403–14. doi: 10.1038/labinvest.3780354 [DOI] [PubMed] [Google Scholar]
- 54.Carmeliet P, Moons L, Lijnen R, Baes M, Lemaitre V, Tipping P, Drew A, Eeckhout Y, Shapiro S, Lupu F and Collen D (1997) Urokinase-generated plasmin activates matrix metalloproteinases during aneurysm formation. Nat Genet 17:439–44. doi: 10.1038/ng1297-439 [DOI] [PubMed] [Google Scholar]
- 55.Pushpakumar SB, Kundu S, Metreveli N and Sen U (2013) Folic acid mitigates angiotensin-II-induced blood pressure and renal remodeling. PLoS ONE 8:e83813. doi: 10.1371/journal.pone.0083813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zakiyanov O, Kalousova M, Zima T and Tesar V (2019) Matrix Metalloproteinases in Renal Diseases: A Critical Appraisal. Kidney Blood Press Res 44:298–330. doi: 10.1159/000499876 [DOI] [PubMed] [Google Scholar]
- 57.Ren J, Zhang J, Rudemiller NP, Griffiths R, Wen Y, Lu X, Privratsky JR, Gunn MD and Crowley SD (2019) Twist1 in Infiltrating Macrophages Attenuates Kidney Fibrosis via Matrix Metallopeptidase 13-Mediated Matrix Degradation. J Am Soc Nephrol 30:1674–1685. doi: 10.1681/ASN.2018121253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cai G, Zhang X, Hong Q, Shao F, Shang X, Fu B, Feng Z, Lin H, Wang J, Shi S, Yin Z and Chen X (2008) Tissue inhibitor of metalloproteinase-1 exacerbated renal interstitial fibrosis through enhancing inflammation. Nephrol Dial Transplant 23:1861–75. doi: 10.1093/ndt/gfm666 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.