

Abstract
Hybrid quantum mechanical/molecular mechanical (QM/MM) methods have become an indispensable tool for the study of biomolecules. In this article, we briefly review the basic methodological details of QM/MM approaches and discuss their applications to various energy transduction problems in biomolecular machines, such as long-range proton transports, fast electron transfers and mechanochemical coupling. We highlight that for these applications it is particularly important to balance computational efficiency and accuracy. Using several recent examples, we illustrate the value and limitations of QM/MM methodologies for both ground and excited states, as well as strategies for calibrating them in specific applications. We conclude with brief comments on several areas that can benefit further efforts to make QM/MM analyses more quantitative and applicable to increasingly complex biological problems.
Keywords: QM/MM, energy transduction, biomolecular machines, proton transfers, electron transfers, nucleotide hydrolysis
1. Introduction
One of the hallmarks of living systems is the efficient transduction of energy among different forms via biomolecular machines.1,2 Remarkable examples include the conversion of light energy (or O2 activation) into proton motive force by bacteriorhodopsin (cytochrome c oxidase), utilization of the proton gradient to synthesize ATP by the F0F1-ATPase and to transport small molecules across the membrane by various transporters, and driving the motion of biomolecular motors with the chemical energy of ATP hydrolysis. The definition of efficiency for these biomolecular machines is somewhat subtle,3 but generally the values are substantially higher compared to those of artificial machines. Therefore, uncovering the physical and chemical principles that govern the working mechanism of naturally evolved systems is not only of fundamental significance, but also valuable for providing guidance to the development of novel molecular machines at the nanoscale.4,5
Since the ultimate driving force for most biomolecular machines involves chemical reactions, such as ATP hydrolysis, coupled proton-electron-transfers or electronic excitation, theoretical and computational analysis of bioenergy transduction necessarily involves treating chemical reactions and dissecting their coupling to other processes, such as protein conformational transitions or translocation of small molecules or ions. Therefore, compared to the problem of enzyme catalysis, which also involves chemical reactions in biomolecules, the topic of bioenergy transduction is arguably even more challenging as it requires methodologies that are able to bridge broader length and time scales. While striking the balance between computational accuracy and efficiency is relevant to most biophysical problems, its central role in the meaningful analysis of bioenergy transduction drives the development and integration of multi-scale computational methodologies, including semi-empirical quantum mechanical/molecular mechanical (QM/MM) methods,6 ab initio QM/MM approaches7 and more recently, machine learning (ML) techniques.
To illustrate the value and limitation of these methodologies, as well as strategies for properly calibrating them in specific systems, we will discuss several recent applications in the area of bioenergy transduction. Due to limited space, we will mainly focus on studies in our own labs, although complementary efforts by others will also be mentioned. The discussions will focus on steps that are most tightly coupled with the chemical reactions, while methodologies that tackle other steps such as large-scale conformational transitions can be found in recent reviews by others,8,9 including general conceptual issues related to the efficiency of energy transduction.4,10,11 We will end with a brief outlook that comments on future developments that will enable the analysis of increasingly complex bioenergy transduction problems.
2. Theory and Methods
To study biological processes driven by chemical reactions, the basic computational frame-work is the hybrid QM/MM approach12,13 pioneered by Warshel, Levitt and Karplus, who were awarded the Nobel Prize in Chemistry for their efforts. At the conceptual level, the approach is intuitive: the reactive region of the system is treated at a QM level, while the environment is described with a more simplified MM model. Through the contributions of many researchers, the QM/MM approach has become an indispensable tool for the analysis of condensed phase problems,14–16 especially biomolecules.17 Recent developments and applications, especially in chemical and enzymatic systems, have been summarized in excellent review articles;7,18 some of the remaining challenges have also been discussed.19 In the following, we briefly touch upon several generally relevant methodological issues.
2.1. QM/MM Methods for Ground Electronic States
In the most standard scheme applied to biomolecules (Fig. 1), the QM/MM energy is given in the additive form,
| (1) |
which indicates that the QM and MM atoms interact through electrostatic, van der Waals and bonded terms; in most implementations, only the QM/MM electrostatic interaction (see below) is included in the self-consistent determination of the QM region wavefunction, Ψ (or the electron density), while the van der Waals and bonded terms are treated at the classical force field level. When the QM/MM partitioning is conducted across a covalent bond (e.g., between Cα and Cβ atoms of an amino acid), link atoms,20,21 frozen orbitals22 or pseudo potentials23 are required to properly treat the boundary QM atoms; care needs to be exercised to avoid artificial polarization of the QM atoms,24,25 and it is generally advised against partitioning across highly polar covalent bonds.
Figure 1:
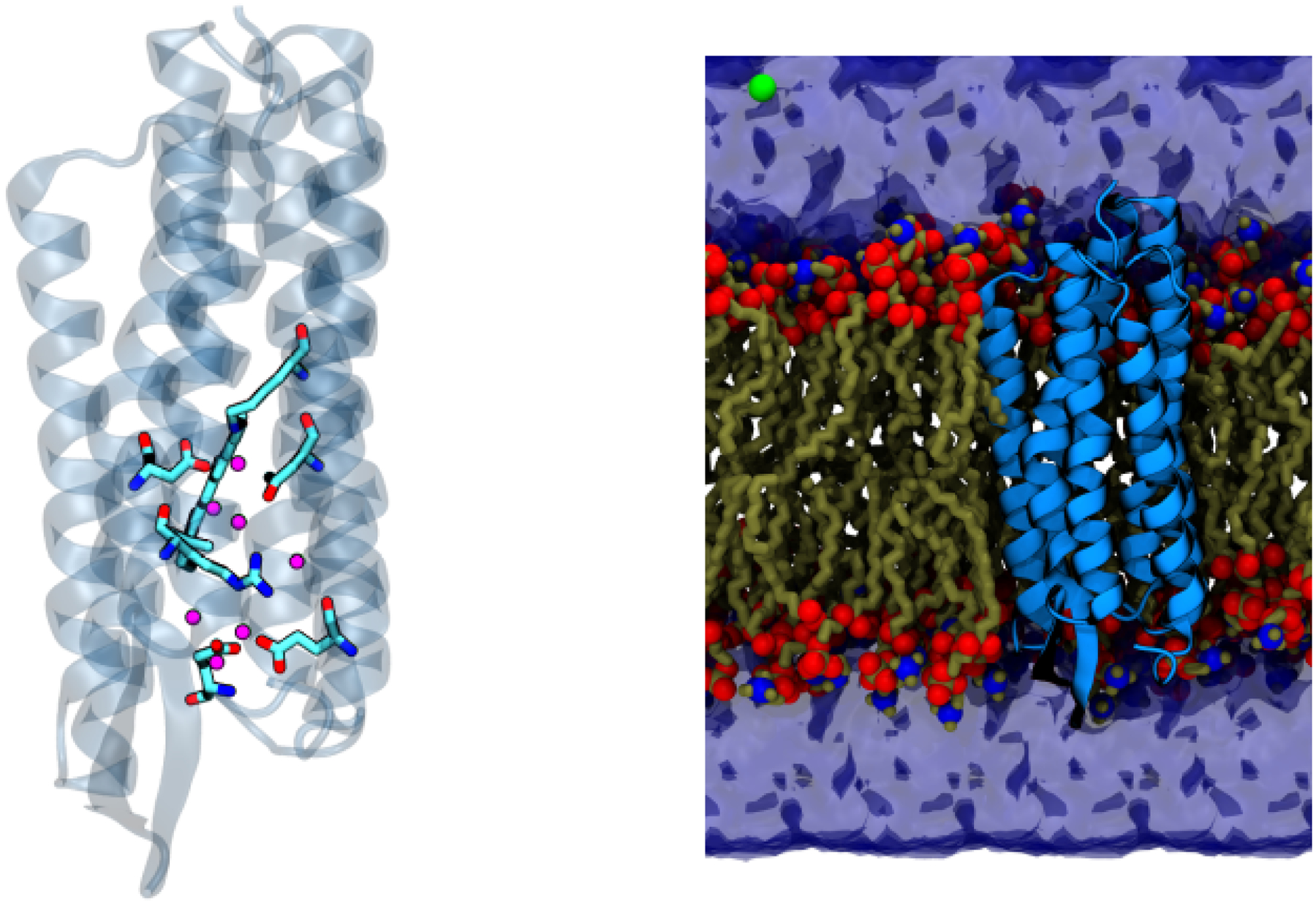
Illustration of a typical QM/MM set-up using the analysis of the O→bR transition in bacteriorhodopsin as an example. In the left panel, the QM region, which includes key amino acids and water molecules, is highlighted in the licorice form, while the rest of the MM protein environment is shown in transparent cartoon. As shown in the right panel, the entire protein is then embedded in a solvated lipid bilayer; the lipids, water molecules and salt ions are also treated at the MM level.
The proper QM level depends on the problem of interest. While ab initio or density functional theory (DFT) are generally more reliable than semi-empirical QM methods, they are computationally expensive. Even with modern hardwares, ab initio or DFT based QM/MM simulations are typically limited to tens to hundreds of picoseconds of sampling,26 which are usually too short for a reliable computation of equilibrium (e.g., free energy) or dynamical properties. Therefore, carefully calibrated semi-empirical QM methods remain an attractive choice, especially for bioenergetics problems; in recent years, density functional tight binding models6,27,28 have emerged as promising choices in many applications.
An issue that has attracted much debate in recent years concerns the appropriate size of the QM region;29–34 with development of efficient algorithms and implementations on modern hardwares, it has become possible to conduct QM/MM simulations with hundreds or thousands of QM atoms,26 or even with the entire system treated at the DFT35 or DFTB36 level. The computational cost associated with such large QM regions, however, limits the amount of conformational sampling. Therefore, depending on the properties of interest, it is important to choose the QM region to best balance computational cost and sampling efficiency. For example, for the analysis of reaction mechanism and free energy profiles, adequate sampling is essential; this is expected to be particularly important to systems involved in bioenergy transduction, as the driving chemical reactions often involve the migration of charged species (e.g., protons, electrons, metal ions) over a long distance, thus there are significant protein and solvent responses that need to be captured with extensive sampling. For properties that are particularly sensitive to electronic structure, such as NMR chemical shifts and hyperfine coupling constants, it is possible that large QM regions are required.32,34,37
Regarding QM/MM interactions, in the simplest model, the electrostatic Hamiltonian involves Coulombic interactions between the QM atoms (nuclei and electrons) and fixed MM partial charges. At short QM-MM distances, however, the point charge models for the MM atoms may lead to overpolarization of the QM atoms. Thus a more physical model is to “blur” the MM charges as spherical Gaussians.21,38
It is increasingly realized that it is valuable to explicitly include electronic polarization at the MM level. In recent years, substantial progress has been made in the systematic parameterization and validation of polarizable MM force field models for biomolecules,39 including, for example, the CHARMM-Drude model,40 AMEOBA,41 and SIBFA.42 Improvements in computational efficiency and implementation on GPUs have made it possible to conduct extensive molecular dynamics simulations for realistic biomolecules, including the ribosome.43 The inclusion of electronic polarization is particularly relevant to a reliable treatment of bioenergy transduction, since many systems involve buried charges or ion-pairs; as shown in a recent analysis,44 electronic polarization is critical to the description of the stabilization, conformational dynamics and hydration levels of these buried charges or dipoles. In this context, while charge-scaling has been advocated as an empirical approach for approximating the effect of electronic polarization,45,46 such phenomenological model may not be able to capture both energetic and dynamic properties correctly,47 thus including the MM polarization explicitly in QM/MM simulations is preferable.48,49
2.2. QM/MM Methods for Excited Electronic States
Many bioenergy transduction processes are initiated by the absorption of photon, thus the description of electronically excited states is required. Often, ground state structures are QM/MM geometry optimized and the excited state can then be treated with a broad set of QM methods that include, for example, time-dependent DFT (TD-DFT) or post-Hartree-Fock methods. The typical errors for TD-DFT are in the range of 0.2–0.4 eV for singly excited states, strongly depending on the functional applied. Pure GGAs tend to have smaller errors, increasing with the amount of exact exchange. For example, an error of 0.26 eV was reported for B3LYP for a set of medium sized molecules of biological relevance;50 since this behavior is often systematic, a simple shift in energies can help facilitate a comparison with experimental data. However, limitations of common functionals and the adiabatic linear-response approximations are well known, especially for charge-transfer and doubly excited states, which are not uncommon in biological systems.51,52 For some problems, range-separated functionals (LC-DFT) provide major improvements, although not completely resolving the issues.53,54
The absorption spectrum is better computed by sampling of the ground state potential energy surface and subsequent vertical excitation energy calculations, when nuclear quantum effects can be neglected. Here, methods with significantly reduced computational cost are needed, since convergence of the spectrum requires the calculation of hundreds or even thousands of snapshots. Semi-empirical methods are therefore suitable for these type of applications, however, the computational challenges put also strong constraints on the choice of methods, as discussed in detail for retinal proteins.55 The Hartree-Fock based OM2 and OM3 methods within a MRCI implementation proved to be quite reliable, while the DFT based linear response TD-DFTB method was not able to treat these types of systems. They show the same failures as the GGA based TD-DFT methods. Long-range (LC) corrected DFT functionals have been shown to ameliorate the problems, and LC-TD-DFTB has been shown to be quite accurate for the calculation of absorption and fluorescence properties.56,57 However, the typical TD-DFT problems discussed for retinal proteins and chlorophylls in detail, are not completely solved.54
Since the ground and excited states usually feature rather different electronic distributions, polarization of the MM environment was found to make a non-negligible contribution to the excitation energy.48,49,58 It is worth noting that in some cases, force fields have intrinsic limitations, such as for strong hydrogen-bonding interactions with the chromophore (see examples below), thus calling for large QM regions in QM/MM simulations.
2.3. Sampling Transitions and Dynamics Relevant to Chemistry in Biomolecules
For most problems in bioenergy transduction, the key quantity of interest is the underlying free energy profile, which can be used to compute rate constants with well-accepted theoretical models such as the transition state theory.59 The rate constants can then be fed into kinetic network models for gaining further mechanistic insights,60 such as dominant kinetic pathway(s) and the overall time-scale and efficiency of energy transduction; in this context, it is important to recall that rate constants depend exponentially on the free energy barrier, thus small errors in the computed barrier can lead to large errors in kinetics, hence effective computational techniques for adjusting computed rate constants based on experimental observables are highly valuable in this regard.60,61 For the computation of free energy profiles, in principle the methodologies commonly used for classical simulations, such as metadynamics62 and finite-temperature string63 methods, are readily adapted. The key difference lies in the choice of collective variables (CVs), which might take unique forms for chemical processes. For example, charge centroids based on either structural,64 charge65,66 or electron density67 have been used to construct CVs for studying long-range proton transfers.
Since QM/MM computations are generally more expensive than classical simulations, it is worthwhile considering multi-level strategies. For example, semi-empirical QM methods are usually more reliable for structural properties than for energetics. Thus, one effective protocol68 is to first sample the reaction pathway with, for example, DFTB/MM string simulations; with configurations from the optimized minimal free energy path as the initial guess, string calculations with a higher-level QM/MM potential are expected to converge more rapidly for more accurate energetics. Alternatively, configurations from semi-empirical QM/MM simulations can be used in conjunction with machine learning techniques to improve energetics; see below for further discussions.
A particularly interest topic concerns the direct simulation of sequential electron transfers (ETs) in proteins, for which two of us have developed the Fragment Orbital Tight Binding Density Functional Theory (FO-DFTB).69,70 In this quantum-chemical calculation, the quantum region is divided into several fragments, with the general idea to reduce the computational cost as well as to allow for an easy and efficient parallelization. The fragments are defined in such a way that any conjugated π-electron systems are kept intact, so the electronic structure of the isolated fragments can be computed straightforwardly. For biological ET, the fragments may be side chains of aromatic amino acids, peptide bond moieties, or nucleic acid bases. Notably, while the electronic structure of a single fragment is calculated, the other fragments as well as the entire condensed phase environment is treated as point charges, so the fragment is polarized properly.
Next, the Hamiltonian (and overlap) matrices are set up in the basis of the computed fragment orbitals, and they are further used to propagate the electron density corresponding to the excess charge (electron or hole), by way of integrating a time-dependent Schrödinger equation. Effectively, the entire molecular system, with the exception of an excess electron or hole, is treated using a classical MM force field. Note that this also includes the ET-active fragments at any moment that they do not carry any excess charge. The actual propagation of the excess charge is performed in every step of the MD simulation, and proceeds as follows:
The Hamiltonian (and overlap) matrices in the basis of frontier orbitals of the individual fragments are obtained with FO-DFTB/MM. Only the orbitals that play a role in the ET process need to be considered; in low-energy biological ET, these include typically one or few frontier orbitals. The diagonal elements, (“site energies” representing the ionization potentials for hole transfer or the electron affinities for excess electron transfer) as well as the off-diagonal elements (“electronic couplings”) were benchmarked, and high accuracy was observed although scaling is required in some cases.71–74
A time-dependent Schrödinger equation in the basis of the fragment molecular orbitals (FMOs) is set up, so that it can be integrated for the expansion coefficients of the individual FMOs. Since the problem at hand clearly possesses a semi-classical character, one of the many available non-adiabatic propagation schemes is used. Most common choices are the trajectory surface hopping and the mean-field Ehrenfest methods. We note that semi-classical non-adiabatic MD was pioneered75,76 and first applied to charge separation in photosynthetic reaction center proteins77 by Warshel et al., and our methodology follows the development of real-time time-dependent DFTB.78
The wave function of the excess electron is projected onto the atomic charges of the ET-active fragments, and their force-field charges are update accordingly. This procedure can be understood as an “on-the-fly” reparametrization of the force-field, driven by the electronic structure of the quantum region subject to the non-adiabatic propagation.
Forces on the atoms are calculated using the force field including the updated charges. At this stage, the dynamics of the system is exclusively governed by the classical equations of motion. Nonetheless, such a scheme is capable of describing the dynamic relaxation of the system in response to any partial or complete electron transfers, which represents the microscopic mechanism of the reorganization processes in the phenomenological Marcus theory (see discussion below).
For the purpose of analysis, the charge occupation (squared expansion coefficient) of each fragment is recorded along the trajectories being performed. This value ranges between 0 (neutral fragment) and 1 (fragment completely occupied by the hole or excess electron being transferred). After averaging over the ensemble of simulations, the time-dependent occupations can be fitted using a kinetic model79 to obtain rate constants of the forward and backward ET events.
2.4. Computation of Experimental Observables
It is critical to compute experimental observables so that computational results can be validated and predictions tested. With QM/MM methods, a broad range of observables can in principle be computed,80 ranging from various spectra through free energy relations to kinetic isotope effects.81 As mentioned above, computed properties such as rate constants, equilibrium constants and spectral densities can be fed into kinetic network models (either classical or quantum59) for probing macroscopic features relevant to the energy transduction process. The convergence of those observables depends on the system and sometimes requires extensive sampling. For example, kinetic isotope effects can be computed with path-integral techniques,82 which may require the equivalence of multiple nanoseconds of sampling,34,83 which is possible only with semi-empirical methods. The computation of infrared spectra,84,85 especially multi-dimensional ones,86 also requires extensive sampling.
For semi-empirical methods, the trade-off between computational efficiency and accuracy is a matter of concern, however, well calibrated semi-empirical methods have been shown to be very useful in interpreting and predicting spectral properies of complex systems. For example, the mid-IR C=O double bond vibration is highly sensitive to the hydrogen bonding environment, showing red-shifts up to 50 cm−1 in strongly hydrogen bonded systems relative to the gas phase, and an reparametrized semi-empirical method has been shown to capture these shifts reliably,87 being able to resolve details of water bridged hydrogen bonds between amino acids, which were experimentally difficult to determine. This helped to differentiate between various intermediate structures in the bR photocycle at different temperatures.88,89
Another frequently computed property for energy transfer (e.g., light-harvesting) systems is the spectral density, which describes the coupling of the nuclear degrees of freedom to the electronic structure. In principle it can be computed from a time series of excitation energies along a classical MD trajectory. However, since classical force fields often do not capture the geometry of the chromophore reliably, spectral density computed using MD trajectories sampled with classical force fields often suffers from the “geometry mismatch” problem.90 Accurate spectral densities compared to experimental data were obtained using a semi-empirical method specifically reparametrized for an accurate desription of vibrational frequencies.90,91
2.5. Integration with Machine Learning
In recent years, machine learning (ML) has become an increasingly powerful tool in computational analysis of molecular systems.92 In the context of QM/MM simulations and bioenergy transduction, ML techniques are uniquely valuable in several areas (Fig. 2). First, ML techniques can be used to enhance the efficiency of free energy sampling, especially when multiple collective variables93 are potentially important to describing the coupling between the reaction and environmental degrees of freedom.94 Second, ML methods can be used to improve the accuracy of semi-empirical QM/MM simulations via iterative “Δ-learning”, in which configurations from “low-level” QM/MM (e.g., DFTB/MM) trajectories are used to learn the differences (Δ) from “high-level” QM/MM energies and forces; the differences are then used to either directly estimate correction of the free energy surface or re-sample the potential energy surface for improved free energies.95,96 These “multi-level” free energy simulation methods have so far been applied to relatively simple solution reactions,97 as learning the Δ reliably for realistic systems with a large QM region is, in fact, not straightforward and requires further developments. Finally, ML can also be used to learn other properties, such as electronic coupling elements98 or vertical excitation energies99 as functions of molecular configurations; training such ML models can substantially reduce the cost of electron transfer and spectra calculations.
Figure 2:
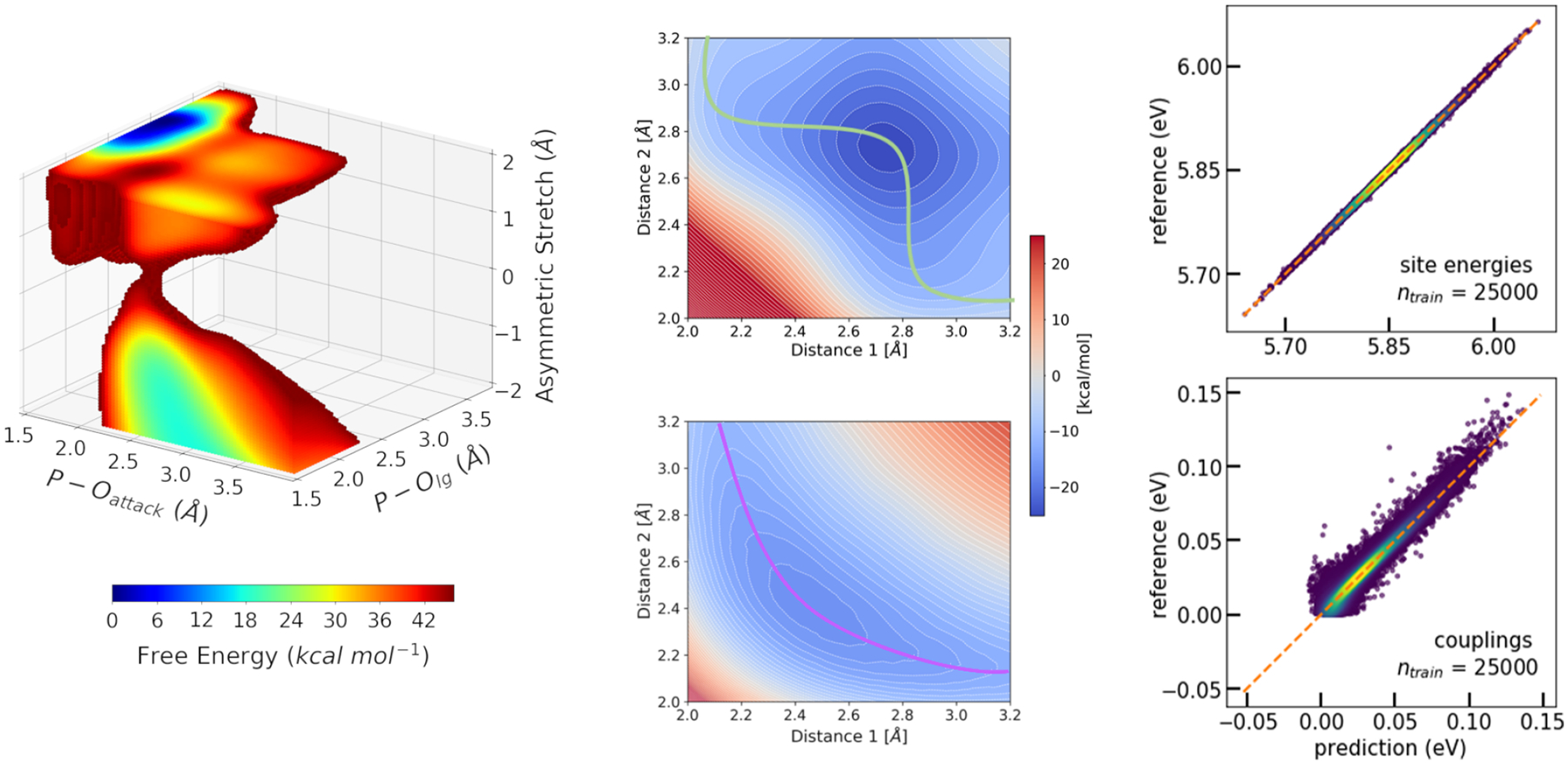
Machine learning (ML) techniques can be integrated with QM/MM simulations in diverse ways. Left: Reinforcement learning and other techniques can be used to accelerate QM/MM free energy simulations. The example shown here is the three-dimensional free energy surface for the hydrolysis of methyl phosphate in solution at the DFTB3/MM level.19 Middle: Neural networks using symmetry function representation of molecular structures are used in a Δ-ML scheme to correct the energies yielded by DFTB3. Here, the qualitatively wrong DFTB3 energy landscape of thiol–disulfide shuffling reaction (top) is corrected to agree with ab inition reference quantitatively (bottom).169 Right: Kernel ridge regression combined with the Coulomb matrix representation was used to model the Hamiltonians describing ET processes in organic semiconductors. Shown is the accuracy of prediction vs. reference for electron transfer site energies (top) and couplings (bottom) for geometries taken from an MD simulation of crystalline anthracene.98
3. Application and Case Studies
In this section, we discuss several recent applications to illustrate the value and limitations of QM/MM based simulations, including calibration of the methods in realistic applications for meaningful mechanistic insights. Due to the sampling requirement of these applications, we focus largely on DFTB/MM studies, although complementary efforts using other QM/MM methods are also mentioned for comparison.
3.1. Long-range Proton Transport in Proteins
3.1.1. Key Mechanistic Questions
Since the proton motive force plays key roles in bioenergetics, long-range proton transports coupled with either photon absorption or electron transfers are richly featured in bioenergy transductions. Representative examples include bacteriorhodopsin (bR), Complex I, Complex IV (cytochrome c oxidase) and the F0F1-ATPase. In terms of the key questions of common interest, these include: (1) What amino acids/co-factors are involved as the proton donor, acceptor and mediating groups? This is difficult to answer based on experiments alone because the positions of hydrogen atoms are usually not visible in crystal or EM structures; mutation experiments and spectroscopic data are potentially informative, although a molecular level interpretation is often not as straightforward as it may appear. An example that we’ll touch upon below concerns the proton storage group (PRG) in bR, which has attracted much attention in the past decades till very recently.100 (2) Is there a single, dominant proton transfer pathway or many pathways are involved with similar fluxes? The typical approach for identifying potential proton transfer pathway(s) is to focus on water wires that mediate proton transfers via the canonical Grotthuss mechanism. As discussed in increasing number of studies,101,102 however, water wires in static crystal structures in the absence of the excess proton may not represent the proton transfer pathway and very often multiple pathways may contribute. (3) Finally, arguably the most puzzling question is what controls the gating of proton transfers;103 the timing and directionality of long-range proton transfers lie at the heart of the efficiency of bioenergy transduction, and understanding the underlying molecular mechanism requires going beyond structural models to evaluate how thermodynamics and kinetics of proton transfers are modulated by other events, such as reduction of nearby co-factors104,105 and change in the local hydration level.101,106–108
3.1.2. Model Validation
Proton transfer energetics depend critically on the pKa values of the donor, acceptor and mediating group(s). Therefore, microscopic pKa calculations are essential validations for both the enzyme model (e.g., titration state of key residues) and the computational (QM/MM) methodology.109 The prediction of reliable microscopic pKa values relies on an accurate treatment of both proton affinity of the relevant titratable group and its interaction with the protein environment while in different protonation states. Moreover, the responses of the protein and internal water molecules to the change of titration state also needs to be properly captured;110 these in general include both dipolar reorientations and electronic polarization, which requires extensive conformational sampling and treatment of electronic polarizability of the environment, respectively. In the study of cytochrome c oxidase, for example, DFTB/MM based thermodynamic integration simulations111,112 were used to probe the microscopic pKa value of the critical Glu286 residue, which was buried in a relatively hydrophobic internal cavity and thus featured a rather shifted experimental pKa value of ~9.7. DFTB/MM free energy simulations with different enzyme models and approximate treatments of electronic polarization112 found that reproducing the experimental pKa value required penetration of water molecules into the cavity, which was coupled with the protonation of a propionate group of heme a3. Therefore, in addition to serving as validation, microscopic pKa calculations can potentially provide important mechanistic insights as well. Along this line, it is worth noting that microscopic pKa calculations have also been used to calibrate reactive force field models (MS-EVB) for studying proton transfers in solution and proteins.113
3.1.3. Example: the O to bR Transition in Bacteriorhodopsin
One prototypical example of proton pumps is bacteriorhodopsin, whose photocycle features several proton transfer reactions coupled to conformational transitions. Some of the few last remaining challenges in the study of these complex processes were the atomic structure of the O state, especially with regard to the hydration of the active site, and the mechanism of the conversion to the ground state of bR, featuring a long-range proton transfer. Application of parallelized DFTB3/MM metadynamics protocols as well as other extended sampling methods made it possible to answer these questions with multi-dimensional free energy surfaces114 (Fig. 3).
Figure 3:
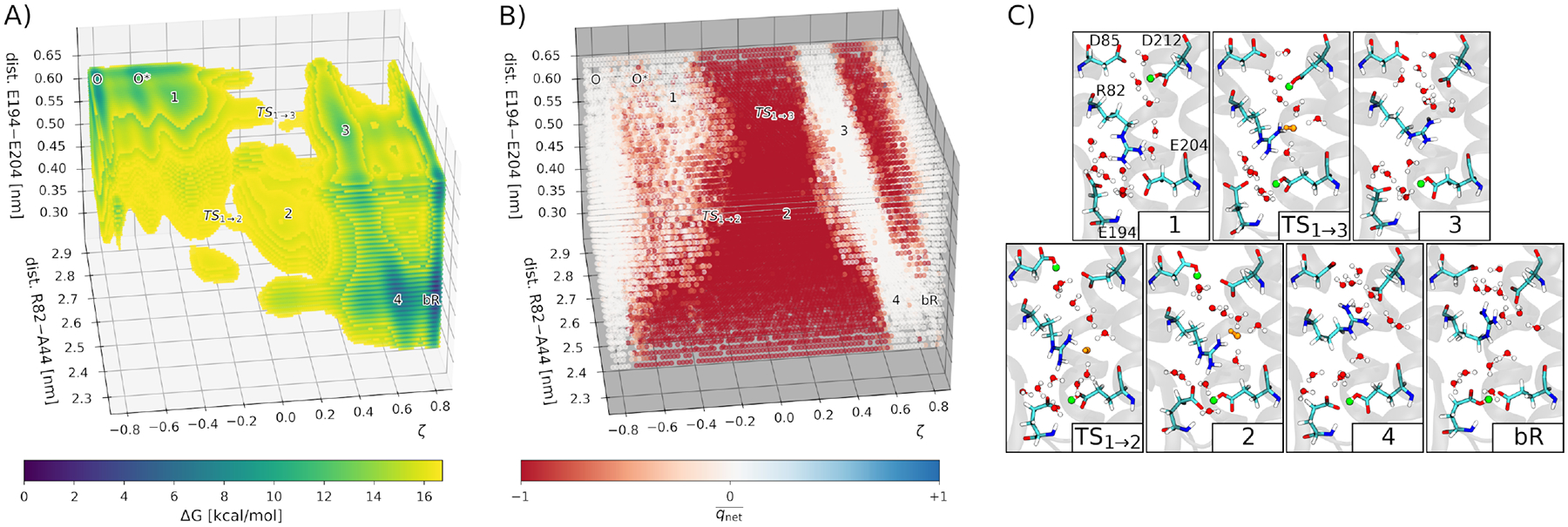
DFTB3/MM free energy simulations reveal a proton hole mechanism for the O→bR transition in bacteriorhodopsin,114 which involves a proton transfer from D85 to the PRG (E194, E204), mediated by D212, cavity water and R82. A: The 3D free energy surface from ~100 ns multi-walker metadynamics simulations. The relevant states are labeled. B: Net charge of QM water molecules. White: , neutral water; red: , OH−; blue: , H3O+. C: Representative structures corresponding to the states identified in the free energy surface. Green: proton bound to an Asp or Glu; orange: charged water species (here, always OH−).
The thermodynamics and kinetics of the proton transfer was obtained in a good agreement with experimental estimates, which underscored the credibility of the methodology used. The reaction energy of −3.6 kcal/mol was consistent with the measure pKa differences between D85 and the PRG. Concerning the kinetics, the obtained energy barrier translated into a time scale of ca. 0.6 ms, which is in the range of experimental estimates.
The study114 provided insight into the microscopic proton transfer mechanism and the key structural patterns, regarding especially charged amino acid side chains and water molecules, coupled to the actual proton transfer. The side chain of R82 sidechain switches between two possible distinct orientations to stabilize specific negatively charged amino acids: D85/D212 in the ground state bR, and PRG in the O state. Linked to that is the control of the hydration level of the active site by means of the motion of the R82 side chain and opening and closing of the PRG. The ground state bR features a low internal hydration level due to the closed PRG, and there is no continuous water wire because of the intervening R82 side chain. The latter feature helps prevent any wasteful back transfer of protons.103 On the other hand, the O state has an elevated level of internal hydration, made possible by the open PRG; a continuous water wire is formed between D85 and the PRG, allowed by the reoriented side chain of R82. From a simple electrostatic perspective, the positive charge of R82 may also aid the generation and transfer of the hydroxide ion, which was observed in DFTB3/MM free energy simulations.114 The observation of the proton hole mechanism corroborates with the chloride pumping ability of the D85S mutant of bR,115 and with the similar mechanisms of bR and halorhodopsin, which feature conserved patterns of electrostatic interactions between the transferred anion and the protein’s amino acid side chains.116
3.1.4. Vibrational Spectra
Two examples concern vibrational bands in bacteriorhodopsin from FT-IR studies by Gerwert and co-workers.117,118 The first example focused on the PRG (see Fig. 3C), whose identity has been debated for many years. Numerous crystal structure, including the very recent high-resolution crystal structures for multiple kinetic states,100 revealed a pair of glutamate residues (Glu 194, Glu204) whose sidechains are extremely close to each other, with carboxylate oxygens merely separated by ~2.4 Å. Such unusual geometry together with mutation data suggested that the pair of glutamates are involved in storing the proton. The FT-IR study of Gerwert and co-workers observed a diffuse band around 2,000 cm−1, which is commonly observed in protonated water clusters. Since a number of water molecules were observed near the pair of glutamates, it was initially suggested that the proton was in fact stored on these water molecules in the form of a “delocalized” proton. This interesting hypothesis stimulated a set of computational studies using different QM/MM methodologies and sizes of the QM region; nuclear quantum effects were also evaluated with ring-polymer molecular dynamics.85 While it remains challenging to exactly match experimental line shape, the latest DFT/MM simulations119 continue to support the model84 in which the excess proton is delocalized between the pair of glutamates, although nearby water molecules are clearly important to provide additional stabilizations.
Another unique infrared signature in bR was observed for a number of water molecules trapped between the protonated retinal Schiff base and deprotonated Asp96 in the N kinetic state.118 The vibrational peaks were observed around 2540–2775 cm−1, which is substantially red-shifted compared to neutral water. DFTB3/MM simulations120 were able to capture the key experimental observation (Fig. 5A) and highlighted how the unique environment of the water cluster led to its significant polarization and therefore dramatic red-shift of the collective O-H vibration. An interesting observation was that the computed line shape was sensitive to the number of water molecules in the “cavity”, since including a larger number of water molecules perturbs the hydrogen bonding network and thus the vibrational coupling between the strongly polarized water molecules. Therefore, by systematically comparing computed and measured vibrational spectra, one is potentially able to characterize the structure and composition of water molecules in protein internal cavities, which are difficult to accomplish otherwise, especially for transient kinetic states.
Figure 5:
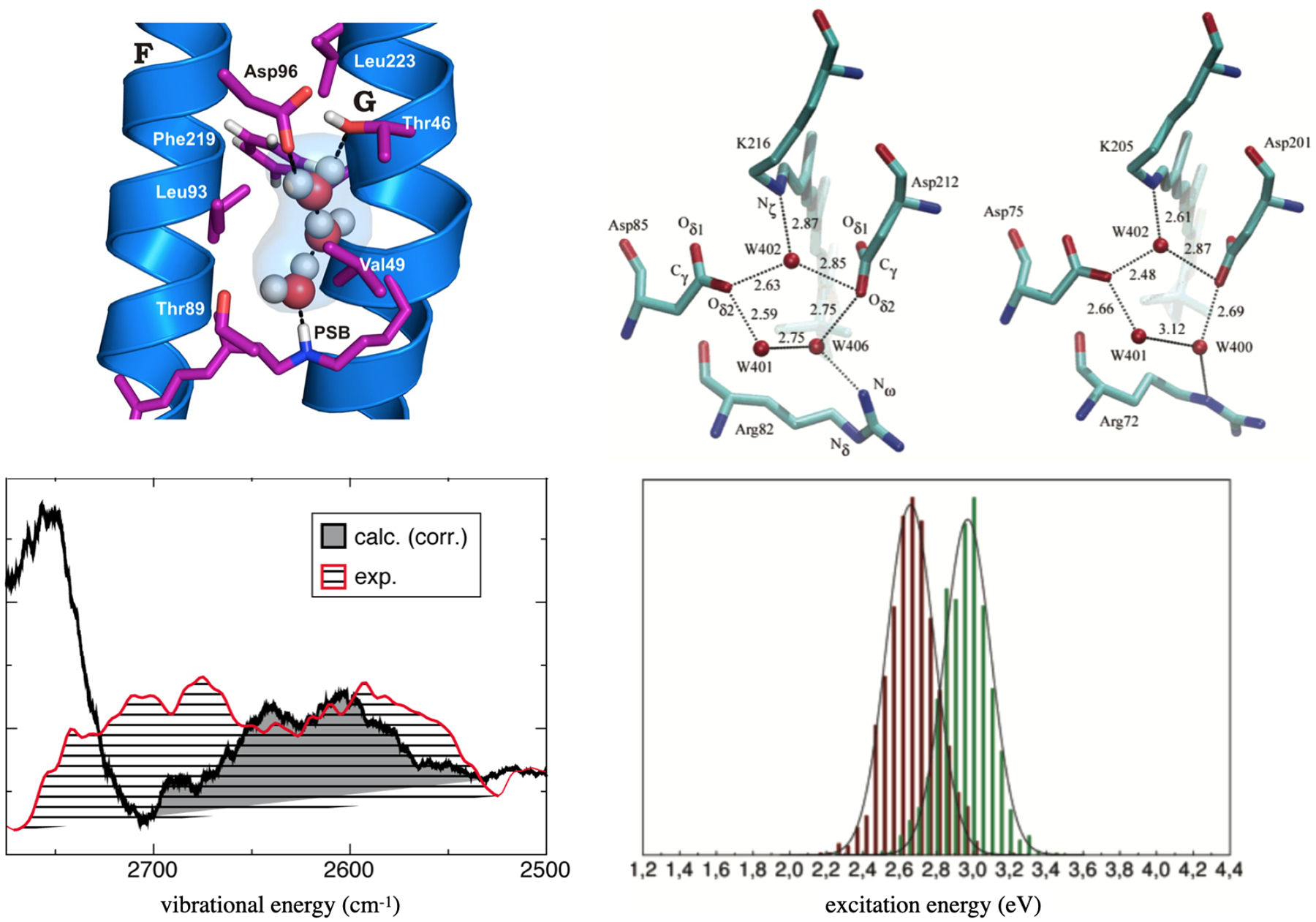
QM/MM calculations help explain shifts in vibrational and electronic spectra in retinal proteins. Left: A water cluster in the N state of bR has a unique local environment that leads to significant red-shifts of the computed (DFTB3/MM) vibrational spectra, in agreement with experimental observations.120 Right: Difference in the hydrogen-bonding networks in the active sites of bR and ppR lead to significant shifts in the computed S0–S1 transitions: the histograms for bR (red) and ppR (green) are based on OM2/MRCI calculations sampled along ground state classical MD trajectories.134
3.2. Fast Electron transfer
3.2.1. General Remarks
Of perhaps even more importance in bioenergetics are electron transfer (ET) processes as the primary “tool” of biological energy transduction. ET taking place between an electron donor and an acceptor in a complex molecular environment may be described with the Marcus theory.59 For instance, Wu & van Voorhis developed a scheme based on constrained DFT the diabatic potential energy surfaces needed in the Marcus theory to express the reaction and reorganization energies.121,122 The application of constrained DFT was crucial as it turned out to effectively avoid the over-delocalization problem of DFT.123
Most biological ETs of interest, however, involve one or several electron relays, and a condition for the applicability of the Marcus theory is that any reorganization processes have finished completely before the subsequent ET event. Thus, the Marcus model works for slow hopping ET but not for fast ones, where “slow” means that the individual events are infrequent rather than that they would take a long time to complete. This was paraphrased by Troisi as a “speed limit” for hopping transfer.124 It appears that numerous ET pathways in bioenergetics exceed this speed limit, and the temporal scales of the reorganization and of the (relatively fast, or frequent) ET itself overlap. On the same note, Matyushov et al. reasoned that the energy barriers to ET in proteins (as a representative of a complex molecular system) are not real equilibrium quantities; rather, they depend on the specific temporal scale of the ET reaction.125 Such cases then need to be described by more flexible methodologies that do not imply any separation of temporal scales.
Biomolecular ET has been a frequent subject of computational studies, and the state of the art in 2015 was reviewed by Blumberger.126 Fast ET has proven challenging to describe, but still it has been investigated using a few different approaches, usually employing more advanced versions of the traditional ET models. For example, the light-driven ET in multiheme cytochrome STC from S. oneidensis in aqueous solution occurs on a scale of several nanoseconds, and it was investigated by a combination of experimental and computational techniques.127 This work used a non-ergodicity correction by Matyushov,128 which removes the contribution of slow motions from the total reorganization energy. Also, the dynamics of ultrafast ET in flavodoxin protein was shown to depend on its coupling to environmental fluctuations.129 This coupling reduces the reaction free energy as well as the reorganization energy. Most recently, the mechanism of the highly efficient ET supported by polymerized cytochrome OmcS in G. sulfurreducens was discovered.130 The process takes place over micrometers, with elementary hopping on sub-nanosecond scale – hence the notion of “protein wire” – and accelerates upon cooling. On the other side of the spectrum of computational methods is the study of sub-picosecond ET in a rhenium complex coupled to the azurin protein from P. aeruginosa.131 Computationally expensive combination of surface hopping and hybrid-DFT based calculations of excited states were feasible given the short temporal scale to cover, making it possible to reveal the mechanism of the reaction.
By comparison, our FO-DFTB approach relies on the simultaneous propagation of the coupled electronic and nuclear degrees of freedom in a semi-classical fashion. This method does not involve any assumption on the relative rates of the ET reaction and the relaxation (reorganization) processes, making it possible to resolve any relevant temporal scales in an unbiased manner. The efficiency of the method makes it possible to explicitly describe ET processes on a nanosecond scale, as illustrated by the examples below.
3.2.2. Examples: Photolyases and Cryptochromes
We applied the FO-DFTB/MM methodology to investigate multi-step ETs in the proteins of the photolyase cryptochrome family (PCF), which are involved in DNA repair and in various signaling pathways. The part of the photoactivation process in E. coli DNA photolyase, which constitutes an ET along three conserved Trp side chains (ET from the protein surface to the FAD cofactor) was investigated in our first study.132 The ET was simulated on the naturally occurring temporal scales without introducing any system-specific parameters, and a kinetic analysis yielded rates in excellent agreement with experimental data. A detailed picture of the ET process emerged, and it became clear that the second ET step may occur before the structural relaxation following the first ET step has completed. This illustrates the flexibility of the non-adiabatic simulation approach, which is applicable even in this case for which the Marcus theory is not applicable, because of the non-equilibrium reaction conditions due to the overlapping temporal scales of the processes involved (ET itself, delocalization of charge, relaxation of protein, reorganization of solvent). As for the features of the ET in that protein and perhaps PCF in general, it was shown that it is the polarization of the solvent at the surface of the protein that makes the process exergonic and thus uni-directional.
Then, we investigated the interesting case of a class III cyclobutane pyrimidine dimer (CPD) photolyase from A. tumefaciens, which features not just one but two different, branching ET pathways containing a total of six Trp side chains, which were both found to play a role in the photoreduction of FAD.133 We discovered an intriguing thermodynamic and kinetic competition between the two pathways: one pathway supports a faster ET while the other pathway stabilizes the final product better. This balance manifested itself in our simulations by the electron hole first transferring along the “fast” Trp triad, before a sequence of backward ET steps led to the transfer of the electron hole to the second pathway. The whole process occurred on a temporal scale of nanosecond, and was accompanied by complex repolarization of the environment (parts of the protein as well as, not least, water molecules), emphasizing the need for a multi-scale computational description (Fig. 4).
Figure 4:
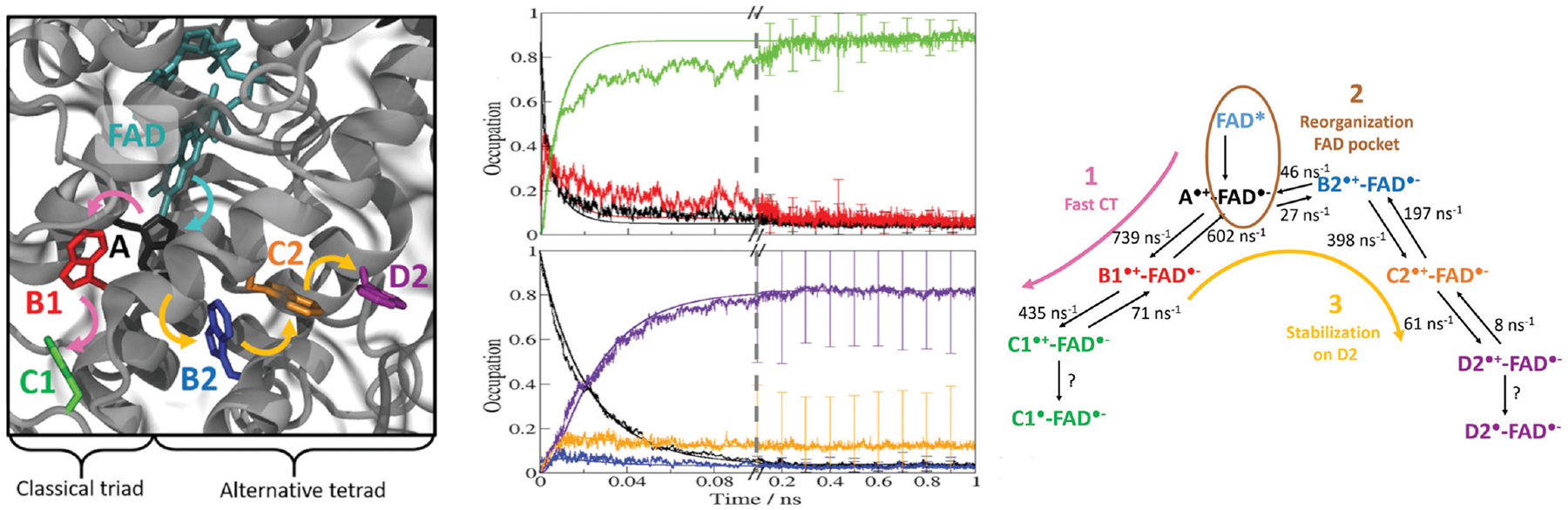
Multi-scale non-adiabatic semi-classical simulations of electron transfer in complex and bio-molecular systems involve a simultaneous propagation of the transferring electron and of the protein dynamics. By doing so, they provide atomic-level insight into the coupling of these processes. The example shown considers two different ET pathways in a photolyase protein PhrA.133 Left: Active site of PhrA, showing the amino acid side chain participating in the branching electron transfer pathways. Center: Temporal course of the averaged occupation of the individual participating amino acid side chains by the transferring electron hole, along the two different pathways. Right: The kinetic scheme inferred from the temporal dependences of the occupations reveals a possible mechanism of the kinetic and thermodynamic control of the ET pathways in PhrA. All panels share the same color code to designate the different amino acid side chain considered as electron relays.
3.3. Electronic Excitation
3.3.1. The challenge of excited states in biomolecules
The computational determination of protein excited states is a significant challenge. The size of the chromophores requires approximate methods like time-dependent density functional theory (TD-DFT) or more approximate post-Hartree Fock methods like CC2 to be applied. In some cases, the determination of an optimized structure using QM/MM geometry minimization techniques may be sufficient when average and optimized structures agree sufficiently well. This is the case for many retinal proteins like bR or ppR, as shown by explicit calculations.134 Here, the active site is characterized by a very stable and strongly hydrogen bonded structure. We note that force fields using fixed point charges, however, may have difficulties in describing strongly hydrogen bonded complexes, therefore equilibrating the protein structure using such force fields may lead to conformations that are not suitable for further QM/MM investigations; this problem was described recently for the ChR2 protein,135 as well as for a Glucose binding protein.136 Due to the long sampling times required, the system then requires using either a polarizable force field136 or a semi-empirical QM/MM approach, which has been shown to retain the active site structure well.135 In the latter case, however, a minimum energy structure may not be adequate, and absorption spectra have to be computed by sampling excited states along QM/MM MD trajectories.54
All approximate quantum methods trade accuracy with computational efficiency, which is particularly challenging for electronically excited states as mentioned in Sect.2.2. We note, however, that the effects of approximations are already visible in the determination of the ground state structures of conjugated electronic systems,54,55 where the bond-alternation (the difference of the bond-length of neighboring single and double bonds) is sensitive to the method applied. This is important since the ground state structure also determines the excited states properties, which calls for a careful choice of methods for both ground and excited states calculations. Similarly, torsional angles and planarity of the chromophore can be dependent on the approximation, which may also affect excited states properties.
The problems of TD-DFT for excited states are nowadays well known, as recently discussed for the case of chlorophylls.137 Important to mention is the overestimation of excited states energies, which leads to a blue shift in the computed spectra and is pronounced for hybrid and long-range corrected functionals. This, however, is less problematic since a systematic blue-shift can be corrected when comparing to experimental data. More problematic is the inability of TD-DFT (or single reference methods in general) to describe doubly excited states, and the problems with describing charge transfer states,52 which have been seen for retinal proteins early on.51,55 For retinal, this can result in a wrong dependence of excitation energies on the chromophore structure, but also on the influence of the protein electrostatic field. The development of long-range corrected (LC) TD-DFT methods could partially resolve many of the TD-DFT problems. However, LC-TD-DFT methods are far from being perfect for color-tuning studies in retinal proteins (see below), still underestimating the response to the protein electrostatic field. Similar effects have been reported for chlorophylls,54 and it can be expected to hold for other color pigments as well.
3.3.2. Color tuning of excited states in biomolecules
Many photoactive biomolecules, like rhodopsins (Rh), green fluorescent proteins (GFP) or Blue light using FAD (BLUF) proteins contain one chromophore bound to a protein matrix. In contrast, light harvesting systems like the Fenna-Matthews-Olson complex (FMO) or light-harvesting complex II (LH2) contain several (chlorophyl) chromophors. The maximum absorption wave-length is determined by several factors, which are: (i) the geometrical and electronic features of the chromophore, (ii) the steric, electrostatic and hydrogen bonding interactions with the environment and (iii) a possible coupling between the chromophores within the protein complexes.
Due to their diverse molecular and electronic structure, the various chromophores have very different absorption maxima in gas phase, but also very different responses to steric and electrostatic interactions with their environments. A steric interaction with nearby protein groups can twist the often planar structure, thereby disturbing the conjugation of the delocalized electronic system. Through hydrogen bonding, as in the case of the retinal Schiff base in retinal proteins, the localization of charge on the chromophore can be strongly affected, leading to large color shifts. Electrostatic interactions with the environment can lead to a further color shift, when ground and excited states have different dipole moments. Polarizable residues, in particular in the immediate vicinity of the chromophore, can lead to a polarization-shift. Retinal proteins, for example, absorb in a range of 300–700 nm, which illustrates the immense tunability due to a very sensitive response to external electrostatic potentials54,134, while it is much less for most other chromophores. For example, in Ref. 134, the color shift of 70 nm between bR and ppR (phoborhodopsin) has been investigated; it was found that the shift was partly due to the different hydrogen bonding networks around the retinal and partly due to the different electrostatic fields from the protein environment (Fig. 5). In multi-chromophoric systems like LH2, close-lying chromophores lead to a coupling of excited states, i.e., a delocalization of the excited states over several chromophores, which results in a red shift of about 50 nm in LH2.54,138–140
The impact of polarization on retinal excited state properties has been emphasized by Warshel and coworkers early on141 and has then been investigated in detail using quantum mechanical and polarizable force field methods.58,142 Including polarization allows to compute accurate absolute excitation energies, when a reliable and well calibrated ab initio method like SORCI is used for the QM region. This allowed to determine protonation states of protein side chains143 and discriminate between different structural models proposed for early intermediates of the bR photocycle.88,89 Several other implementations of QM/MM with polarizable force fields have been reported so far.48,49 For chlorophylls144 in LH2, interestingly, direct electrostatic interactions seem to cancel the polarization response, therefore it was suggested to rather neglect the direct electrostatic interaction when not including polarization.145
3.4. Mechanochemical Coupling
3.4.1. Key Mechanistic Questions
Many bioenergy transduction processes in cells are driven by the binding and hydrolysis of NTP (e.g., ATP or GTP). While in many cases, large-scale conformational transitions have been established to be coupled directly to the binding of NTP and/or dissociation of hydrolysis products, it is an intrinsically interesting question how the hydrolysis of NTP is coupled with the conformational state of biomolecule. Indeed, if the coupling were weak, futile nucleotide hydrolysis would occur, leading to reduced thermodynamic efficiency of the energy transduction.146 In addition to the more visible conformational transitions, other more subtle changes may make notable contributions to the chemical step. For example, an emerging theme in nucleic acid enzymes such as DNA polymerases and RNase H is that transient cation recruiting into the active site may play a major role in activating the chemical step.147,148 For example, in the DNA polymerase η, time-resolved crystallography has captured the presence of the third Mg2+ ion during the nucleotide addition process.149 Whether the third Mg2+ is essential to lowering the activation barrier of nucleotide addition or its main role is to stabilize the product has been debated. Nevertheless, the importance of such transiently recruited metal ion (i.e., not highly populated in the ground state structure) to the chemical activity has been well received.150,151 After all, it is common to invoke change of protonation state of active site residues for efficient catalysis in enzymes, despite the fact that the concentration of protons is often substantially lower than those for common metal ions under physiological conditions.
In short, the key mechanistic questions commonly encountered in biomolecular machines using NTP as “fuels” include: (1) In what conformational state does the chemical step (e.g., ATP hydrolysis) occur? (2) What are the roles of the key regulatory events, which are likely local in nature, such as closure of the nucleotide binding pocket, reduction of the level of local hydration, and recruitment of additional metal ion(s)? (3) How are these local regulatory transitions coupled to the more global conformational transitions, so as to ensure a tight mechanochemical coupling and therefore a high thermodynamic efficiency of the bioenergy transduction process?
3.4.2. Model Validation
Since nucleotide hydrolysis is the driving reaction for many biomolecular machines, it is important to calibrate the QM/MM methodology for phosphoryl transfer chemistry.152 This has been a challenging task due to the highly charged nature of phosphates and existence of multiple competing mechanisms,153,154 which place major demands in both computational accuracy and sampling efficiency. In addition to energetic properties, kinetic isotope effects are valuable for confirming that the nature of transition state is adequately captured.83,153
3.4.3. Competing Pathways and Mechanochemical Coupling
To illustrate the value of QM/MM simulations to the analysis of chemical steps in biomolecular machines, we briefly discuss two recent examples (Fig. 6). The first is a classical biomolecular motor, myosin II, in which the hydrolysis of ATP requires closure of the active site, which in turn is coupled with the remarkable rotation of the converter domain more than 40 Å away.155 Since our QM/MM analysis of myosin156 has been summarized in several recent articles,19,152 we only highlight two key points here. First, even with the crystal structure that features a fully closed active site (pre-powerstroke state), DFTB3/MM simulations were able to identify several reaction pathways for the hydrolysis of ATP that have rather similar rate-limiting free energy barriers. These pathways differ in terms of the mechanism through which the lytic water deprotonates to generate the strong nucleophile (OH−) and the routes that the ionized proton takes to reach the hydrolysis product, the inorganic phosphate and ADP. The observation highlights that even in enzyme active sites that have been “optimized” by evolution, competing pathways exist, thus it is essential to develop (almost) automated methodologies for the exploration of reaction pathways with little prior human biases. Along this line, the computational efficiency of DFTB3/MM approach was essential to the successful integration between metadynamics and finite temperature string methods for the exploration of alternative reaction mechanisms.156
Figure 6:
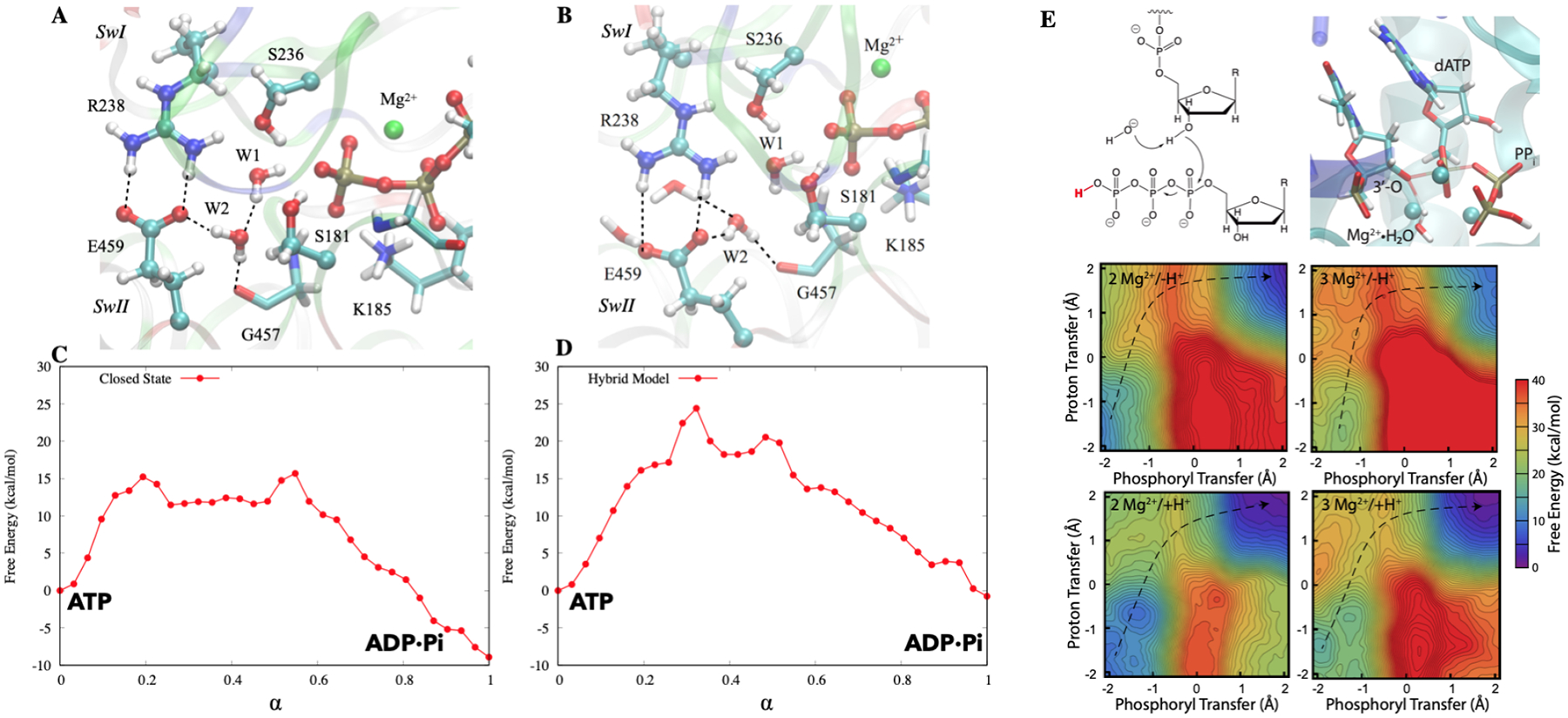
QM/MM studies of reaction mechanisms in biomolecular motors myosin156 (A–D) and DNA polymerase η160 (E). A. The pre-powerstroke state based on the crystal structure of the myosin motor domain complexed with ADP·vanadate (PDB code: 1VOM); B. A computational “hybrid” model, in which the active-site protein residues are restrained to adopt the structure of the pre-powerstroke state, while the rest adopts the post-rigor crystal structure (PDB code: 1FMW). C–D. Free energy profiles computed for the two structural models based on DFTB3/MM string calculations. Note that although the active-site structures are very similar in the two models, the hydrolysis free energy profiles are significantly different. E. Mechanism, transition state structure, and free-energy surfaces for a mechanism with a Mg2+-coordinated hydroxide as the base under 4 different conditions: 2 Mg2+ and deprotonated leaving group, 3 Mg2+ and deprotonated leaving group, 2 Mg2+ and protonated leaving group, 3 Mg2+ and protonated leaving group.
Another unique piece of insight was obtained by studying the hydrolysis energetics in an artificial “hybrid” construct that featured a closed active site in a post-rigor state of the myosin motor domain; this model was established to explicitly probe the coupling between ATP hydrolysis and distal conformational transitions. As shown in Fig. 6A–D, although the hybrid and pre-powerstroke active sites feature essentially the same set of first coordination shell interactions for the lytical water and the γ-phosphate, the computed free energy profiles differ significantly in terms of both the rate-limiting barrier height and the overall exergonicity; both quantities are less favorable by ~ 9 kcal/mol for the hybrid structural model. This result clearly underscores the notion that reaction free energy profiles in enzyme active sites are not solely determined by first-shell interactions. In particular, in the absence of conformational transitions beyond the active site, the reactive moieties and catalytic motifs (e.g., Switch II) in the hybrid model are surrounded by a cluster of water molecules, which are not observed in the pre-powerstroke state. As a result, the active site is less pre-organized in the hybrid model, leading to less favorable reaction energetics. This observation is reminiscent of the results from recent computational analysis of loop closure in triose phosphate isomerase;157 it was found that the proton transfer energetics remained largely similar to the bulk values as far as a few water molecules were trapped in the active site, even with the “lid” loop adopting essentially the fully closed configuration. Evidently, establishing a well pre-organized active site for efficient catalysis requires conformational rearrangements beyond the nearest neighbors of the catalytic motifs, which together with cooperative conformational transitions form the basis of mechanochemical coupling in biomolecular machines.
As a second example, we briefly discuss DNA polymerase η, especially the role of the third Mg2+ ion in nucleotide addition. While energy transduction is not the primary function of DNA and RNA polymerases, they are no doubt sophisticated biomolecular machines in that their functional cycles involve complex and coordinated structural transitions at different spatial and temporal scales; the goal of many mechanistic studies is to establish the driving force and functional significance of these structural transitions.158 For DNA polymerase η, a much debated issue in recent years concerns the role of the third Mg2+ ion identified in time-resolved crystallography studies;149 a closely related question concerns the timing of the deprotonation of the 3’-OH in the terminal base, for which multiple mechanisms have been put forward.159 To shed light onto these issues, we have conducted extensive free energy simulations using DFTB3/MM following model calibration, including microscopic pKa calculations for both model compounds and enzyme active sites.160 By systematically comparing free energy profiles for ten plausible mechanistic models, we found that the lowest activation barrier occurred for a reaction where a Mg2+-coordinated water deprotonates the nucleophilic 3’-OH in concert with the phosphoryl transfer step. The presence of a third Mg2+ in the active site was observed to indeed lower the activation barrier for the water-as-base mechanism, as did protonation of the pyrophosphate leaving group. This mechanistic model, which does not invoke any specific protein residue as the catalytic base to activate 3’-OH, is consistent with a recent study161 that systematically removed potential hydrogen-bonding partners of the 3’-OH; it was observed that no single or combined perturbations eliminated the catalytic activity; i.e., neither the proton acceptor nor the departure path of the nucleophile deprotonation is fixed. These observations supported a model in which the 3’-OH deprotonation does not require a specific general base and it is readily activated by the three Mg2+ ions with flexible proton exit routes.
DFT/MM simulations have also been employed to probe various mechanistic issues in related systems,94,159,162–167 sometimes reaching similar conclusions, such as the key features of ATP/GTP hydrolysis transition states in biomolecular motors162–164,167 and the role of the third Mg2+ ion in DNA polymerase η.165 In other cases, different conclusions were drawn, such as the mechanism of 3’-OH deprotonation in DNA polymerase.159 With substantial differences in the time scale of sampling and general computational accuracy between DFT/MM and DFTB/MM simulations, it remains difficult to resolve discrepancies based solely on computational results; as emphasized in many studies, it is imperative to view the computational results in the context of available experimental data, and only by combining information obtained from experiments with that obtained from simulation can we obtain microscopic answers to questions of chemical reactivity in complex settings.
4. Conclusions and Outlooks
In this review article, we have discussed QM/MM methods and their applications to various bioenergy transduction processes, such as long-range proton transport, fast electron transfers and nucleotide hydrolysis. These applications require balancing computational efficiency and accuracy for the system of interest, making semi-empirical QM/MM models particularly valuable, although calibration and comparison to higher-level QM/MM methods is indispensable. Looking forward, there are several areas that can benefit further efforts to make such QM/MM analyses more quantitative and applicable to increasingly complex problems.
First, further improvements in both semi-empirical and ab initio QM methods, especially for transition metal ions and non-covalent interactions, are of major significance, since many systems in bioenergetics involve metal co-factors and require treating a large number of atoms at the QM level for the description of long-range proton/electron transfers or mechanochemical coupling; further integration with machine learning techniques is likely required to reach quantitative accuracy for these challenging problems.
Second, for the MM region, it is important to further enhance not only the accuracy (e.g., an explicit consideration of electronic polarization) but also complexity that better represents the working environment of the biomolecular machine(s) under investigation. Examples for the latter includes representation of the transmembrane potential, pH gradient and molecular crowding. In other words, the general aim is to develop truly multi-scale computational models that enable the analysis of energy transduction under realistic cellular conditions.
Finally, while providing a better understanding of experimental observations remains a major goal for computational studies, we believe “blind” predictions will play an important role in critically evaluating different QM/MM methodologies, similar to such efforts in the areas of protein structure prediction (CASP, CAPRI) and ligand binding (SAMPLX). As QM/MM calculations become increasingly affordable and standardized, along with advances in high-throughput experiments168 that are able to efficiently generate a large amount of data, the time might be ripe for initiating such efforts in the QM/MM community.
References
- (1).Nicholls DG; Ferguson SJ Bioenergetics, 3rd ed.; Academic Press: New York, 2002. [Google Scholar]
- (2).Roux B, Ed. Molecular Machines; World Scientific: Singapore, 2011. [Google Scholar]
- (3).Bustamante C; Keller D; Oster G The physics of molecular motors. Acc. Chem. Res 2001, 34, 412–420. [DOI] [PubMed] [Google Scholar]
- (4).Astumian RD; Mukherjee S; Warshel A The Physics and Physical Chemistry of Molecular Machines. Phys. Chem. Chem. Phys 2016, 17, 1719–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Stoddart JF Mechanically Interlocked Molecules (MIMs)-Molecular Shuttles, Switches, and Machines (Nobel Lecture). Angew. Chem. Int. Ed 2017, 56, 11094–11125. [DOI] [PubMed] [Google Scholar]
- (6).Christensen AS; Kubar T; Cui Q; Elstner M, Semi-empirical Quantum Mechanical Methods for Non-covalent Interactions for Chemical and Biochemical Applications. Chem. Rev 2016, 116, 5301–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Brunk E; Rothlisberger U Mixed Quantum Mechanical/Molecular Mechanical Molecular Dynamics Simulations of Biological Systems in Ground and Electronically Excited States. Chem. Rev 2015, 115, 6217–6263. [DOI] [PubMed] [Google Scholar]
- (8).Wang W; Cao SQ; Zhu LZ; Huang XH Constructing Markov State Models to elucidate the functional conformational changes of complex biomolecules. WIREs Comput. Mol. Sci 2018, 8, e1343. [Google Scholar]
- (9).Thirumalai D; Hyeon C; Zhuravlev PI; Lorimer GH Symmetry, Rigidity, and Allosteric Signaling: From Monomeric Proteins to Molecular Machines. Chem. Rev 2019, 119, 6788–6821. [DOI] [PubMed] [Google Scholar]
- (10).Sivak DA; Crooks GE Thermodynamic Metrics and Optimal Paths. Phys. Rev. Lett 2012, 108, 190602. [DOI] [PubMed] [Google Scholar]
- (11).Mugnai ML; Hyeon C; Hinczewski M; Thirumalai D Theoretical perspectives on biological machines. Rev. Mod. Phys 2020, 92, 025001. [Google Scholar]
- (12).Warshel A; Levitt M Theoretical Studies of Enzymic Reactions - Dielectric, Electrostatic and Steric Stabilization of Carbonium-Ion in Reaction of Lysozyme. J. Mol. Biol 1976, 103, 227–249. [DOI] [PubMed] [Google Scholar]
- (13).Field MJ; Bash PA; Karplus M A Combined Quantum-Mechanical and Molecular Mechanical Potential for Molecular-Dynamics Simulations. J. Comput. Chem 1990, 11, 700–733. [Google Scholar]
- (14).Lipkowitz KB, Boyd DB, Eds. J. Gao, In Reviews in Computational Chemistry VII; VCH: New York, 1995; p 119. [Google Scholar]
- (15).Hu H; Yang WT Free Energies of Chemical Reactions in Solution and in Enzymes with Ab Initio Quantum Mechanics/Molecular Mechanics Methods. Annu. Rev. Phys. Chem 2008, 59, 573–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Chung LW; Sameera WMC; Ramozzi R; Page AJ; Hatanaka M; Petrova GP; Harris TV; Li X; Ke ZF; Liu FY et al. , The ONIOM Method and Its Applications. Chem. Rev 2015, 115, 5678–5769. [DOI] [PubMed] [Google Scholar]
- (17).Senn HM; Thiel W QM/MM methods for biomolecular systems. Angew. Chem. Int. Ed 2009, 48, 1198–1229. [DOI] [PubMed] [Google Scholar]
- (18).Lonsdale R; Mulholland A QM/MM Modelling of Drug-Metabolizing Enzymes. Curr. Top. Med. Chem 2014, 14, 1339–1347. [DOI] [PubMed] [Google Scholar]
- (19).Cui Q; Pal T; Xie L Perspective: Biomolecular QM/MM Simulations: What are some of the “Burning Issues”? J. Phys. Chem. B 2021, 125, 689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Reuter N; Dejaegere A; Maigret B; Karplus M Frontier bonds in QM/MM methods: A comparison of different approaches. J. Phys. Chem. A 2000, 104, 1720–1735. [Google Scholar]
- (21).Das D; Eurenius KP; Billings EM; Sherwood P; Chatfield DC; Hodoscek M; Brooks BR Optimization of quantum mechanical molecular mechanical partitioning schemes: Gaussian delocalization of molecular mechanical charges and the double link atom method. J. Chem. Phys 2002, 117, 10534–10547. [Google Scholar]
- (22).Gao JL; Amara P; Alhambra C; Field MJ, A generalized hybrid orbital (GHO) method for the treatment of boundary atoms in combined QM/MM calculations. J. Phys. Chem. A 1998, 102, 4714–4721. [Google Scholar]
- (23).Zhang Y Pseudobond ab initio QM/MM approach and its applications to enzyme reactions. Theor. Chem. Acc 2006, 116, 43–50. [Google Scholar]
- (24).Antes I; Thiel W Adjusted connection atoms for combined quantum mechanical and molecular mechanical methods. J. Phys. Chem. A 1999, 103, 9290–9295. [Google Scholar]
- (25).König PH; Hoffmann M; Frauenheim T; Cui Q A critical evaluation of different QM/MM frontier treatments with SCC-DFTB as the QM method. J. Phys. Chem. B 2005, 109, 9082–9095. [DOI] [PubMed] [Google Scholar]
- (26).Vennelakanti V; Nazemi A; Mehmood R; Steeves AH; Kulik HJ Harder, better, faster, stronger: large-scale QM and QM/MM for predictive modeling in enzymes and proteins. Curr. Opin. Struct. Biol 2022, 72, 9–17. [DOI] [PubMed] [Google Scholar]
- (27).Gaus M; Cui Q; Elstner M Density Functional Tight Binding (DFTB): Application to organic and biological molecules. WIREs Comput. Mol. Sci 2014, 4, 49–61. [Google Scholar]
- (28).Bannwarth C; Caldeweyher E; Ehlert S; Hansen A; Pracht P; Seibert J; Spicher S; Grimme S Extended tight-binding quantum chemistry methods. WIREs Comput. Mol. Sci 2020, e01493. [Google Scholar]
- (29).Kulik HJ; Zhang J; Klinman JP; Martínez TJ How Large Should the QM Region Be in QM/MM Calculations? The Case of Catechol O-Methyltransferase. J. Phys. Chem. B 2016, 120, 11381–11394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Mehmood R; Kulik HJ Both Configuration and QM Region Size Matter: Zinc Stability in QM/MM Models of DNA Methyltransferase. J. Chem. Theory Comput 2020, 16, 3121–3134. [DOI] [PubMed] [Google Scholar]
- (31).Jindal G; Warshel A Exploring the Dependence of QM/MM Calculations of Enzyme Catalysis on the Size of the QM Region. J. Phys. Chem. B 2016, 120, 9913–9921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Flaig D; Beer M; Ochsenfeld C Convergence of Electronic Structure with the Size of the QM Region: Example of QM/MM NMR Shieldings. J. Chem. Theory Comput 2012, 8, 2260–2271. [DOI] [PubMed] [Google Scholar]
- (33).Das S; Nam K; Major DT Rapid Convergence of Energy and Free Energy Profiles with Quantum Mechanical Size in Quantum Mechanical–Molecular Mechanical Simulations of Proton Transfer in DNA. J. Chem. Theory Comput 2018, 14, 1695–1705. [DOI] [PubMed] [Google Scholar]
- (34).Demapan D; Kussman J; Ochsenfeld C; Cui Q Factors that Determine the Variation of Equilibrium and Kinetic Properties of QM/MM enzyme simulations: QM region, Conformation and Boundary Condition. J. Chem. Theory Comput 2022, 18, 2530–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Weber C; Cole DJ; O’Regan DD; Payne MC Renormalization of myoglobinligand binding energetics by quantum many-body effects. Proc. Natl. Acad. Sci. U.S.A 2014, 111, 5790–5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Nishimura Y; Nakai H Dcdftbmd: Divide-and-Conquer Density Functional TighBinding Program for Huge-System Quantum Mechanical Molecular Dynamics Simulations. J. Comput. Chem 2019, 40, 1538–1549. [DOI] [PubMed] [Google Scholar]
- (37).Schulz CE; van Gastel M; Pantazis DA; Neese F Converged Structural and Spectroscopic Properties for Refined QM/MM Models of Azurin. Inorg. Chem 2021, 60, 7399–7412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hou G; Zhu X; Elstner M,; Cui Q A modified QM/MM Hamiltonian with the Self-Consistent-Charge Density-Functional-Tight-Binding Theory for highly charged QM regions. J. Chem. Theory Comp 2012, 8, 4293–4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Inakollu VSS; Geerke DP; Rowley CN; Yu H Polarisable force fields: what do they add in biomolecular simulations? Curr. Opin. Struct. Biol 2020, 61, 182–190. [DOI] [PubMed] [Google Scholar]
- (40).Lemkul JA; Huang J; Roux B; MacKerell AD Jr., An Empirical Polarizable Force Field Based on the Classical Drude Oscillator Model: Development History and Recent Applications. Chem. Rev 2016, 116, 4983–5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Jing Z; Liu C; Cheng SY; Qi R; Walker BD; Piquemal J-P; Ren P Polarizable Force Fields for Biomolecular Simulations: Recent Advances and Applications Annual Review of Biophysics. Annu. Rev. Biophys 2019, 48, 371–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Naseem-Khan S; Lagardere L; Narth C; Cisneros GA; Ren P; Gresh N; Piguemal J-P Development of the Quantum-Inspired SIBFA Many-Body Polarizable Force Field: Enabling Condensed-Phase Molecular Dynamics Simulations. J. Chem. Theory Comput 2022, In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Lagardere L; Jolly LH; Lipparini F; Aviat F; Stamm B; Jing ZFF; Harger M; Torabifard H; Cisneros GA; Schnieders MJ; Gresh N; Maday Y; Ren PY; Ponder JW; Piquemal J-P Tinker-HP: a massively parallel molecular dynamics package for multiscale simulations of large complex systems with advanced point dipole polarizable force fields. Chem. Sci 2018, 9, 956–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Deng J; Cui Q Electronic Polarization is Essential for the Stabilization and Dynamics of Buried Ion Pairs in Staphylococcal Nuclease Mutant. J. Am. Chem. Soc 2022, 144, 4594–4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Leontyev IV; Stuchebrukhov AA Accounting for electronic polarization in non-polarizable force fields. Phys. Chem. Chem. Phys 2011, 13, 2613–2626. [DOI] [PubMed] [Google Scholar]
- (46).Duboue-Dijon E; Javanainen M; Delcroix P; Jungwirth P; Martinez-Seara H A practical guide to biologically relevant molecular simulations with charge scaling for electronic polarization. J. Chem. Phys 2020, 153, 050901. [DOI] [PubMed] [Google Scholar]
- (47).Cui K; Yethiraj A; Schmidt JR Influence of Charge Scaling on the Solvation Properties of Ionic Liquid Solutions. J. Phys. Chem. B 2019, 123, 9222–9229. [DOI] [PubMed] [Google Scholar]
- (48).Loco D; Lagardere L; Adjoua O; Piquemal JP Atomistic Polarizable Embeddings: Energy, Dynamics, Spectroscopy, and Reactivity. Acc. Chem. Res 2021, 54, 2812–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Bondanza M; Nottoli M; Cupellini L; Lipparini F; Mennucci B Polarizable embedding QM/MM: the future gold standard for complex (bio)systems? Phys. Chem. Chem. Phys 2020, 22, 14433–14448. [DOI] [PubMed] [Google Scholar]
- (50).Parac M; Grimme S Comparison of Multireference Møller-Plesset Theory and Time-Dependent Methods for the Calculation of Vertical Excitation Energies of Molecules. J. Phys. Chem. A 2002, 106, 6844–6850. [Google Scholar]
- (51).Wanko M; Garavelli M; Bernardi F; Niehaus T; Frauenheim T; Elstner M A global investigation of excited state surfaces within time-dependent density-functional response theory. J. Chem. Phys 2004, 120, 1674–1692. [DOI] [PubMed] [Google Scholar]
- (52).Dreuw A; Head-Gordon M Single-Reference ab Initio Methods for the Calculation of Excited States of Large Molecules. Chem. Rev 2005, 105, 4009–4037. [DOI] [PubMed] [Google Scholar]
- (53).Jacquemin D; Perpète EA; Scuseria GE; Ciofini I; Adamo C TD-DFT Per-formance for the Visible Absorption Spectra of Organic Dyes: Conventional versus Long-Range Hybrids. J. Chem. Theory Comput 2008, 4, 123–135. [DOI] [PubMed] [Google Scholar]
- (54).Bold BM; Sokolov M; Maity S; Wanko M; Dohmen PM; Kranz JJ; Kleinekathöfer U; Höfener S; Elstner M Benchmark and performance of long-range corrected time-dependent density functional tight binding (LC-TD-DFTB) on rhodopsins and light-harvesting complexes. Phys. Chem. Chem. Phys 2020, 22, 10500–10518. [DOI] [PubMed] [Google Scholar]
- (55).Wanko M; Hoffmann M; Strodel P; Koslowski A; Thiel W; Neese F; Frauenheim T; Elstner M Calculating absorption shifts for retinal proteins: Computational challenges. J. Phys. Chem. B 2005, 109, 3606–3615. [DOI] [PubMed] [Google Scholar]
- (56).Kranz JJ; Elstner M; Aradi B; Frauenheim T; Lutsker V; Garcia AD; Niehaus TA Time-Dependent Extension of the Long-Range Corrected Density Functional Based Tight-Binding Method. J. Chem. Theory Comput 2017, 13, 1737–1747. [DOI] [PubMed] [Google Scholar]
- (57).Sokolov M; Bold BM; Kranz JJ; Höfener S; Niehaus TA; Elstner M Analytical Time-Dependent Long-Range Corrected Density Functional Tight Binding (TD-LC-DFTB) Gradients in DFTB+: Implementation and Benchmark for Excited-State Geometries and Transition Energies. J. Chem. Theory Comput 2021, 17, 2266–2282. [DOI] [PubMed] [Google Scholar]
- (58).Wanko M; Hoffmann M; Frahmcke J; Frauenheim T; Elstner M Effect of polarization on the opsin shift in rhodopsins. 2. empirical polarization models for proteins. J. Phys. Chem. B 2008, 112, 11468–11478. [DOI] [PubMed] [Google Scholar]
- (59).Nitzan A Chemical Dynamics in Condensed Phases: Relaxation, Transfer, and Reactions in Condensed Molecular Systems; Oxford University Press: Oxford, UK, 2014. [Google Scholar]
- (60).Swanson JMJ Multiscale kinetic analysis of proteins. Curr. Opin. Struct. Biol 2022, 72, 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Reinhardt CR; Konstantinovsky D; Soudackov AV; Hammes-Schiffer S Kinetic model for reversible radical transfer in ribonucleotide reductase. Proc. Natl. Acad. Sci. U.S.A 2022, 119, e2202022119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Valsson O; Tiwary P; Parrinello M Enhancing Important Fluctuations: Rare Events and Metadynamics from a Conceptual Viewpoint. Annu. Rev. Phys. Chem 2016, 67, 159–184. [DOI] [PubMed] [Google Scholar]
- (63).E W; Vanden-Eijnden E Transition-Path Theory and Path-Finding Algorithms for the Study of Rare Events. Annu. Rev. Phys. Chem 2010, 61, 391–420. [DOI] [PubMed] [Google Scholar]
- (64).Koenig P; Ghosh N; Hoffman M; Elstner M,; Tajkhorshid E; Frauenheim T; Cui Q Towards theoretical analysis of long-range proton transfer kinetics in biomolecular pumps. J. Phys. Chem. A 2006, 110, 548–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Gillet N; Elstner M; Kubař T Coupled-perturbed DFTB-QM/MM metadynamics: Application to proton-coupled electron transfer. J. Chem. Phys 2018, 149, 072328. [DOI] [PubMed] [Google Scholar]
- (66).Maag D; Böser J; Hourahine B; Elstner M; Witek HA; Kubař T Mechanism of proton-coupled electron transfer resolved with QM/MM implementation of coupled-perturbed density-functional tight-binding manuscript submitted 2022, [DOI] [PubMed] [Google Scholar]
- (67).Li CH; Voth GA Using Constrained Density Functional Theory to Track Proton Transfers and to Sample Their Associated Free Energy Surface. J. Chem. Theory Comput 2021, 17, 5759–5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Yagi K; Ito S; Sugita Y Exploring the Minimum-Energy Pathways and Free-Energy Profiles of Enzymatic Reactions with QM/MM Calculations. J. Phys. Chem. B 2021, 125, 4701–4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Kubar T; Elstner M, A hybrid approach to simulation of electron transfer in complex molecular systems. J. Royal Soc. Interf 2013, 10, 20130415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Kubar T; Elstner M, Efficient algorithms for the simulation of non-adiabatic electron transfer in complex molecular systems: application to DNA. Phys. Chem. Chem. Phys 2013, 15, 5794–5813. [DOI] [PubMed] [Google Scholar]
- (71).Kubar T; Woiczikowski PB; Cuniberti G; Elstner M, Efficient calculation of charge-transfer matrix elements for hole transfer in DNA. J. Phys. Chem. B 2008, 112, 7937–7947. [DOI] [PubMed] [Google Scholar]
- (72).Kubas A; Hoffmann F; Heck A; Oberhofer H; Elstner M,; Blumberger J Electronic couplings for molecular charge transfer: Benchmarking CDFT, FODFT, and FODFTB against high-level ab initio calculations. J. Chem. Phys 2014, 140, 104105. [DOI] [PubMed] [Google Scholar]
- (73).Kubas A; Gajdos F; Heck A; Oberhofer H; Elstner M,; Blumberger J Electronic couplings for molecular charge transfer: Benchmarking CDFT, FODFT and FODFTB against high-level ab initio calculations. II. Phys. Chem. Chem. Phys 2015, 17, 14342–14354. [DOI] [PubMed] [Google Scholar]
- (74).Gillet N; Berstis L; Wu XJ; Gajdos F; Heck A; de la Lande A; Blumberger J; Elstner M, Electronic Coupling Calculations for Bridge-Mediated Charge Transfer Using Constrained Density Functional Theory (CDFT) and Effective Hamiltonian Approaches at the Density Functional Theory (DFT) and Fragment-Orbital Density Functional Tight Binding (FODFTB) Level. J. Chem. Theory Comput 2016, 12, 4793–4805. [DOI] [PubMed] [Google Scholar]
- (75).Warshel A Bicycle-pedal model for the first step in the vision process. Nature 1976, 260, 679–683. [DOI] [PubMed] [Google Scholar]
- (76).Warshel A; Hwang JK Simulation of the dynamics of electron transfer reactions in polar solvents: Semiclassical trajectories and dispersed polaron approaches. J. Chem. Phys 1986, 84, 4938–4957. [Google Scholar]
- (77).Creighton S; Hwang JK; Warshel A; Parson WW; Norris J Simulating the dynamics of the primary charge separation process in bacterial photosynthesis. Biochem 1988, 27, 774–781. [Google Scholar]
- (78).Niehaus TA; Heringer D; Torralva B; Frauenheim T Importance of electronic self-consistency in the TDDFT based treatment of nonadiabatic molecular dynamics. Eur. Phys. J. D 2005, 35, 467–477. [Google Scholar]
- (79).Lüdemann G; Solov’yov I; Kubař T; Elstner M Solvent driving force ensures fast formation of a persistent and well-separated radical pair in plant cryptochrome. J. Am. Chem. Soc 2015, 137, 1147–1156. [DOI] [PubMed] [Google Scholar]
- (80).Cui Q Quantum Mechanical Methods in Biochemistry and Biophysics. J. Chem. Phys 2016, 145, 140901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Gao JL; Truhlar DG Quantum mechanical methods for enzyme kinetics. Annu. Rev. Phys. Chem 2002, 53, 467–505. [DOI] [PubMed] [Google Scholar]
- (82).Major DT; Gao J An Integrated Path Integral and Free-Energy Perturbation-Umbrella Sampling Method for Computing Kinetic Isotope Effects of Chemical Reactions in Solution and in Enzymes. J. Chem. Theory Comput 2007, 3, 949–960. [DOI] [PubMed] [Google Scholar]
- (83).Roston D; Cui Q Substrate and Transition State Binding in Alkaline Phosphatase Exhibited by Computational Analysis of Isotope Effects. J. Am. Chem. Soc 2016, 138, 11946–11957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Phatak P; Ghosh N; Yu H; Cui Q; Elstner M Amino acids with an intermolecular proton bond as the proton storage site in bacteriorhodopsin. Proc. Acad. Natl. Sci. U.S.A 2008, 105, 19672–19677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Goyal P; Ghosh N; Phatak P; Clemens M; Gaus M; Elstner M,; Cui Q Proton storage site in bacteriorhodopsin: new insights from QM/MM simulations of microscopic pKa and infrared spectra. J. Am. Chem. Soc 2011, 133, 14981–14997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Baiz CR; Blasiak B; Bredenbeck J; Cho M; Choi JH; Corcelli SA; Dijkstra AG; Feng CJ; Garrett-Roe S; Ge NH; Hanson-Heine MWD; Hirst JD; Jensen TLC; Kwac K; Kubarych KJ; Londergan CH; Maekawa H; Reppert M; Saito S; Roy S; Skinner JL; Stock G; Straub JE; Thielges MC; Tominaga K; Tokmakoff A; Torii H; Wang L; Webb LJ; Zanni MT Vibrational Spectroscopic Map, Vibrational Spectroscopy, and Inter-molecular Interaction. Chem. Rev 2020, 120, 7152–7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Welke K; Watanabe HC; Wolter T; Gaus M; Elstner M QM/MM simulations of vibrational spectra of bacteriorhodopsin and channelrhodopsin-2. Phys. Chem. Chem. Phys 2013, 15, 6651–6659. [DOI] [PubMed] [Google Scholar]
- (88).Wolter T; Welke K; Phatak P; Bondar A-N; Elstner M Excitation energies of a water-bridged twisted retinal structure in the bacteriorhodopsin proton pump: a theoretical investigation. Phys. Chem. Chem. Phys 2013, 15, 12582–12590. [DOI] [PubMed] [Google Scholar]
- (89).Wolter T; Elstner M; Fischer S; Smith JC; Bondar A-N Mechanism by which Untwisting of Retinal Leads to Productive Bacteriorhodopsin Photocycle States. J. Phys. Chem. B 2015, 119, 2229–2240. [DOI] [PubMed] [Google Scholar]
- (90).Maity S; Bold BM; Prajapati JD; Sokolov M; Kubař T; Elstner M; Kleinekathöfer U DFTB/MM Molecular Dynamics Simulations of the FMO Light-Harvesting Complex. J. Phys. Chem. Lett 2020, 11, 8660–8667. [DOI] [PubMed] [Google Scholar]
- (91).Maity S; Daskalakis V; Elstner M; KleinekathÃfer U Multiscale QM/MM molecular dynamics simulations of the trimeric major light-harvesting complex II. Phys. Chem. Chem. Phys 2021, 23, 7407–7417. [DOI] [PubMed] [Google Scholar]
- (92).Noe F; Tkatchenko A; Müller KR; Clementi C Machine Learning for Molecular Simulation. Annu. Rev. Phys. Chem 2020, 71, 361–390. [DOI] [PubMed] [Google Scholar]
- (93).Zhang LF; Wang H; E WN Reinforced dynamics for enhanced sampling in large atomic and molecular systems. J. Chem. Phys 2018, 148, 124113. [DOI] [PubMed] [Google Scholar]
- (94).Berta D; Buigues PJ; Badaoui M; Rosta E Cations in motion: QM/MM studies of the dynamic and electrostatic roles of H+ and Mg2+ ions in enzyme reactions. Curr. Opin. Struct. Biol 2020, 61, 198–206. [DOI] [PubMed] [Google Scholar]
- (95).Shen L; Yang WT Molecular Dynamics Simulations with Quantum Mechanics/Molecular Mechanics and Adaptive Neural Networks. J. Chem. Theory. Comput 2018, 14, 1442–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Rizzi A; Carloni P; Parrinello M Targeted Free Energy Perturbation Revisited: Accurate Free Energies from Mapped Reference Potentials. J. Phys. Chem. Lett 2021, 12, 9449–9454. [DOI] [PubMed] [Google Scholar]
- (97).Barros EP; Ries B; Boselt L; Champion C; Riniker S Recent developments in multiscale free energy simulations. Curr. Opin. Struct. Biol 2022, 72, 55–62. [DOI] [PubMed] [Google Scholar]
- (98).Krämer M; Dohmen PM; Xie WW; Holub D; Christensen AS; Elstner M Charge and Exciton Transfer Simulations Using Machine-Learned Hamiltonians. J. Chem. Theory Comput 2021, 16, 4061–4070. [DOI] [PubMed] [Google Scholar]
- (99).Chen MS; Zuehlsdorff TJ; Morawietz T; Isborn CM; Markland TE Ex-ploiting Machine Learning to Efficiently Predict Multidimensional Optical Spectra in Complex Environments. J. Phys. Chem. Lett 2020, 11, 7559–7568. [DOI] [PubMed] [Google Scholar]
- (100).Borshchevskiy V; Kovalev K; Round E; Efremov R; Astashkin R; Bourenkov G; Bratanov D; Balandin T; Chizhov I; Baeken C; Gushchin I; Kuzmin A; Alekseev A; Rogachev A; Willbold D; Engelhard M; Bamberg E; Buldt G; Gordeliy V True-atomic-resolution insights into the structure and functional role of linear chains and low-barrier hydrogen bonds in proteins. Nat. Struct. Mol. Biol 2022, 29, 440. [DOI] [PubMed] [Google Scholar]
- (101).Liang RB; Swanson JMJ; Peng YX; Wikström M; Voth GA Multiscale simulations reveal key features of the proton-pumping mechanism in cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A 2016, 113, 7420–7425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Cai XH; Haider K; Lu JX; Radik S; Son CY; Cui Q; Gunner M Network Analysis of a Proposed Exit Pathway for Protons to the P-side of Cytochrome c Oxidase. BBA Bioener 2018, 1859, 997–1005. [DOI] [PubMed] [Google Scholar]
- (103).Gunner MR; Amin M; Zhu X; Lu J Molecular mechanisms for generating transmembrane proton gradients. Biochim. Biophys. Acta - Bioeng 2013, 1827, 892–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Goyal P; Yang S; Cui Q Microscopic basis for kinetic gating in Cytochrome c oxidase: insights from QM/MM analysis. Chem. Sci 2015, 6, 826–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Kaila VRI Resolving Chemical Dynamics in Biological Energy Conversion: Long-Range Proton-Coupled Electron Transfer in Respiratory Complex I. Acc. Chem. Res 2021, 54, 4462–4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Pisliakov AV; Sharma PK; Chu ZT; Haranczyk M; Warshel A Electrostatic basis for the unidirectionality of the primary proton transfer in cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A 2008, 105, 7726–7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Son CY; Yethiraj A; Cui Q Cavity Hydration Dynamics in Cytochrome c Oxidase and Functional Implications. Proc. Natl. Acad. Sci. U.S.A 2017, 114, E8830–E8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Muhlbauer ME; Saura P; Nuber F; Di Luca A,; Friedrich T; Kaila VRI Water-Gated Proton Transfer Dynamics in Respiratory Complex I. J. Am. Chem. Soc 2020, 142, 13718–13728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Riccardi D; Schaefer P; Cui Q pKa calculations in solution and proteins with QM/MM free energy perturbation simulations. J. Phys. Chem. B 2005, 109, 17715–17733. [DOI] [PubMed] [Google Scholar]
- (110).Zheng YQ; Cui Q Microscopic Mechanisms that Govern the Titration Response and pKa Values of Buried Residues in Staphylococcal Nuclease Mutants. Proteins: Struct., Funct., & Bioinf 2017, 85, 268–281. [DOI] [PubMed] [Google Scholar]
- (111).Ghosh N; Prat-Resina X; Gunner M; Cui Q Microscopic pKa analysis of Glu286 in Cytochrome c Oxidase (Rhodobacter sphaeroides): Toward a Calibrated Molecular Model. Biochem 2009, 48, 2468–2485. [DOI] [PubMed] [Google Scholar]
- (112).Goyal P; Lu J; Yang S; Gunner MR; Cui Q Changing hydration level in an internal cavity modulates the proton affinity of a key glutamate in Cytochrome c Oxidase. Proc. Natl. Acad. Sci. U.S.A 2013, 110, 18886–18891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Lee S; Liang RB; Voth GA; Swanson JMJ Computationally Efficient Multiscale Reactive Molecular Dynamics to Describe Amino Acid Deprotonation in Proteins. J. Chem. Theory Comput 2016, 12, 879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Maag D; Mast T; Elstner M; Cui Q; Kubar T O to bR Transition in Bacteriorhodopsin Occurs Through a Proton Hole Mechanism. Proc. Natl. Acad. Sci. U.S.A 2021, 118, e2024803118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Rouhani S; Cartailler JP; Facciotti MT; Walian P; Needleman R; Lanyi JK; Glaeser RM; Luecke H Crystal structure of the D85S mutant of bacteriorhodopsin: Model of an O-like photocycle intermediate. J. Mol. Biol 2001, 313, 615–628. [DOI] [PubMed] [Google Scholar]
- (116).Song Y; Gunner MR Halorhodopsin pumps Cl− and bacteriorhodopsin pumps protons by a common mechanism that uses conserved electrostatic interactions. Proc. Natl. Acad. Sci. U.S.A 2014, 111, 16377–16382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Garczarek F; Gerwert K Functional waters in intraprotein proton transfer monitored by FTIR difference spectroscopy. Nature 2006, 439, 109–112. [DOI] [PubMed] [Google Scholar]
- (118).Freier E; Wolf S; Gerwert K Proton transfer via a transient linear water-molecule chain in a membrane protein. Proc. Natl. Acad. Sci. U.S.A 2011, 108, 11435–11439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Tripathi R; Forbert H; Marx D Settling the Long-Standing Debate on the Proton Storage Site of the Prototype Light-Driven Proton Pump Bacteriorhodopsin. J. Phys. Chem. B 2019, 123, 9598–9608. [DOI] [PubMed] [Google Scholar]
- (120).Wolf S; Freier E; Cui Q; Gerwert K Infrared spectral marker bands characterizing a transient water wire inside a hydrophobic membrane protein. J. Chem. Phys 2015, 141, 22D524. [DOI] [PubMed] [Google Scholar]
- (121).Wu Q; Van Voorhis T Direct Calculation of Electron Transfer Parameters through Constrained Density Functional Theory. J. Phys. Chem. A 2006, 110, 9212–9218. [DOI] [PubMed] [Google Scholar]
- (122).Wu Q; Van Voorhis T Constrained Density Functional Theory and Its Application in Long-Range Electron Transfer. J. Chem. Theory Comput 2006, 2, 765–774. [DOI] [PubMed] [Google Scholar]
- (123).Wu Q; Van Voorhis T Direct optimization method to study constrained systems within density-functional theory. Phys. Rev. A 2005, 72, 024502. [Google Scholar]
- (124).Troisi A The speed limit for sequential charge hopping in molecular materials. Org. Electron 2011, 12, 1988–1991. [Google Scholar]
- (125).LeBard DN; Martin DR; Lin S; Woodbury NW; Matyushov DV Protein dynamics to optimize and control bacterial photosynthesis. Chem. Sci 2013, 4, 4127–4136. [Google Scholar]
- (126).Blumberger J Recent Advances in the Theory and Molecular Simulation of Biological Electron Transfer Reactions. Chem. Rev 2015, 115, 11191–11238. [DOI] [PubMed] [Google Scholar]
- (127).van Wonderen JH; Hall CR; Jiang X; Adamczyk K; Carof A; Heisler I; Piper SEH; Clarke TA; Watmough NJ; Sazanovich IV; Towrie M; Meech SR; Blumberger J; Butt JN Ultrafast Light-Driven Electron Transfer in a Ru(II)tris(bipyridine)-Labeled Multiheme Cytochrome. J. Am. Chem. Soc 2019, 141, 15190–15200. [DOI] [PubMed] [Google Scholar]
- (128).Matyushov DV Protein electron transfer: Dynamics and statistics. J. Chem. Phys 2013, 139, 025102. [DOI] [PubMed] [Google Scholar]
- (129).Lu Y; Kundu M; Zhong D Effects of nonequilibrium fluctuations on ultrafast short-range electron transfer dynamics. Nat. Comm 2020, 11, 2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Dahl PJ; Yi SM; Gu Y; Acharya A; Shipps C; Neu J; O’Brien JP; Morzan UN; Chaudhuri S; Guberman-Pfeffer MJ; Vu D; Yalcin SE; Batista VS; Malvankar NSA 300-fold conductivity increase in microbial cytochrome nanowires due to temperature-induced restructuring of hydrogen bonding networks. Sci. Adv 2022, 8, eabm7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Mai S; Menger MFSJ; Marazzi M; Stolba DL; Monari A; González L Competing ultrafast photoinduced electron transfer and intersystem crossing of [Re(CO)3(Dmp)(His124)(Trp122)]+ in Pseudomonas aeruginosa azurin: a nonadiabatic dynamics study. Theor. Chem. Acc 2020, 139, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Woiczikowski PB; Steinbrecher T; Kubař T; Elstner M Nonadiabatic QM/MM simulations of fast charge transfer in Escherichia coli DNA photolyase. J. Phys. Chem. B 2011, 115, 9846–9863. [DOI] [PubMed] [Google Scholar]
- (133).Holub D; Lamparter T; Elstner M; Gillet N Biological relevance of charge transfer branching pathways in photolyases. Phys. Chem. Chem. Phys 2019, 21, 17072–17081. [DOI] [PubMed] [Google Scholar]
- (134).Hoffmann M; Wanko M; Strodel P; König PH; Frauenheim T; Schulten K; Thiel W; Tajkhorshid E; Elstner M Color Tuning in Rhodopsins: The Mechanism for the Spectral Shift between Bacteriorhodopsin and Sensory Rhodopsin II. J. Am. Chem. Soc 2006, 128, 10808–10818. [DOI] [PubMed] [Google Scholar]
- (135).Guo Y; Beyle FE; Bold BM; Watanabe HC; Koslowski A; Thiel W; Hegemann P; Marazzi M; Elstner M Active site structure and absorption spectrum of channelrhodopsin-2 wild-type and C128T mutant. Chem. Sci 2016, 7, 3879–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Pang Z; Sokolov M; Kubar T; Elstner M Unravelling the mechanism of glucose binding in a protein-based fluorescence probe: molecular dynamics simulation with a tailor-made charge model. Phys. Chem. Chem. Phys 2022, 24, 2441–2453. [DOI] [PubMed] [Google Scholar]
- (137).Richter R; Foerster JM; Schelter I; Kümmel S Self-interaction correction, electrostatic, and structural influences on time-dependent density functional theory excitations of bacteriochlorophylls from the light-harvesting complex 2. J. Chem. Phys 2020, 153, 2441–2453. [DOI] [PubMed] [Google Scholar]
- (138).List NH; Curutchet C; Knecht S; Mennucci B; Kongsted J Toward Reliable Prediction of the Energy Ladder in Multichromophoric Systems: A Benchmark Study on the FMO Light-Harvesting Complex. J. Chem. Theory Comput 2013, 9, 4928–4938. [DOI] [PubMed] [Google Scholar]
- (139).Lahav Y; Noy D; Schapiro I Spectral tuning of chlorophylls in proteins: Bacteriorhodopsin vs. ring deformation. Phys. Chem. Chem. Phys 2021, 23, 6544–6551. [DOI] [PubMed] [Google Scholar]
- (140).Sirohiwal A; Pantazis DA Electrostatic profiling of photosynthetic pigments: implications for directed spectral tuning. Phys. Chem. Chem. Phys 2021, 23, 24677–24684. [DOI] [PubMed] [Google Scholar]
- (141).Warshel A; Chu ZT Nature of the Surface Crossing Process in Bacteriorhodopsin: Computer Simulations of the Quantum Dynamics of the Primary Photochemical Event. J. Phys. Chem. B 2001, 105, 9857–9871. [Google Scholar]
- (142).Wanko M; Hoffmann M; Frauenheim T; Elstner M Effect of polarization on the opsin shift in rhodopsins. 1. A Combined QM/QM/MM Model for Bacteriorhodopsin and Pharaonis Sensory Rhodopsin II. J. Phys. Chem. B 2008, 112, 11462–11467. [DOI] [PubMed] [Google Scholar]
- (143).Fr”ahmke J; Wanko M; Phatak P; Mroginski A; Elstner M The Protonation State of Glu181 in Rhodopsin Revisited: Interpretation of Experimental Data on the Basis of QM/MM Calculations. J. Phys. Chem. B 2010, 114, 11338–11352. [DOI] [PubMed] [Google Scholar]
- (144).Loco D; Polack E; Capresa S; Lagardere L; Lipparini F; Piquemal JP; Menucci B A QM/MM Approach Using the AMOEBA Polarizable Embedding: From Ground State Energies to Electronic Excitations. J. Chem. Theory Comput 2016, 12, 36543661. [DOI] [PubMed] [Google Scholar]
- (145).Cignoni E; Cupellini L; Menucci B A fast method for electronic couplings in embedded multichromophoric systems. J. Phys.: Condens. Matt 2022, 34, 304004. [DOI] [PubMed] [Google Scholar]
- (146).Hill TL Free energy transduction in biology; Academic Press: New York, 1977. [Google Scholar]
- (147).Samara NL; Yang W Cation traffcking propels RNA hydrolysis. Nat. Struct. Mol. Biol 2018, 25, 715–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Wu WJ; Yang W; Tsai MD How DNA polymerases catalyze replication and repair with contrasting fidelity. Nat. Rev. Chem 2017, 1, 0068. [Google Scholar]
- (149).Gao Y; Yang W Capture of a third Mg2+ is essential for catalyzing DNA synthesis. Science 2016, 352, 1334–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Raper AT; Reed AJ; Suo ZC Kinetic Mechanism of DNA Polymerases: Contributions of Conformational Dynamics and a Third Divalent Metal Ion. Chem. Rev 2018, 118, 6000–6025. [DOI] [PubMed] [Google Scholar]
- (151).Manigrasso J; De Vivo M; Palermo G Controlled Trafficking of Multiple and Diverse Cations Prompts Nucleic Acid Hydrolysis. ACS Cata 2021, 11, 8786–8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Roston D; Lu X; Fang D; Demapan D; Cui Q Analysis of Phosphoryl Transfer Enzymes with QM/MM Free Energy Simulations. Methods in Enzymol 2018, 607, 53–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Lassila JK; Zalatan JG; Herschlag D Biological phosphoryl transfer reactions: Understanding mechanism and catalysis. Annu. Rev. Biochem 2011, 80, 669–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Kamerlin SCL; Sharma PK; Prasad RB; Warshel A Why nature really chose phosphate. Q. Rev. Biophys 2013, 46, 1–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Sweeney HL; Houdusse A Structural and Functional Insights into the Myosin Motor Mechanism. Annu. Rev. Biophys 2010, 39, 539–557. [DOI] [PubMed] [Google Scholar]
- (156).Lu X; Ovchinnikov V; Roston DR; Demapan D; Cui Q Regulation and Plasticity of Catalysis in Enzymes: Insights from Analysis of Mechanochemical Coupling in Myosin. Biochem 2017, 56, 1482–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Liao QH; Kulkarni Y; Sengupta U; Petrovic D; Mulholland AJ; van der Kamp MW,; Strodel B; Kamerlin SCL Loop Motion in Triosephosphate Isomerase Is Not a Simple Open and Shut Case. J. Am. Chem. Soc 2018, 140, 15889–15903. [DOI] [PubMed] [Google Scholar]
- (158).Unarta IC; Zhu LZ; Tse CKM; Cheung PPH; Yu J; Huang XH Molecular mechanisms of RNA polymerase II transcription elongation elucidated by kinetic network models. Curr. Opin. Struct. Biol 2018, 49, 54–62. [DOI] [PubMed] [Google Scholar]
- (159).Genna V; Vidossich P; Ippoliti E; Carloni P; De Vivo M A Self-Activated Mechanism for Nucleic Acid Polymerization Catalyzed by DNA/RNA Polymerases. J. Am. Chem. Soc 2016, 138, 14592–14598. [DOI] [PubMed] [Google Scholar]
- (160).Roston D; Demapan D; Cui Q Extensive Free Energy Simulations Identify Water as the Base in Nucleotide Addition by DNA Polymerase. Proc. Natl. Acad. Sci. U.S.A 2019, 116, 25048–25056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Gregory MT; Gao Y; Cui Q; Yang W Multiple deprotonation paths of the nucleophile 3’-OH in the DNA synthesis reaction. Proc. Natl. Acad. Sci. U.S.A 2021, 118, e2103990118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).McCullagh M; Saunders MG; Voth GA Unraveling the mystery of ATP hydrolysis in actin filaments. J. Am. Chem. Soc 2014, 136, 13053–13058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Sun R; Sode O; Dama JF; Voth GA Simulating Protein Mediated Hydrolysis of ATP and Other Nucleoside Triphosphates by Combining QM/MM Molecular Dynamics with Advances in Metadynamics. J. Chem. Theory Comput 2017, 13, 2332–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Tripathi R; Glaves R; Marx D The GTPase hGBP1 converts GTP to GMP in two steps via proton shuttle mechanisms. Chem. Sci 2017, 8, 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Stevens DR; Hammes-Schiffer S Exploring the Role of the Third Active Site Metal Ion in DNA Polymerase η with QM/MM Free Energy Simulations. J. Am. Chem. Soc 2018, 140, 8965–8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Donati E; Genna V; De Vivo M Recruiting Mechanism and Functional Role of a Third Metal Ion in the Enzymatic Activity of 5’ Structure-Specific Nucleases. J. Am. Chem. Soc 2020, 142, 2823–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Priess M; Goddeke H; Groenhof G; Schaefer LV Molecular Mechanism of ATP Hydrolysis in an ABC Transporter. ACS Central Sci 2018, 4, 1334–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Mokhtari DA; Appel MJ; Fordyce PM; Herschlag D, High throughput and quantitative enzymology in the genomic era. Curr. Opin. Struct. Biol 2021, 71, 259–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Gómez-Flores CL; Maag D; Kansari M; Vuong V-Q; Irle S; Gräter F; Kubař T; Elstner M Accurate Free Energies for Complex Condensed-Phase Reactions Using an Artificial Neural Network Corrected DFTB/MM Methodology. J. Chem. Theory Comput 2022, 18, 1213–1226. [DOI] [PubMed] [Google Scholar]