

Abstract
Biomolecular condensates are proposed to cause diseases such as cancer and neurodegeneration by concentrating proteins at abnormal subcellular loci. Imaging-based compound screens have been used to identify small molecules reversing or promoting biomolecular condensates. However, limitations of conventional imaging-based methods restrict the screening scale. Here we used a graph convolutional network (GCN)-based computational approach and identified small molecule candidates that reduce the nuclear LLPS of TDP-43, an essential protein that phase transits in neurodegenerative diseases. We demonstrated that the GCN-based deep learning algorithm is suitable for spatial information extraction from the molecular graph. Thus, it is a promising method to identify small molecule candidates with novel scaffolds. Furthermore, we validated that these candidates do not affect the normal splicing function of TDP-43. Taken together, a combination of an imaging-based screen and a GCN-based deep learning method dramatically improves the speed and accuracy of the compound screen for biomolecular condensates.
Graphical Abstract
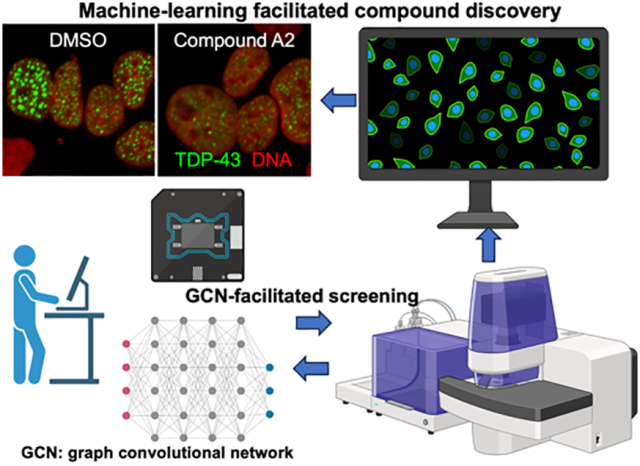
Introduction
Proteins form biomolecular condensates through a biophysical mechanism named liquid-liquid phase separation (LLPS). Biomolecular condensates are believed as a cause of cancer or neurodegeneration by concentrating proteins at abnormal subcellular loci to form large protein assemblies at the micrometer scale. They have been attractive drug targets since the initial discovery of protein LLPS(or called de-mixing) in 20091, because compelling evidence suggests their essential role in causing major diseases such as cancer and neurodegeneration. Top examples are chimeric transcription factors that cause cancer and RNA-binding proteins that form aggregates in neurodegeneration diseases. Ewing Sarcoma is caused by fusion of EWS low complexity domain and the DNA binding motif of FLI12. Various leukemia can be driven by the fusion of NUP98 and HOX transcription factors3. Nuclear RNA binding proteins TDP-43 and FUS forms protein aggregates in neurons in neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD)4. However, although the interest in exploring condensate-modifying compounds has been growing in drug discovery, these proteins are often described as “undruggable” because of lacking measurable enzymatic activity5. The lack of quantifiable parameters also leads to difficulties in high throughput screens. For example, the LLPS of TDP-43 or FUS is a transition from a diffused phase to a concentrated phase6, 7. Thus, there are not many parameters for distinguishing the concentrated phase from the diffused phase other than an increase in protein density. Therefore, new screening strategies will promote the efficiency of identifying new compounds for modulating LLPS.
For compound screening experiments, cellular models of LLPS mimic diseases in vivo better than in vitro models that require purified proteins and specific biophysical conditions. However, some widely used methods that induce LLPS in cells cannot be used for compound screens because they use toxic compounds, such as stress granules induced by sodium arsenite, or other extreme conditions, such as 42°C heat shock or severe osmotic change. Secondary effects from these stress inducers cannot be ruled out8. Thus, we cannot conclude that cellular phenotypes observed on the screen are solely caused by the compounds. The TDP-43 anisosome, a unique LLPS phenomenon in living cells, overcomes most of these problems. RNA-binding deficient TDP-43 faithfully exhibit LLPS property in almost every cell type examined7. When TDP-43 loses its RNA-binding activity caused by post-translational acetylation or disease-causing mutations, it forms intranuclear liquid droplets exhibiting a core-shell structure, named anisosomes (“uneven body” in Greek). The anisosomal core enriches HSP70 chaperones. The anisosome-like structures have also been observed in animals in diseases, providing its relevance to disease7. TDP-43 forms protein aggregates in over 90% of ALS cases, over 40% of FTD cases9. TDP-43 pathology correlates with cognitive decline in Alzheimer’s Disease10. Intranuclear TDP-43 anisosomes are recognized as an LLPS state proceeding the cytoplasmic aggregates observed in the postmortem tissue at the end stage of neurodegeneration7. Thus, developing an antagonist for TDP-43 anisosomes is a promising drug development strategy for ALS and FTD.
We designed an imaging-based compound screening system and performed a classic screening experiment for 7000 FDA-approved compounds based on the size and number of TDP-43-containing anisosomes in DLD1 cells. However, we cannot perform the same screen for diverse compound libraries, which contains 170,000 compounds in total, because it exceeds the capacity of cell culture, image collection, and data analysis. To overcome this problem, we employed a graph convolutional network (GCN)-based computational strategy to virtually identify candidate compounds. GCN uses molecular graphs composed of nodes and edges to extract chemical information and then map the obtained spatial information to generate accurate molecular classifications.11, 12 Compared to molecular dynamics (MD) or quantum mechanics (QM) based methods, the most important advantage of deep learning based method lies in the fact that the required computation cost can be substantially reduced.13 This novel method enabled us to rapidly screen a large number of compounds that are from NCATS in-house diverse library collection. Among the 170,000(give the number) screened compounds, GCN identified ~1100 candidates (0.65%) that were predicted to show biological activity of inhibiting anisosomes. We validated the top five candidates and found that two of them reduce the number and size of TDP-43 anisosomes. This demonstrated that GCN-based machine learning process is sufficient to facilitate identification of new compounds of similar biological activity that modulates LLPS in living cells. These compounds might lead to development of new small molecules for neurodegenerative diseases by antagonizing LLPS of TDP-43. In addition, MD simulations with metadynamics analysis were conducted to predict insights in predicting the interaction conformations of these promising candidate compounds within possible targets. Our findings show the high applicability and promising prospect of graph-based deep learning methods in neuroscience-related drug discovery. The investigations conducted in this study as well as the combinational methodology will be highly instructive for future studies.
Results
Fluorescence imaging-based compound screen identifies compounds antagonizing anisosome formation in cells
In the RNA-unbound state, TDP-43 form nuclear condensates named “anisosomes” through liquid-liquid phase separation7. Anisosomes (Figure 1A) are hypothesized as the cellular phenotype proceeding aggregation. We previously found that VER155008, an ATP competitive antagonist that inhibits the ATPase activity of HSP70, modulates anisosomes in vivo (Figure 1A)7. We sought to find more small molecules that modulate the size or the number of anisosomes in living cells. Thus, we conducted a fluorescence imaging-based screening to identify compounds that can reduce the number of anisosomes (Figure 1B) and performed the first screening experiment with a library of ~5000 NCATS in-house compounds14. The TDP-43 anisosomes were induced by adding doxycycline to a clone of the DLD1 cell line. The single clone engineered DLD1 cell line allows low dose doxycycline (1ug/mL) to induce the expression of carboxyl-terminal GFP-tagged acetylation-mimic TDP-43 (TDP-432KQ-clover), which abolishes RNA-binding. We chose the DLD1 cell line for our screening because of its genome integrity. Phenotypes among cells are consistent because of the genome stability. RNA-binding deficient TDP-43 forms anisosomes highlighted by the white dot because their intensity is much higher than the diffused signal (Figure 1C). Compounds that reduced the number of dots per cell by 50% or more were selected. To prepare the initial training set for computational screening, we initially selected the active compounds from the first experimental results, based on the metrics of efficacy and IC50 values; the organized data is provided in our GitHub page for reference. However, it is worth noting that this pipeline required tremendous effort, data storage, and computing time. If we had planned to use the fluorescence imaging-based screening method for LLPS to screen for ~200,000 novel compounds, we would need 6 months of uninterrupted machine time for the imaging and data analyses, and culture 2 billion cells (~500 10cm plates), assuming everything works successfully as planned. This workload is beyond our capacity. Thus, we need to come up with this novel virtual screening strategy to identify promising candidates for imaging-based analyses.
Figure 1. Imaging-based screening identifies small molecules modulating TDP-43 anisosomes.
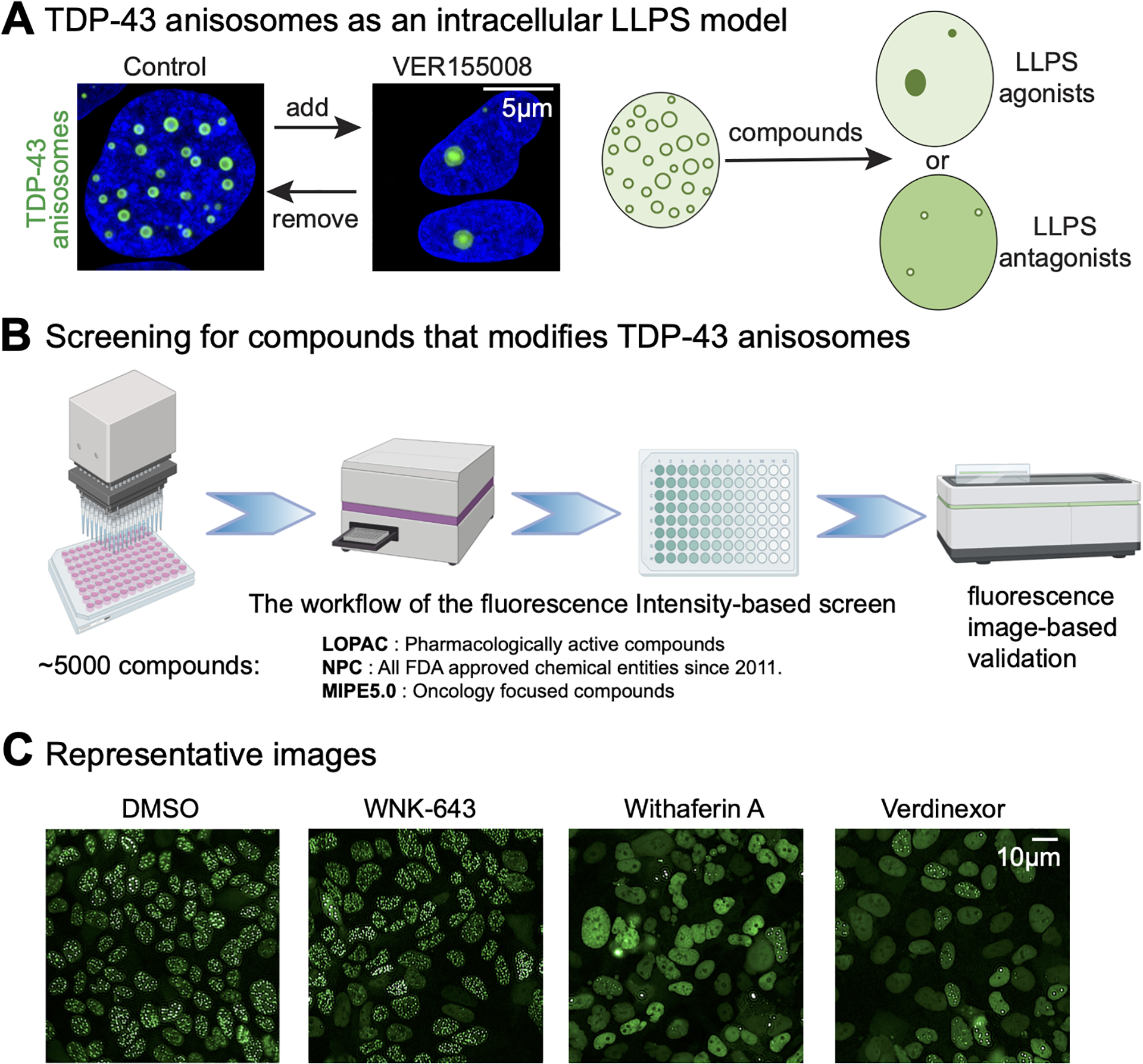
(A) HSP70 inhibitor can modulate TDP-43 anisosomes, which encourage us to find more small molecules that can modulate anisosomes. (B) The workflow of conventional imaging-based compound screen. (C) Identified compounds can modulate TDP-43 anisosomes. Anisosomes are highlighted with white dots in the nucleus. Compared to DMSO, WNK-643 treated cells show increased number of anisosomes, while Withaferin A and Verdinexor decrease the number of anisosomes.
GCN model validation
In this study, we proposed and verified a high-performance GCN screening model that is suitable for different kinds of structural assignments. It is worth pointing out that for large-scale virtual screening tasks, the cost of conventional QM computation methods is very expensive; and for structure-based methods, they are difficult to be applied without target information, they are difficult to be applied. It is notable that the inhibitory effects of the small molecules are mainly dependent on their structural flexibility and functional groups; GCN can encode the molecular graphs of both active and inactive compounds, and furthermore extract the common features in structure. We trained our model upon the collected experimental data. Before model development, the similarity analysis of the active compounds was performed using Ward method to ensure there is low risk of overfitting; and from the results shown in Figure S1 (more details can be found in our GitHub page), we noticed that the molecular diversity of the active compounds is considerable. In this study, upon the task for identification of anisosome modulators through virtual screening, our proposed model performs robustly with a validation accuracy of higher than 0.90 (its mechanism is presented in Figure 2, and the screening workflow is described in Figure 3, more technical evaluations are provided in our GitHub page for reference). It was noting that the proposed GCN model performs better than other machine learning based methods, especially upon the few-shot learning cases, such a benchmarking result is consistent with the conclusion of our previous study focusing on COVID19 inhibitors screening using endocytosis assay11. This training model was further applied in the screening of ~170,000 compounds from NCATS diverse chemical libraries that had never been screened in anisosome assays, we identified ~1100 compounds with top ranking predicted activity and tested in fluorescence imaging-based assay. There are ~300 compounds confirmed to be active, indicating that such a tool could be adopted as a powerful complementary approach for future experimental screening. Among these active compounds, 5 candidates (A1 to A5) displayed high potency, further indicating that such a tool will become a powerful complementary for future experiment work. We had conducted a comparative study with respect to other popular machine learning (ML) based algorithms to demonstrate that the GCN architecture is highly suitable for few-shot learning cases, especially in which the ratio of active to inactive compounds is low11. However, it is worth noting that ML based methods are robust for overall molecular features extraction, thus can be employed for a secondary screening of GCN results to save experimental cost.
Figure 2. The mechanism of GCN architecture for drug screenings.
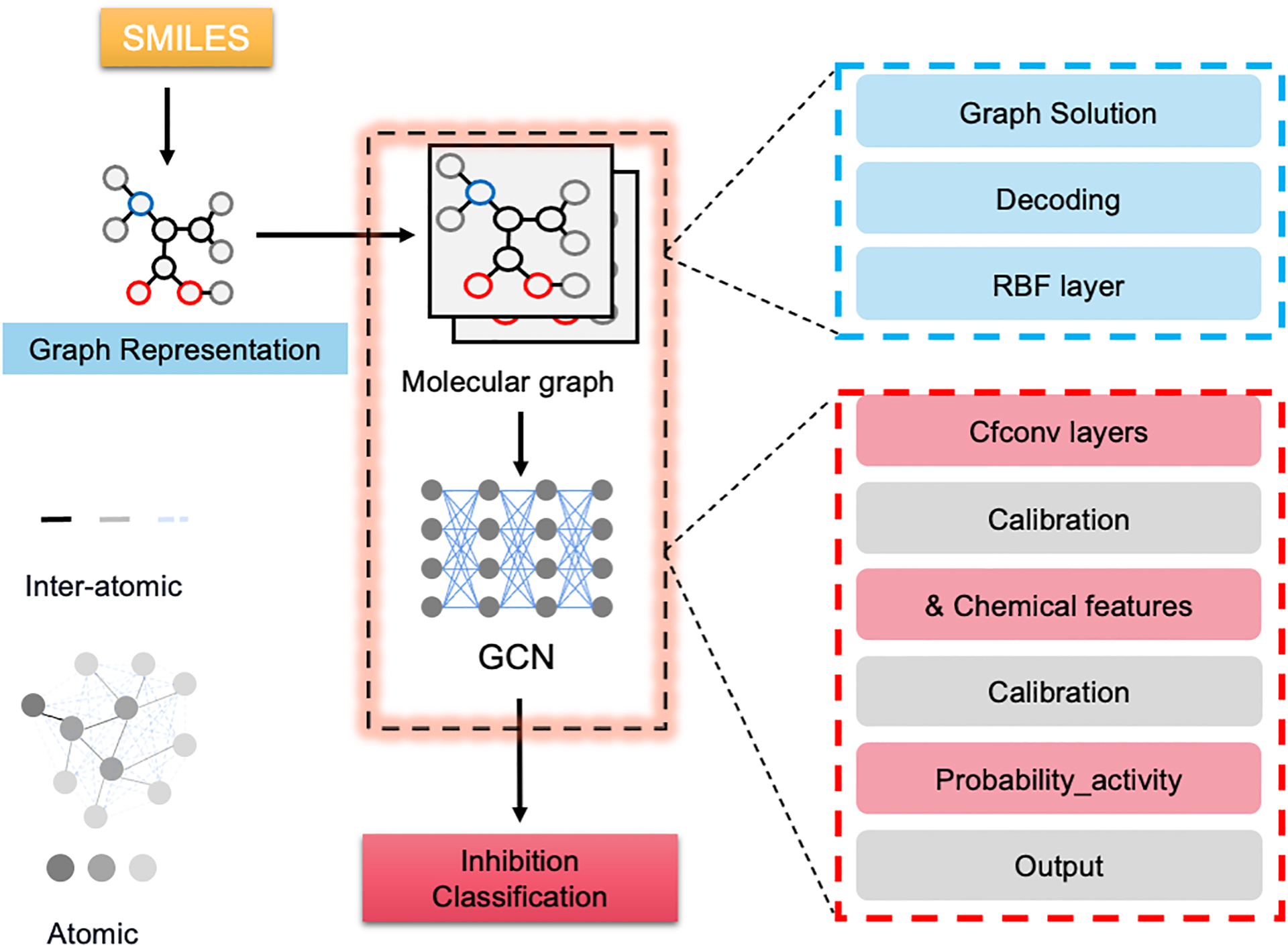
The input simplified molecular-input line-entry system (SMILES) string is translated into molecular graphs that are composed of nodes and edges representing atomic and inter-atomic features, respectively; the structural information is extracted via encoding the input molecular graph with assistance of chemical descriptors, and the continuous-filter convolutions (CFCONV) layers are included for inter-atomic information processing.
Figure 3. The workflow of molecular graph based screening model upon anisosome related drug discovery task.
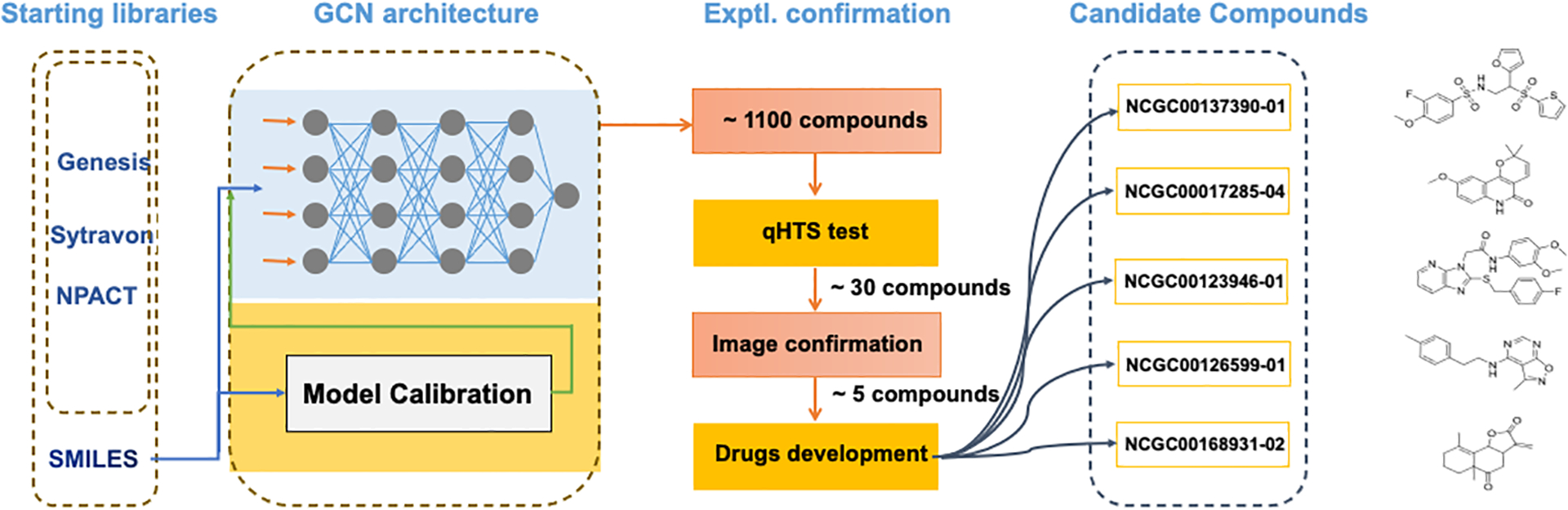
The original model was trained on collected experimental data, and the well-trained model was applied to screen NIH in-house libraries (Genesis, Sytravon, NPACT). The top ranked candidates are experimentally validated by qHTS, and the identified potent compounds are further verified by image-based assay for activity confirmation.
Identification of two novel candidates
Using imaging analysis, we have demonstrated that, among the 5 potent candidates (Figure 3), compound A2 and A5 show strong biological activity by inhibiting the TDP-43 anisosome formation. They not only work in the DLD1 cells (an epithelial cell line), but also in U2OS cells (human sarcoma cells), as shown in Figure 4A.
Figure 4. Biological validation of compound A1–A5.
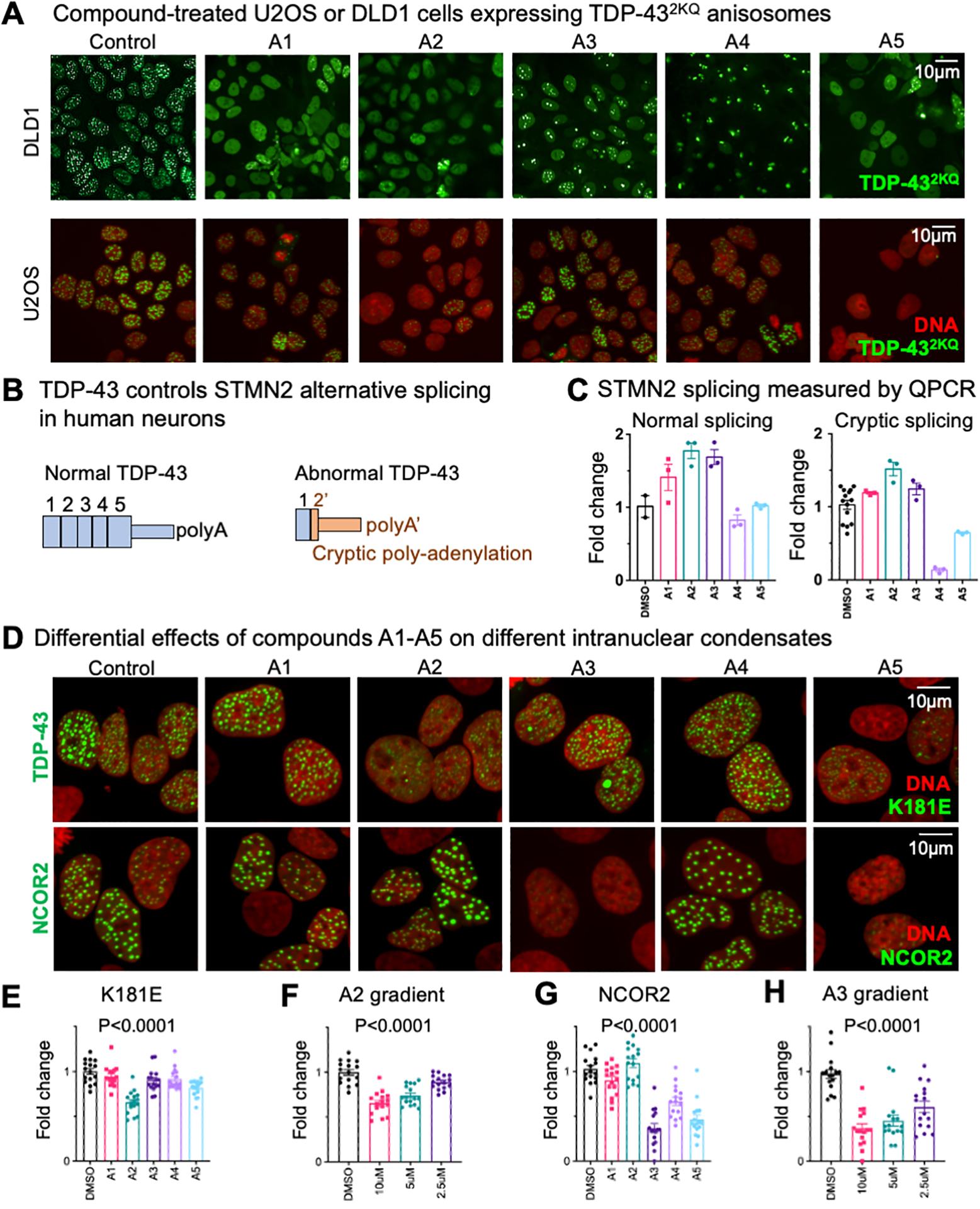
(A) DLD1 and U2OS cells expressing TDP-43 anisosomes were treated with 10uM compounds A1~A5. (B) A scheme of how STMN2 mRNA splicing patterns regulated by TDP-43. (C) Normal and Cryptic spliced mRNA quantified by real time PCR. (D) U2OS cells expressing ALS-causing TDP-43K181E or NCOR2 anisosomes treated with 10uM A2 and A5 compounds. Statistical analyses of demixing cells treated by A1–A5 are shown in panels E-H. (E) shows demixing of TDP-43K181E treated by 10uM A1–A5. (F) shows TDP-43K181E demixing under different concentration of compound A2. (G) shows demixing of NCOR2 treated by 10uM A1–A5. (H) shows NCOR2 demixing under different concentration of compound A3. Statistical analysis: one-way ANOVA.
For therapeutic purpose, the anisosome inhibitors should not inhibit the normal function of TDP-43. The primary biological function of TDP-43 is to maintain normal RNA splicing. STMN2 mRNA is very sensitive to the concentration and activity of TDP-43 (Figure 4B)15. Abnormal TDP-43 produces over 10 times more cryptic STMN2 mRNA than the normal condition15. We used quantitative PCR to determine the STMN2 splicing pattern, which indicates the normal biological function of TDP-43. Because STMN2 is a neuron-specific gene, we used neuron-like SH-SY5Y cell line to determine the splicing changes in the presents of A1 ~ A515. None of the five compounds show significant inhibitory effect on the normal function of TDP-43 (Figure 4C). Interestingly, compound A4 suppressed the cryptic splicing but did not affect the normal splicing. However, A4 promoted aggregation of TDP-43 anisosomes due to fewer but larger TDP-43 fluorescent granules in treated cells (Figure 4A). Because the regulation of STMN2 alternative splicing is not fully elucidated, this data suggest that A4 might affect another protein that regulate alternative splicing. Thus, we concluded that A4 might not be suitable for further drug development. We have identified previously that an ALS-causing TDP-43 mutation (TDP-43K181E) promotes phase separation in cells. To test whether these compounds show potential therapeutic value, we used cells expressing this disease-causing mutation and test whether compounds A1–A5 inhibit LLPS of TDP-43K181E. Consistent with the TDP-432KQ variant, compound A2 shows the strongest effect in suppressing TDP-43 phase separation (Figures 4D and 4E). Importantly, the suppression effect was concentration-dependent (Figure 4F).
We also tested the specificity of compounds A1–A5. A transcription co-suppressor protein NCOR2 can also form anisosome-like nuclear droplets16. Using a U2OS cell line that produces NCOR2 nuclear droplets, we confirmed that compound A2 does not change the morphology of NCOR2 droplets (Figure 4D), suggesting that compound A2 might directly interact with TDP-43. However, although compound A5 reduces TDP-43 anisosomes, it also reduces the NCOR2 nuclear droplets, suggesting that it may targeting a common mechanism of anisosome formation. Surprisingly, compound A3 suppressed the LLPS of NCOR2 but not TDP-43 (Figures 4D–4H), suggesting that A3 specifically inhibit LLPS of NCOR2 and there might be fundamental differences among anisosomes formed by different proteins. Thus, our compound screening and validation effort also provided additional chemical tool for studying LLPS in the nucleus. In addition, we also tested these compounds by using a cytoplasmic LLPS compartment, the stress granule, and a naturally existed LLPS compartment, the nucleolus. Normal DLD1 cells were treated with compound A1–A5 overnight, followed by 1 hour incubation of 250μM sodium arsenite. Stress granule and nucleolus were visualized by immunostaining of G3BP1 (stress granule) and NPM1 (nucleolus). Compounds A1–A4 showed no effect on stress granule formation (Figure S2). Consistently, A5-treated cells exhibited reduction of stress granules, demonstrating is non-selective inhibition of LLPS compartments. However, A5 slightly changed the morphology of the nucleolus, which requires further studies on identifying the target of A5.
MD simulation proposes that Compound A2 may disrupt the dimerization interface of the N-terminal domain of TDP-43
Based on our observations, the compound A2 might specifically interact with TDP-43 in the cell-based assays. To provide insights for future target deconvolution, we performed molecular docking and MD simulations for compound A2 and TDP-43. We used the folded domains of TDP-43, whose structures have been deposited in the RCSB-PDB (PDB code: 6B1G and 4BS2). We performed MD simulations for the two hybrid systems to generate the binding hypotheses, A2–6B1G (N-terminal domain) and A2–4BS2 (RNA-binding motifs), respectively (Figure 5A–B). Simulation results showed that A2 may interact with the amino acid residue E17 and surrounding residues of the TDP-43 N-terminus (Figure 5A), which is consistent with our experimental observations7. Althought LLPS is sought to mediated by the interaction of the unstructured, low complexity region, for which developing small molecules is hard, other interactions in the folded region of the N-terminal domain are also essential for TDP-43 LLPS7. The free binding energies within A2–6B1G and A2–4BS2 were presented in Figure 5C; and metadynamics analysis was employed to identify the most stable conformations for these two simulated systems. The MD simulation predicted that compound A2 bound more likely to the N-terminal domain than the RNA-recognition motifs of TDP-43. Interestingly, the E17 residue form salt bridges at the dimerization interface, which are crucial for the anisosome formation7.The physical chemistry insights provided by MD simulations will be highly instructive for future biophysics investigation.
Figure 5. Molecular simulation predicts compound A2 interacting at the dimer interface of TDP-43 N-terminal domain.
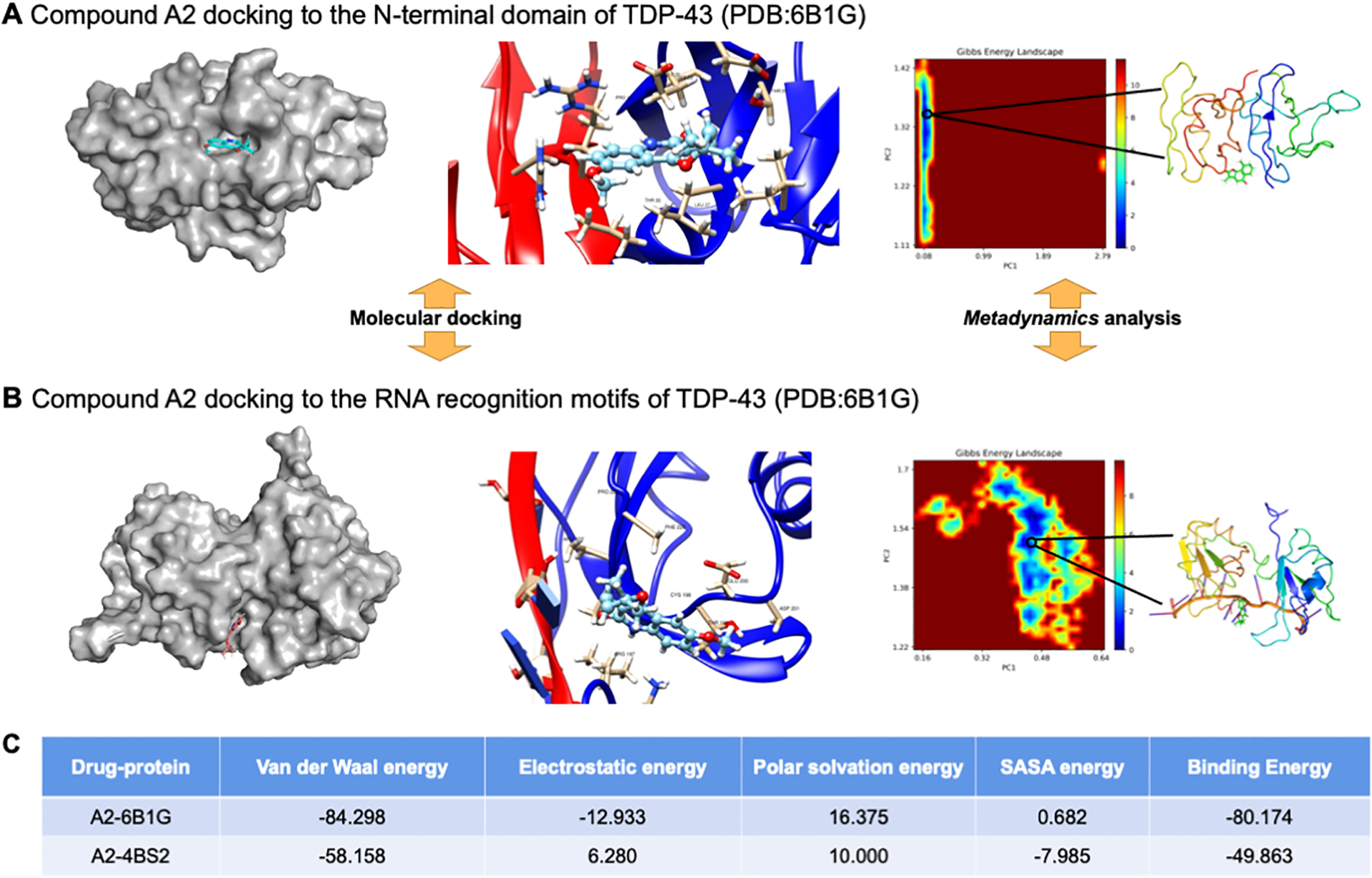
(A) and (B) The molecular docking and MD simulation results of A2–6B1G and A2–4BS2 hydrid systems, metadynamics analysis is applied for conformation searching. (C) The calculated free energy items (in kJ·mol−1) by MD simulations for these two hybrid systems are summarized.
Discussion and Conclusions
A big challenge in developing inhibitors for liquid-liquid phase separation is that the measurable parameters are usually not suitable for scaling up assays. TDP-43, an ideal target for neurodegeneration, only shows small differences by forming anisosomes in the nucleus during cellular stress. This difference can only be captured by detailed imaging analyses. For screening millions of new compounds, an imaging-based strategy takes years of experiment, quadrillion bytes of data storage, and thousands of hours of CPU time for data processing. Here, we developed a deep learning-based method to facilitate the screening tasks and reduced the experimental and computing task by over 10,000 times. Our research demonstrated the power of graph based deep learning technologies on drug discovery for liquid-liquid phase separation for the first time. This strategy can be widely used for screening for other types of LLPS phenomena, if a small pilot screen has been successfully conducted.
In this study, we made an initial yet fundamental trial of molecular graph-based virtual screening technology for in silico drug discovery; we had demonstrated that such a novel architecture performs well in few-shot learning case, especially upon the ones with biased distribution. The results of MD simulations further confirmed the inhibition effects of the identified compounds and provide valuable instructions for chemical optimization. However, it is worth noting that, the accuracy of this model may be largely limited by the overall molecular diversity of the original training data; thus, we anticipate that the data sampling could be further enhanced with experimental collection of new compounds. Although we have not clearly identified the biological targets of these compounds, we have demonstrated that they share common molecular moieties that could potentially target the same biological pathways. We demonstrated the specificity of compound A2 by showing they do not inhibit the NCOR2 droplets, and the therapeutic potential by showing that they can also inhibit another mutant form of TDP-43. The compound A5 can inhibit anisosomal formation in general, providing a useful lead compound for identifying the common mechanism of anisosome formation. Doxycycline, an FDA-approved antibiotic, has been widely used for inducing gene expression, because it has been safely used at a similar concentration for treating millions of patients over many decades. However, we cannot rule out that the presence of doxycycline might cause some cellular stress that might affect the screening outcome, although this possibility is very low. Future directions will focus on the mechanistic insight, including 1) discover the biological targets of the top candidates, 2) determine the binding affinity of the compounds, and 3) determine the molecular mechanisms on how the compounds change the LLPS behavior of TDP-43. Because we are aiming to identify compounds of therapeutic potential in the central nervous system, future ex vivo experiments should be conducted in neuron-like cells such as SH-SY5Y cells or induced pluripotent stem cell-derived neurons. Taken together, we strongly believe that deep learning-based technology will substantially promote compound screen for liquid-liquid phase separation.
Experimental Section
Plasmids
The TDP-432KQ-clover expression plasmid and the inducible expressing system (rtTA3G) was previous published7. NCOR2-clover expressing plasmid was generated by inserting the cDNA sequence of mouse NCOR2 aa1032–1731 to replace the TDP-432KQ sequence in the TDP-432KQ-clover expression plasmid.
Cell culture, lentiviral transduction and inducible expression of anisosomes
Cell lines used in this paper are: HEK293T (ATCC: CRL-11268), U2OS (ATCC: HTB-96), SH-SY5Y (ATCC: CRL-2266), and DLD1 (ATCC: CCL-221). Routine maintenance of these model cell lines follows the standard protocol. In brief, U2OS and HEK293T cells were cultured in complete DMEM supplemented with 10% Fetal bovine serum (FBS). SH-SY5Y cells were cultured in DMEM/F12 supplemented with 10% FBS. HEK293T (TDP-43KO) cell line17 were cultured in DMEM supplemented with NEAA (Gibco, 11140050, 100X), Sodium Pyruvate (Gibco, 11360070, 100X) and 10% FBS.
To package lentivirus, a second-generation packaging system was used. Briefly, 0.5 million 293T cells were seeded per well in a 6-well plate. For lentiviral transfection, 2.5 μg of the lentiviral plasmid, 1.25 μg of pMD2.G and 0.625 μg of psPAX2 were inoculated to each well using the transIT-X2 transfection reagent. Culture medium was changed to fresh medium at 12~24 hours post transfection. Two days after transfection, the culture medium was filtered through a 0.45 μm syringe filter to generate the viral stock. 10–50 μg/mL protamine sulfate was added to the viral stock for transduction of U2OS or DLD1 cells. After 24 hours of incubation with cells, the virus-containing media were removed, and cells were passaged once before selection. Transduced cells are selected based on the selection marker encoded by the lentivirus. For U2OS, the concentrations of the antibiotics used for selection were 200 μg/mL for neomycin (G418, Gibco 10131035), 20 μg/mL for blasticidin (Gibco A1113903), and 1 μg/mL for puromycin (Gibco A1113803). For DLD1 cells, the concentrations were 500 μg/mL for neomycin, 20 μg/mL for blasticidin and 2 μg/mL for puromycin. Detailed guides and protocols posted can be found on the Addgene website:
After stable cells were generated, DLD1 cells were sorted based on the fluorescence to generate clones from a single cell. Then, a clone was selected for the initial compound screen. Doxycycline-inducible U2OS stable cell lines were used as polyclonal populations because their genome is not stable. To induce TDP-43 and NCOR2 anisosomes formation, we used low dose doxycycline (1ug/mL) to treat stable DLD1 or U2OS cell lines. After 24 hours, anisosomes start to form in the nucleus. Then compound treatment or imaging experiments starts between 24 and 48 after doxycycline induction.
Imaging-based compound screening
Imaging-based screening was performed by using a DLD1 clone expressing TDP-43 anisosomes. DLD1 cells were induced by adding 1 μg/mL doxycycline (Sigma Aldrich D5207) to the culture medium 24 hours before plating cells. Cells were plated in 384-well plate at 4000 cells/well density by using Thermo Fisher Multidrop Combi liquid dispenser. Cells were plated overnight before compound treatment. For each compound, three concentrations were applied, 1uM, 5uM and 10uM. After 24 hour compound treatment, cells were then fixed and imaged. ImagExpress HTai was used to capture fluorescent anisosomes. Exported fluorescent images were used to quantify anisosome numbers per cell. The purity of all compounds used in this manuscript, including lead compounds is >95%.
Stress granule induction and immunostaining
DLD1 cells were plated in 96 well plate with 1.5HN glass bottom. Cells were treated with different compounds (A1~A5) at 10μM for 24 hours. Then 250uM sodium arsenite (Sigma #106277) were added for 1 hour to induce stress granule in compound-treated cells. DMSO was used as control. After arsenite treatment, cells were fixed by treated cells with 100uL 4% paraformaldehyde (PFA) in PBS per well for 10 minutes at room temperature. Standard immunostaining was performed on fixed cells. Fluorescent images were taken after the staining by spinning-disc confocal and maximum intensity projection images were analyzed.
Standard immunostaining was performed on fixed cells. After removing fixative buffer, cells were rinsed with 200uL PBS twice to remove residue PFA. Then cells were treated with 1% Triton X-100 in PBS buffer to permeable the cell membrane. Then cells were treated in the blocking buffer (5% BSA, 0.1% Trinton X-100 in PBS) for 1 hour. Blocking buffer were also used for primary and secondary antibody incubation. Primary antibodies (G3BP1: Proteintech 13057–2-AP, and NPM1: Thermo Fisher FC-61991) were diluted to 1:100 and applied to cells for 1 hour at room temperature, then primary antibody buffer were removed by aspiration. Three washes with the washing buffer (0.1% Trinton X-100 in PBS) were introduced before incubation of secondary antibody. Alexa488-conjugated anti-rabbit antibody and Cy3-conjugated anti-mouse antibody were used as secondary (1:1000). Cells were incubated with secondary antibody for 30min. Then 4 washes were conducted to remove residual unbound antibodies. Then cells were stained with DAPI for the nuclear DNA.
Detailed protocol can be found on Abcam webpage:
Quantitative real-time PCR
Total RNA was harvested from compound-treated Sh-Sy5y neuron-like cells. Cells were treated with compounds at 10uM final concentration for 24 hours before harvesting. The cDNAs were generated by using standard protocol recommended by the High-Capacity cDNA Reverse Transcription Kit (ThermoFisher Catlog # 4368813). Quantitative real-time PCR was carried out in triplicates, using iTaq Universal SYBR green (Bio-Rad) in a CFX Opus 96 real-time PCR system. Primer sets for STMN splicing: full length STMN2 forward, F: 5’-AGCTGTCCATGCTGTCACTG-3’; full length STMN2 reverse, 5’-GGTGGCTTCAAGATCAGCTC-3’; Truncated STMN2 forward, 5’-GGACTCGGCAGAAGACCTTC-3’; Truncated STMN2 reverse, 5’-GCAGGCTGTCTGTCT CTCTC-3’; HRPS18 forward, 5’-GCAGAATCCACGCCAGTACAA G-3’; HRPS18 reverse, 5’-GCTTGTTGTCCAGACCATTGGC-3’.
Graph convolutional network as the deep learning method for in silico screening
Molecular graphs-based screening approaches have been proven to own robustness for challenging structural assignments,11, 18 as these kinds of methods could directly extract structure information from drug compounds to realize accurate classification instead of utilizing external descriptors.19 However, it is flexible to include different chemical knowledge as collected descriptors for specific screening tasks.12, 13, 20 In addition, the computational cost of GCN-dominant methods can be substantially reduced compared to conventional computations. In this study, we applied our self-developed GCN model for drug screenings with a focus on anisosome inhibitors, and the SchNet architecture was employed. The schematics of the developed GCN model can be found in Figure 1. For any target compound, its structure information that is translated from the generated graphs of molecules (molecular graphs) can be obtained from its simplified molecular-input line-entry system (SMILES) string, such a translation can be conducted with Deep Graph Library (DGL) library. Then within the framework of GCN, the molecular graph will be further encoded into numerical descriptors for specific processing.
It is notable that the generated molecular graph is mainly composed of nodes and edges. The nodes represent atom points, while the edges are corresponding to inter-atomic connections, like bonds and etc. And with these features, the correlation between structure similarity and drug properties (e.g., inhibition effects) can be initialized. The most important advantage of GCN architecture lies in the fact that, within any molecular graph, all the connections between every two atoms can be fully utilized for structural information extraction. The numerical descriptors obtained from the graphs are recorded in a distance tensor, within the RBF (radial basis function) layer.Moreover, to decently encode these generated molecular graphs at the atomic level, several CFCONV (continuous-filter convolutions) layers that are used for process local correlation were added to record and optimize the inter-atomic information during evolution, and the chemical descriptors (features) generated by RDKit package are also used to enhance the screening accuracy. For example, within the layer, the th atom’s (N is the total number of atoms) evolution can be presented by the equation below:
where, indicates the filter generation that can map the atoms’ descriptions to the filtration; and represents element-wise multiplication. To intelligently manage the evolution accuracy for specific tasks, the Gaussian-type based function, , is adopted for error control, which can be written as below:
in which, is a pre-defined cutoff, and indicates the bonding connection between the th and th atoms. The represent hyperparameters, and in this study, it is set to 0.1.
For any specific classification task, the predicted target value, , by GCN model should be verified with respect to a reference value (e.g. experimental measurement), , and the accuracy can be output with a squared loss function:
In this study, we applied the proposed GCN model to screening anisosome modulators. We first trained the model on the collected qHTS data, composed of ~3000 compounds. In our experiment, ~300 compounds show inhibition activities and the rest are inactive (more technical details can be found in our GitHub page: https://github.com/tcsnfrank0177/Molecular_Intelligence_DrugDiscovery.git). The original data set was randomly divided into training and test sets by a ratio of 9:1. The model’s accuracy was calibrated with the results of the compounds that are contained in the test set. Some parameterized ML models were also added for secondary screening of GCN prediction results. Then we employed the well-trained model to screen the three independent NCATS in-house libraries, Genesis, Sytravon, and NCATS Pharmacologically Active Chemical Toolbox (NPACT), for new candidates identification (the workflow can be found in Figure 2); it is worth pointing out that all the three libraries had not been experimentally tested upon this assay.
Molecular docking and MD simulation
To verify the binding interactions of identified compounds with the possible targets: 4bs2 and 6g1b, molecular docking was first conducted with AutoDock Vina 1.1.221 to obtain the initial conformation. To decently solve the drugs’ interaction, MD simulations were further conducted with GROMACS 2019.6 upon the protein-drug hybrid system under the temperature of 310K, Amber 99 force field was applied.22 The solubility of NaCl is set to 0.9%. The total simulation time is 100 ns, and time-step is 1 fs. We employed V-rescale approach for temperature control;23 and Berendsen method is adopted for pressure control.24 Energy minimization was conducted with the Steep method for the first 10,000 steps; and after 10 ns’ NVT simulations, then NPT simulations were started. The electrostatic interactions were described by Particle Mesh Ewald (PME) method. For metadynamics analysis upon protein backbone, the system is described by the collective variables (CV) of RMSD and gyrate. The gmx sham is used for free energy landscape(FEL) analysis; and g_mmpbsa is employed for binding energy calculations for drug-protein hybrid systems.
Supplementary Material
Acknowledgments
The work was supported by the National Center for Advancing Translational Sciences (NCATS) in the National Institutes of Health (NIH). P.G. is the receptor of Opportunity Award Grant by NCATS (NIH). H.Y. is supported by NINDS R00 grant #NS114162, the Junior Investigator Award from TARCC, and Endowed Scholarship from UT Southwestern Medical Center. D.W.C is supported by NINDS R01 grant #NS121604. Lastly, we would like to thank NIH Biowulf for providing computational resource.
Abbreviations and Acronyms:
- CFCONV
continuous-filter convolutions
- CPU
central processing unit
- DLD1
A colorectal adenocarcinoma cell line name
- EWS
Ewing’s Sarcoma RNA Binding Protein (a protein)
- FLI1
Friend leukemia integration 1 (a protein name)
- FTD
frontotemporal dementia
- FUS
Fused in sarcoma (a protein name)
- G3BP1
Ras GTPase-activating protein-binding protein 1 (a protein name)
- GCN
graph convolutional network
- HOX
Homobox (a protein name)
- HSP70
Heat Shock Protein 70 (a protein name)
- LLPS
Liquid-liquid phase separation
- NCATS
The National Center for Advancing Translational Sciences
- NCOR2
nuclear receptor corepressor 2 (a protein name)
- NUP98
Nuclear pore protein 98 (a protein name)
- SH-SY5Y
A neuroblastoma cell line name
- STMN2
stathmin2 (a protein name)
- TDP-43
TAR DNA-binding protein 43 (a protein name)
Footnotes
Ancillary Information
The clustering results of the qHTS screened compounds, the effect of compound A1–A5 on other cellular LLPS compartments, and QC reports of A1–A5 can be found in Supporting Information. Molecular Formula Strings and PDB files are included in the supplementary files.
References
- (1).Brangwynne CP; Eckmann CR; Courson DS; Rybarska A; Hoege C; Gharakhani J; Julicher F; Hyman AA Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324 (5935), 1729–1732. DOI: 10.1126/science.1172046. [DOI] [PubMed] [Google Scholar]
- (2).Boulay G; Sandoval GJ; Riggi N; Iyer S; Buisson R; Naigles B; Awad ME; Rengarajan S; Volorio A; McBride MJ; et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 2017, 171 (1), 163–178 e119. DOI: 10.1016/j.cell.2017.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ahn JH; Davis ES; Daugird TA; Zhao S; Quiroga IY; Uryu H; Li J; Storey AJ; Tsai YH; Keeley DP; et al. Phase separation drives aberrant chromatin looping and cancer development. Nature 2021, 595 (7868), 591–595. DOI: 10.1038/s41586-021-03662-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Taylor JP; Brown RH Jr.; Cleveland DW Decoding ALS: from genes to mechanism. Nature 2016, 539 (7628), 197–206. DOI: 10.1038/nature20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mitrea DM; Mittasch M; Gomes BF; Klein IA; Murcko MA Modulating biomolecular condensates: a novel approach to drug discovery. Nat Rev Drug Discov 2022, 21 (11), 841–862. DOI: 10.1038/s41573-022-00505-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Patel A; Lee HO; Jawerth L; Maharana S; Jahnel M; Hein MY; Stoynov S; Mahamid J; Saha S; Franzmann TM; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162 (5), 1066–1077. DOI: 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]; Lu S; Hu J; Arogundade OA; Goginashvili A; Vazquez-Sanchez S; Diedrich JK; Gu J; Blum J; Oung S; Ye Q; et al. Heat-shock chaperone HSPB1 regulates cytoplasmic TDP-43 phase separation and liquid-to-gel transition. Nat Cell Biol 2022, 24 (9), 1378–1393. DOI: 10.1038/s41556-022-00988-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gasset-Rosa F; Lu S; Yu H; Chen C; Melamed Z; Guo L; Shorter J; Da Cruz S; Cleveland DW Cytoplasmic TDP-43 De-mixing Independent of Stress Granules Drives Inhibition of Nuclear Import, Loss of Nuclear TDP-43, and Cell Death. Neuron 2019, 102 (2), 339–357 e337. DOI: 10.1016/j.neuron.2019.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Yu H; Lu S; Gasior K; Singh D; Vazquez-Sanchez S; Tapia O; Toprani D; Beccari MS; Yates JR 3rd; Da Cruz S; et al. HSP70 chaperones RNA-free TDP-43 into anisotropic intranuclear liquid spherical shells. Science 2021, 371 (6529). DOI: 10.1126/science.abb4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Markmiller S; Soltanieh S; Server KL; Mak R; Jin W; Fang MY; Luo EC; Krach F; Yang D; Sen A; et al. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 2018, 172 (3), 590–604 e513. DOI: 10.1016/j.cell.2017.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]; Molliex A; Temirov J; Lee J; Coughlin M; Kanagaraj AP; Kim HJ; Mittag T; Taylor JP Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163 (1), 123–133. DOI: 10.1016/j.cell.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ling SC; Polymenidou M; Cleveland DW Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 2013, 79 (3), 416–438. DOI: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Amador-Ortiz C; Lin WL; Ahmed Z; Personett D; Davies P; Duara R; Graff-Radford NR; Hutton ML; Dickson DW TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 2007, 61 (5), 435–445. DOI: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Gao P; Xu M; Zhang Q; Chen CZ; Guo H; Ye Y; Zheng W; Shen M Graph Convolutional Network-Based Screening Strategy for Rapid Identification of SARS-CoV-2 Cell-Entry Inhibitors. Journal of Chemical Information and Modeling 2022, 62 (8), 1988–1997. DOI: 10.1021/acs.jcim.2c00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Schütt KT; Sauceda HE; Kindermans PJ; Tkatchenko A; Müller KR SchNet – A deep learning architecture for molecules and materials. The Journal of Chemical Physics 2018, 148 (24), 241722. DOI: 10.1063/1.5019779 (acccessed 2023/01/11). [DOI] [PubMed] [Google Scholar]
- (13).Gao P; Zhang J; Peng Q; Zhang J; Glezakou V-A General Protocol for the Accurate Prediction of Molecular 13C/1H NMR Chemical Shifts via Machine Learning Augmented DFT. Journal of Chemical Information and Modeling 2020, 60 (8), 3746–3754. DOI: 10.1021/acs.jcim.0c00388. [DOI] [PubMed] [Google Scholar]; Gao P; Zhang J; Qiu H; Zhao S A general QSPR protocol for the prediction of atomic/inter-atomic properties: a fragment based graph convolutional neural network (F-GCN). Physical Chemistry Chemical Physics 2021, 23 (23), 13242–13249, 10.1039/D1CP00677K. DOI: 10.1039/D1CP00677K. [DOI] [PubMed] [Google Scholar]
- (14).Huang R; Southall N; Wang Y; Yasgar A; Shinn P; Jadhav A; Nguyen D-T; Austin CP The NCGC Pharmaceutical Collection: A Comprehensive Resource of Clinically Approved Drugs Enabling Repurposing and Chemical Genomics. Science Translational Medicine 2011, 3 (80), 80ps16–80ps16. DOI: doi: 10.1126/scitranslmed.3001862. [DOI] [PMC free article] [PubMed] [Google Scholar]; Huang R; Zhu H; Shinn P; Ngan D; Ye L; Thakur A; Grewal G; Zhao T; Southall N; Hall MD; et al. The NCATS Pharmaceutical Collection: a 10-year update. Drug Discovery Today 2019, 24 (12), 2341–2349. DOI: 10.1016/j.drudis.2019.09.019. [DOI] [PubMed] [Google Scholar]
- (15).Melamed Z; Lopez-Erauskin J; Baughn MW; Zhang O; Drenner K; Sun Y; Freyermuth F; McMahon MA; Beccari MS; Artates JW; et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci 2019, 22 (2), 180–190. DOI: 10.1038/s41593-018-0293-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hoshino H; Nishino TG; Tashiro S; Miyazaki M; Ohmiya Y; Igarashi K; Horinouchi S; Yoshida M Co-repressor SMRT and class II histone deacetylases promote Bach2 nuclear retention and formation of nuclear foci that are responsible for local transcriptional repression. J Biochem 2007, 141 (5), 719–727. DOI: 10.1093/jb/mvm073. [DOI] [PubMed] [Google Scholar]
- (17).Schmidt HB; Barreau A; Rohatgi R Phase separation-deficient TDP43 remains functional in splicing. Nat Commun 2019, 10 (1), 4890. DOI: 10.1038/s41467-019-12740-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).St. John PC; Guan Y; Kim Y; Kim S; Paton RS Prediction of organic homolytic bond dissociation enthalpies at near chemical accuracy with sub-second computational cost. Nature Communications 2020, 11 (1), 2328. DOI: 10.1038/s41467-020-16201-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kwon Y; Lee D; Choi Y-S; Kang M; Kang S Neural Message Passing for NMR Chemical Shift Prediction. Journal of Chemical Information and Modeling 2020, 60 (4), 2024–2030. DOI: 10.1021/acs.jcim.0c00195. [DOI] [PubMed] [Google Scholar]; Gerrard W; Bratholm LA; Packer MJ; Mulholland AJ; Glowacki DR; Butts CP IMPRESSION – prediction of NMR parameters for 3-dimensional chemical structures using machine learning with near quantum chemical accuracy. Chemical Science 2020, 11 (2), 508–515, 10.1039/C9SC03854J. DOI: 10.1039/C9SC03854J. [DOI] [PMC free article] [PubMed] [Google Scholar]; Scarselli F; Gori M; Tsoi AC; Hagenbuchner M; Monfardini G The Graph Neural Network Model. IEEE Transactions on Neural Networks 2009, 20 (1), 61–80. DOI: 10.1109/TNN.2008.2005605. [DOI] [PubMed] [Google Scholar]
- (19).Gao P; Zhang J; Sun Y; Yu J Accurate predictions of aqueous solubility of drug molecules via the multilevel graph convolutional network (MGCN) and SchNet architectures. Physical Chemistry Chemical Physics 2020, 22 (41), 23766–23772, 10.1039/D0CP03596C. DOI: 10.1039/D0CP03596C. [DOI] [PubMed] [Google Scholar]
- (20).Behler J Perspective: Machine learning potentials for atomistic simulations. The Journal of Chemical Physics 2016, 145 (17), 170901. DOI: 10.1063/1.4966192 (acccessed 2023/01/11). [DOI] [PubMed] [Google Scholar]; Behler J First Principles Neural Network Potentials for Reactive Simulations of Large Molecular and Condensed Systems. Angewandte Chemie International Edition 2017, 56 (42), 12828–12840, 10.1002/anie.201703114. DOI: 10.1002/anie.201703114 (acccessed 2023/01/11). [DOI] [PubMed] [Google Scholar]; Gao P; Zhang J; Sun Y; Yu J Toward Accurate Predictions of Atomic Properties via Quantum Mechanics Descriptors Augmented Graph Convolutional Neural Network: Application of This Novel Approach in NMR Chemical Shifts Predictions. The Journal of Physical Chemistry Letters 2020, 11 (22), 9812–9818. DOI: 10.1021/acs.jpclett.0c02654. [DOI] [PubMed] [Google Scholar]; Wang J; Olsson S; Wehmeyer C; Pérez A; Charron NE; de Fabritiis G; Noé F; Clementi C Machine Learning of Coarse-Grained Molecular Dynamics Force Fields. ACS Central Science 2019, 5 (5), 755–767. DOI: 10.1021/acscentsci.8b00913. [DOI] [PMC free article] [PubMed] [Google Scholar]; Meldgaard SA; Kolsbjerg EL; Hammer B Machine learning enhanced global optimization by clustering local environments to enable bundled atomic energies. The Journal of Chemical Physics 2018, 149 (13), 134104. DOI: 10.1063/1.5048290 (acccessed 2023/01/11). [DOI] [PubMed] [Google Scholar]; Ouyang R; Xie Y; Jiang D.-e . Global minimization of gold clusters by combining neural network potentials and the basin-hopping method. Nanoscale 2015, 7 (36), 14817–14821, 10.1039/C5NR03903G. DOI: 10.1039/C5NR03903G. [DOI] [PubMed] [Google Scholar]; Sørensen KH; Jørgensen MS; Bruix A; Hammer B Accelerating atomic structure search with cluster regularization. The Journal of Chemical Physics 2018, 148 (24), 241734. DOI: 10.1063/1.5023671 (acccessed 2023/01/11). [DOI] [PubMed] [Google Scholar]; Lu C; Liu Q; Wang C; Huang Z; Lin P; He L Molecular Property Prediction: A Multilevel Quantum Interactions Modeling Perspective. Proceedings of the AAAI Conference on Artificial Intelligence 2019, 33 (01), 1052–1060. DOI: 10.1609/aaai.v33i01.33011052 (acccessed 2023/01/11). [DOI] [Google Scholar]; Wexler RB; Martirez JMP; Rappe AM Chemical Pressure-Driven Enhancement of the Hydrogen Evolving Activity of Ni2P from Nonmetal Surface Doping Interpreted via Machine Learning. Journal of the American Chemical Society 2018, 140 (13), 4678–4683. DOI: 10.1021/jacs.8b00947. [DOI] [PubMed] [Google Scholar]; Mansouri Tehrani A; Oliynyk AO; Parry M; Rizvi Z; Couper S; Lin F; Miyagi L; Sparks TD; Brgoch J Machine Learning Directed Search for Ultraincompressible, Superhard Materials. Journal of the American Chemical Society 2018, 140 (31), 9844–9853. DOI: 10.1021/jacs.8b02717. [DOI] [PubMed] [Google Scholar]; Panapitiya G; Avendaño-Franco G; Ren P; Wen X; Li Y; Lewis JP Machine-Learning Prediction of CO Adsorption in Thiolated, Ag-Alloyed Au Nanoclusters. Journal of the American Chemical Society 2018, 140 (50), 17508–17514. DOI: 10.1021/jacs.8b08800. [DOI] [PubMed] [Google Scholar]; Rupp M; Ramakrishnan R; von Lilienfeld OA Machine Learning for Quantum Mechanical Properties of Atoms in Molecules. The Journal of Physical Chemistry Letters 2015, 6 (16), 3309–3313. DOI: 10.1021/acs.jpclett.5b01456. [DOI] [Google Scholar]; Bai Y; Wilbraham L; Slater BJ; Zwijnenburg MA; Sprick RS; Cooper AI Accelerated Discovery of Organic Polymer Photocatalysts for Hydrogen Evolution from Water through the Integration of Experiment and Theory. Journal of the American Chemical Society 2019, 141 (22), 9063–9071. DOI: 10.1021/jacs.9b03591. [DOI] [PMC free article] [PubMed] [Google Scholar]; Mater AC; Coote ML Deep Learning in Chemistry. Journal of Chemical Information and Modeling 2019, 59 (6), 2545–2559. DOI: 10.1021/acs.jcim.9b00266. [DOI] [PubMed] [Google Scholar]
- (21).Trott O; Olson AJ AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry 2010, 31 (2), 455–461. DOI: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Meagher KL; Redman LT; Carlson HA Development of polyphosphate parameters for use with the AMBER force field. Journal of Computational Chemistry 2003, 24 (9), 1016–1025, 10.1002/jcc.10262. DOI: 10.1002/jcc.10262 (acccessed 2023/01/11). [DOI] [PubMed] [Google Scholar]
- (23).Bussi G; Donadio D; Parrinello M Canonical sampling through velocity rescaling. The Journal of Chemical Physics 2007, 126 (1), 014101. DOI: 10.1063/1.2408420 (acccessed 2023/01/11). [DOI] [PubMed] [Google Scholar]
- (24).Berendsen HJC; Postma JPM; van Gunsteren WF; DiNola A; Haak JR Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics 1984, 81 (8), 3684–3690. DOI: 10.1063/1.448118 (acccessed 2023/01/11). [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.