

Abstract
Introduction:
Glucocorticoids (GCs) have unique actions in their combined anti-inflammatory and immunosuppressive activities and are among the most commonly-prescribed drugs, particularly for inflammatory conditions. They are often used clinically to treat inflammatory eye diseases like uveitis, optic neuritis, conjunctivitis, keratitis and others, but are often accompanied by side effects, like ocular hypertension that can be vision threatening.
Areas covered:
The review will focus on the complex molecular mechanism of action of GCs that involve both transactivation and transrepression and their use therapeutically that can cause significant systemic side effects, particularly ocular hypertension that can lead to glaucoma.
Expert Opinion:
While we are still unclear as to all the mechanisms responsible for GC-induced ocular hypertension, however, there are potential novel therapies that are in development that can separate some of the anti-inflammatory therapeutic efficacy from their ocular hypertension side effect. This review provides some insight into these approaches.
Keywords: Glucocorticoids; Ocular hypertension, Glaucoma; Glucocorticoid molecular mechanisms; SEGRAs
1. Introduction
Since their initial discoveries, the corticosteroids have received increased attention particularly related to their role in development, inflammation, metabolism, cognition and immunosuppression[1]. Specifically, corticosteroids (having both glucocorticoid and mineralocorticoid activity) have a vast array of actions including modifying carbohydrate, protein, and lipid metabolism, electrolyte and fluid balance, as well as preserving normal function of the cardiovascular system, kidney function, endocrine system and the nervous system. Glucocorticoids (GCs), which lack mineralocorticoid activity, have been a primary source of therapy for suppressing inflammation for a variety of inflammatory diseases, including: hyper-reactive airway disease, autoimmune diseases, Graves’ disease, systemic lupus erythematosus, and others. GCs are a major therapy for treating inflammatory eye diseases like uveitis, optic neuritis, conjunctivitis, keratitis and others (see Table 1). These ocular GCs vary in their anti-inflammatory potency as well as their ocular penetration (Table 2). This review will focus on GC actions in the eye with specific reference to their major side effect of elevating intraocular pressure and induction of glaucoma. While GCs are administered clinically, the natural occurring GCs (cortisol in humans and corticosterone in rodents) are steroid hormones that are released in response to stress and can regulate a number of functions including carbohydrate, lipid, and protein metabolism, development, homeostasis, cognition, and inflammation[2]. The endogenous synthesis and secretion of GCs are regulated by hypothalamus-pituitary (HPA) axis and adrenal glands in a circadian manner as well as in response to various stimuli[3] However, GC availability is further regulated at the cellular and tissue levels which adds further complexities on how GC signaling may occur and is tissue specific. Most secreted GCs are sequestered and remain inactive with 80% to 90% of circulating GCs bound to corticosteroid-binding globulin (CBG) and 5% to 15% bound to albumin. Therefore, only about 5% of natural systemic GC remains free and active, thus accessibility is regulated by the CBG and albumin concentrations[1,2,4].
Table 1.
Ophthalmic Steroids
| Steroid | Commercial Name | Dose | Indication |
|---|---|---|---|
| TopIcal Ocular Steroids | |||
| Dexamethasone | Decadron® Maxidex® |
0.1% solution 0.1%suspension |
Inflammatory eye diseases, cornea injury, foreign bodies |
| Difluprednate ophthalmic | Durezol® | 0.05% emulsion | Anterior uveitis, inflammation affecting from of eye, pain following ocular surgery |
| Fluorometholone ophthalmic | Flarex®, Fluor-op® FML, FML liquifilm, FML Forte |
0.1% ointment 0.1% suspension 0.25% suspension |
Inflammatory ocular conditions and injury |
| Loteprednol etabonate ophthalmic | Alrex® Lotemax® Inveltys ® |
0.5% gel 0.1% suspension 0.25% suspension |
Inflammation following ocular surgery, seasonal allergy conjunctivitis, ocular inflammation |
| Medrysone | HMS® (no longer available in the US, generic versions may be available) | 1% suspension | Superficial ocular inflammation, allergic conjunctivitis |
| Prednisolone acetate ophthalmic | Omnipred® PredForte® PredMild® |
1.0% solution 1.0% suspension 0.12% suspension |
Corneal injury, foreign bodies, ocular inflammation |
| Prednisolone sodium phosphate ophthalmic | 0.1% solution | Corneal injury, foreign bodies, ocular inflammation | |
| Rimexolone ophthalmic | Vexol® | 1% solution | Anterior uveitis, post-operative inflammation following ocular surgery |
| Intraocular Steroids | |||
| Dexamethasone |
Ozurdex ® Dexycu ® |
0.7 mg implant Injectable suspension (5 uL of 9% suspension) |
Posterior uveitis Cataract surgery |
| Triamcinolone acetonide | Triesence ® Trivaris ® |
Injectable suspensions 40 or 80 mg/mL |
Intraocular inflammation; Vitrectomy |
| Fluocinolone acetonide | Iluvien®, Yutiq ®, Retisert ® | 0.59, 0.19, 0.18 mg implant | Macular edema, uveitis |
Table 2.
Comparisons of Anti-inflammatory Potency and Ocular Penetration of Topical Ocular Glucocorticoids
| Glucocorticoid | Relative Anti-inflammatory Activity | Dose | Peak Aqueous Humor Concentration (ng/mL) | Time to Peak (minutes) | IOP Rise (mmHg) |
|---|---|---|---|---|---|
| Cortisol | 1 | 0.5% | 3.2 | ||
| Prednisolone | 4 | 1% | 7650 | 30 | 10.0 |
| Prednisolone Acetate | 4 | 1% | 670–1130 | 45–120 | |
| Prednisolone Phosphate | 4 | 1% | 26 | 90–240 | |
| Fluorometholone | 40 | 0.1% | 5.1 | 30–60 | 6.1 |
| Dexamethasone | 25 | 0.1% | 31 | 90–120 | 22.0 |
| Dexamethasone Phosphate | 25 | 0.1% | 31 | 55 | |
| Medrysone | 4 | 1% | 1.0 | ||
| Loteprednol etabonate | 25 | 0.5% | * | * | |
| Rimexolone | 25 | 1% | ~6 | ||
| Difluprednate emulsion | 60 | 0.05% | 30 | 60 |
Rapidly metabolized and inactivated in cornea (Druzgala P et al. PMID 1959381)[124]
Data from: Murdick PW, Keates RH, Donovan EF et al. (PMID: 5928150)[125]
Awan MA, Agarwal PK, Watson DG et al (PMID: 19293163)[126]
Razeghinejad MR, Katz L (PMID: 21757964)[127]
Yamaguchi M, Yasueda S et al (PMID: 16023810)[128]
Tissue-specific metabolizing enzymes 11β-hydroxysteroid dehydrogenases (11β-HSDs) further regulate GC bioavailability at the cellular level[5]. 11β-HSDs catalyze the interconversion of active GCs. 11β-HSD2 acts as potent dehydrogenase and inactivates GCs by converting cortisol to cortisone. The 11β-HSD1 acts as reductase that facilitates conversion of inactive cortisone to active cortisol. These enzymes are mostly present in GC target tissues including liver, adipose tissue, brain and other tissues[4,6]. Thus, these two 11β-HSDs (1&2) regulate GC bioavailability at the cellular level. Various inhibitors of 11β-HSD1 have also been designed to regulate GC availability in various tissues[6]. Early studies have reported altered activity of other cortisol metabolizing enzymes in primary open angle glaucoma (POAG) patient’s trabecular meshwork (TM) cells and peripheral blood[7,8]. Stokes et al[9] reported the presence of 11β-HSD1 in human trabecular meshwork cells and 11β-HSD2 in non-pigmented ciliary epithelium and corneal epithelium and endothelium. Later, two studies by the same group reported the presence of 11β-HSD1 in non-pigmented ciliary epithelium and corneal epithelium[10,11]. They further showed that carbenoxolone (an inhibitor of 11β-HSD1) was able to lower intraocular pressure (IOP) in ocular hypertension patients by 10%[11]. The expression of these isoenzymes (particularly 11β-HSD1 in trabecular meshwork) may alter sensitivity to GCs and hence may play an important role in steroid-induced ocular hypertension and POAG. Recently, a new 11β-HSD1 inhibitor, KR-6707, was found to reduce cortisol levels in mouse eyes and it provided a protective effect against ischemia-reperfusion-induced eye injury and reversed the elevation in IOP normally seen accompanying ischemia-reperfusion[12]. Such observations suggest a role for endogenous GCs in regulating IOP.
The major actions of glucocorticoids are derived from the multiple signaling pathways linked to the glucocorticoid receptor (GR). These mechanisms, while important for anti-inflammatory actions, can also result in a plethora of other deleterious effects deemed as side effects (Table 3). These actions include immunosuppression, inhibition of wound repair, osteoporosis, metabolic disturbances and ocular hypertension. For treating chronic ocular inflammatory diseases, GC-induced ocular hypertension represents a major problem as it can lead to iatrogenic open-angle glaucoma. It is interesting that not all patients receiving GCs will see an elevation in intraocular pressure; in fact, only about 35 to 40% of the adult population is susceptible to developing ocular hypertension following glucocorticoid treatment. Interestingly, children appear to be more susceptible to increases in IOP following GC treatment, with > 60% of children found to be steroid-responders[13–15] The reason for this potential difference in responsiveness to GCs is not well understood. There have been a number of proposed mechanisms to account for responders versus non-responders, including genetic differences and differences in receptor subtype expression. In this review we will provide insight into some of these potential mechanisms that may help explain these differences in ocular responsiveness, and also we will explore the relationship between GC-induced ocular hypertension and glaucoma.
Table 3.
Side Effects of Glucocorticoid Therapy
| Systemic |
| • Cushing Syndrome |
| • Osteoporosis & osteonecrosis |
| • Diabetes mellitus |
| • Limb muscle atrophy |
| • Edema |
| • Suppression of HPA axis |
| • Immunosuppression and infections |
| • Hypertension |
| • Retardation of growth |
| • Impaired healing |
| • Thinning of skin & increased brusibility |
| • Peptic ulcer |
| • Psychoses |
| • Increased abdominal fat |
| Ocular |
| • Ocular hypertension and iatrogenic glaucoma |
| • Posterior subcapsular cataracts |
| • Infections |
| • Impaired wound healing |
The GR is ubiquitously expressed in almost all cell types with the same GC response elements on GC responsive genes. However, there are different responses to GCs within different tissues. All of these varied effects depends on numerous genomic and non-genomic mechanisms responsible for altering GR signaling, including different GR-binding sites and availability and chromatin state and accessibility, all of which are specific for each tissue and each cell type[16,17]. GCs enter cell by diffusion and interact with cytosolic glucocorticoid receptors. The classical GC genomic actions are mediated through the GR isoform GRα (Figure 1). In the absence of ligand (i.e GCs), GRα resides mostly in the cytoplasm of cells as part of a large multiprotein complex that includes chaperone proteins (hsp90, hsp70, and p23)[18] and immunophilins (FKBP51 and FKBP52), that are responsible for maintaining the high-affinity ligand binding GR confirmation[19]. In the presence of a GC, GRα undergoes a conformational change, resulting in the dissociation of the multi-protein complex. As a result, a conformational change of the GRα protein exposes nuclear localization signals, and the ligand-bound GRα is rapidly translocated by microtubule motor proteins along the microtubules through pores in the nuclear envelope to enter the nucleus. Once inside the nucleus, GRα can activate or repress specific genes by either transactivation or transrepression (Figure 2). During transactivation, the ligand activated GRα isoform homodimerizes and acts as a transcription factor by binding to specific DNA sequences on genes carrying GC response elements (GREs) to alter gene expression. Currently, there are number of models proposed for transactivation of GR. Ligand-bound GRα can activate gene expression[17] by: a) GRα homodimers binding directly to GREs, b) monomeric GRα binding to GRE and interacting with a neighboring transcription factor, known as a composite mechanism, c) monomeric GRα interacting with transcription factor (TF) in absence of DNA binding, known as a tethering mechanism, and d) monomeric GRα binding to half site GREs[20]. The consensus GRE sequence is GAGAACAnnnTGTTCT, an imperfect palindromic sequence comprised of two half sites separated by a three base pair spacer[1,2,21,22]. When GR binds to GREs as a dimer, with each GR occupied by one half a GRE site, respectively with the spacer required for GR-DNA interaction. The conformational changes resulting from such binding leads to recruitment of chromatin-remodeling complexes and co-regulators facilitating transcriptional activation or repression of genes by influencing activity of RNA polymerase II. However, there are also reports of negative GREs (nGREs) that mediate GC-dependent repression of target genes[4,23]. These nGREs differ from classical GREs in having an inverted palindromic sequence, CTCC(n)0–2GGAGA and variable spacer length ranging from zero to two nucleotides. This variable short length spacer does not allow GR homodimerization. The GR binding to nGREs promotes formation of repressing complex with corepressors SMRT/NCoR and histone deacetylase[23], which repress gene transcription. Recently it was shown that exogenous GCs mediate gene expression by favoring GR homodimer formation and utilizing classic two GRE half sites[20]. All of these models of transactivation and GRE binding sites adds another level of complexity in our understanding of how GC functions within cells and how such diverse complex interactions occurring in different tissues could explain some of the observed tissue specific side effects as perhaps what is seen in the trabecular meshwork (TM) that leads to elevation in intraocular pressure.
Figure 1. Alternatively splicing of the human glucocorticoid receptor gene (NR3C1) leads to isoforms GRα and GRβ.
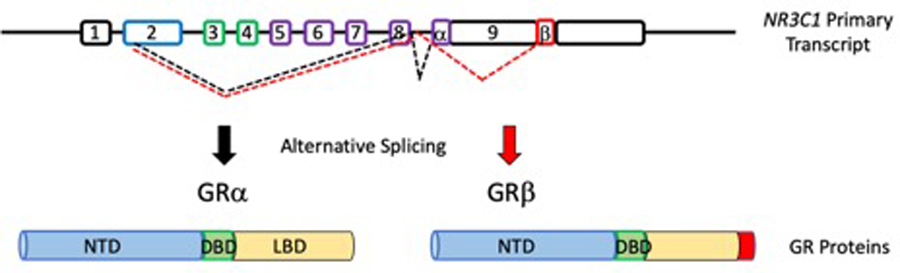
NR3C1 contains 9 exons with 8 introns. The primary transcription start site is within exon 2 which encodes the N-terminal transactivation domain (NTD). Exons 3–8 are also incorporated with exons 3–4 encoding the DNA binding domain (DBD) and exons 5–8 encoding the ligand binding domain (LBD). The only difference between GRα and GRβ is the alternative splicing of exon 9α in GRα and splicing of exon 9β in GRβ. 9α encodes a portion of the LBD, which is missing in exon 9β, which prevents GC binding in GRβ, making this shorter isoform a dominant negative regulator of GR activities. Modified from Oakley & Cidlowski (PMID: 24084075)
Figure 2. Molecular mechanisms of action of glucocorticoid receptors.
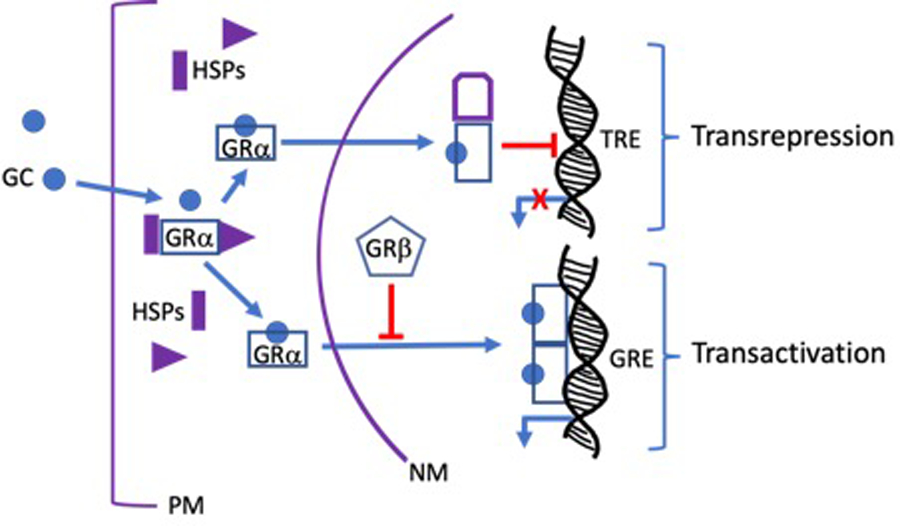
Glucocorticoids (GCs) are lipophilic compounds that freely penetrate the plasma membrane (PM). Upon entering the cytosol, GCs bind to the high affinity ligand binding site on the cytoplasmic GRα complex that also contains several heat shock proteins (HSPs) and immunophilins (not shown). The ligand bound GRα, undergoes a conformational change that dissociates the other proteins in the complex to form activated GRα monomers. The activated monomers are actively transported along microtubules to the nuclear membrane (NM), where they enter the nucleus through nuclear pores. Once inside the nucleus, GRα monomers dimerize and bind to palindromic GC response elements (GREs) on GC regulated genes to turn on transcription in a process known as transactivation. Activated nuclear GRα monomers can also bind to several other transcription factors (TF) including AP1 and NFkB, thereby preventing these TFs from binding and activating their own response elements (TREs) in a process known as transrepression. Many, but not all, of the anti-inflammatory activities of GCs are mediated by transrepression.
In contrast to transactivation, during transrepression, activated GRα monomers bind to other transcription factors (TFs) (e.g. AP-1, NF-κB) independently of direct DNA contact to block them from activating expression of genes that these transcription factors regulate [2,4](Figure 2). This type of protein-protein interaction between monomeric GRα and TFs bound to DNA is also an important mechanism of GR signaling. Moreover, during transrepression, GRα can also bind to TFs not already bound to DNA or it can compete with overlapping DNA binding sites[17], and can also repress gene transcription by tethering or through composite mechanisms. Most of the anti-inflammatory activities of GCs are due to transrepression mechanism in the absence of GRα DNA binding[24–26]. GRα inhibits the activities of inflammatory cytokines AP-1 and NF-κB by binding to the Jun subunit of AP-1[27] and the p65 subunit of NF-κB[28], thereby interfering with activation of these proteins.
GC-induced gene expression is cell-type and tissue-type dependent, which is dependent on chromatin accessibility and GR binding site availability. Furthermore, co-activators that influence posttranslational modifications of histones (acetylation and methylation), act as integrators of transcriptional regulation of GRα[4,29]. The steroid receptor co-activator (SRC) family of proteins contribute to GR transactivation. These co-activators alter chromatin structure making it more accessible to general TFs and the RNA polymerase complex, further stabilizing the transcriptional machinery leading to enhanced transcription of steroid-activated genes. Some of the effects of GCs on the TM are likely due to varying expression of these co-activators. In a recent study, Bermudez et al[30] used RNA sequencing to identify 93 and 606 differentially expressed genes in different expression groups between GC responder and non-responder in bovine TM cells encompassing 35 pathways associated with these differentially expressed genes using RNA sequencing. Since, human and bovine have similar GC responder rate[31], these genes and pathways might explain differential GC responsiveness in the human eye. However, we still need to know the exact function of each gene/pathway in GC-induced OHT and GC-induced glaucoma and its potential correlation to human studies.
Overall GC responsiveness is also regulated by alternative splicing of the GR. The biologically active GRα isoform contains the terminal exon 9α, which encodes the ligand binding domain. Alternatively splicing within intron 8 can cause cutting out exon 9α and replacing with exon 9β to encode the terminal domain that causes a truncated isoform GRβ that lacks the classical ligand binding domain. GRβ acts as a dominant negative regulator of GC activity by inhibiting GRα binding to and activating GREs[32]. GRβ does not appear to prevent GRα transrepression[33] suggesting that GRβ would not suppress the major anti-inflammatory activities of GRα mediated via transrepression. Human TM cells express both GRα and GRβ isoforms, but TM cells isolated from POAG donors have lower levels of GRβ, making these GTM cells much more susceptible to GC-induced gene expression[34–37]. This may explain why the vast majority of POAG patients are GC responders compared to controls[38] and also may explain the differences in high and low GC responsiveness in normals.
In addition to GC effects on gene expression, there also are rapid responses seen within seconds or minutes after GC administration. These rapid responses are due to non-genomic mechanisms of action of cytosolic or membrane bound GRs. In contrast to genomic effects of GR, non-genomic actions of GCs do not require RNA or protein synthesis[39]. A number of studies have reported the non-genomic functions of GR. After GC binds to the inactive GR complex in the cytoplasm and activates GR, the conformational change releases the accessory molecules from GC-GR complex, and these molecules are believed to participate in the secondary non-genomic signaling of GR. One example is c-Src release from inactive GC-GR protein complex. c-Src activates signaling cascades that inhibit phospholipase A2 activity and impair the release of arachidonic acid[40,41]. They also utilize activity of various protein kinases such as phosphoinositide 3-kinase, AKT, and mitogen-activated protein kinases[42] by rapidly increasing concentrations of secondary intracellular messengers such as calcium ions treated with high doses of GCs[43–45]. Some non-genomic effects of GR might be of therapeutic value as GCs induce short-term therapeutic benefits. Although other steroid hormones have membrane receptors, little is known about membrane-bound GRs or any non-genomic actions of GCs in the TM. There is one report showing rapid increase in IOP in POAG patients following dexamethasone treatment[46]. However, whether this effect was genomic or non-genomic needs further confirmation. It would be worth exploring functional and clinical relevance of this non-genomic effects of GR or membrane bound GRs in the TM, as they may be capable of mediating therapeutically relevant GC effects.
2. Glucocorticoid-Induced Ocular Hypertension and Glaucoma
It has been more than fifty years since the first report of steroid-induced ocular hypertension (OH), In the normal population approximately 5% of individual will respond to glucocorticoid (GC) treatment with a high intraocular pressure (IOP), whereas 33% with a moderate increase in IOP to GC treatment. Therefore, not all patients respond to glucocorticoid (GC) administration with elevated intraocular pressure (IOP). Initially it was thought that responders fell into two groups, those showing a significant increase in pressure and those patients with no increase. Subsequently, it was determined that the variability in responsiveness can be separated into three groups; those with high IOP responses (>15 mmHg); those with moderate increases (>6 to 15 mmHg) and those with a low response < 6 mmHg)[47]. It was originally thought that these differences have a genetic linkage with only a variation in a single gene[48] and therefore high responders may have been homozygotes and moderates heterozygotes[48]. Others, however, could not confirm this observation particularly when comparisons were made of IOP responsiveness in monozygotic and dizygotic twin studies[49,50]. In recent genome wide association studies (GWASs) that examined millions of single nucleotide polymorphisms (SNP), a novel orphan G protein coupled receptor (GPCR), GPR158, was identified and this gene was expressed in trabecular meshwork cells and was increased following GC treatment[51]. However, further analysis revealed that this SNP did not meet the level of significance for the GWAS study. This group followed up on this suggestion and tested the effects of overexpression of GPR158 in cultured trabecular meshwork cells and found it stimulated an increase in the levels of tight junction proteins ZO-1 and occludin[52]. These authors suggested that such changes in these tight junction proteins are similar to that seen following GC treatment[52].
In another pharmacogenomic cohort study, a common SNP emerged that obtained GWAS significance that was located within a group of three mucin genes including MUC21, MUC22 and HCG22[53]. RNA expression studies revealed that only MUC22 and HCG22 were found in trabecular meshwork cells and that HCG22 was stimulated by IL-1 and inhibited by TGF-β and GC treatment[53]. In this report, it was suggested that HCG22 may play a role in regulating the ECM of the trabecular meshwork and that mutations in this gene disrupt the binding of GCs and result in a loss of GC-inhibition of HCG22 expression allowing accumulation of ECM components[53]. Other studies, focused on identifying polymorphisms that can account for steroid responsiveness, have been undertaken[54,55]; however, none of these studies found significant polymorphisms that were statistically different between steroid responders and non-responders.
Fingert et al. studied a large number of steroid responders and controls but were unable to detect polymorphisms in those genes closely associated with GC responses, including the GC receptor (NR2C1), the immunophilins (FKP51 and FKP52), and in the spliceosome proteins that regulate expression of the GC receptors[56]. While no direct genetic linkages have been identified to account for responders and non-responders, there has been research centered on the role of GC receptors and their regulation and relative expression may differentiate responders and non-responders.
3. Clinical Ocular Glucocorticoids and Glaucoma Risk
The ocular use of glucocorticoids has traditionally been associated with the treatment of inflammatory conditions and involves administering GCs by topical, periocular or intravitreal injections, or systemic application. During chronic administration there is a real risk of developing secondary iatrogenic open angle glaucoma and/or posterior subcapsular cataracts. The increased risk is dependent on the dose and duration of administration as well as route of administration and individual susceptibility. For instance, it has been reported that the risk of glaucoma increases with doses >7.5 mg of prednisone per day taken for ≥ 6 months[57]. Another study demonstrated an almost 2-fold increase in steroid glaucoma for patients receiving higher doses of hydrocortisone (>40mg per day). The incidence of secondary glaucoma increases for patients with uveitis who are treated long-term with GCs and occurs in greater numbers of patients with chronic uveitis[58]. The incidence of subcapsular cataracts also appears to increase with long-term GC treatment. Patients receiving prednisone of >10 mg per day for ≥ one year have a 39% chance of developing cataracts. While increased risk is associated with prolonged use, the occurrence of cataracts appears to occur only with high doses of GCs[59]. A known family history for cataracts and/or glaucoma may help determine a possible treatment regimen with GCs. If one withdraws the GC, IOP tends to return to pretreated levels. However, if the IOP elevation is prolonged, loss of retinal ganglion cells and any changes in optic nerve head tissue remodeling are not reversed when GC therapy is terminated. Typically, the increase in IOP following GC use tends to return to normal when GC therapy is less than one-year duration. The IOP returns to normal following withdrawal of GCs as a result of a release of GC-inhibition of matrix metalloproteinase gene expression and an enhance turnover of extracellular matrix protein deposits. In some cases, GC-induced ocular hypertension is concomitantly treated with glaucoma medications or with the use of laser trabeculoplasty. Ultimately removal of GC therapy may be warranted to prevent glaucomatous damage. In the Diabetic Macular Edema Trial (MEAD), it was found that following thirty-six months of intravitreal dexamethasone implant (IDI) treatment, a significant rise in IOP of greater that 10 mmHg occurred in one-third of the patients and greater than 40% required drug treatment to lower the IOP[60]. In a twenty-four-month follow-up study it was also shown that the probability of the IDI in elevating IOP was 50 to 60% but that for most patients this was mild and was restored with topical IOP lowering medication[61]. While some patients required surgery, this was not a common occurrence. However, for those glucocorticoid responders continuous monitoring is recommended for the rest of the patient’s life.
In treating some ocular inflammatory conditions, such as allergic conjunctivitis, acne rosacea, superficial punctate keratitis, herpes zoster keratitis, iritis and uveitis, topical administration of GCs is preferred over systemic administration to minimize systemic side effects. Higher GC concentrations can be administered topically to the eye to enhance local effectiveness. Ophthalmic steroids are available in various formulations including solutions, emulsions, ointments, suspensions and depot preparations (Table 1). There also are multiple GCs available for topical ocular administration that vary in their indication and dose. For some posterior inflammatory segment diseases like noninfectious uveitis, central and branch retinal vein occlusion, and macula edema, intravitreal GCs are administered. Because of the disease location, the most effective treatment approach has been to deliver GCs into the vitreous cavity. This can be accomplished through direct injection through the pars plana, use of sustained-release, or biodegradable implants, or using conjugated GC compounds. Different GCs have varying degrees of efficacy depending on their structure, bioavailability and anti-inflammatory potency (Table 2). Triamcinolone acetonide is one of the more common steroids used for intravitreal administration to treat several retinal diseases because of its potent anti-inflammatory actions and long duration (months). These retinal diseases are usually treated with long term therapy with GCs, for months or sometimes years. This may involve either repeated intravitreal injections of steroids or the use of sustained-release implants. Because of the delivery of long-term therapy with steroids, the incidence of steroid-induced glaucoma increases. Patients with a baseline IOP’s greater than 16 mmHg had a higher risk of developing ocular hypertension after a single intravitreal GC injection (53% of patients developed increased IOP) and this risk increased following the second injection (65%)[62]. These patients had a variety of retinal diseases, including non-proliferative as well as proliferative diabetic retinopathy, while others had branch retinal rein occlusion or central retinal vein occlusion. There was a similar profile of IOP elevation commonly seen with intravitreal administration of GCs. Such elevations could, if prolonged, result in glaucoma pathology. Unfortunately, with cases of inflammation it is often difficult to separate GC effects from that seen with prolonged inflammation that may be the result of damage to the normal ocular drainage system that could result in increased IOP.
Early studies have shown an important relationship between GCs and POAG. Levels of cortisol are reported to be elevated in the plasma and aqueous humor of POAG patients[63–67]. In addition, altered cortisol metabolism has been reported in TM cells[68,69] and in peripheral blood cells[70] obtained from POAG patients. Patients responsive to GCs (“steroid responders”) develop ocular hypertension with open gonioscopic angles and increased outflow resistance in the trabecular meshwork outflow pathway, similar to that seen in POAG. This elevated IOP, if persists, can lead to progressive optic nerve damage, optic disc cupping and glaucomatous visual field defects. While glucocorticoid-induced ocular hypertension is considered a drug-induced iatrogenic effect, the common features with POAG exist.
Some of the features of GC-induced ocular hypertension that resemble those seen in POAG patients include common morphological and biochemical responses (see Table 4). Most common are the findings that increases in IOP are due to impaired aqueous humor outflow through the trabecular meshwork (TM) and increased deposition of extracellular matrix material[71] including increased fibronectin deposition in TM JCT region of GC treated eyes[72]. Some other features include reorganization of TM actin cytoskeleton and formation of cross-linked actin networks (CLANS)[69]. Recently it was shown that CLANS can be induced by both GCs and TGFβ2 in confluent TM cells and TM tissues[73] and that TGFβ2 concentrations are elevated in the aqueous humor and in TM of POAG patients[74,75]. CLANS can also be found in POAG TM cells[71,76]. Common cellular and biochemical changes can be seen with POAG cells, TGFβ2-treated TM cells as well as those seen following GC administration. A recent study has shown that GC treatment increased TGFβ2 expression in the TM and that GC-OHT in mice was dependent on TGFβ2 Smad3 signaling[77]. These common pathways may reveal mechanisms responsible for the development of changes in aqueous humor outflow resistance and ultimately in the rise in IOP which, if sustained, can lead to the development of glaucoma.
Table 4.
Potential Treatment Strategies for GC-induced Ocular Hypertension
| • Cortisol Derivatives |
| ∘ Tetrahydrocortisol |
| ∘ Anecortave acetate |
| • Tissue Plasminogen Activator |
| • Gene Therapy |
| ∘ GRE-MMP1 |
| ∘ GRβ |
4. Selective glucocorticoid receptor agonists (SEGRAs)
The broad-spectrum anti-inflammatory and immunosuppressive properties of GCs (Figure 3) makes them one of the most widely prescribed medications worldwide. However, their use is limited by number of side effects associated with high dose and long-term GC therapy. Thus, understanding molecular mechanisms of GC action and glucocorticoid receptor (GR) signaling will help us tailor individual treatment plans for GC therapies to optimize GC benefits while minimizing harmful side effects. GRs influence transcription both through DNA binding dependent (transactivation) and by binding to and inhibiting the activities of other transcription factors (transrepression). It has been assumed that the undesirable side effects of GC therapy require GR transactivation activities, while the anti-inflammatory activities are due to transrepression mechanisms[24–26]. In order to design novel therapeutic targets and strategies, it is also important to very precisely understand receptor biology. The complexity in GR mechanisms has been a driving force for the discovery of novel selective glucocorticoid receptor agonists (SEGRAs) or modulators (SEGRMs). SEGRAs are compounds which promote transrepression but do not mediate transactivation[24]. Clinical enthusiasm for design of SEGRAs is based on observation that mutated GR with altered DNA-binding capacity (transactivation activity) can still show anti-inflammatory activities by binding to and inhibiting pro-inflammatory TFs[78,79]. However, some side effects such as GC-induced osteoporosis are mediated by both transactivation and transrepression, so SEGRAs do not eliminate all undesirable side effects. In addition, some of the GC mediated immunosuppressive activities also involve both GR mediated pathways[24]. Nevertheless, most of the SEGRAs developed have better safety profiles and have shown promise by selectively inducing transrepression without significant transactivation, thus reducing the risk of many systemic side effects, while maintaining anti-inflammatory activities[24]. Many SEGRAs developed have shown promise in in vitro and animal models of inflammation but are still in clinical trials[80]. Over the years there has been more research on SEGRAs, along with development of SEGRA’s for treating ocular diseases. CpdA and ZK-216346 were among the first developed and widely used SEGRAs, which favored GR transrepression and failed to induce GRα transactivation of reporter genes[81–85]. In addition, CpdA promoted the interactions between GRα and NF-κB and attenuated nuclear factor (NF)-κB, indicating induction of GRα- mediated transrepression[86]. However, CpdA was not able to induce RFP-GRα nuclear translocation in TM5 cells as reported by Dibas & Yorio[87]. This could also be due to varying binding affinity between cell lines and levels of GR in these cells[82]. Mapracorat (previously ZK-245186, and subsequently BOL-303242-X) showed full anti-inflammatory efficacy by suppressing inflammation in rabbit models of dry eye and paracentesis-induced inflammation but did not affect body weight. Although not as effective as DEX in inducing OHT, mapracorat still increased IOP compared to vehicle treated rabbits[88]. ZK209614 in form of eye drops suppressed carrageenan- and allergic-conjunctivitis, and did not raise cat IOP to the same degree as the potent GC betamethasone[89]. There was a better therapeutic index showing both anti-allergic and anti-inflammatory effects without unwanted IOP elevation. Mapracorat and BOL-303242 suppressed cytokine production in a number of different ocular cell types and appeared to have several anti-inflammatory activities that were different from traditional GCs[90]. In another study, investigators evaluated myocilin expression in cultured monkey TM cells by BOL-303242-X compared with DEX and prednisolone acetate[91]. BOL-303242-X did not induce myocilin expression (at both mRNA and protein levels) to the extent of DEX or prednisolone acetate, suggesting that reduced transactivation may limit effects on conventional outflow pathway. The relatively new SEGRA GW870086X demonstrated full transrepressive activity, and its transactivation was dramatically diminished compared to DEX[92] in cellular and murine model systems of lung and delayed-type hypersensitivity induced inflammation. Stamer and colleagues evaluated the effect of GW870086X on cultured human TM cells and showed that this SEGRA induced fibronectin production (although less compared to DEX and prednisolone acetate) but did not induce myocilin (similar to BOL-303242 study[91]) or affect hydraulic conductivity[93]. This compound was not entirely devoid of transactivation activity with respect to gene expression regulation. Because of variable responses to these different SEGRAs compared to traditional GCs, gene expression profiling of novel SEGRAs compared to GCs will be required in order to fully understand GR-related pharmacology. More pre-clinical and clinical studies are needed to determine the role of SEGRAs in IOP and outflow facility regulation. The GR mechanisms (transactivation and/or transrepression) regulating GC-induced OHT was previously unknown. There are also different transcriptional and translational isoforms of GR with varying ratios of GR isoforms in different cell types, so the final effects of SEGRAs may also differ. Whether SEGRAs can modulate levels of GR isoforms is also not yet known. Although results have not been disclosed, a Phase III trial (NCT00905450) to evaluate an ophthalmic suspension of BOL-303242-X (Mapracorat) for the treatment of inflammation following cataract surgery is ongoing (sponsored by Bausch & Lomb). At the moment, no SEGRA has been approved for clinical use. Most of the SEGRA’s developed and used preclinically have been applied topically for treating skin or eye disorders[24,88,90] with minimal to no side-effects, their systemic use may add another approach in managing side-effects.
Figure 3. Glucocorticoid anti-inflammatory and immunosuppressive mechanisms of action.
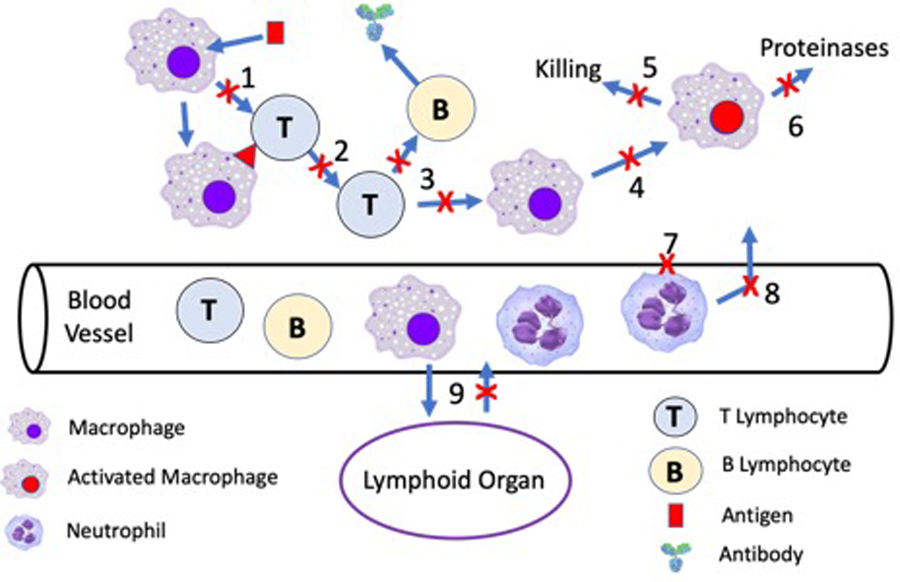
A major reason for the unparalleled anti-inflammatory and immunosuppressive activity of GCs is due to multiple sites of action at many different sites of the inflammatory cascade. GCs inhibit the production of inflammatory cytokines required for T cell maturation (1–2), B cell maturation (3), and macrophage activation (4). Prevention of macrophage activation inhibits macrophage cytotoxicity (5) and elaboration of proteinases (6). GCs tighten vascular endothelial cell junctions (7) which inhibits neutrophil adhesion to vascular endothelial cells and extravasation from the blood vessels to the site of inflammation (8). In addition, GCs stimulate migrations of inflammatory cells into lymphoid organs and inhibit their homing to sites of inflammation (9).
5. GR-dimerization defective (GRdim) transgenic mice
To assist in identifying what GR actions play a role in GC-induced side effects, new approaches are needed to identify components and the precise roles of GR-mediated mechanisms in the pathophysiology of GC-induced ocular hypertension. The lack of good models hinders development of therapeutic approaches that minimize the undesirable side effects of the GCs, while preserving or maximizing their beneficial actions. GRα transactivation requires binding of receptor homodimers to specific GREs. The D loop of the second zinc finger in DBD of GR is crucial for GR dimerization and GR binding to GREs in target genes[94–96]. As mentioned earlier, many of the undesirable side effects of GC therapy require the transactivation functions of GR, while anti-inflammatory activities are due to transrepression[25,97,98]. Therefore, altering the DNA binding domain might separate these two of GR functions[99]. Reichert and colleagues[100] successfully developed a GRdim transgenic mouse model carrying the point mutation A458T in the GR-DBD. These mice represent an important research tool to dissect different GR functions. GRdim mice remain viable and appear normal[100]. In contrast, full GR-/GR- knockout mice had developmental anomalies and died shortly after birth, demonstrating that both transactivation and transrepression functions of GR are essential for survival[101]. Interestingly, in GRdim mice GRE-dependent gene transcription of GRα is diminished, while the transrepression function (cross-talk with AP-1 and NF-κB) of GRα still remains intact[100,102–104]. Although the exact molecular characteristics of the GRdim mutation are debatable[105,106], this mouse line has become important tool to study the mode of GC actions in different diseases. The studies using GRdim mice will not only serve as a genetic model for a novel class of potential selective GR modulators [26,107] but also will provide a model to study the mechanisms whereby GR mediates its effects on target genes and cellular pathways, in particular, pathways associated with the therapeutically undesirable side effects of GR activation. Initial studies performed with GRdim mutant mice demonstrate that this single point mutation reduces dimerization and can be an important tool to dissect the importance of GR functions dependent on DNA binding and those mediated by protein–protein interactions. In addition, studies using GRdim mice will also elucidate GR mechanisms, either transactivation or transrepression, responsible for regulating GC induced OHT and GC responsiveness in the TM. Recently, Patel and colleagues used GRdim mice to provide the first evidence that GR transactivation is responsible for the GC-induced ocular hypertension as well as GC-induction of fibronectin and CLANs in TM cells[108]. This discovery provides insight into how one may target inflammation with GC treatment without the OHT and potential to develop glaucoma. SEGRAs are a start but more selective transrepressor drugs are needed to eliminate this OHT side effect of GCs.
6. THERAPEUTIC APPROACHES TO TREAT GC-OHT
There are a number of diseases that require chronic GC therapies to prevent inflammatory/immune disease associated tissue damage. For example, many patients with chronic uveitis must continue anti-inflammatory therapy to prevent uveitis damage to the retina leading to vision loss and blindness. Local and systemic GCs are commonly used to prevent this vision loss; however, close to half of these patients develop GC-induced ocular hypertension that leads to iatrogenic open-angle glaucoma. While this form of glaucoma initially can be treated by topical ocular glaucoma medications, many of these patients also require glaucoma filtration surgery, including less invasive GATT surgery[109], in attempt to prevent vision loss from steroid-glaucoma. This presents a therapeutic conundrum.
There have been three major experimental approaches to inhibit or block IOP elevation in animal models of GC-OHT (Table 4). The first approach has used concomitant administration of ocular steroid analogs along with the anti-inflammatory GC. Topical ocular administration of the natural cortisol metabolite tetrahydrocortisol reversed DEX-induced ocular hypertension in rabbits[110]. It is interesting to note that this compound also blocked DEX-induced CLAN formation in cultured human TM cells[111], suggesting a potential mechanism by which this metabolite may work. Another cortisol analog, anecortave acetate, was designed as an IOP lowering cortisene. In an open label clinical trial, subTenon’s injection of an anecortave acetate suspension lowered IOP in GC-induced OHT patients[112]. The average starting IOP was over 31 mmHg, and anecortave acetate provided a sustained (up to 6 months) 30% IOP lowering. It is important to note that these 2 steroid analogs are not GC receptor antagonists, so they should not interfere with the anti-inflammatory effects of GCs[113].
Danias, Candia and colleagues have shown that either intravitreal or intracameral injection of tissue plasminogen activator (tPA) significantly lowered IOP in prednisolone-induced ocular hypertensive sheep[114,115]. Sheep receiving topical ocular prednisolone developed an approximately 10 mmHg increase in IOP. Administering tPA both reversed and prevented this GC-induced ocular hypertension. GC-OHT is associated with increased deposition of extracellular matrix (ECM) proteins in the TM. Plasminogen activators are able to degrade ECM proteins, but more importantly, the plasminogen activators cleave pro-matrix metalloproteinase (proMMP) zymogens to activate MMPs, which are more effective in degrading ECM proteins.
The third unique approach to treat GC-OHT involves gene therapy. As previously mentioned, GCs increase ECM deposition in the TM that increases aqueous outflow resistance and elevates IOP. Borras and colleagues developed a viral vector that drives the matrix metalloproteinase-1 (MMP1) under the control of a glucocorticoid response element (GRE). They transduced cultured TM cells and showed that this vector is silent with little MMP1 expressed until the cells were exposed to DEX[116]. This vector was also used to transduce the TM of sheep eyes that had prednisolone-induced ocular hypertension, and GC activation MMP1 both prevented and reversed this steroid-induced ocular hypertension[117,118]. A different gene therapy approach has also been used to suppress DEX-induced ocular hypertension in mice. There are two major alternatively spliced isoforms of the glucocorticoid receptor, GRα and GRβ. The GRβ isoform acts as a dominant regulator of GC activity and suppresses GC effects in cultured TM cells[119]. Recently, we developed a mouse model of DEX-induced OHT in mice by weekly posterior fornix periocular injections of a suspension of DEX-acetate[120]. Transduction of the mouse TM with Ad5.GRβ totally reversed DEX-OHT, and the IOPs remained at baseline levels despite continued DEX injections[121]. Since Ad5 vectors have selective tropism for the TM[122,123], selective expression of GRβ in the TM should block GC-OHT but not the anti-inflammatory effects of GCs in other ocular tissues.
It is anticipated that one or more of these approaches will be further tested clinically in steroid responder patients so that these patients can continue to receive the unsurpassed anti-inflammatory actions of GCs without manifestation of the serious and vision threating side effect of GC-induced OHT.
7. Conclusions
GCs have both anti-inflammatory and immunosuppressive activities and are among the most commonly prescribed drugs.
GC molecular mechanisms of action are complex, involving transactivation, transrepression, and alternative splicing for the GR
GC therapy can cause significant systemic side effects
GC therapy can cause ocular side effects, including posterior subcapsular cataracts and iatrogenic ocular hypertension and glaucoma, both of which can be vision threatening
There are a number of novel approaches that need to be tested in the clinic that may limit GC-OHT and prevent potential iatrogenic glaucoma
8. Expert Opinion
Glucocorticoids continue to be a major therapy for treating inflammatory eye diseases like uveitis, optic neuritis, conjunctivitis, keratitis and others. Unfortunately, the continued use of GCs results in unwanted ocular side effects, the most important of which is elevation of intraocular pressure and induction of glaucoma. A better understanding of the molecular mechanisms responsible for GC anti-inflammatory and side effect activities will allow dissection of the desired vs undesired activities of GCs. The GCs mechanism of action mediated through the GR initiates transrepression and transactivation. In transrepression, the ligand bound glucocorticoid receptor can inhibit the transcriptional activity of other transcription factors and this is responsible for much of the anti-inflammatory activities of GCs. In contrast, GR transactivation specifically increases the rate of gene expression and increases multiple proteins when GR binds to GC responsive elements to promote gene expression. A number of these expressed proteins contribute to increased intraocular pressure mediated by GCs. This understanding will likely lead to the design of anti-inflammatory agents that don’t have major GC side effects. For example, identifying specific molecules that mimic GC transrepression activity without transactivation activity will be a major breakthrough for eliminating the unwanted ocular hypertensive response while retaining the anti-inflammatory activity. An example of this approach is evident in the development of selective glucocorticoid receptor agonists (SEGRAs) that were the first attempt at separating the GR transrepressor from the transactivation activities These compounds were synthetic steroids that showed some promise. More recently selective glucocorticoid receptor modulators (SEGRAMS) having nonsteroidal structures were developed that appear to retain anti-inflammatory activity with minimal transactivation noted. Some of these agents are in clinical trials and awaiting further development. The recent use of mouse models with point mutations to eliminate the interaction of GCs with glucocorticoid response elements on DNA provides a tool to separate the transrepressor from the transactivation activity of GCs. Recent studies demonstrate that the glucocorticoid receptor transactivation is responsible for the GC-induced ocular hypertension as well as some of the extracellular matrix effects seen that is responsible for the decrease in aqueous humor outflow. Such a tool will be helpful in identifying potential new molecules that can be tested to provide the needed ocular anti-inflammatory activity but eliminate the ocular hypertensive side effect associated with GC administration. However, not all anti-inflammatory/immunosuppressive actions of GCs are mediated via transrepression, so it is likely that these new agents will not be more efficacious than GCs with their wide spectrum of activities. While there is much promise in the development of such molecules, there is considerable amount of in vivo and in vitro testing that needs to occur prior to their evaluation in clinical trials. While some of these agents are being evaluated there are no compounds yet available clinically that limit the actions of GC-like agents to their anti-inflammatory actions.
Other therapeutic approaches endeavor to modify the molecular pathogenic processes associated with the development of GC-OHT. Intraocular administration of tPA is efficacious in lowering IOP in an animal model of GC-OHT, most likely through degradation of pathological deposits of ECM in the aqueous outflow pathway. Viral vector transduction of the TM using a GRE regulated MMP (Ad5.GRE.MMP1) also has been used to lower IOP in an animal model of GC-OHT. This IOP lowering activity also presumably is through the degradation of outflow ECM.
Article Highlights:
It has been over fifty years since the first report of steroid-induced ocular hypertension resulting from chronic glucocorticoid (GC) administration, particularly from their use in ocular inflammation.
Glucocorticoid ocular hypertension responsiveness varies within the normal population with 5% of individuals having a high IOP following GC administration and 33% with a moderate response.
Primary open-angle glaucoma patients almost all respond with an elevation in intraocular pressure following ocular delivery of GCs.
Present genome wide association studies (GWAS data) on GC responsiveness have not identified specific genes responsible for this difference in responsiveness. However, research efforts have identified the subtype of glucocorticoid receptor, GRβ, may play a role in GC responsiveness. GC OH is dependent on GC potency, route of administration, duration of therapy and individual susceptibility.
Novel approaches to separate the anti-inflammatory action from its effects on intraocular pressure resulted in a new class of Selective Glucocorticoid Receptor Agonists (SEGRAs) that differentiate the transactivation from transrepression effects of GCs.
Funding:
Manuscript has been funded in part by National Institutes of Health (grant 2 R01 EY016242).
Footnotes
Declaration of Interest:
Thomas Yorio and Abbot F. Clark have received previous funding from National Institutes of Health (NIH) for this study. Guarang C. Patel is also an employee of Regeneron. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References:
Papers of special note have been highlighted as either of interest (*) or of considerable interest (**) to readers.
- 1.Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids: new mechanisms for old drugs. N Engl J Med 2005; 353:1711–3. [DOI] [PubMed] [Google Scholar]
- 2. Ramamoorthy S, Cidlowski JA: Corticosteroids: Mechanisms of Action in Health and Disease. Rheum Dis Clin North Am 2016, 42:15–31, vii. **General Review of GC Mechanism of Action. Provides information regarding transactivation and transrepressor mechanisms of GCs.
- 3.Webster JI, Tonelli L, Sternberg EM: Neuroendocrine regulation of immunity. Annu Rev Immunol 2002, 20:125–63. [DOI] [PubMed] [Google Scholar]
- 4. Wordinger RJ, Clark AF: Effects of glucocorticoids on the trabecular meshwork: towards a better understanding of glaucoma. Progress in retinal and eye research 1999, 18:629–67. *Excellent review of actions of GCs and their effects in raising IOP.
- 5.Seckl JR: 11beta-hydroxysteroid dehydrogenases: changing glucocorticoid action. Curr Opin Pharmacol 2004, 4:597–602. [DOI] [PubMed] [Google Scholar]
- 6.Cooper MS, Stewart PM: 11Beta-hydroxysteroid dehydrogenase type 1 and its role in the hypothalamus-pituitary-adrenal axis, metabolic syndrome, and inflammation. J Clin Endocrinol Metab 2009, 94:4645–54. [DOI] [PubMed] [Google Scholar]
- 7.Weinstein BI, Iyer RB, Binstock JM, Hamby CV, Schwartz IS, Moy FH, Wandel T, Southren AL: Decreased 3 alpha-hydroxysteroid dehydrogenase activity in peripheral blood lymphocytes from patients with primary open angle glaucoma. Exp Eye Res 1996, 62:39–45. [DOI] [PubMed] [Google Scholar]
- 8.Weinstein BI, Munnangi P, Gordon GG, Southren AL: Defects in cortisol-metabolizing enzymes in primary open-angle glaucoma. Invest Ophthalmol Vis Sci 1985, 26:890–3. [PubMed] [Google Scholar]
- 9.Stokes J, Noble J, Brett L, Phillips C, Seckl JR, O’Brien C, Andrew R: Distribution of glucocorticoid and mineralocorticoid receptors and 11beta-hydroxysteroid dehydrogenases in human and rat ocular tissues. Invest Ophthalmol Vis Sci 2000, 41:1629–38. [PubMed] [Google Scholar]
- 10.Onyimba CU, Vijapurapu N, Curnow SJ, Khosla P, Stewart PM, Murray PI, Walker EA, Rauz S: Characterisation of the prereceptor regulation of glucocorticoids in the anterior segment of the rabbit eye. J Endocrinol 2006, 190:483–93. [DOI] [PubMed] [Google Scholar]
- 11.Rauz S, Cheung Cm Fau - Wood PJ, Wood Pj Fau - Coca-Prados M, Coca-Prados M Fau - Walker EA, Walker Ea Fau - Murray PI, Murray Pi Fau - Stewart PM, Stewart PM: Inhibition of 11beta-hydroxysteroid dehydrogenase type 1 lowers intraocular pressure in patients with ocular hypertension. QJM 2003; 96(7):481–90. [DOI] [PubMed] [Google Scholar]
- 12.Kj Choi, Na Y-J, Jung WH, et al. Protective effect of a novel selective 11β-HSD1 inhibitor on eye ischemis-reperfusion induced glaucoma. Biochem Pharmacol 2019; 169 113632: 1–11. [DOI] [PubMed] [Google Scholar]
- 13.Kwok AK, Lam DS, Ng JS, et al. Ocular hypertensive response to topical steroids in children, Ophthalmology 1997; 104:2112–16. [DOI] [PubMed] [Google Scholar]
- 14.Ng JS, Fan DS, Young AL, Yip NK et al. Ocular hypertensive response to topical dexamethasone in children: a dose-dependent phenomenon. Ophthalmol 2000; 107:2097–100. [DOI] [PubMed] [Google Scholar]
- 15.Lam DS, Fan DS, Ng JS, Yu CB et al. Ocular hypertensive and anti-inflammatory responses to different dosages of topical dexamethasone in children: a randomized trial. Clin Exp. Ophthalmol 2005. 33:252–8. [DOI] [PubMed] [Google Scholar]
- 16.Evans RM: The steroid and thyroid hormone receptor superfamily. Science 1988; 240:889–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scheschowitsch K, Leite J, Assreuy J: New insights in glucocorticoid receptor signaling – more than just a ligand binding receptor. Frontiers in Endocrinology 2017, 8:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grad I, Picard D: The glucocorticoid responses are shaped by molecular chaperones. Molecular and Cellular Endocrinology 2007, 275:2–12. [DOI] [PubMed] [Google Scholar]
- 19.Pratt WB, Toft DO: Steroid Receptor Interactions with Heat Shock Protein and Immunophilin Chaperones*. Endocrine Reviews 1997, 18:306–60. [DOI] [PubMed] [Google Scholar]
- 20.Lim HW, Uhlenhaut NH, Rauch A, Weiner J, Hubner S, Hubner N, Won KJ, Lazar MA, Tuckermann J, Steger DJ: Genomic redistribution of GR monomers and dimers mediates transcriptional response to exogenous glucocorticoid in vivo. Genome Res 2015, 25:836–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beato M: Gene regulation by steroid hormones. Cell 1989, 56:335–44. [DOI] [PubMed] [Google Scholar]
- 22.Beato M, Chalepakis G, Schauer M, Slater EP: DNA regulatory elements for steroid hormones. Journal of steroid biochemistry 1989, 32:737–47. [DOI] [PubMed] [Google Scholar]
- 23.Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, Li M, Chambon P: Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell 2011, 145:224–41. [DOI] [PubMed] [Google Scholar]
- 24.Sundahl N, Bridelance J, Libert C, De Bosscher K, Beck IM: Selective glucocorticoid receptor modulation: New directions with non-steroidal scaffolds. Pharmacology & therapeutics 2015, 152:28–41. [DOI] [PubMed] [Google Scholar]
- 25.Cain DW, Cidlowski JA: Specificity and sensitivity of glucocorticoid signaling in health and disease. Best practice & researchClinical endocrinology & metabolism 2015, 29:545–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleiman A, Tuckermann JP: Glucocorticoid receptor action in beneficial and side effects of steroid therapy: lessons from conditional knockout mice. Molecular and cellular endocrinology 2007, 275:98–108. [DOI] [PubMed] [Google Scholar]
- 27.Yang-Yen H-F, Chambard J-C, Sun Y-L, Smeal T, Schmidt TJ, Drouin J, Karin M: Transcriptional interference between c-Jun and the glucocorticoid receptor: Mutual inhibition of DNA binding due to direct protein-protein interaction. Cell 1990, 62:1205–15. [DOI] [PubMed] [Google Scholar]
- 28.Nissen RM, Yamamoto KR: The glucocorticoid receptor inhibits NFkappaB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev 2000, 14:2314–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lonard DM, O’Malley BW: Expanding functional diversity of the coactivators. Trends in Biochemical Sciences 2005, 30:126–32. [DOI] [PubMed] [Google Scholar]
- 30.Bermudez JY, Webber HC, Brown B, Braun TA, Clark AF, Mao W: A Comparison of Gene Expression Profiles between Glucocorticoid Responder and Non-Responder Bovine Trabecular Meshwork Cells Using RNA Sequencing. PLoS ONE 2017, 12:e0169671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mao W, Tovar-Vidales T, Yorio T, Wordinger RJ, Clark AF: Perfusion-Cultured Bovine Anterior Segments as an Ex Vivo Model for Studying Glucocorticoid-Induced Ocular Hypertension and Glaucoma. Investigative Ophthalmology & Visual Science 2011, 52:8068–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oakley RH, SAR M, Cidlowski JA: The human glucocorticoid receptor beta isoform, Expression, biochemical properties, and putative function. J Biol Chem 1996, 271(16):9550–9. [DOI] [PubMed] [Google Scholar]
- 33.Gougat C, Jaffuel D, Gagliardo R, Henriquet C, Bousquet J, Demoly P, Mathieu M: Overexpression of the human glucocorticoid receptor alpha and beta isoforms inhibits AP-1 and NF-kappaB activities hormone independently. J Mol Med 2002, 80(5):309–18. [DOI] [PubMed] [Google Scholar]
- 34. Zhang X, Clark AF, Yorio T: Regulation of glucocorticoid responsiveness in glaucomatous trabecular meshwork cells by glucocorticoid receptor-beta. Invest Ophthalmol Vis Sci 2005, 46(120):4607–16. **First report of the GC subtype receptor responses and the role of GRβ in GC ocular hypertension response.
- 35.Zhang X, Ognibene CM, Clark AF, Yorio T: Dexamethasone inhibition of trabecular meshwork cell phagocytosis and its modulation by glucocorticoid receptor beta. Exp Eye Res 2007, 84(2):275–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jain A, Wordinger TJ, Yorio T, Clark AF: Spliceosome protein (SRp) regulation of glucocorticoid receptor isoforms and glucocorticoid response in human trabecular meshwork cells. Invest Ophthalmol Vis Sci 2012, 53(2):857–66. **Studies demonstrating how GC subtype receptors may be regulated.
- 37. Jain A, Wordinger RJ, Yorio T, Clark AF: Role of the alternatively spliced glucocorticoid receptor isoform GRβ in steroid responsiveness and glaucoma. J Ocul Pharmacol Ther 2014, 30(2–3):121–7. **Study demonstrating the role of GRβ in steroid responsiveness and glaucoma.
- 38.Becker B, Mills DW: Coricosteroids and Intraocular Pressure. Arch Ophthal 1963, 70:500–7. [DOI] [PubMed] [Google Scholar]
- 39.Groeneweg FL, Karst H, de Kloet ER, Joëls M: Mineralocorticoid and glucocorticoid receptors at the neuronal membrane, regulators of nongenomic corticosteroid signalling. Molecular and Cellular Endocrinology 2012, 350:299–309. [DOI] [PubMed] [Google Scholar]
- 40.Solito E, Mulla A, Morris JF, Christian HC, Flower RJ, Buckingham JC: Dexamethasone induces rapid serine-phosphorylation and membrane translocation of annexin 1 in a human folliculostellate cell line via a novel nongenomic mechanism involving the glucocorticoid receptor, protein kinase C, phosphatidylinositol 3-kinase, and mitogen-activated protein kinase. Endocrinology 2003, 144:1164–74. [DOI] [PubMed] [Google Scholar]
- 41.Croxtall JD, Choudhury Q, Flower RJ: Glucocorticoids act within minutes to inhibit recruitment of signalling factors to activated EGF receptors through a receptor-dependent, transcription-independent mechanism. British Journal of Pharmacology 2000, 130:289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Samarasinghe RA, Witchell SF, DeFranco DB: Cooperativity and complementarity: synergies in non-classical and classical glucocorticoid signaling. Cell Cycle 2012, 11:2819–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lowenberg M, Stahn C, Hommes DW, Buttgereit F: Novel insights into mechanisms of glucocorticoid action and the development of new glucocorticoid receptor ligands. Steroids 2008, 73:1025–9. [DOI] [PubMed] [Google Scholar]
- 44.De Bosscher K, Haegeman G: Minireview: latest perspectives on antiinflammatory actions of glucocorticoids. Mol Endocrinol 2009, 23:281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Norman AW, Mizwicki MT, Norman DP: Steroid-hormone rapid actions, membrane receptors and a conformational ensemble model. Nat Rev Drug Discov 2004, 3:27–41. [DOI] [PubMed] [Google Scholar]
- 46.Weinreb RN, Polansky JR, Kramer SG, Baxter JD: Acute effects of dexamethasone on intraocular pressure in glaucoma. Invest Ophthalmol Vis Sci 1985, 26:170–5. [PubMed] [Google Scholar]
- 47.Becker B Intraocular pressure response to topical corticosteroids. Invest. Ophthalmol 1965; 4:198–205. [PubMed] [Google Scholar]
- 48.Becker B, Hahn KA. Topical corticosteroids and heredity in primary open-angle glaucoma. Am J Ophthalmol 1964; 57:543–51. [DOI] [PubMed] [Google Scholar]
- 49.Armaly MF, Statistical attrubutes of the steroid hypertensive response in the clinically normal eye. I. The demonstration of three levels of response. Invest Ophthalmol 1965; 4:187–97. [PubMed] [Google Scholar]
- 50.Schwartz JT, Reuling FH, Feinleib M, Garrison RJ, Collie DJ. Twin study on ocular pressure following topically applied dexamethasone. II. Inheritance of variation in pressure response. Arch Ophthalmol 1973; 90:281–6. [DOI] [PubMed] [Google Scholar]
- 51.Fini ME, Schwartz SG, Gao X, Jeong S, Patel N, Itakura T, Price MO, Price FW Jr, Varma R, Stamer WD. Steroid-induced ocular hypertension/glaucoma: Focus on pharmacogenomics and implications for precision medicine. Prog Retinal Eye Res 2017; 56:58–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patel N, Itakura T, Gonzalez JM Jr, Schwartz SG, Fini ME. GPR158, an orphan member of G protein-coupled receptor family C: glucocorticoid-stimulated expression and novel nuclear role. PloS One 2015; 10(2): e0117758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jeong S, Patel N, Edlund CK, Hartialal J, Hazelett DJ, Itakura T, Wu PC, Avery RL, Davis JL, Flynn HW, Lalwani G, Puliafito CA, Wafapoor H, Hijikata M, Keicho N, Gao X, Argueso P, Allayee H, Coetzee GA, Pletcher MT, Conti DV, Schwartz SG, Eaton AM, Fini ME. Identification of a novel mucin gene HCG22 associated with steroid-induced ocular hypertension. Investig Ophthalmol Vis Sci 2015; 56:2737–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hogewind BF, Micheal S, Bakker B, Hoyng CB, den Hollander AI. Analysis of single nucleotide polymorphisms in the SFRS3 and FKBP4 genes in corticosteroid-induced ocular hypertension. Ophthalmic Genet 2012; 33:221–4. [DOI] [PubMed] [Google Scholar]
- 55.Hogewind BF, Micheal S, Schoenmaker-Koller FE, Hoyng CB, den Hollander AI. Analyses of sequence variants in the MYOC gene and of single nucleotide polymorphisms in the NR3C1 and FKBP5 genes in corticosteroid-induced ocular hypertension. Ophthalmic Genet 2015; 36:299–302. [DOI] [PubMed] [Google Scholar]
- 56.Fingert JH, Alward WL, Wang K, Yorio T, Clark AF. Assessmen of SNPs associated with the human glucocorticoid receptor in primary open-angle glaucoma and steroid responders. Mol Vis 2010; 16:596–601. [PMC free article] [PubMed] [Google Scholar]
- 57.Huscher D, Thiele K, Gromnica-Ihle E, Hein G, Demary W, Demary W, Dreher R, Zink A, Buttgereit F. Dose-related patterns of glucocorticoid-induced side effects. Ann Rheum Dis 2009; 68:1119–24. [DOI] [PubMed] [Google Scholar]
- 58.Herbert HM, Viswanathan A, Jackson H, Lightman SI. Risk factors for elevated intraocular pressure in uveitis. J Glaucoma 2004; 13:96–9. [DOI] [PubMed] [Google Scholar]
- 59.Caplan A, Fett N, Rosenbach M, Werth VP, Micheletti RG. Prevention and management of glucocorticoid-induced side effects: A comprehensive review. J Am Acad Dermatol 2017; 76:201–7. [DOI] [PubMed] [Google Scholar]
- 60.Boyer DS, Yoon YH, Belfort R Jr, Bandello F et al. Three-year randomized, sham-controlled trial of dexamethasone intravitreal implant in patients with diabetic macular edema. Ophthalmology 2014; 121:1904–14. [DOI] [PubMed] [Google Scholar]
- 61.Zarranz-Ventura J, Sala-Puigdollers A, Velazquez-Villoria D et al. Long-term probability of intraocular pressure elevation with intravitreal dexamethasone implant in the real-world. PLOS ONE 2019; 14(1):e0209997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rhee DJ, Peck RE, Belmont J, Martidis A, Liu M et al. Intraocular pressure alterations following intravitreal triamcinolone acetonide. Br J Ophthalmol 2006; 90:999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ray S, Mehra KS, Misra S, Singh R. Plasma cortisol in glaucoma. Ann Opthalmol 1977; 9:1151–4. [PubMed] [Google Scholar]
- 64.Rozsival P, Hampl R, Obenberger J, Starka L, Rehak S. Aqueous humor and plasma cortisol levels in glaucoma and cataract patients. Curr Eye Res. 1081; 1:391–6. [DOI] [PubMed] [Google Scholar]
- 65.Meredig WE, Jentzen F, Hartmann F. Systemic side effects of topically applied corticosteroid medication (author’s translation). Klinische Monatsb;atter fur Augenheilkunde 1980; 176:907–10. [DOI] [PubMed] [Google Scholar]
- 66.Schwartz B, Seddon JM. Increased plasma cortisol levels in ocular hypertension. Arch. Ophthalmol 1981; 99:1791–1794. [DOI] [PubMed] [Google Scholar]
- 67.McCarty GR, Schwartz B. Increased plasma noncortisol glucocorticoid activity in open-angle glaucoma. Invest Ophthalmol Vis Sci 1991; 32:1600–8. [PubMed] [Google Scholar]
- 68.Southren AL, Gordon GG, Munnangi PR et al. Altered cortisol metabolism in cells cultured from trabecular meshwork specimens obtained from patients with primary open-angle glaucoma. Invest Ophthalmol Vis Sci 1983; 24:1413–7. [PubMed] [Google Scholar]
- 69.Weinstein BI, Munnangi P, Gordon GG, Southren AL. Defects in cortisol-metabolizing enzymes in primary open-angle glaucoma. Invest Ophthalmol Vis Sci 1985; 26:890–3. [PubMed] [Google Scholar]
- 70.Weinsten BI, Iyer RB, Binstock JM, Hamby CV et al. Decreased 3 alpha-hysroxysteroid dehydrogenase activity in peripheral blood lymphocytes from patients with primary open angle glaucoma. Exp Eye Res 1996; 62:29–45. [DOI] [PubMed] [Google Scholar]
- 71.Clark AF. Steroids, ocular hypertension and glaucoma. J Glaucoma 1995; 4:354–69. [PubMed] [Google Scholar]
- 72.Clark AF, Wordinger RJ. The role of steroids in outflow resistance. Exp Eye Res 2009; 88:752–9. [DOI] [PubMed] [Google Scholar]
- 73.Bermudez JY, Montecchi-Palmer M, Mao W, Clark AF. Cross-linked actin networks (CLANs) in glaucoma. Exp Eye Res 2017; 159:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tripathi RC, Li J, Chan WF, Tripathi BJ. Aqueous humor in glaucomatous eyes contains an increased level of TGF-beta2. Exp Eye Res 1994; 59:723–7. [DOI] [PubMed] [Google Scholar]
- 75.Inatani M, Tannihara H, Katsuta H, Honjo M et al. Transforming growth factor-beta 2 levels in aqueous humor of glaucomatous eyes. Graefes Arch Clin Exp Ophthalmol 2001; 239:109–13. [DOI] [PubMed] [Google Scholar]
- 76.Hoare MJ, Grierson I, Brotchie D, Pollock N, Cracknell K, Clark AF. Cross-linked actin networks (CLANs) in the trabecular meshwork of the normal and glaucomatous human eye in situ. Invest Ophthalmol Vis Sci 2009; 50:1244–63. [DOI] [PubMed] [Google Scholar]
- 77.Kesetti RB, Maddineni P, Patel PD, Searby C, Sheffield VC, Zode GS: Transforming growth factor β2 (TGFβ2) signaling plays a key role in glucocorticoid-induced ocular hypertension. J Biol Chem 2018, 293(25):9854–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reichardt SD, Weinhage T, Rotte A, Foller M, Oppermann M, Luhder F, Tuckermann JP, Lang F, van den Brandt J, Reichardt HM: Glucocorticoids induce gastroparesis in mice through depletion of l-arginine. Endocrinology 2014, 155:3899–908. [DOI] [PubMed] [Google Scholar]
- 79.Quinn M, Ramamoorthy S, Cidlowski JA: Sexually dimorphic actions of glucocorticoids: beyond chromosomes and sex hormones. Annals of the New York Academy of Sciences 2014, 1317:1–6. [DOI] [PubMed] [Google Scholar]
- 80.Gessi S, Merighi S, Borea PA: Glucocorticoid’s pharmacology: past, present and future. Current pharmaceutical design 2010, 16:3540–53. [DOI] [PubMed] [Google Scholar]
- 81.De Bosscher K, Haegeman G, Elewaut D: Targeting inflammation using selective glucocorticoid receptor modulators. Curr Opin Pharmacol 2010, 10:497–504. [DOI] [PubMed] [Google Scholar]
- 82.De Bosscher K, Vanden Berghe W, Beck IM, Van Molle W, Hennuyer N, Hapgood J, Libert C, Staels B, Louw A, Haegeman G: A fully dissociated compound of plant origin for inflammatory gene repression. Proc Natl Acad Sci U S A 2005, 102:15827–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Robertson S, Allie-Reid F, Vanden Berghe W, Visser K, Binder A, Africander D, Vismer M, De Bosscher K, Hapgood J, Haegeman G, Louw A: Abrogation of glucocorticoid receptor dimerization correlates with dissociated glucocorticoid behavior of compound a. J Biol Chem 2010, 285:8061–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lesovaya E, Yemelyanov A, Swart AC, Swart P, Haegeman G, Budunova I: Discovery of Compound A--a selective activator of the glucocorticoid receptor with anti-inflammatory and anti-cancer activity. Oncotarget 2015, 6:30730–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jaroch S, Berger M, Huwe C, Krolikiewicz K, Rehwinkel H, Schäcke H, Schmees N, Skuballa W: Discovery of quinolines as selective glucocorticoid receptor agonists. Bioorganic & Medicinal Chemistry Letters 2010, 20:5835–8. [DOI] [PubMed] [Google Scholar]
- 86.Zheng Y, Ishiguro H, Ide H, Inoue S, Kashiwagi E, Kawahara T, Jalalizadeh M, Reis LO, Miyamoto H: Compound A Inhibits Bladder Cancer Growth Predominantly via Glucocorticoid Receptor Transrepression. Mol Endocrinol 2015, 29:1486–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dibas A, Yorio T: Glucocorticoid therapy and ocular hypertension. Eur J Pharmacol 2016, 787:57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shafiee A, Bucolo C, Budzynski E, Ward KW, Lopez FJ: In vivo ocular efficacy profile of mapracorat, a novel selective glucocorticoid receptor agonist, in rabbit models of ocular disease. Invest Ophthalmol Vis Sci 2011, 52:1422–30. [DOI] [PubMed] [Google Scholar]
- 89.Kato M, Hagiwara Y, Oda T, Imamura-Takai M, Aono H, Nakamura M: Beneficial pharmacological effects of selective glucocorticoid receptor agonist in external eye diseases. J Ocul Pharmacol Ther 2011, 27:353–60. [DOI] [PubMed] [Google Scholar]
- 90.Spinelli SL, Xi X, McMillan DH, Woeller CF, Richardson ME, Cavet ME, Zhang JZ, Feldon SE, Phipps RP: Mapracorat, a selective glucocorticoid receptor agonist, upregulates RelB, an anti-inflammatory nuclear factor-kappaB protein, in human ocular cells. Exp Eye Res 2014, 127:290–8. [DOI] [PubMed] [Google Scholar]
- 91.Pfeffer BA, DeWitt CA, Salvador-Silva M, Cavet ME, Lopez FJ, Ward KW: Reduced myocilin expression in cultured monkey trabecular meshwork cells induced by a selective glucocorticoid receptor agonist: comparison with steroids. Invest Ophthalmol Vis Sci 2010, 51:437–46. [DOI] [PubMed] [Google Scholar]
- 92.Uings IJ, Needham D, Matthews J, Haase M, Austin R, Angell D, Leavens K, Holt J, Biggadike K, Farrow SN: Discovery of GW870086: a potent anti-inflammatory steroid with a unique pharmacological profile. Br J Pharmacol 2013, 169:1389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stamer WD, Hoffman EA, Kurali E, Krauss AH: Unique response profile of trabecular meshwork cells to the novel selective glucocorticoid receptor agonist, GW870086X. Invest Ophthalmol Vis Sci 2013, 54:2100–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Alroy I, Freedman LP: DNA binding analysis of glucocorticoid receptor specificity mutants. Nucleic Acids Res 1992, 20:1045–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schena M, Freedman LP, Yamamoto KR: Mutations in the glucocorticoid receptor zinc finger region that distinguish interdigitated DNA binding and transcriptional enhancement activities. Genes Dev 1989. Oct;3(10):1590–601. [DOI] [PubMed] [Google Scholar]
- 96.Beck IM, De Bosscher K, Haegeman G: Glucocorticoid receptor mutants: man-made tools for functional research. Trends Endocrinol Metab 2011, 22:295–310. [DOI] [PubMed] [Google Scholar]
- 97.Kassel O, Herrlich P: Crosstalk between the glucocorticoid receptor and other transcription factors: molecular aspects. Molecular and cellular endocrinology 2007, 275:13–29. [DOI] [PubMed] [Google Scholar]
- 98.Karin M: New Twists in Gene Regulation by Glucocorticoid Receptor: Is DNA Binding Dispensable? Cell 1998, 93:487–90. [DOI] [PubMed] [Google Scholar]
- 99.Heck S, Kullmann M, Gast A, Ponta H, Rahmsdorf HJ, Herrlich P, Cato AC: A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. The EMBO journal 1994, 13:4087–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, Gass P, Schmid W, Herrlich P, Angel P, Schutz G: DNA binding of the glucocorticoid receptor is not essential for survival. Cell 1998, 93:531–41. [DOI] [PubMed] [Google Scholar]
- 101.Cole TJ, Blendy JA, Monaghan AP, Krieglstein K, Schmid W, Aguzzi A, Fantuzzi G, Hummler E, Unsicker K, Schutz G: Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes & development 1995, 9:1608–21. [DOI] [PubMed] [Google Scholar]
- 102.Reichardt HM, Tuckermann JP, Göttlicher M, Vujic M, Weih F, Angel P, Herrlich P, Schutz G: Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. The EMBO journal 2001, 20:7168–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Frijters R, Fleuren W, Toonen EJ, Tuckermann JP, Reichardt HM, van der Maaden H, van Elsas A, van Lierop MJ, Dokter W, de Vlieg J, Alkema W: Prednisolone-induced differential gene expression in mouse liver carrying wild type or a dimerization-defective glucocorticoid receptor. BMC genomics 2010, 11:359-2164-11-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tuckermann JP, Reichardt HM, Arribas R, Richter KH, Schütz G, Angel P: The DNA Binding-Independent Function of the Glucocorticoid Receptor Mediates Repression of Ap-1–Dependent Genes in Skin. 1999. pp. 1365–70. [DOI] [PMC free article] [PubMed]
- 105.Jewell CM, Scoltock AB, Hamel BL, Yudt MR, Cidlowski JA: Complex Human Glucocorticoid Receptor dim Mutations Define Glucocorticoid Induced Apoptotic Resistance in Bone Cells. Molecular Endocrinology 2012, 26:244–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Roohk DJ, Mascharak S, Khambatta C, Leung H, Hellerstein M, Harris C: Dexamethasone-mediated changes in adipose triacylglycerol metabolism are exaggerated, not diminished, in the absence of a functional GR dimerization domain. Endocrinology 2013, 154:1528–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schäcke H, Schottelius A, Döcke W-D, Strehlke P, Jaroch S, Schmees N, Rehwinkel H, Hennekes H, Asadullah K: Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. 2004. pp. 227–32. [DOI] [PMC free article] [PubMed]
- 108.Patel GC, Millar JC, Clark AF: Glucocorticoid receptor transactivation is required for glucocorticoid-induced ocular hypertension and glaucoma. Ines Ophthalmol Vis Sci 2019:1967–1978. [DOI] [PMC free article] [PubMed]
- 109.Boese EA, Shah M: Gonioscopy-assisted transluminal trabeculotomy (GATT) is an effective procedure for steroid-induced glaucoma. J Galucoma 2019, doi:1097/IJG [Epub ahead of print]. [DOI] [PubMed]
- 110.Southren AL, L’Hommedieu D, Gordon GG, Weinstein BI: Intraocular hypotensive effect of a topically applied cortisol metabolite: 3 alpha, 5 beta tetrahydrocortisol. Invest Ohthalmol Vis Sci 1987: 28(5):901–903. [PubMed] [Google Scholar]
- 111.Clark AF, Lane D, Wilson K, Miggans ST, McCartney MD: Inhibition of dexamethasone-induced cytoskeletal changes in cultured human trabecular meshwork cells by tetrahydocortisol. Invest Ophthalmol Vis Sci 1996; 37(5):805–813. [PubMed] [Google Scholar]
- 112.Robin AL, Suan EP, Sjaarda RN et al. : Reduction of intraocular pressure with anecortave acetate in eyes with ocular steroid injection-related glaucoma. Arch Ophthalmol 2009; 127(2):173–178. [DOI] [PubMed] [Google Scholar]
- 113.McNatt LG, Weimer L, Yanni J, Clark AF: Angiostatic activity of steroids in the chick embryo CAM and rabbit cornea models of neovascularization. J Ocul Pharmacol Ther 1999; 15(5):413–423. [DOI] [PubMed] [Google Scholar]
- 114.Gerometta R, Kumar S, Shah S, Alvarez L, Candia O, Danias J: Reduction of steroid-induced intraocular pressure elevation in sheep by tissue plasminogen activator. Invest Ophthalmol Vis Sci 2013; 54(13):7903–7909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Candia OA, Gerometta RM, Danias J: Tissue plasminogen activator reduces the elevated intraocular pressure induced by prednisolone in sheep. Exp Eye Res 2014; 128:114–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Spiga MG, Borras T: Development of a gene therapy virus with a glucocorticoid-inducible MMP1 for the treatment of steroid glaucoma. Invest Ophthalmol Vis Sci 2010; 51(6):3029–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gerometta R, Spiga MG, Borras T, Candia OA: Treatment of sheep steroid-induced ocular hypertension with a glucocorticoid-inducible MMP1 gene therapy virus. Invest Ophthalmol Vis Sci 2010. 51(6):3042–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Borras T, Buie LK, Spiga MG: Inducible scAAV2.GRE.MMP1 lowers IOP long-term in a large animal model for steroid-induced glaucoma gene therapy. Gene Ther 2016, 23:438–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang X, Clark AF, Yorio T: Regulation of glucocorticoid responsiveness in glaucomatous trabecular meshwork cells by glucocorticoid receptor-beta. Investigative Ophthalmology Vis Sci 2005, 46:4607–4616. [DOI] [PubMed] [Google Scholar]
- 120.Patel GC, Phan TN, Maddineni P, Kasetti RB, Millar JC, Clark AF, Zode GS. Dexamethasone-induced ocular hypertension in Mice: Effects of myocilin and route of administration. Am J Pathol 2016; 187:713–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Patel GC, Liu Y, Millar JC, Clark AG: Glucocorticoid receptor GRβ regulates glucocorticoid-induced ocular hypertension in mice. Sci Rep 2018; 8(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shepard AR, Millar JC, Pang IH, Jacobson N, Wang WH, Clark AF: Adenoviral gene transfer of active transforming growth factor-{beta}2 elevates intraocular pressure and reduces outflow facility in rodent eyes. Invest Ophthalmol Vis Sci 2010: 51(4):2067–2076. [DOI] [PubMed] [Google Scholar]
- 123.Budenz DL, Bennett J, Alonso L, Maguire A: In vivo gene transfer into murine corneal endothelial and trabecular meshwork cells. Invest Ophthalmol Vis Sci 1995. 36(11):2211–2215. [PubMed] [Google Scholar]
- 124.Druzgala P, Wu WM, Bodor N. Ocular absorption and distribution of loteprednol etabonate, a soft steroid, in rabbit eyes. Curr Eye Res 1991; 10(10):933–937. [DOI] [PubMed] [Google Scholar]
- 125.Murdick PW, Keates RH, Donovan EF et al. Ocular penetration studies. II. Topical administration of prednisolone. Arch ophthalmol 1966; 76(4):602–603. [DOI] [PubMed] [Google Scholar]
- 126.Awan MA, Agarwal PK, Watson DG et al. Penetration of topical and subconjunctival corticosteroids into human aqueous humour and its therapeutic significance. Br J Ophthalmol 2009; 93(6):708–713. [DOI] [PubMed] [Google Scholar]
- 127.Razeghinejad MR, Katz L. 2012; Ophthalmic Res 47(2):66–80. [DOI] [PubMed] [Google Scholar]
- 128.Yamaguchi M, Yasueda S et al. Formulation of an ophthalmic lipid emulsion containing anti-inflammatory steroidal drug, difluprednate. Int J Pharm 2005: 301(1–2):121–128. [DOI] [PubMed] [Google Scholar]