

Dear Editor,
Palmoplantar keratoderma (PPK) comprises a heterogeneous group of disorders characterized by hyperkeratosis of the palms and soles. It can be hereditary or acquired. Punctate PPK is rare. We report an elderly man with a 50-year history of persistent slowly progressing lesions on both hands and soles affecting many of his family members.
A 68-year-old farmer presented with eruptions on palms and soles of 50 years’ duration. On examination, multiple well-circumscribed oval to circular dark brown heaped-up keratotic lesions were seen on the palms and soles. The intervening skin appeared normal. They started between 15 and 20 years of age, first on the palms and then gradually progressed, covering almost the entire palms and soles up to the subungual area [Figure 1]. In some of the lesions, particularly over pressure sites, the scales had fallen leaving conspicuous concave pits; interdigital spaces were relatively spared. Throughout, the patient remained asymptomatic with no itching or hyperhidrosis and managed his routine work. Past medical history and examination of other systems were unremarkable. Family history disclosed that his mother had similar lesions, and eruptions in various stages of evolution were seen in the younger brother and two children with onset of disease in the second decade [Figure 2], all remaining asymptomatic. The grandchildren, who were of prepubertal age, had not developed the disease. Routine blood and urine investigations, pelvis ultrasound, chest X-ray, and computed tomography of the chest, abdomen, and pelvis were normal.
Figure 1.
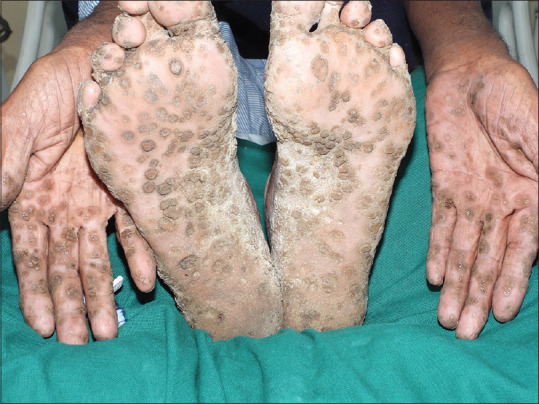
Circular to oval keratotic eruptions on palms and soles with intervening normal skin
Figure 2.
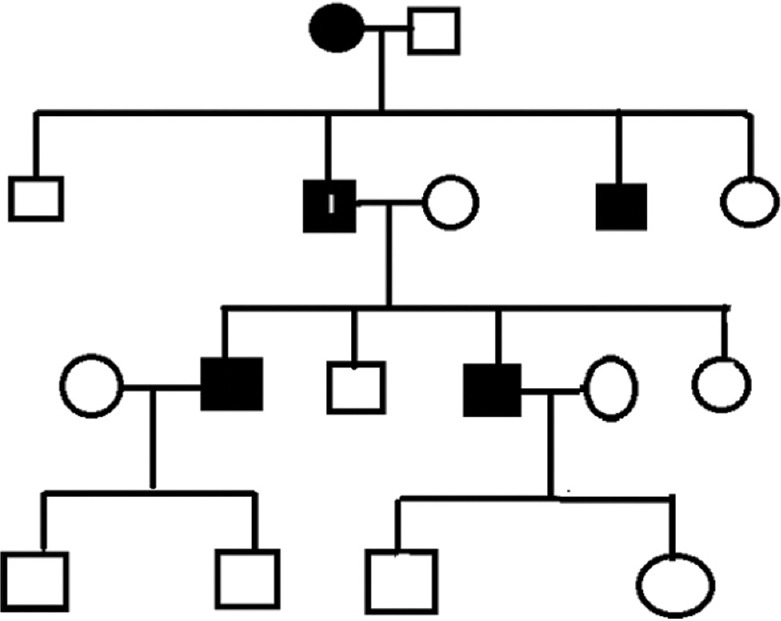
Chart showing affected members in three generations; I—index patient
Histopathology [Figure 3] revealed a broad almost rectangular cornoid lamella occupying a saucer-like depression in the epidermis with some zones of parakeratosis. The stratum granulosum was normal and intact. A diagnosis of PPK type 1 (Buschke–Fischer–Brauer disease) was made, and the patient was started on 40% urea and 20% salicylic acid ointment with little improvement.
Figure 3.
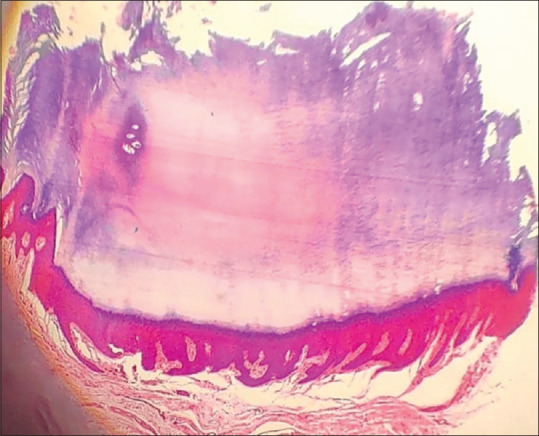
Cornoid lamella in a saucer-like epidermal pit (H&E 4x)
Hereditary PPK can be further classified into three major categories: diffuse, focal, and punctate PPK (PPPK). Most of the types of PPK are inherited as autosomal dominant or autosomal recessive disorders. The prevalence of PPPK is estimated to be 1.17/100,000.[1] It is believed that both genetic and environmental factors play a role in the etiology. PPPK displays an autosomal dominant pattern of inheritance and has been linked with two loci on chromosomes 15q22–15q24 and 8q24.13–8q24.21.[2] This autosomal dominant condition tends to occur much later than other hereditary keratodermas, usually between 20 and 30 years of age. Males tend to be more commonly affected. On palms, the lesions are usually scattered. On soles, they are usually coalesced but in our patient it was discrete. The lesions may evolve over time, becoming translucent, opaque, or verrucous. Some papules may form a keratotic core and detachment, which may lead to a characteristic central depression. Patients generally remain asymptomatic enabling them to work, but occasionally pain can be caused by pressure. Genetic factors in association with hard manual work have been said to precipitate hereditary PPK.[3] Clinical differentials include punctate porokeratosis and arsenical keratosis. Smaller keratotic lesions with fine pits and a typical cornoid lamella composed of a parakeratotic column arising from a dyskeratotic base are seen on histopathology. Arsenicosis is often accompanied by the dyspigmentation of the trunk in addition to the involvement of palms and soles, affecting many in the family. The management of PPPK includes topical keratolytics, liquid nitrogen, psoralen plus ultraviolet A (PUVA), systemic or topical retinoids, systemic acitretin, etretinate or alitretinoin, and topical steroid.[4]
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Stanimirović A, Kansky A, Basta-Juzbasić A, Skerlev M, Beck T. Hereditary palmoplantar keratoderma, type papulosa, in Croatia. J Am Acad Dermatol. 1993;29:435–7. doi: 10.1016/0190-9622(93)70207-a. [DOI] [PubMed] [Google Scholar]
- 2.Gao M, Yang S, Li M, Yan KL, Jiang YX, Cui Y, et al. Refined localization of a punctate palmoplantar keratoderma gene to a 5.06-cM region at 15q22.2-15q22.31. Br J Dermatol. 2005;152:874–8. doi: 10.1111/j.1365-2133.2005.06488.x. [DOI] [PubMed] [Google Scholar]
- 3.Mittal RR, Jha A. Hereditary punctate palmoplantar keratoderma – A cinical study. Indian J Dermatol Venereol Leprol. 2003;69:90–1. [PubMed] [Google Scholar]
- 4.Pai VV, Kikkeri NN, Athanikar SB, Sori T, Rao R. Type I punctate palmoplantar keratoderma (Buschke-Fisher-Brauer disease) in a family—A report of two cases. Foot. 2012;22:240–2. doi: 10.1016/j.foot.2012.03.004. [DOI] [PubMed] [Google Scholar]