

Abstract
Rodent models of breast cancer have played critical roles in our understanding of breast cancer development and progression as well as preclinical testing of cancer prevention and therapeutics. In this chapter, we first review the values and challenges of conventional genetically engineered mouse (GEM) models and newer iterations of these models, especially those with inducible or conditional regulation of oncogenes and tumor suppressors. Then, we discuss non-germline (somatic) genetically engineered mouse models of breast cancer with temporospatial control, made possible by intraductal injection of viral vectors to deliver oncogenes or to manipulate the genome of mammary epithelial cells. Next, we introduce the latest development in precision editing of endogenous genes using in vivo CRISPR-CAS9 technology. We conclude with the recent development in generating somatic rat models for modeling estrogen receptor-positive breast cancer, something that has been difficult to accomplish in mice.
Introduction
Rodent models have been highly valuable for understanding how breast cancer initiates and progresses. These models are also critical tools for preclinical testing of therapeutics for treatment and prevention. For most of the 20th century, rodent models relied on chemical carcinogens, physical agents (such as gamma irradiation), and retroviruses (e.g., mouse mammary tumor virus) for insertional mutagenesis, as well as oncogenic viruses (such as Rous-sarcoma virus). While valuable at that time and still used to date, the lack of defined oncogenic drivers as well as the lack of tissue and/or cell type specificity has greatly limited the utility of these models.
Alongside these early models, cultured human breast tumor cell lines were also engrafted into immunodeficient mice (usually in the fat-pad, but also subcutaneously and occasionally in the kidney capsule). These xenograft models offered the opportunity to investigate human breast tumor progression in vivo and to test therapeutic agents on human tumor cells in vivo. Since the cells can be genetically manipulated in various ways in culture and then xenografted in mice, these models have been used widely to elucidate genetic factors controlling breast cancer progression and spread (Wu et al. 2009). Some of these genetic factors influence signaling networks within human cancer cells, while others impact secreted molecules and receptors that interact with cells in the microenvironment. Along the same line, various stromal cells can be genetically manipulated and co-transplanted with human breast tumor cells to investigate molecular interactions between tumor and stromal cells (Kuperwasser et al. 2004). Alternatively, these human tumor cells can be engrafted into mice defective in different cellular components of the immune system and/or with altered stromal cells to investigate tumor cell and microenvironment interactions. These cell line xenograft models are reproducible and highly accessible to anyone with a vivarium, but cell lines often have drifted far from human tumors and cannot recapitulate the complexity and large intratumor heterogeneity in patient tumors. Some of the drawbacks of using cell line models can be overcome by using grafts from patient specimens (patient-derived xenografts or PDXs), which is discussed in detail in the chapter by Drs. Caldas and Lewis. However, a critical limitation of these xenograft models is the use of immunodeficient hosts, which largely prevents the study of the immune cell contribution. The immune system is known to have a fundamental contribution to tumor formation, progression and therapeutic response as discussed in detail in the chapters by Drs. Welm and Park, and by Drs. Emens and Loi. Many of cancer therapeutic drugs, antiestrogens included, target both tumor cells themselves and immune cells (Bottos et al. 2016; Chakraborty et al. 2021). Some of them may exert efficacy primarily via targeting non-cancerous cells such as myeloid cells (Goswami et al. 2022) and cancer-associated fibroblasts (Chen et al. 2021) while immune checkpoint inhibitors (ICIs) target the cytotoxic interaction between T cells and tumor cells (Sharma and Allison 2020). Progress is being made in engrafting human haemopoietic stem cells into special mouse recipients to “humanize” the immune system of the mice, so that human tumor cells can survive in mice for an extended window of time, but the engrafted human immune cells can only restore part of the human immune system and inevitably attack the host (Jin et al. 2021).
Genetic engineering of mice can overcome the drawbacks of undefined genetic alterations and the lack of immune cell participation or incompatible interactions of mutated mammary cells and stromal cells. Hundreds of genetically engineered mouse (GEM) models have been created, and these preclinical rodent models of cancer play critical roles in investigating genes in cancer development and progression as well as preclinical testing of cancer prevention and therapeutics. The following sections will discuss these genetically engineered rodent models and the latest developments in this area.
Value and challenges of germline genetically engineered mouse models and their newer iterations.
GEM models have greatly advanced our understanding of breast cancer initiation and progression as well as therapeutic response and resistance. Individual models have also been bred together to generate compound mutant mouse lines to study oncogenic collaboration in driving tumor formation and progression as well as therapeutic resistance (Sinn et al. 1987; Donehower et al. 1995; Podsypanina et al. 2004). As these models are immunocompetent, they are especially valuable for studying the involvement of the immune system (Welte et al. 2016; Kim et al. 2019), a key advantage over xenograft models. Breeding tumor models into mouse lines whose immune and other stromal cell types have been genetically modified has led to an ever-expanding pool of models for studying cancer cell-immune cell interactions, epithelial-stromal interactions, and cancer cell-metastatic niche interactions. Commonly used and well-characterized GEM models often use MMTV LTR to drive the expression of oncogenes. Examples include MMTV-c-Myc (the first mammary transgenic model) (Stewart et al. 1984), MMTV-PyMT (popular due to its speed to develop tumors and high metastatic burden) (Guy et al. 1992), MMTV-ErbB2 (Neu) (reviewed in (Ursini-Siegel et al. 2007)), and MMTV-Wnt1 (reviewed in (Li et al. 2000)). These models have been instrumental in understanding breast cancer formation and progression as well as cancer cell heterogeneity and therapeutic resistance.
However, there are some challenges with these transgenic models. The MMTV LTR is expressed in a subset of luminal epithelial cells. Depending on the insertion site of the MMTV LTR, the expression intensity and distribution within the luminal cell layer can vary significantly. Therefore, multiple founder lines need to be screened to obtain useful lines to model breast cancer. In addition, beyond the mammary epithelia, the MMTV LTR also has activity in salivary gland, kidney, lung, and some hematopoietic cells (Stocklin et al. 1993; Godley et al. 1996). Therefore, tumors sometimes appear in other sites besides the mammary glands. Systemic factors from these developing tumors may also affect the interpretation of mammary gland phenotypes. Furthermore, transgenic expression in non-mammary tissues may complicate the study of mammary tumor metastasis. For example, finding tumor cells in distant organs may not be the result of metastasis but rather the emergence of new primary tumors in these organs. This leakiness of transgene expression in other organs can potentially complicate the interpretation of studies to characterize the pre-metastatic niche. For example, microenvironment changes in lungs in these mice before the appearance of metastatic tumors may be the outcome of leaky transgene expression in lungs but not due to the lungs receiving signals from the primary site.
Besides these conventional transgenic promoters, other promoters have been used to express transgenes in selected cell subsets within the luminal or the basal/cap cell populations, since distinct cell subsets within the mammary ductal tree may contribute to different breast cancer subtypes. The whey acidic protein (Andres et al. 1987; Pittius et al. 1988) and β-lactoglobulin (BLG) gene promoters (Whitelaw et al. 1992; Selbert et al. 1998), which can be stimulated by lactation, have been used to direct transgene expression to differentiated alveolar and secretory luminal cells. Not surprisingly, when different promoters are used to regulate the expression of the same oncogenic driver, tumors with different characteristics can develop (Herschkowitz et al. 2007), reflecting the impact of cell of origin on breast cancer phenotypes.
We have reported that cytokeratin 6 (Krt6) marks a luminal cell sub-population with progenitor features and targeting PyMT into this cell population leads to papillary tumors that resemble the normal-like breast cancer subtype in patients (Bu et al. 2011). On the other hand, multiple transgenic lines have been reported using Krt5 or Krt14 to express oncogenes and other genes in the basal cell population (Kuraguchi et al. 2009; Bowman-Colin et al. 2013; Bao et al. 2019). One caveat is that these promoters are also not limited to the mammary epithelial population. These keratin promoters are active in epithelial cells in other non-mammary organs such as skin, salivary gland, and ovary (Vassar et al. 1989; Jonkers et al. 2001; Orsulic et al. 2002; Grimm et al. 2006). WAP is known to be expressed in brain, testes, and even muscle (Wagner et al. 1997), and the BLG promoter has activity in salivary gland (Whitelaw et al. 1992). Of note, a given oncogene may exhibit significant differences in potency in driving tumor formation in different cell types or organs (Leder et al. 1986).
Transgenic models have been used widely as preclinical models to test therapeutics. These immunocompetent models have certainly helped develop new and better therapeutics including immunotherapeutic agents, and they have helped uncover new mechanisms in drug resistance. However, the MMTV LTR is known to be responsive to steroid hormones including glucocorticoid, progesterone, and androgens (Cato et al. 1987). The WAP promoter is highly responsive to steroid and peptide hormones and is transcriptionally regulated by STAT5, as is MMTV LTR (Hobbs et al. 1982). Therefore, therapeutic agents that impact the production and signaling of these hormones may incidentally impact transgene expression levels and complicate the interpretation of the results from these preclinical studies. For example, ruxolitinib is an FDA-approved small molecule inhibitor of Janus kinase (JAK), which phosphorylates and activates STAT5, but ruxolitinib treatment would shut down expression of transgenic oncogenes in WAP or MMTV transgenic models; therefore, these transgenic models should not be used for testing therapeutic efficacy of ruxolitinib or any drugs that target JAK-STAT signaling.
Transgenic and knockout models generally introduce genetic alterations into the germline. These genetic alterations can affect normal mammary gland development. For example, tumors begin to appear in MMTV-PyMT mice before puberty; widespread hyperplasia is present in MMTV-Wnt1 mice even before puberty (Lin et al. 1992; Cunha and Hom 1996); ductal development is impaired in MMTV-ErbB2 mice (Mukherjee et al. 2000). On the other hand, germline knockout leads to loss of gene function in all cells starting from embryogenesis, often causing embryogenic defects, impaired organ development, and tumor development in non-mammary tissue, impeding the study of gene functions in mammary tumorigenesis. For example, Tp53 knockout mice usually succumb to lymphomas and sarcomas before mammary tumors appear (Donehower et al. 1992). To overcome these drawbacks, mammary tissues from knockout mice can be transplanted into wild type mice so that the specific impact on mammary development and tumorigenesis can be studied. For example, transplantation of Tp53 knockout mammary gland pieces into cleared fat-pad wild type mice allowed the study of the impact of Tp53 on mammary tumor development without the interference of other tumors in Tp53-null mice (Jerry et al. 2000). Interestingly, the mammary gland has potent reprograming capacity – when mixed with mammary epithelial cells and engrafted into a cleared fat pad, even neural stem cells (tagged by LacZ) have been reported to contribute to the outgrowth of the mammary ductal tree (Booth et al. 2008).
Another approach to study the impact of gene deletion selectively in the mammary gland is to use a conditional knockout method. Cre is a bacterial recombinase that catalyzes recombination between a pair of DNA recognition sites termed LoxP sites (Sauer and Henderson 1988; Lewandoski 2001). Breeding a mouse line carrying Cre under the control of a mammary-specific promoter with a mouse line that has part or a whole tumor suppressor flanked by LoxP sites allows the deletion of the tumor suppressor in mammary epithelia. For example, MMTV-Cre/BRCA1loxp/loxp mice develop normally but exhibit blunted ductal morphogenesis and eventually tumors (Xu et al. 1999). Of note, the use of MMTV-Cre vs. WAP-Cre can also lead to tumors of different properties (Lin et al. 2004; Herschkowitz et al. 2007; Jiang et al. 2010).
A Cre transgene may also be used to turn on the expression of a transgenic oncogene. For example, a loxP-flanked DNA fragment harboring transcriptional stop signals may be inserted between a transgenic promoter and the transgenic oncogene, preventing transgenic expression unless in cells expressing transgenic Cre (Andrechek et al. 2000). This hybrid system can be used to achieve more fine tuning in cell specificity of transgenic expression since the cell has to support both the promoter driving Cre and the promoter driving the floxed STOP oncogene in order to turn on the gene of interest. Alternatively, this method can be used to conditionally turn on an activated version of a proto-oncogene engineered into the endogenous locus (i.e. knock-in), so that the activated oncogene can be under the control of the endogenous promoter (Andrechek et al. 2000). Besides this Cre-LoxP system, the FLP-FRT recombination system is also commonly used and sometimes combined with the Cre-LoxP to achieve more sophisticated conditional deletion/activation of one or more genes (Dymecki and Tomasiewicz 1998; Branda and Dymecki 2004).
To overcome the drawback of premature oncogene expression before completion of organ development, inducible promoters have been used to drive oncogenic expression. These promoters are silent until induced by a drug, providing a temporal control to GEM models. While several methods can be used to achieve inducible expression (No et al. 1996; Cronin et al. 2001), the most common method is the tet-operon (tetO)/repressor bi-transgenic system, in which TRE (tetO + a minimum promoter) is used to control the gene of interest while a mammary cell-selective promoter drives the expression of tTA, which is a transcriptional activator resulting from the fusion of tet repressor and the VP16 transcription activator and can bind and activate TRE under the tight control of tetracycline or doxycycline. The original tTA is active in the absence of tetracycline or doxycycline, while a later developed variant version (also called reverse tTA, or rtTA) requires tetracycline or doxycycline for transcriptional activation. Both Tet-Off and Tet-On systems are widely used to control oncogene and tumor suppressor gene expression in cancer modeling as well as other genes involved in development and diseases. A tetracycline-regulated vector can also be used to delete a tumor suppressor gene at a selected time. For example, Tet-On can be used to turn on Cre at a selected time to delete part or a whole tumor suppressor to investigate its impact on cancer development and progression.
Besides inducible gene promoters, another commonly used approach to temporal control of gene expression is the use of a transgene (CreERT or CreERT2) that fuses Cre to a mutated ligand-binding domain of estrogen receptor (ER), which does not bind to estrogen but binds to tamoxifen with high affinity (Littlewood et al. 1995; Feil et al. 1996; Indra et al. 1999). Tamoxifen is a selective estrogen receptor modifier and is commonly used to suppress estrogen receptor activity of breast cancer. In the absence of tamoxifen, this fusion protein is complexed with HSP90 and other intracellular proteins, masking the Cre recombinase activity, but tamoxifen binding releases Cre from this inhibitory complex (Whitfield et al. 2015). Therefore, tamoxifen administration can be used to unleash the Cre activity to delete a tumor suppressor gene. Likewise, this inducible Cre can also be used to conditionally activate a transgenic oncogene that is separated from its promoter by a floxed STOP nucleotide sequence. Besides controlling Cre, this ER gene fragment can also be fused with an oncogenic transcription factor (such as c-Myc) so that tamoxifen can be used to control its transcriptional activity (Littlewood et al. 1995).
Besides inducibly expressing an oncogene or deleting a tumor suppressor to initiate mammary tumors, inducible gene expression tools have also been used to test whether an oncogene is required for maintenance, continued growth, progression, and metastasis of breast cancer. For example, following doxycycline administration to induce transgenic c-Myc to cause mammary tumors, withdrawal of doxycycline caused half of the tumors to rapidly regress, indicating that continued high levels of c-Myc are needed for tumor maintenance; therefore, these tumors are addicted to transgenic c-Myc (i.e., oncogenic addiction) (D’Cruz et al. 2001). However, the remaining half of the tumors gained activating mutations in Kras, causing them to become independent of the initiating c-Myc oncogene (D’Cruz et al. 2001). These data have implications in cancer treatment, as they suggest that patients with c-Myc-overexpressed tumors may need to be treated with drugs targeting both Myc and Ras signaling for eradication.
While these GEM models have greatly advanced research in breast cancer initiation, progression, and treatment, conventional GEM models and even conditional and inducible models usually cause a tissue-wide impact, often leading to early stages of tumorigenesis throughout the whole organ. If the initiating oncogenic event is a potent oncogene, multi-focal tumors rapidly emerge, as in the case of MMTV-PyMT. In most cases, stochastic tumors form after a much longer tumor latency because the mutated epithelia still need additional genetic (and epigenetic) changes to progress to cancer, and only a small subset of these precancerous cells gain sporadic secondary or tertiary genetic lesions, leading to clonally derived cancer. While these models are suitable for studying tumor progression, they are not highly relevant for the study of tumor initiation, especially the initiation of sporadic breast cancer, which arises from one or a few mutated cells in a normally developed ductal tree. These singly mutated cells then evolve into a cancer after additional rounds of mutations and clonal expansion. The next section will discuss models that are made to better mimic sporadic cancer formation.
Intraductal delivery of viral vectors to manipulate somatic mammary cells to model sporadic breast cancer.
Most human breast tumors arise sporadically as a result of somatic mutations in a few cells that clonally expand into a cancer. To closely model the formation of these sporadic breast tumors in patients, we and others have used retrovirus and lentivirus to carry protooncogenes and their various mutants into a small number of mammary epithelial cells after the mammary gland is fully developed (Fig 1) (Du and Li 2007; Siwko et al. 2008). The mammary gland is uniquely amenable to this type of genetic manipulation since these viruses can be delivered by injection into the nipple duct (i.e., up-the-teat injection) (Yang et al. 1995). While a human nipple harbors the openings of several lactiferous ducts, which branch into segmental ducts and then subsegmental ducts which further expand into ductile and acini (alveoli) forming lobules, a mouse teat is home to a single major duct that branches out to form the entire ductal tree. This duct is sealed off by keratinized skin cells in virgin mice, but can be easily exposed by transection using surgical scissors. Then a Hamilton syringe fitted with a blunt needle can be used to inject up to 30 μl of virus (greater than 30 μl can lead to duct rupture; Fig 1A). Viruses injected into the nipple duct can travel up the entire ductal tree, infecting any cell along the route (Fig 1B). Depending on the titer of the virus injected, a few to thousands of cells can be infected. Since the luminal cells are “chained’ tightly to each other by tight junctions, desmosomes, and gap junctions, and since this epithelial layer is also covered by the underneath myoepithelial layer and further sealed off from the stroma by basement membrane, viruses flushed into the lumen appear to infect luminal cells only, rarely infect myoepithelial cells, and generally do not infect cells beyond the basement membrane (Bu et al. 2009). Retroviruses generally require cell division for integration into the genome, but lentivirus can infect all cells in contact and therefore have a broader target population. Besides oncogenes, viruses can also be used to carry Cre into mammary cells to delete loxP-marked tumor suppressor genes to initiate cancer. For deleting a floxed gene, Cre is needed only for a brief time; therefore adenovirus, which is a DNA virus and does not integrate into genome, can substitute for retrovirus and lentivirus for a transient expression of Cre (Visbal et al. 2011). On the other hand, for inducible expression, an rtTA or CreERT needs to be delivered by a retroviral or lentiviral vector so that these genes are present when inducers are administered.
Fig 1:
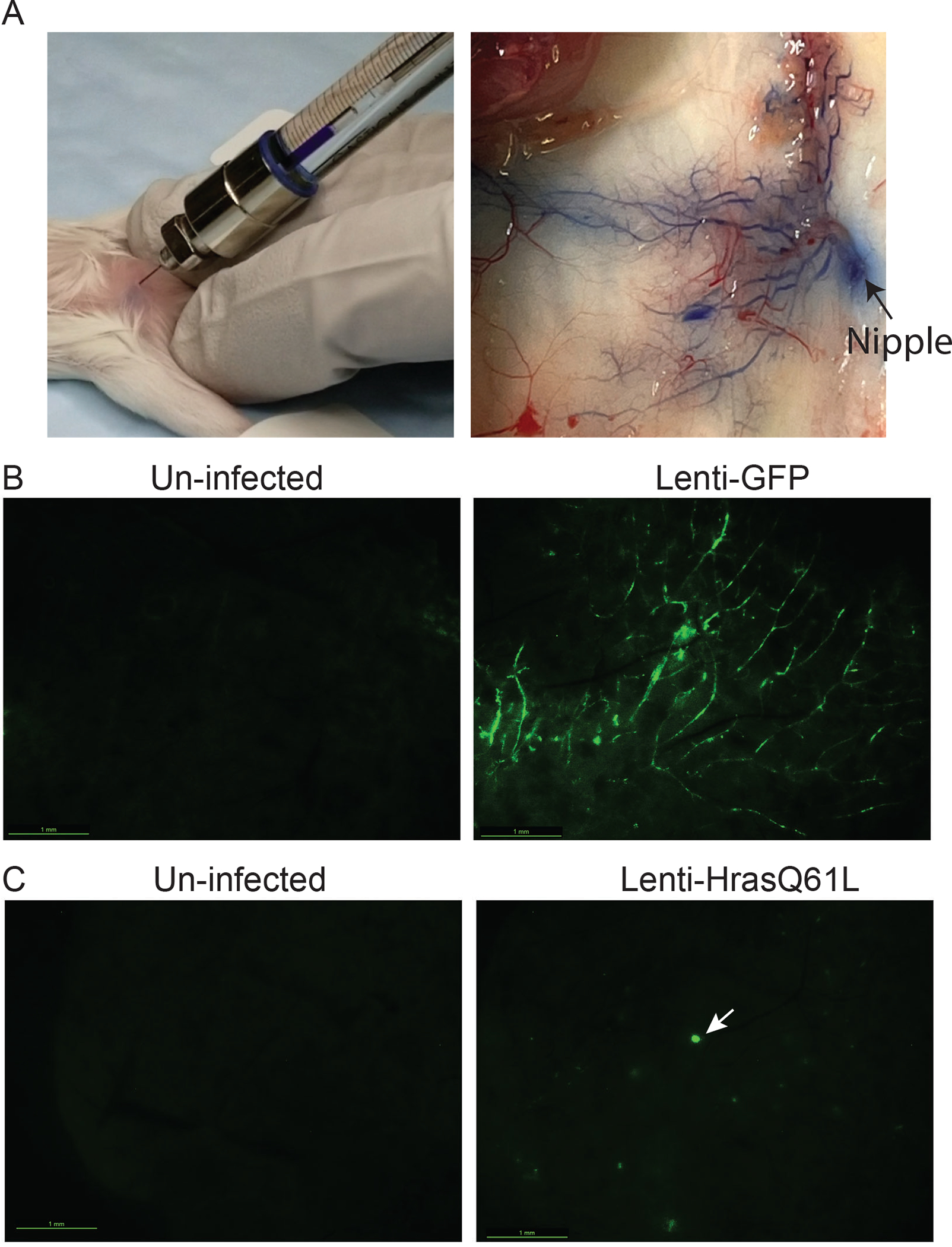
Intraductal injection of lentivirus to engineer genetic alterations in mammary cells in mice.
(A) Photos showing a successful intraductal injection. Bromophenol blue spreads throughout the ductal tree.
(B) Photos showing whole-mount GFP imaging of a control gland un-injected with virus and a gland injected by Lenti-GFP (3.6×106 IUs) three days earlier.
(C) Photos showing whole-mount GFP imaging of a control gland un-injected with virus and a gland injected by Lenti-HrasQ61L-GFP (2.4×106 IUs) eight days earlier. Several GFP+ precancerous early lesions can be seen, and the largest one is noted by an arrow.
Lenti- and retrovirus-mediated expression can be highly valuable for studying the impact of reproduction on mammary tumorigenesis compared to conventional transgenic models. This is because mammary-selective promoters such as MMTV or WAP have the progesterone receptor and STAT5-binding sites (Chalepakis et al. 1988; Préfontaine et al. 1999) and are induced during pregnancy and lactation when high progesterone and prolactin levels activate progesterone receptor and STAT5. As a result, they transcribe higher levels of MMTV or WAP-driven transgene during reproduction. These fluctuations of transgene expression can significantly complicate any study of reproduction on mammary tumors and progression. In contrast, ubiquitous promoters used in viral vectors do not become more transcriptionally active during reproduction; thus, viral vectors can generate more suitable models for studying the impact of reproduction on mammary tumor formation and progression. By using a retrovirus to deliver ErbB2 and Wnt1 and then impregnating the mice, we found that reproduction instigates precancerous lesions to advance to cancer (Haricharan et al. 2013). We further showed that this accelerated initiation is due to precancerous cells activating the prolactin-JAK2-STAT5 signaling in late pregnancy and lactation, leading to a weakened apoptosis anticancer barrier and thus accelerated progression to cancer (Haricharan et al. 2013). In addition, while involution normally deactivates STAT5 and turns on STAT3 to activate apoptosis and clear excess alveolar cells gained during reproduction, it cannot timely distinguish the prolactin receptor-JAK2-STAT5 signaling in these precancerous cells; therefore, this involution-associated protection against cell expansion is also weakened in these precancerous lesions, leading to accelerated progression to cancer (Haricharan et al. 2013). Since precancerous lesions are more likely to be found in older than in younger women, this finding provides an explanation for why a late-age childbirth increases breast cancer risk. Using a similar approach, we showed that hyperprolactinemia-inducing antipsychotics also stimulate precancerous early lesions to progress to cancer via activation of STAT5, exposing a potential breast cancer risk of using this group of antipsychotics in women who have developed atypia (Johnston et al. 2018).
Since tumor initiation in these somatic models closely mimics human breast cancer initiation, we have also used these models to study breast cancer chemoprevention, a field that has not received its deserved attention as discussed in the chapter by Dr. Brown and Heckman-Stoddard. Using a viral vector to initiate tumorigenesis and then administering therapeutic agents to test their efficacy on the progression of these precancerous lesions to cancer, we have found that even a short-term treatment with small molecular inhibitors against JAK1/2 (ruxolitinib) or the STAT protein family (C188–9) or the key anti-apoptosis protein Bcl2 (venetoclax) can restore the apoptosis anticancer barrier and prevent breast cancer (Haricharan et al. 2013; Young et al. 2022). Based on these preclinical data, we have already initiated a multi-center window-of-opportunity clinical trial to test the impact of ruxolitinib on apoptosis and biomarkers including pSTAT5 and its selected targets in premalignant lesions (Translational BCa Research Consortium TBCRC042; ClinicalTrials.gov Identifier: NCT02928978).
However, this intraductal viral method of model generation has some drawbacks. (1) Each experimental animal needs to be intraductally injected, which is time-consuming and not trivial technically, while transgenic and knockout models need only animal breeding to generate new experimental mice. (2) Another concern is that the use of foreign genes or reporters or epitope tags (e.g., HA, FLAG, Cre, rtTA, GFP, and luciferase) in these vectors may unnaturally provoke the immune system because they are neoantigens (Gambotto et al. 2000; Han et al. 2008; Limberis et al. 2009; Petkov et al. 2013). Luciferase is an especially potent neoantigen (Petkov et al. 2013). These neoantigens may both cause immune clearance of the infected cells and complicate the interpretation of the data. To avoid these complications, several tolerized models have been made (Day et al. 2014; Aoyama et al. 2018). For example, “Glowing Head” mice are transgenic for brain-specific expression of GFP and luciferase and they were reported to be peripherally tolerant to both luciferase and GFP (Day et al. 2014), although recent studies suggest that even the “Glowing Head” mice are not completely immune-tolerized (Grzelak et al. 2022). (3) This method also lacks specificity to infect a selected subset of the luminal layer, such as luminal stem cells, progenitor cells, ductal luminal cells, or alveolar cells. Cell type-specific expression of an oncogene is important in understanding the cancer cell of origin and cancer heterogeneity, since it is becoming increasingly clear that the cell of origin may also contribute to the heterogeneity observed in human breast cancer (as reviewed by (Visvader 2011)).
Intraductal injection of RCAS virus into Tva-transgenic mice for introducing genetic materials into specific subsets of mammary epithelial cells.
To achieve cell-type specificity, we have adapted the RCAS-TVA method (Fig 2) (Federspiel et al. 1994). RCAS is a vector derived from Rous sarcoma virus (Hughes 2004). This virus does not infect mammalian cells since the gene encoding its receptor TVA is present only in avian species. Ectopic expression of Tva sensitizes any cell to RCAS infection. Transgenic expression of Tva under the control of a cell type-specific promoter thus introduces cell-type specificity to RCAS infection. The RCAS-TVA method was initially used to introduce oncogenes into specific cells in brain to model gliomas (Holland et al. 2000), and has subsequently been used to model cancer in many other tissues including the ovary (Orsulic et al. 2002), liver (Lewis et al. 2005), and pancreas (Lewis et al. 2003). We have created several Tva transgenic lines driven off the MMTV promoter (Du et al. 2006), WAP promoter (Haricharan et al. 2014), Krt6 (Bu et al. 2011), and TOP (a synthetic promoter that is active in cells with active Wnt signaling) (Bu et al. 2013) for RCAS infection of general luminal cells, alveolar cells, a luminal progenitor, and Wnt signaling-activated cells, respectively. Taking advantage of the cell-specific infection feature in these Tva transgenic lines, we have made a number of observations: (1) ErbB2 activation in Wnt-activated cells fails to cause tumors since the infected cells appear to undergo cell death rather than rapid tumorigenesis (Bu et al. 2013), potentially explaining why Wnt signaling is less often observed in HER2+ breast cancer than in basal-like breast cancer (BLBC); (2) STAT5 activity in WAP+ alveolar cells can stimulate tumor initiation (Haricharan et al. 2013); (3) Krt6-positive mammary epithelial progenitors are not at increased vulnerability to tumorigenesis initiated by ErbB2 compared to the bulk luminal population (Holloway et al. 2015), suggesting that another cell subset in the luminal epithelia may be more susceptible to ErbB2-driven tumorigenesis.
Fig 2.

RCAS-TVA technology for delivering genetic alterations into selected mammary cells in mice. In a Tva-transgenic mouse line, intraductal injection delivers RCAS to mammary epithelial cells expressing TVA. The number of infected cells varies from a few (Bu et al. 2013; Bu et al. 2019) to tens of thousands (Du et al. 2006) depending upon both the TVA+ cells available for infection and the amount of virus injected. Stable expression of an oncogene from LTR following RCAS integration causes infected cells to expand to form atypical lesions, which evolve into cancer.
We also used RCAS-TVA to study the cell subset in precancerous lesions that evolves into cancer, since precancerous lesions are heterogeneous in cell subsets just like the normal ductal epithelia. While the cell of origin in the normal epithelia has been shown by us and others to affect the characteristics of the resulting mammary tumors, it was previously unknown whether one or multiple cell subsets in precancerous lesions contribute to the eventual cancer. In MMTV-Wnt1 transgenic mice, we identified a Krt6a-expressing precancerous stem cell (PcSC) subset and a more differentiated whey acidic protein-positive (WAP) cell subset. RCAS-mediated introduction of constitutively active versions of either Hras or Braf into these two cell types (made possible through intraductal injection of RCAS into Krt6a-Tva and WAP-Tva bred with MMTV-Wnt1) led to rapid tumorigenesis (Bu et al. 2019). However, the resulting tumors were dramatically different in protein profiles and histopathology (Bu et al. 2019). These observations indicate that different subsets of precancerous cells can contribute to the cancer etiology, but their differentiation status may impact the resulting cancer characteristics.
Besides using RCAS to deliver oncogenic mutations to selected cell subsets, we have also used this retroviral vector system more generally to study mammary tumor initiation and progression. For example, we found that RCAS-mediated expression of a constitutively activated ErbB2 in somatic mammary cells leads to the formation of an ATM/p53/ARF-dependent senescence barrier and an ATM-dependent but p53/ARF-independent apoptosis barrier (Reddy et al. 2010; Sinha et al. 2015). And our subsequent studies showed that among these ErbB2-activated cells, the loss of ER leads to the formation of a highly aggressive tumor subtype (Ding et al. 2021). In addition, by injecting RCAS to express both Cre and PyMT into mice transgenic for Tva (K19-Tva) and floxed for Fgfr1, we deleted Fgfr1 selectively in PyMT-activated mammary cells, and showed that Fgfr1 was required for both primary tumor growth and metastasis (Wang et al. 2017).
We have also used this RCAS-TVA method to trace the infected cell population. For example, using RCAS to deliver an HA-tagged endogenous gene to trace the infected cell population, we found that the half-life of mammary epithelial cells as a group is approximately 31 days (Dong et al. 2010). Using intraductal injection of a lentivirus to deliver both ErbB2 and floxed GFP in mice that carried a Cre knocked into the PR locus, we were able to ask whether PR was ever expressed in the evolution to cancer. We found that 75% of tumors retained GFP in all tumor cells (Dong et al. 2016), suggesting that throughout the evolution of a majority of the tumors initiated by ErbB2, PR was never expressed.
There are some additional advantages in using RCAS as a viral vector. This virus is replication competent in avian cells; therefore, large amounts of high titer virus can be produced in an avian cell line such as the chicken fibroblast line DF1 by simply expanding the infected cell culture rather than using large scale transfection of lentiviral DNA constructs mixed together with ancillary vectors which are usually needed in making replication-deficient lentiviruses. However, in mammalian cells, this virus does not make any appreciable amounts of viral proteins such as Gag, Pol, or Env for reasons that are not clear (Li et al. 2011); therefore, it does not elicit immune rejection of infected cells. RCAS is also much safer to handle than lentivirus – it does not infect mammals, so that concerns of accidental infection of the experimenter are minimal, while accidental human infection by lentivirus is a non-negligible risk to the experimenter – even though replication-deficient vectors cannot spread, the oncogene carried by these vectors can nevertheless pose a significant cancer risk.
RCAS-TVA technology also has a few drawbacks. Shared with the lentiviral and other retroviral vectors are the time-consuming intraductal injection of each experimental mouse, and concern of immune response to any foreign antigen introduced by a viral vector. A technical drawback of RCAS is that this vector can accommodate only a gene less than 3.0kb. Large inserts lead to diminished levels of expression and spontaneous truncation (Li et al. 2011). This weakness could be overcome using lentivirus that is pseudo-typed with the RSV env gene (Lewis et al. 2001; Siwko et al. 2008), so that it can accommodate large inserts and can integrate into the genomes of both dividing and non-dividing cells.
Furthermore, all of these viral vector approaches to introducing oncogenes into somatic cells in mice suffer one common drawback. Following infection and integration into the genome, provirus expresses the gene of interest under the control of exogenous promoters (e.g., viral LTR or a housekeeping gene); therefore, these virus-delivered ectopic genes lack the physiological control of the oncogene in its native locus. Furthermore, retrovirus and lentivirus integrate largely randomly into the genome of the infected cells, as one or multiple copies (although usually preferentially in regions of active chromatin). Depending on the integration site and the number of integrated copies, the expression levels of the oncogene could vary greatly among the infected cells. Moreover, while the chance of a lentivirus/retrovirus landing in transcribed region of the chromatin of a given cell and disrupting transcription of the critical gene is relatively small, when many cells are infected in multiple mice, a potential tumorigenic impact of virus-caused host gene disruption cannot be totally dismissed.
CRISPR-mediated editing of endogenous genes in somatic mammary cells.
To overcome the drawbacks of viral vector approaches including non-physiologically relevant promoters, insertional effects, and potential immune responses to ectopic genes and tags, the CRISPR/Cas9 system has been adapted in animal models to edit endogenous genes in their native loci (Fig 3) (Anzalone et al. 2020), eliminating the drawbacks associated with the use of viral vector-mediated oncogene expression. CRISPR editing can introduce both indels and point mutations (Anzalone et al. 2020). gRNA-directed, Cas9-catalyzed double-strand breaks (DSBs) at specific genomic locations are repaired by non-homologous end joining (NHEJ) repair, leading to insertions and deletions and subsequent loss of gene function (Fig 3A). Multiple mouse models have been produced by using virus carrying gRNA to somatically indel-edit tumor suppressor genes in mice transgenic for CAS9 (Platt et al. 2014; Sanchez-Rivera et al. 2014; Xue et al. 2014; Annunziato et al. 2016; Oldrini et al. 2018; Annunziato et al. 2019; Annunziato et al. 2020; Teng et al. 2021).
Fig 3:

The CRISPR method for introducing indels and point mutations into endogenous genes in their native loci. (A) In a Cas9 transgenic mouse line, intraductal injection of AAV carrying gRNA causes DSBs at the targeted loci, which are repaired by non-homologous end joining (NHEJ), leading to small insertions and deletions (indels). This method is most commonly used for loss of function studies, such as tumor suppressor genes. (B) We have found that a modified AAV vector carrying both gRNA and HDR can lead to precise point mutations. This approach can be used to test gain of function mutations of proto-oncogenes. Of note, indels still occur in this approach, but cells that suffered gain of function changes have competitive advantages and are thus selected for in tumor evolution. (C) Example of efficient infection of mammary epithelial cells by serotype 9 of AAV.
Precise missense and activating mutations in proto-oncogenes can be achieved by a deaminase-mediated base-editing approach, which uses a nuclease-deficient Cas9-fused with a cytidine or adenine deaminase transition mutation (C:G to T:A base editing [CBE] or A:T to G:C base editing [ABE]) (Anzalone et al. 2020). The C:G to G:C transversion mutation has also been reported (Kurt et al. 2021; Yuan et al. 2021). But only the CBE technique has been used for tumor modeling in vivo (Annunziato et al. 2020), and this editing method is restricted by the availability of the protospacer adjacent motif (PAM) sequence close to the editing candidate, and currently can perform only 6 out of 12 possible point mutations. Furthermore, as a specific deaminase cannot differentiate between the intended base and others in close proximity, this method often introduces unintended mutations proximal to the targeted nucleotide within the base-editing window (Annunziato et al. 2020). Prime editor is another approach to introduce mutations (Anzalone et al. 2020). It is comprised of a Cas9 nickase fused with a reverse transcriptase and a pegRNA, which provides both gRNA and a template for reverse transcription. This method has been used to introduce an activating point mutation into Ctnbb1 to initiate liver tumors in mice, but there are still technical challenges in vivo (Liu et al. 2021).
On the other hand, coupling homology-directed repair (HDR) to gRNA/Cas9 can introduce all 12 point mutations (Anzalone et al. 2020). The HDR donor sequence is a short DNA fragment matching the target sequence but with pre-designed point mutations so that homologous recombination at the Cas9 cleavage site leads to precision mutations although indels also occur. This method is mature for in vitro experiments, but employing it to somatically edit proto-oncogenes in vivo for cancer modeling to date has not been very successful (Platt et al. 2014; Winters et al. 2017; Oldrini et al. 2018). For example, when Platt et al (Platt et al. 2014) constructed an AAV vector to both indel-edit Tp53 and Lkb1 and introduce an activating point mutation in the Kras proto-oncogene in respiratory epithelium to initiate lung cancer, they detected robust indel mutations of Tp53 and Lkb1 in the resulting tumors, but the KrasG12D signal was barely above the baseline even though cells with edited KrasG12D were predicted to be highly selected for in lung tumorigenesis (Ji et al. 2007). We confirmed that serotype 9 of AAV can efficiently infect mammary epithelial cells in vivo (Fig 3C). And by modifying the original targeting vector (Platt et al. 2014) to remove reporter genes that may cause immune rejection and/or interfere with HDR function, we have achieved high efficiency precision editing of proto-oncogenes in mammary glands (Fig 3B; Bu et al, submitted). Somatic editing of two common proto-oncogenes, Kras and Pik3ca, in either normal or hyperplastic mammary glands led to swift mammary tumor development (Bu et al, submitted). The resulting tumors overall showed less inter-tumor variation than tumors induced by lentivirus, likely due to the steadier expression levels of the edited genes across infected cells than with virus-expressed oncogenes. These CRISPR-edited tumor models therefore offer more consistent models for cancer biology studies and therapeutic development.
Rat models of breast cancer: can we finally model all aspects of ER+ breast cancer?
Rats are the first mammalian species domesticated for laboratory research (before mid-19th century) (Gill et al. 1989). They are not large mice – they are 5 million evolution years closer to humans than mice (Zhao et al. 2004). Rats are much more social than mice, can learn complex cognitive tasks, and are well suited for behavioral, psychological, and neuroscience studies (Jacob and Kwitek 2002). They are also closer pharmacologically to human than mice and are preferred over mice for toxicology studies (Kratchman, 2018)(Jacob and Kwitek 2002). Apc mutant mice develop tumors in the small intestine, but Apc mutant rats develop colorectal cancer (Irving et al. 2014) just like human carriers of an APC mutant allele, suggesting that rats are more similar to humans with respect to tumor susceptibility than mice in some cases. Rats have 6 pairs of mammary glands, which form lobules as humans do while the mouse ductal tree largely lacks acini except during pregnancy and lactation (Cardiff et al. 2018). These lobules are also wrapped in modest amounts of connective tissue fibroblasts as lobules in human are, although the involvement of connective tissue and fibroblasts in rat mammary glands is much less than in human breasts (Masso-Welch et al. 2000).
While hundreds of transgenic and knockout mouse models of breast cancer exist and many more exist for development and disease in general, there are far fewer transgenic and knockout rat lines. Though pronuclear microinjection can be used to generate transgenic mouse with high efficacy, the application in rats is met with low embryo survival. Likewise, while mouse embryonic stem cells can be maintained in vitro with relative ease and genetically manipulated before implantation into pseudo-pregnant hosts, rat embryonic stem cells lose multipotency fast. These technical hurdles plus the per-diem cost have contributed to the general shortage of genetically engineered rat lines. But technological improvements including improved rat ES cell culturing, genetic manipulation of the spermatogonial stem cells in vitro, and use of CRISPR have lessened the difficulty in generating transgenic and knockout rats (Tong et al. 2011; Remy et al. 2014; Chapman et al. 2015; Back et al. 2019). On the other hand, intraductal injection of viruses for genetic manipulation was first established in rats, not mice (Wang et al. 1991), since the teat opening of rats is much larger and can be more easily injected. Besides retrovirus and lentivirus, we have found that intraductal injection of AAV can also be used to deliver gRNA to edit endogenous genes in situ in rats with similar ease as in mice (Bu et al, unpublished observations).
While only a few genetically engineered mouse models develop ER+ mammary tumors and none appears to be estrogen-dependent (Lin et al. 2004; Zhang et al. 2005; Meyer et al. 2011; Tikoo et al. 2012; Chan et al. 2014; Dabydeen and Furth 2014; Van Keymeulen et al. 2015; Ando et al. 2017; Ozdemir et al. 2018) (with the possible exception of Stat1 KO(Chan et al. 2012)), rats readily develop estrogen-dependent mammary tumors following treatment with carcinogens such as 3-methylcholanthrene (MCA), N-methyl-N-nitrosourea (NMU), or 7,12-dimethybenz[a]anthracene (DMBA) (Huggins et al. 1959; Huggins et al. 1961; McCormick et al. 1981), at the ages of approximately 45–60 days, which is a stage of sexual maturation and rapid mammary gland development (Russo et al. 1990). High proportions (50% or more) of these tumors harbor an activation mutation in Hras (Sukumar et al. 1983; Zarbl et al. 1985; Kumar et al. 1990), although mutations in Pik3ca, the most commonly mutated protooncogene in human ER+ breast cancer, appear to be modest in carcinogen-induced tumors (Showler et al. 2017; Gil Del Alcazar et al. 2022). Intraductal injection of retrovirus carrying HrasG12E and KrasG12E also led to ovarian hormone-dependent ER+ tumors (Wang et al. 1991; Thompson et al. 1998b), and further studies showed that the transforming potential of mutated RAS is mostly transmitted via the effector protein RAF (Showler et al. 2017). We found that intraductal injection of lentivirus carrying Hras mutated at codon 61 (Q61L) into rats also led to hormone-dependent ER+/PR+ adenocarcinoma (Bu and Li 2020), while the same virus in mice led to metaplastic carcinoma with heavy squamous differentiation (Fig 4), a very rare histological subtype in women, further illustrating differences of rats vs. mice in breast cancer modeling. Furthermore, intraductal injection into rats of retrovirus carrying a constitutively activated version of ErbB2 also led to ER+ tumors (Wang et al. 1992), and we have reported that intraductal injection of lentivirus carrying PIK3CAH1047R likewise led to ER+/PR+ mammary tumors in rats (Bu and Li 2020). Moreover, Steensma and colleagues showed that germline Nf1 knockout, which derepresses both Ras signaling and ER transcription, also led to ER+/PR+ mammary tumors in rats (Dischinger et al. 2018). In addition, we found that intraductal injection of AAV carrying gRNA targeting Nf1 also led to ER+/PR+ mammary tumors in rats (Fig 5). Together, these data indicate that rats have a strong tendency to develop ER+ tumors regardless of carcinogen-mediated gene mutations or virus-mediated oncogene delivery/editing.
Fig 4.

HRASQ61L causes ovarian hormone-dependent ER+ adenocarcinoma in rats but metaplastic carcinoma with heavy squamous differentiation in mice. Lenti-HrasG61L was intraductally injected into adult FVB/n mice and Sprague Dawley rats. Mammary tumors appear within a few weeks in both groups of animals. Tumors in mice are metaplastic carcinoma with heavy squamous differentiation (also ER-negative). However, tumors in rats are adenocarcinoma with no evidence of metaplasia, but with strong positivity for both ERα and PR (not shown)(Bu and Li 2020). These ER+/PR+ tumors are also ovarian hormone dependent (Bu and Li 2020).
Fig 5.

The Nf1 CRISPR KO rat model of ER+/PR+ breast cancer. AAV9 carrying gRNA targeting Nf1 was intraductally injected into Cas9 rats. Tumors were palpated within two weeks to three months. These tumors are adenocarcinomas positive for both ERα and PR.
It is unclear at this time why rats are highly prone to ER+ mammary tumors. It is possible that compared to the ER- cells, the ER+ subset of mammary epithelial population in rats is highly vulnerable to transformation by carcinogens and oncogenic mutations (at the least what we have tested so far). Alternatively, the stromal microenvironment or systemic factors in rats may also contribute to this difference, and it has been reported that the tumor immune environment of rats appears to bear similarity to human breast cancer’s (Gil Del Alcazar et al. 2022) although rigorous comparisons between, rats, mice and humans are yet to be done. In addition, since even in mouse, ER is often expressed in precancerous lesions before it is downregulated in progression to cancer, it is possible that ER expression is controlled differently in tumor evolution in rats versus mice.
Rat models may provide a valuable tool to investigate the progression from ductal carcinoma in situ (DCIS) to invasive cancer. DCIS is detected in high frequency in women – approximately 50 thousand cases a year in the U.S. It is considered to be the immediate precursor to invasive cancer. As detailed in the chapter by Drs. Behbod and Thompson, there is an intense interest in the breast cancer community to understand mechanisms controlling DCIS progression to invasive cancer, but DCIS is rarely reported in GEM models, hampering this area of research. However, DCIS appears to be a common occurrence in rats either treated with carcinogen (Middleton 1965; Russo and Russo 1978; Russo and Russo 1987; Thompson et al. 1995; Thompson et al. 1998a; Russo and Russo 2000; Gil Del Alcazar et al. 2022) or intraductally injected with viruses to deliver oncogenic changes (Wang et al. 1991) (Fig 6).
Fig 6:

ER+/PR+ DCIS, invasive adenocarcinoma in a rat model of breast cancer. Lenti-PIK3CAH1047R was intraductally injected into adult Sprague Dawley rats. These rats develop ER+/PR+ DCIS, followed by invasive ER+/PR+ adenocarcinoma with a median latency of approximately 3.5 months (Bu and Li 2020).
Rat models may also offer a new tool to investigate ER+ breast cancer bone metastasis. Human ER+ breast cancer metastasizes primarily to bone, but none of the genetic models in mice to date have been reported to develop spontaneous bone metastasis although a few mouse and rat tumor cell lines implanted into fat pad can sometimes form bone metastasis (Simmons et al. 2015). In rats bearing tumors induced by carcinogens, two groups detected hypercalcemia (Gullino et al. 1975; Stoica et al. 1983; Stoica et al. 1984), and one group even observed potential tumor cells in bone marrow although adenocarcinomatous or papillary features were absent, raising concerns about their mammary origin (Gullino et al. 1975). Our preliminary characterization indicates that some of these rat models of ER+ breast cancer develop bone metastases that can be confirmed by X-ray and histology (Bu et al, unpublished observations). These models may provide a much-needed tool to address multiple critical questions in ER+ breast cancer research: (1) What are the mechanisms controlling metastatic growth in bone vs visceral organs? (2) What are the mechanisms of endocrine resistance in bone? (3) Are immune cells involved in metastatic progression in bone? (4) Why do ER+ breast cancer bone metastases present as immune cold when they are immersed in an immune rich bone microenvironment? (5) Could ER+ bone metastases be converted to immune hot and then treated with anti-immune checkpoint therapy?
The rat’s larger size also offers benefit for metastasis studies (1) since primary tumors can be allowed to develop larger, seeding more metastatic cells in distant organs, and (2) since metastatic lesions may also be allowed to expand to a larger size than in mice. Furthermore, laboratory rats can live 3–5 years or even longer depending on the strain, while mice typically live no more than 2 years. Therefore, there is more time to observe metastatic development or residual disease development and recurrence following primary tumor resection or therapeutic interventions.
Concluding remarks
Many rodent models have been developed in the past, but improved models are emerging due to technological advancement in genetic manipulation in rodents. Immunocompetence is a critical advantage of these models, but they also have some model-dependent drawbacks. Potential pitfalls and limitations should be thoroughly considered so that the most appropriate models are selected for the specific questions to be addressed. While the number of mouse models far exceeds that of rat models and mice cost much less to purchase and to house, rats do fill a critical gap – ER+ breast cancer and bone metastasis.
Acknowledgements:
We thank Drs. Jeffrey Rosen and Gary Chamness for critical review of this manuscript. This work was supported by DOD CDMRP BC191649 & BC191646 as well as NIH R01CA271498.
References:
- Ando S, Malivindi R, Catalano S, Rizza P, Barone I, Panza S, Rovito D, Emprou C, Bornert JM, Laverny G et al. 2017. Conditional expression of Ki-Ras(G12V) in the mammary epithelium of transgenic mice induces estrogen receptor alpha (ERalpha)-positive adenocarcinoma. Oncogene 36: 6420–6431. [DOI] [PubMed] [Google Scholar]
- Andrechek ER, Hardy WR, Siegel PM, Rudnicki MA, Cardiff RD, Muller WJ. 2000. Amplification of the neu/erbB-2 oncogene in a mouse model of mammary tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America 97: 3444–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres AC, Schonenberger CA, Groner B, Hennighausen L, LeMeur M, Gerlinger P. 1987. Ha-ras oncogene expression directed by a milk protein gene promoter: tissue specificity, hormonal regulation, and tumor induction in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 84: 1299–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziato S, de Ruiter JR, Henneman L, Brambillasca CS, Lutz C, Vaillant F, Ferrante F, Drenth AP, van der Burg E, Siteur B et al. 2019. Comparative oncogenomics identifies combinations of driver genes and drug targets in BRCA1-mutated breast cancer. Nature communications 10: 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziato S, Kas SM, Nethe M, Yucel H, Del Bravo J, Pritchard C, Bin Ali R, van Gerwen B, Siteur B, Drenth AP et al. 2016. Modeling invasive lobular breast carcinoma by CRISPR/Cas9-mediated somatic genome editing of the mammary gland. Genes & development 30: 1470–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziato S, Lutz C, Henneman L, Bhin J, Wong K, Siteur B, van Gerwen B, de Korte-Grimmerink R, Zafra MP, Schatoff EM et al. 2020. In situ CRISPR-Cas9 base editing for the development of genetically engineered mouse models of breast cancer. EMBO J 39: e102169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone AV, Koblan LW, Liu DR. 2020. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nature biotechnology 38: 824–844. [DOI] [PubMed] [Google Scholar]
- Aoyama N, Miyoshi H, Miyachi H, Sonoshita M, Okabe M, Taketo MM. 2018. Transgenic mice that accept Luciferase- or GFP-expressing syngeneic tumor cells at high efficiencies. Genes Cells 23: 580–589. [DOI] [PubMed] [Google Scholar]
- Back S, Necarsulmer J, Whitaker LR, Coke LM, Koivula P, Heathward EJ, Fortuno LV, Zhang Y, Yeh CG, Baldwin HA et al. 2019. Neuron-Specific Genome Modification in the Adult Rat Brain Using CRISPR-Cas9 Transgenic Rats. Neuron 102: 105–119 e108. [DOI] [PubMed] [Google Scholar]
- Bao J, Di Lorenzo A, Lin K, Lu Y, Zhong Y, Sebastian MM, Muller WJ, Yang Y, Bedford MT. 2019. Mouse Models of Overexpression Reveal Distinct Oncogenic Roles for Different Type I Protein Arginine Methyltransferases. Cancer Res 79: 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth BW, Mack DL, Androutsellis-Theotokis A, McKay RD, Boulanger CA, Smith GH. 2008. The mammary microenvironment alters the differentiation repertoire of neural stem cells. Proceedings of the National Academy of Sciences of the United States of America 105: 14891–14896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottos A, Gotthardt D, Gill JW, Gattelli A, Frei A, Tzankov A, Sexl V, Wodnar-Filipowicz A, Hynes NE. 2016. Decreased NK-cell tumour immunosurveillance consequent to JAK inhibition enhances metastasis in breast cancer models. Nature communications 7: 12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman-Colin C, Xia B, Bunting S, Klijn C, Drost R, Bouwman P, Fineman L, Chen X, Culhane AC, Cai H et al. 2013. Palb2 synergizes with Trp53 to suppress mammary tumor formation in a model of inherited breast cancer. Proceedings of the National Academy of Sciences of the United States of America. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branda CS, Dymecki SM. 2004. Talking about a revolution: The impact of site-specific recombinases on genetic analyses in mice. Dev Cell 6: 7–28. [DOI] [PubMed] [Google Scholar]
- Bu W, Chen J, Morrison GD, Huang S, Creighton CJ, Huang J, Chamness GC, Hilsenbeck SG, Roop DR, Leavitt AD et al. 2011. Keratin 6a marks mammary bipotential progenitor cells that can give rise to a unique tumor model resembling human normal-like breast cancer. Oncogene 30: 4399–4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu W, Li Y. 2020. Intraductal Injection of Lentivirus Vectors for Stably Introducing Genes into Rat Mammary Epithelial Cells in Vivo. J Mammary Gland Biol Neoplasia 25: 389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu W, Liu Z, Jiang W, Nagi C, Huang S, Edwards DP, Jo E, Mo Q, Creighton CJ, Hilsenbeck SG et al. 2019. Mammary Precancerous Stem and Non-Stem Cells Evolve into Cancers of Distinct Subtypes. Cancer Res 79: 61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu W, Xin L, Toneff M, Li L, Li Y. 2009. Lentivirus vectors for stably introducing genes into mammary epithelial cells in vivo. J Mammary Gland Biol Neoplasia 14: 401–404. [DOI] [PubMed] [Google Scholar]
- Bu W, Zhang X, Dai H, Huang S, Li Y. 2013. Mammary cells with active Wnt signaling resist ErbB2-induced tumorigenesis. PLoS One 8: e78720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardiff RD, Jindal S, Treuting PM, Going JJ, Gusterson B, Thompson HJ. 2018. Mammary Gland. in Comparative Anatomy and Histology (eds. Treuting P, Dintzis S, Montine KS), pp. 487–509. Academic Press. [Google Scholar]
- Cato AC, Henderson D, Ponta H. 1987. The hormone response element of the mouse mammary tumour virus DNA mediates the progestin and androgen induction of transcription in the proviral long terminal repeat region. Embo J 6: 363–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty B, Byemerwa J, Shepherd J, Haines CN, Baldi R, Gong W, Liu W, Mukherjee D, Artham S, Lim F et al. 2021. Inhibition of estrogen signaling in myeloid cells increases tumor immunity in melanoma. The Journal of clinical investigation 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalepakis G, Arnemann J, Slater E, Brüller HJ, Gross B, Beato M. 1988. Differential gene activation by glucocorticoids and progestins through the hormone regulatory element of mouse mammary tumor virus. Cell 53: 371–382. [DOI] [PubMed] [Google Scholar]
- Chan SR, Rickert CG, Vermi W, Sheehan KC, Arthur C, Allen JA, White JM, Archambault J, Lonardi S, McDevitt TM et al. 2014. Dysregulated STAT1-SOCS1 control of JAK2 promotes mammary luminal progenitor cell survival and drives ERalpha(+) tumorigenesis. Cell death and differentiation 21: 234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SR, Vermi W, Luo J, Lucini L, Rickert C, Fowler AM, Lonardi S, Arthur C, Young LJ, Levy DE et al. 2012. STAT1-deficient mice spontaneously develop estrogen receptor alpha-positive luminal mammary carcinomas. Breast cancer research : BCR 14: R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman KM, Medrano GA, Jaichander P, Chaudhary J, Waits AE, Nobrega MA, Hotaling JM, Ober C, Hamra FK. 2015. Targeted Germline Modifications in Rats Using CRISPR/Cas9 and Spermatogonial Stem Cells. Cell reports 10: 1828–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, McAndrews KM, Kalluri R. 2021. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nature reviews Clinical oncology 18: 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin CA, Gluba W, Scrable H. 2001. The lac operator-repressor system is functional in the mouse. Genes & development 15: 1506–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha GR, Hom YK. 1996. Role of mesenchymal-epithelial interactions in mammary gland development. J Mammary Gland Biol Neoplasia 1: 21–35. [DOI] [PubMed] [Google Scholar]
- D’Cruz CM, Gunther EJ, Boxer RB, Hartman JL, Sintasath L, Moody SE, Cox JD, Ha SI, Belka GK, Golant A et al. 2001. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nature medicine 7: 235–239. [DOI] [PubMed] [Google Scholar]
- Dabydeen SA, Furth PA. 2014. Genetically engineered ERalpha-positive breast cancer mouse models. Endocrine-related cancer 21: R195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day CP, Carter J, Weaver Ohler Z, Bonomi C, El Meskini R, Martin P, Graff-Cherry C, Feigenbaum L, Tuting T, Van Dyke T et al. 2014. “Glowing head” mice: a genetic tool enabling reliable preclinical image-based evaluation of cancers in immunocompetent allografts. PLoS One 9: e109956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Liu Y, Lee DK, Tong Z, Yu X, Li Y, Xu Y, Lanz RB, O’Malley BW, Xu J. 2021. Cell lineage tracing links ERalpha loss in Erbb2-positive breast cancers to the arising of a highly aggressive breast cancer subtype. Proceedings of the National Academy of Sciences of the United States of America 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dischinger PS, Tovar EA, Essenburg CJ, Madaj ZB, Gardner EE, Callaghan ME, Turner AN, Challa AK, Kempston T, Eagleson B et al. 2018. NF1 deficiency correlates with estrogen receptor signaling and diminished survival in breast cancer. NPJ breast cancer 4: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower LA, Godley LA, Aldaz CM, Pyle R, Shi YP, Pinkel D, Gray J, Bradley A, Medina D, Varmus HE. 1995. Deficiency of p53 accelerates mammary tumorigenesis in Wnt-1 transgenic mice and promotes chromosomal instability. Genes & development 9: 882–895. [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr., Butel JS, Bradley A. 1992. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356: 215–221. [DOI] [PubMed] [Google Scholar]
- Dong J, Tong T, Reynado AM, Rosen JM, Huang S, Li Y. 2010. Genetic manipulation of individual somatic mammary cells in vivo reveals a master role of STAT5a in inducing alveolar fate commitment and lactogenesis even in the absence of ovarian hormones. Dev Biol 346: 196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Zhao W, Shi A, Toneff M, Lydon J, So D, Li Y. 2016. The PR status of the originating cell of ER/PR-negative mouse mammary tumors. Oncogene 35: 4149–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Li Y. 2007. RCAS-TVA in the mammary gland: an in vivo oncogene screen and a high fidelity model for breast transformation? Cell Cycle 6: 823–826. [DOI] [PubMed] [Google Scholar]
- Du Z, Podsypanina K, Huang S, McGrath A, Toneff MJ, Bogoslovskaia E, Zhang X, Moraes RC, Fluck M, Allred DC et al. 2006. Introduction of oncogenes into mammary glands in vivo with an avian retroviral vector initiates and promotes carcinogenesis in mouse models. Proceedings of the National Academy of Sciences of the United States of America 103: 17396–17401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dymecki SM, Tomasiewicz H. 1998. Using Flp-recombinase to characterize expansion of Wnt1-expressing neural progenitors in the mouse. Dev Biol 201: 57–65. [DOI] [PubMed] [Google Scholar]
- Federspiel MJ, Bates P, Young JA, Varmus HE, Hughes SH. 1994. A system for tissue-specific gene targeting: transgenic mice susceptible to subgroup A avian leukosis virus-based retroviral vectors. Proceedings of the National Academy of Sciences of the United States of America 91: 11241–11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil R, Brocard J, Mascrez B, LeMeur M, Metzger D, Chambon P. 1996. Ligand-activated site-specific recombination in mice. Proceedings of the National Academy of Sciences of the United States of America 93: 10887–10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambotto A, Dworacki G, Cicinnati V, Kenniston T, Steitz J, Tüting T, Robbins PD, DeLeo AB. 2000. Immunogenicity of enhanced green fluorescent protein (EGFP) in BALB/c mice: identification of an H2-Kd-restricted CTL epitope. Gene therapy 7: 2036–2040. [DOI] [PubMed] [Google Scholar]
- Gil Del Alcazar CR, Trinh A, Aleckovic M, Rojas Jimenez E, Harper NW, Oliphant MUJ, Xie S, Krop ED, Lulseged B, Murphy KC et al. 2022. Insights into Immune Escape During Tumor Evolution and Response to Immunotherapy Using a Rat Model of Breast Cancer. Cancer immunology research 10: 680–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill TJ 3rd, Smith GJ, Wissler RW, Kunz HW. 1989. The rat as an experimental animal. Science 245: 269–276. [DOI] [PubMed] [Google Scholar]
- Godley LA, Kopp JB, Eckhaus M, Paglino JJ, Owens J, Varmus HE. 1996. Wild-type p53 transgenic mice exhibit altered differentiation of the ureteric bud and possess small kidneys. Genes & development 10: 836–850. [DOI] [PubMed] [Google Scholar]
- Goswami S, Anandhan S, Raychaudhuri D, Sharma P. 2022. Myeloid cell-targeted therapies for solid tumours. Nature reviews Immunology. [DOI] [PubMed] [Google Scholar]
- Grimm SL, Bu W, Longley MA, Roop DR, Li Y, Rosen JM. 2006. Keratin 6 is not essential for mammary gland development. Breast cancer research : BCR 8: R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzelak CA, Goddard ET, Lederer EE, Rajaram K, Dai J, Shor RE, Lim AR, Kim J, Beronja S, Funnell APW et al. 2022. Elimination of fluorescent protein immunogenicity permits modeling of metastasis in immune-competent settings. Cancer Cell 40: 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullino PM, Pettigrew HM, Grantham FH. 1975. N-nitrosomethylurea as mammary gland carcinogen in rats. Journal of the National Cancer Institute 54: 401–414. [PubMed] [Google Scholar]
- Guy CT, Cardiff RD, Muller WJ. 1992. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol 12: 954–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han WGH, Unger WWJ, Wauben MHM. 2008. Identification of the immunodominant CTL epitope of EGFP in C57BL/6 mice. Gene therapy 15: 700–701. [DOI] [PubMed] [Google Scholar]
- Haricharan S, Dong J, Hein S, Reddy JP, Du Z, Toneff M, Holloway K, Hilsenbeck SG, Huang S, Atkinson R et al. 2013. Mechanism and preclinical prevention of increased breast cancer risk caused by pregnancy. eLife 2: e00996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haricharan S, Hein SM, Dong J, Toneff MJ, Aina OH, Rao PH, Cardiff RD, Li Y. 2014. Contribution of an alveolar cell of origin to the high-grade malignant phenotype of pregnancy-associated breast cancer. Oncogene 33: 5729–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, Rasmussen KE, Jones LP, Assefnia S, Chandrasekharan S et al. 2007. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol 8: R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs AA, Richards DA, Kessler DJ, Rosen JM. 1982. Complex hormonal regulation of rat casein gene expression. J Biol Chem 257: 3598–3605. [PubMed] [Google Scholar]
- Holland EC, Li Y, Celestino J, Dai C, Schaefer L, Sawaya RA, Fuller GN. 2000. Astrocytes give rise to oligodendrogliomas and astrocytomas after gene transfer of polyoma virus middle T antigen in vivo. The American journal of pathology 157: 1031–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway KR, Sinha VC, Toneff MJ, Bu W, Hilsenbeck SG, Li Y. 2015. Krt6a-positive mammary epithelial progenitors are not at increased vulnerability to tumorigenesis initiated by ErbB2. PLoS One 10: e0117239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins C, Briziarelli G, Sutton H, Jr. 1959. Rapid induction of mammary carcinoma in the rat and the influence of hormones on the tumors. The Journal of experimental medicine 109: 25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins C, Grand LC, Brillantes FP. 1961. Mammary cancer induced by a single feeding of polymucular hydrocarbons, and its suppression. Nature 189: 204–207. [DOI] [PubMed] [Google Scholar]
- Hughes SH. 2004. The RCAS vector system. Folia Biol (Praha) 50: 107–119. [PubMed] [Google Scholar]
- Indra AK, Warot X, Brocard J, Bornert JM, Xiao JH, Chambon P, Metzger D. 1999. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic acids research 27: 4324–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving AA, Yoshimi K, Hart ML, Parker T, Clipson L, Ford MR, Kuramoto T, Dove WF, Amos-Landgraf JM. 2014. The utility of Apc-mutant rats in modeling human colon cancer. Dis Model Mech 7: 1215–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob HJ, Kwitek AE. 2002. Rat genetics: attaching physiology and pharmacology to the genome. Nature reviews Genetics 3: 33–42. [DOI] [PubMed] [Google Scholar]
- Jerry DJ, Kittrell FS, Kuperwasser C, Laucirica R, Dickinson ES, Bonilla PJ, Butel JS, Medina D. 2000. A mammary-specific model demonstrates the role of the p53 tumor suppressor gene in tumor development. Oncogene 19: 1052–1058. [DOI] [PubMed] [Google Scholar]
- Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA et al. 2007. LKB1 modulates lung cancer differentiation and metastasis. Nature 448: 807–810. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Deng T, Jones R, Li H, Herschkowitz JI, Liu JC, Weigman VJ, Tsao MS, Lane TF, Perou CM et al. 2010. Rb deletion in mouse mammary progenitors induces luminal-B or basal-like/EMT tumor subtypes depending on p53 status. The Journal of clinical investigation 120: 3296–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin KT, Du WL, Lan HR, Liu YY, Mao CS, Du JL, Mou XZ. 2021. Development of humanized mouse with patient-derived xenografts for cancer immunotherapy studies: A comprehensive review. Cancer science 112: 2592–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston AN, Bu W, Hein S, Garcia S, Camacho L, Xue L, Qin L, Nagi C, Hilsenbeck SG, Kapali J et al. 2018. Hyperprolactinemia-inducing antipsychotics increase breast cancer risk by activating JAK-STAT5 in precancerous lesions. Breast cancer research : BCR 20: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. 2001. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nature genetics 29: 418–425. [DOI] [PubMed] [Google Scholar]
- Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, Sheng K, Niu Y, Goldstein A, Zhao N et al. 2019. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nature cell biology 21: 1113–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Sukumar S, Barbacid M. 1990. Activation of ras oncogenes preceding the onset of neoplasia. Science 248: 1101–1104. [DOI] [PubMed] [Google Scholar]
- Kuperwasser C, Chavarria T, Wu M, Magrane G, Gray JW, Carey L, Richardson A, Weinberg RA. 2004. Reconstruction of functionally normal and malignant human breast tissues in mice. Proceedings of the National Academy of Sciences of the United States of America 101: 4966–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraguchi M, Ohene-Baah NY, Sonkin D, Bronson RT, Kucherlapati R. 2009. Genetic mechanisms in Apc-mediated mammary tumorigenesis. PLoS genetics 5: e1000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurt IC, Zhou R, Iyer S, Garcia SP, Miller BR, Langner LM, Grunewald J, Joung JK. 2021. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nature biotechnology 39: 41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leder A, Pattengale PK, Kuo A, Stewart TA, Leder P. 1986. Consequences of widespread deregulation of the c-myc gene in transgenic mice: multiple neoplasms and normal development. Cell 45: 485–495. [DOI] [PubMed] [Google Scholar]
- Lewandoski M 2001. Conditional control of gene expression in the mouse. Nature reviews Genetics 2: 743–755. [DOI] [PubMed] [Google Scholar]
- Lewis BC, Chinnasamy N, Morgan RA, Varmus HE. 2001. Development of an avian leukosis-sarcoma virus subgroup A pseudotyped lentiviral vector. J Virol 75: 9339–9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BC, Klimstra DS, Socci ND, Xu S, Koutcher JA, Varmus HE. 2005. The absence of p53 promotes metastasis in a novel somatic mouse model for hepatocellular carcinoma. Mol Cell Biol 25: 1228–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BC, Klimstra DS, Varmus HE. 2003. The c-myc and PyMT oncogenes induce different tumor types in a somatic mouse model for pancreatic cancer. Genes & development 17: 3127–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Ferris A, Lewis BC, Orsulic S, Williams BO, holland EC, Hughes SH. 2011. The RCAS/TVA somatic gene transfer method in modeling human cancer. in Genetically-Engineered Mice for Cancer Research: Design, Analysis, Pathways, Validation and Pre-clinical Testing (eds. Green JE, Ried T), pp. 83–111. Springer [Google Scholar]
- Li Y, Hively WP, Varmus HE. 2000. Use of MMTV-Wnt-1 transgenic mice for studying the genetic basis of breast cancer. Oncogene 19: 1002–1009. [DOI] [PubMed] [Google Scholar]
- Limberis MP, Bell CL, Wilson JM. 2009. Identification of the murine firefly luciferase-specific CD8 T-cell epitopes. Gene therapy 16: 441–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SC, Lee KF, Nikitin AY, Hilsenbeck SG, Cardiff RD, Li A, Kang KW, Frank SA, Lee WH, Lee EY. 2004. Somatic mutation of p53 leads to estrogen receptor alpha-positive and -negative mouse mammary tumors with high frequency of metastasis. Cancer Res 64: 3525–3532. [DOI] [PubMed] [Google Scholar]
- Lin TP, Guzman RC, Osborn RC, Thordarson G, Nandi S. 1992. Role of endocrine, autocrine, and paracrine interactions in the development of mammary hyperplasia in Wnt-1 transgenic mice. Cancer Res 52: 4413–4419. [PubMed] [Google Scholar]
- Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI. 1995. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic acids research 23: 1686–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Liang SQ, Zheng C, Mintzer E, Zhao YG, Ponnienselvan K, Mir A, Sontheimer EJ, Gao G, Flotte TR et al. 2021. Improved prime editors enable pathogenic allele correction and cancer modelling in adult mice. Nature communications 12: 2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masso-Welch PA, Darcy KM, Stangle-Castor NC, Ip MM. 2000. A developmental atlas of rat mammary gland histology. J Mammary Gland Biol Neoplasia 5: 165–185. [DOI] [PubMed] [Google Scholar]
- McCormick DL, Adamowski CB, Fiks A, Moon RC. 1981. Lifetime dose-response relationships for mammary tumor induction by a single administration of N-methyl-N-nitrosourea. Cancer Res 41: 1690–1694. [PubMed] [Google Scholar]
- Meyer DS, Brinkhaus H, Muller U, Muller M, Cardiff RD, Bentires-Alj M. 2011. Luminal expression of PIK3CA mutant H1047R in the mammary gland induces heterogeneous tumors. Cancer Res 71: 4344–4351. [DOI] [PubMed] [Google Scholar]
- Middleton PJ. 1965. The histogenesis of mammary tumours induced in the rat by chemical carcinogens. British journal of cancer 19: 830–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Louie SG, Campbell M, Esserman L, Shyamala G. 2000. Ductal growth is impeded in mammary glands of C-neu transgenic mice. Oncogene 19: 5982–5987. [DOI] [PubMed] [Google Scholar]
- No D, Yao TP, Evans RM. 1996. Ecdysone-inducible gene expression in mammalian cells and transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 93: 3346–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldrini B, Curiel-Garcia A, Marques C, Matia V, Uluckan O, Grana-Castro O, Torres-Ruiz R, Rodriguez-Perales S, Huse JT, Squatrito M. 2018. Somatic genome editing with the RCAS-TVA-CRISPR-Cas9 system for precision tumor modeling. Nature communications 9: 1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsulic S, Li Y, Soslow RA, Vitale-Cross LA, Gutkind JS, Varmus HE. 2002. Induction of ovarian cancer by defined multiple genetic changes in a mouse model system. Cancer Cell 1: 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir BC, Sflomos G, Brisken C. 2018. The challenges of modeling hormone receptor-positive breast cancer in mice. Endocrine-related cancer 25: R319–R330. [DOI] [PubMed] [Google Scholar]
- Petkov SP, Heuts F, Krotova OA, Kilpelainen A, Engström G, Starodubova ES, Isaguliants MG. 2013. Evaluation of immunogen delivery by DNA immunization using non-invasive bioluminescence imaging. Hum Vaccin Immunother 9: 2228–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittius CW, Hennighausen L, Lee E, Westphal H, Nicols E, Vitale J, Gordon K. 1988. A milk protein gene promoter directs the expression of human tissue plasminogen activator cDNA to the mammary gland in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 85: 5874–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M et al. 2014. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159: 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podsypanina K, Li Y, Varmus HE. 2004. Evolution of somatic mutations in mammary tumors in transgenic mice is influenced by the inherited genotype. BMC medicine 2: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Préfontaine GG, Walther R, Giffin W, Lemieux ME, Pope L, Haché RJ. 1999. Selective binding of steroid hormone receptors to octamer transcription factors determines transcriptional synergism at the mouse mammary tumor virus promoter. J Biol Chem 274: 26713–26719. [DOI] [PubMed] [Google Scholar]
- Reddy JP, Peddibhotla S, Bu W, Zhao J, Haricharan S, Du YC, Podsypanina K, Rosen JM, Donehower LA, Li Y. 2010. Defining the ATM-mediated barrier to tumorigenesis in somatic mammary cells following ErbB2 activation. Proceedings of the National Academy of Sciences of the United States of America 107: 3728–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy S, Tesson L, Menoret S, Usal C, De Cian A, Thepenier V, Thinard R, Baron D, Charpentier M, Renaud JB et al. 2014. Efficient gene targeting by homology-directed repair in rat zygotes using TALE nucleases. Genome research 24: 1371–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo IH, Russo J. 1978. Developmental stage of the rat mammary gland as determinant of its susceptibility to 7,12-dimethylbenz[a]anthracene. Journal of the National Cancer Institute 61: 1439–1449. [PubMed] [Google Scholar]
- Russo J, Gusterson BA, Rogers AE, Russo IH, Wellings SR, van Zwieten MJ. 1990. Comparative study of human and rat mammary tumorigenesis. Lab Invest 62: 244–278. [PubMed] [Google Scholar]
- Russo J, Russo IH. 1987. Biological and molecular bases of mammary carcinogenesis. Lab Invest 57: 112–137. [PubMed] [Google Scholar]
- –. 2000. Atlas and histologic classification of tumors of the rat mammary gland. J Mammary Gland Biol Neoplasia 5: 187–200. [DOI] [PubMed] [Google Scholar]
- Sanchez-Rivera FJ, Papagiannakopoulos T, Romero R, Tammela T, Bauer MR, Bhutkar A, Joshi NS, Subbaraj L, Bronson RT, Xue W et al. 2014. Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature 516: 428–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer B, Henderson N. 1988. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proceedings of the National Academy of Sciences of the United States of America 85: 5166–5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbert S, Bentley DJ, Melton DW, Rannie D, Lourenço P, Watson CJ, Clarke AR. 1998. Efficient BLG-Cre mediated gene deletion in the mammary gland. Transgenic Res 7: 387–396. [DOI] [PubMed] [Google Scholar]
- Sharma P, Allison JP. 2020. Dissecting the mechanisms of immune checkpoint therapy. Nature reviews Immunology 20: 75–76. [DOI] [PubMed] [Google Scholar]
- Showler K, Nishimura M, Daino K, Imaoka T, Nishimura Y, Morioka T, Blyth BJ, Kokubo T, Takabatake M, Fukuda M et al. 2017. Analysis of genes involved in the PI3K/Akt pathway in radiation- and MNU-induced rat mammary carcinomas. J Radiat Res 58: 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons JK, Hildreth BE 3rd, Supsavhad W, Elshafae SM, Hassan BB, Dirksen WP, Toribio RE, Rosol TJ. 2015. Animal Models of Bone Metastasis. Veterinary pathology 52: 827–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha VC, Qin L, Li Y. 2015. A p53/ARF-dependent anticancer barrier activates senescence and blocks tumorigenesis without impacting apoptosis. Molecular cancer research : MCR 13: 231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P. 1987. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell 49: 465–475. [DOI] [PubMed] [Google Scholar]
- Siwko SK, Bu W, Gutierrez C, Lewis B, Jechlinger M, Schaffhausen B, Li Y. 2008. Lentivirus-mediated oncogene introduction into mammary cells in vivo induces tumors. Neoplasia 10: 653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart TA, Pattengale PK, Leder P. 1984. Spontaneous mammary adenocarcinomas in transgenic mice that carry and express MTV/myc fusion genes. Cell 38: 627–637. [DOI] [PubMed] [Google Scholar]
- Stocklin E, Botteri F, Groner B. 1993. An activated allele of the c-erbB-2 oncogene impairs kidney and lung function and causes early death of transgenic mice. J Cell Biol 122: 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica G, Koestner A, Capen CC. 1983. Characterization of N-ethyl-N-nitrosourea--induced mammary tumors in the rat. The American journal of pathology 110: 161–169. [PMC free article] [PubMed] [Google Scholar]
- –. 1984. Neoplasms induced with high single doses of N-ethyl-N-nitrosourea in 30-day-old Sprague-Dawley rats, with special emphasis on mammary neoplasia. Anticancer research 4: 5–12. [PubMed] [Google Scholar]
- Sukumar S, Notario V, Martin-Zanca D, Barbacid M. 1983. Induction of mammary carcinomas in rats by nitroso-methylurea involves malignant activation of H-ras-1 locus by single point mutations. Nature 306: 658–661. [DOI] [PubMed] [Google Scholar]
- Teng K, Ford MJ, Harwalkar K, Li Y, Pacis AS, Farnell D, Yamanaka N, Wang YC, Badescu D, Ton Nu TN et al. 2021. Modeling High-Grade Serous Ovarian Carcinoma Using a Combination of In Vivo Fallopian Tube Electroporation and CRISPR-Cas9-Mediated Genome Editing. Cancer Res 81: 5147–5160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson HJ, McGinley JN, Rothhammer K, Singh M. 1995. Rapid induction of mammary intraductal proliferations, ductal carcinoma in situ and carcinomas by the injection of sexually immature female rats with 1-methyl-1-nitrosourea. Carcinogenesis 16: 2407–2411. [DOI] [PubMed] [Google Scholar]
- Thompson HJ, McGinley JN, Wolfe P, Singh M, Steele VE, Kelloff GJ. 1998a. Temporal sequence of mammary intraductal proliferations, ductal carcinomas in situ and adenocarcinomas induced by 1-methyl-1-nitrosourea in rats. Carcinogenesis 19: 2181–2185. [DOI] [PubMed] [Google Scholar]
- Thompson TA, Kim K, Gould MN. 1998b. Harvey ras results in a higher frequency of mammary carcinomas than Kirsten ras after direct retroviral transfer into the rat mammary gland. Cancer Res 58: 5097–5104. [PubMed] [Google Scholar]
- Tikoo A, Roh V, Montgomery KG, Ivetac I, Waring P, Pelzer R, Hare L, Shackleton M, Humbert P, Phillips WA. 2012. Physiological levels of Pik3ca(H1047R) mutation in the mouse mammary gland results in ductal hyperplasia and formation of ERalpha-positive tumors. PLoS One 7: e36924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong C, Huang G, Ashton C, Li P, Ying QL. 2011. Generating gene knockout rats by homologous recombination in embryonic stem cells. Nature protocols 6: 827–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursini-Siegel J, Schade B, Cardiff RD, Muller WJ. 2007. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nature reviews Cancer 7: 389–397. [DOI] [PubMed] [Google Scholar]
- Van Keymeulen A, Lee MY, Ousset M, Brohee S, Rorive S, Giraddi RR, Wuidart A, Bouvencourt G, Dubois C, Salmon I et al. 2015. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 525: 119–123. [DOI] [PubMed] [Google Scholar]
- Vassar R, Rosenberg M, Ross S, Tyner A, Fuchs E. 1989. Tissue-specific and differentiation-specific expression of a human K14 keratin gene in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 86: 1563–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visbal AP, LaMarca HL, Villanueva H, Toneff MJ, Li Y, Rosen JM, Lewis MT. 2011. Altered differentiation and paracrine stimulation of mammary epithelial cell proliferation by conditionally activated Smoothened. Dev Biol 352: 116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE. 2011. Cells of origin in cancer. Nature 469: 314–322. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Wall RJ, St-Onge L, Gruss P, Wynshaw-Boris A, Garrett L, Li M, Furth PA, Hennighausen L. 1997. Cre-mediated gene deletion in the mammary gland. Nucleic acids research 25: 4323–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Kennan WS, Yasukawa-Barnes J, Lindstrom MJ, Gould MN. 1992. Difference in the response of neu and ras oncogene-induced rat mammary carcinomas to early and late ovariectomy. Cancer Res 52: 4102–4105. [PubMed] [Google Scholar]
- Wang BC, Kennan WS, Yasukawa-Barnes J, Lindstrom MJ, Gould MN. 1991. Carcinoma induction following direct in situ transfer of v-Ha-ras into rat mammary epithelial cells using replication-defective retrovirus vectors. Cancer Res 51: 2642–2648. [PubMed] [Google Scholar]
- Wang W, Meng Y, Dong B, Dong J, Ittmann MM, Creighton CJ, Lu Y, Zhang H, Shen T, Wang J et al. 2017. A Versatile Tumor Gene Deletion System Reveals a Crucial Role for FGFR1 in Breast Cancer Metastasis. Neoplasia 19: 421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte T, Kim IS, Tian L, Gao X, Wang H, Li J, Holdman XB, Herschkowitz JI, Pond A, Xie G et al. 2016. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nature cell biology 18: 632–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitelaw CB, Harris S, McClenaghan M, Simons JP, Clark AJ. 1992. Position-independent expression of the ovine beta-lactoglobulin gene in transgenic mice. The Biochemical journal 286 ( Pt 1): 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield J, Littlewood T, Evan GI, Soucek L. 2015. The estrogen receptor fusion system in mouse models: a reversible switch. Cold Spring Harb Protoc 2015: 227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters IP, Chiou SH, Paulk NK, McFarland CD, Lalgudi PV, Ma RK, Lisowski L, Connolly AJ, Petrov DA, Kay MA et al. 2017. Multiplexed in vivo homology-directed repair and tumor barcoding enables parallel quantification of Kras variant oncogenicity. Nature communications 8: 2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Jung L, Cooper AB, Fleet C, Chen L, Breault L, Clark K, Cai Z, Vincent S, Bottega S et al. 2009. Dissecting genetic requirements of human breast tumorigenesis in a tissue transgenic model of human breast cancer in mice. Proceedings of the National Academy of Sciences of the United States of America 106: 7022–7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T, Hennighausen L, Wynshaw-Boris A, Deng CX. 1999. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nature genetics 22: 37–43. [DOI] [PubMed] [Google Scholar]
- Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS, Cai W, Yang G, Bronson R, Crowley DG et al. 2014. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 514: 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Tsukamoto T, Popnikolov N, Guzman RC, Chen X, Yang JH, Nandi S. 1995. Adenoviral-mediated gene transfer into primary human and mouse mammary epithelial cells in vitro and in vivo. Cancer letters 98: 9–17. [PubMed] [Google Scholar]
- Young A, Bu W, Jiang W, Ku A, Kapali J, Dhamne S, Qin L, Hilsenbeck SG, Du YN, Li Y. 2022. Targeting the Pro-survival Protein BCL-2 to Prevent Breast Cancer. Cancer prevention research 15: 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan T, Yan N, Fei T, Zheng J, Meng J, Li N, Liu J, Zhang H, Xie L, Ying W et al. 2021. Optimization of C-to-G base editors with sequence context preference predictable by machine learning methods. Nature communications 12: 4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarbl H, Sukumar S, Arthur AV, Martin-Zanca D, Barbacid M. 1985. Direct mutagenesis of Ha-ras-1 oncogenes by N-nitroso-N-methylurea during initiation of mammary carcinogenesis in rats. Nature 315: 382–385. [DOI] [PubMed] [Google Scholar]
- Zhang X, Podsypanina K, Huang S, Mohsin SK, Chamness GC, Hatsell S, Cowin P, Schiff R, Li Y. 2005. Estrogen receptor positivity in mammary tumors of Wnt-1 transgenic mice is influenced by collaborating oncogenic mutations. Oncogene 24: 4220–4231. [DOI] [PubMed] [Google Scholar]
- Zhao S, Shetty J, Hou L, Delcher A, Zhu B, Osoegawa K, de Jong P, Nierman WC, Strausberg RL, Fraser CM. 2004. Human, mouse, and rat genome large-scale rearrangements: stability versus speciation. Genome research 14: 1851–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]