

Abstract
Objective
Heterozygous mutations within the voltage-gated sodium channel α subunit (SCN1A) are responsible for the majority of cases of Dravet syndrome (DS), a severe developmental and epileptic encephalopathy. Development of novel therapeutic approaches is mandatory in order to directly target the molecular consequences of the genetic defect. The aim of the present study was to investigate whether cis-acting long non-coding RNAs (lncRNAs) of SCN1A are expressed in brain specimens of children and adolescent with epilepsy as these molecules comprise possible targets for precision-based therapy approaches.
Methods
We investigated SCN1A mRNA expression and expression of two SCN1A related antisense RNAs in brain tissues in different age groups of pediatric non-Dravet patients who underwent surgery for drug resistant epilepsy. The effect of different antisense oligonucleotides (ASOs) directed against SCN1A specific antisense RNAs on SCN1A expression was tested.
Results
The SCN1A related antisense RNAs SCN1A-dsAS (downstream antisense, RefSeq identifier: NR_110598) and SCN1A-usAS (upstream AS, SCN1A-AS, RefSeq identifier: NR_110260) were widely expressed in the brain of pediatric patients. Expression patterns revealed a negative correlation of SCN1A-dsAS and a positive correlation of lncRNA SCN1A-usAS with SCN1A mRNA expression. Transfection of SK-N-AS cells with an ASO targeted against SCN1A-dsAS was associated with a significant enhancement of SCN1A mRNA expression and reduction in SCN1A-dsAS transcripts.
Conclusion
These findings support the role of SCN1A-dsAS in the suppression of SCN1A mRNA generation. Considering the haploinsufficiency in genetic SCN1A related DS, SCN1A-dsAS is an interesting target candidate for the development of ASOs (AntagoNATs) based precision medicine therapeutic approaches aiming to enhance SCN1A expression in DS.
Keywords: Dravet syndrome, long non coding RNA, regulatory RNA, precision medicine, epilepsy
Abbreviations
- ASM
antiseizure medication
- ASO
antisense oligonucleotide
- dsAS
downstream antisense
- DS
Dravet syndrome
- FCD
focal cortical dysplasia
- GG
ganglioglioma
- HS
hippocampal sclerosis
- KD
ketogenic diet
- LGT
low grade tumor
- lncRNA
long non-coding RNA
- MCD
malformation of cortical development
- MOGHE
mild malformation of cortical development with oligodendroglial hyperplasia
- mMCD
mild malformation of cortical development with heterotopic neurons in the white matter
- MVNT
multinodular and vacuolating neuronal tumor
- PMG
polymicrogyria
- SCN1A
sodium channel 1 α gene
- TPB
TATA-Box binding protein
- usAS
upstream antisense
1. Introduction
Dravet syndrome (DS) is a severe developmental and epileptic encephalopathy (DEE) with manifestation in early infancy. The incidence in the US is estimated to be 1/150,000 [1]. In the majority of cases, DS is caused by heterozygous loss-of-function mutations within the voltage-gated sodium channel α subunit (SCN1A), which leads to haploinsufficiency of the type I voltage-gated sodium channel NaV1.1 [2]. The latter is primarily expressed on axons of fast spiking GABAergic inhibitory interneurons. The SCN1A mutations result in impaired synaptic Na+ currents causing an impairment of GABAergic inhibition of downstream neurons [3,4]. The clinical hallmarks of SCN1A related DS are multiple seizure types, cognitive deterioration, behavioral disturbances, and ataxia [5]. Furthermore, the risk of sudden unexpected death in epilepsy is about 15-fold higher in DS compared to other epilepsies [6,7]. Although most patients receive polytherapy with antiseizure medications, only a minority of patients will become seizure free. In addition, relief of cognitive, behavioral, and motor disturbances is usually not achieved by treatment with current available Antiseizure medication (ASMs). Eventually, epilepsy specific parameters (i.e., as frequency of status epilepticus) seem to have only a minor predictive value for severity of cognitive decline [8,9]. Thus, future treatment approaches should go beyond mere seizure suppression and aim to ameliorate the entire spectrum of symptoms in DS patients. In this context, the development of approaches directly targeting the molecular consequences of the genetic SCN1A defect are of particular interest. In DS patients with SCN1A deficiency, the haploinsufficiency opens opportunities for molecular targeting approaches aiming to upregulate SCN1A.
Long non-coding RNAs (lncRNAs) have been identified as a novel subcategory of regulatory RNA molecules. Especially, lncRNAs, oriented in the opposite direction to protein-coding genes and defined as antisense RNAs, exert a pronounced effect on gene expression with a specific impact on the neighboring gene [10]. In humans, the SCN1A transcription locus is surrounded by gene sequences encoding two antisense RNAs (Figure 1). One transcript with the RefSeq identifier NR_110598 is located downstream to SCN1A, and therefore further defined as SCN1A downstream AS (SCN1A-dsAS). The other transcript with the identifier NR_110260 is located upstream to SCN1A and downstream to SCN9A and referred to as SCN1A upstream AS (SCN1A-usAS, Figure 1).
Figure 1.

Human locus of SCN1A and its antisense transcripts SCN1A-dsAS (NR_110598) and SCN1A-usAS (NR_110260) located downstream to SCN9A on chromosome 2 [11]. Black arrows around transcripts indicate primer location (top: forward primer, bottom: reverse primer).
Recently, it was shown that expression of SCN1A could be enhanced by targeting SCN1A-dsAS with antisense oligonucleotides (ASOs) on both mRNA and protein level [12]. Therefore, SCN1A-dsAS appears to be an interesting transcript as a target for precision medicine approaches in SCN1A related DS. However, further development of this approach for neuropediatric patients requires information about the expression of this potential target antisense RNA during human brain development and across the age range, and about its regional distribution in different brain areas.
In the present study, we therefore investigated the expression of SCN1A related antisense RNAs in surgical brain tissue specimen from pediatric patients. The ratio between antisense RNAs and SCN1A mRNA expression rates were analyzed in order to gain knowledge about the regulatory impact of antisense RNAs SCN1A-dsAS and -usAS on SCN1A expression.
2. Methods
2.1. Patient selection
Patients were selected from two tertiary epilepsy centers (Munich and Vogtareuth, Germany). All patients suffered from medically refractory epilepsy defined as resistant to at least two common ASMs appropriate for the underlying epilepsy syndrome. All included patients underwent a thorough presurgical diagnostic evaluation including a long-term EEG/video monitoring and high-resolution MRI as reported previously [13]. The decision for surgery was taken in an interdisciplinary case conference involving neurologists, pediatric neurologists, neurosurgeons, neuropsychologists, and neurophysiologists.
2.2. Specimen collection
Three to four biopsy specimens (maximum about 5 × 5 × 5 mm/biopsy) per patient were collected and immediately frozen on dry ice. Specimens were either directly transferred for further processing to the laboratory (Munich) or stored at −70°C for further transport and processing (Vogtareuth). A detailed protocol including documentation of all steps was applied for all specimens in order to ensure an accurate way of transportation and continuous cooling.
2.3. Histopathological evaluation
Routine histological evaluation of brain specimens was performed by the Neuropathological Reference Center for Epilepsy surgery of the University of Erlangen.
2.4. RNA-isolation
100 mg tissue was grounded in 900 µL of Isol-RNA Lysis Reagent (VWR, Germany) and incubated at room temperature for 10 min. The suspension was cleared at 17.000 g and 4°C for 5 min and treated with 200 µL chloroform (Roth, Germany) for 5 min under continuous shaking. The aqueous phase was separated from the organic phase by spinning at 17.000g and 4°C for 20 min. RNA was precipitated from the aqueous phase with 650 µL isopropanol (Roth) at −20°C for at least 1 h. RNA was pelleted at 17.000g and 4°C for 35 min and then washed twice with 75% ethanol (Roth). The RNA pellet was air-dried and dissolved in 15–35 µL nuclease-free water. For re-purification, RNA was treated with 350 µL of lysis buffer (Qiagen, Germany) and 100 µL ethanol and loaded on RNAeasy® spin column (Qiagen, Germany). Further purification was performed according to the manufacturer’s instructions. RNA quality was assessed on an analytical agarose gel or with the qubitTM RNA IQ assay kit (Thermo Fischer Scientific, MA, USA).
2.5. First-strand cDNA synthesis (reverse transcription)
0.375 µg Oligo-dT12-18 (Thermo Fischer Scientific, MA, USA) were annealed to 500–1,000 ng bulk RNA together with 7.5 nmol of each dNTPs in a 9 µL reaction scale. Thereafter, the RNA was first denatured at 65°C for 5 min and subsequently cooled down to 37°C. Afterwards, 3 µL 5× First-strand buffer (Thermo Fischer Scientific, MA, USA), 1.5 µL DTT (100 mM, Thermo Fischer Scientific, MA, USA), 30 U murine RNAse Inhibitor (New England Biolabs, USA), and 150 U M-MLV reverse transcriptase (Thermo Fischer Scientific, MA, USA) were added to the reaction. After 45 min of incubation at 37°C, the reaction was stopped at 70°C for 10 min. For expression analysis by qPCR, the cDNA was diluted with 185 µL of nuclease-free water.
2.6. Quantitative RT-PCR analysis
2 µL of cDNA was amplified in a 10 µL 1× Fast SYBR® Green Master Mix (Thermo Fischer Scientific, MA, USA) in the presence of 2.5 pmol of each primer. Three technical replicates were run per primer pair. The reaction was run in an Applied Biosystems StepOneTM or Biorad CFX-384 cycler with the following settings: 95°C, 3 min for initial denaturing and 40 cycles with 95°C, 3 s and 60°C, 30 s. Typically, we designed primers annealing either to two subsequent exons or at least to the border between both exons to avoid the amplification of genomic DNA (Table 1). For normalization and quantification using the ∆Ct method, TBP served as a housekeeping gene. Two to four biopsy specimens per patient were analyzed to calculate the mean expression 2(ΔCt) value.
Table 1.
Primer sequence information for SCN1A, SCN1A-dsAS, and SCN1A-usAS
| Target | Primer | 5′→ 3′ sequence |
|---|---|---|
| TBP | TBP-fw | CGGTTTGCTGCGGTAATCAT |
| TBP-rv | GGTGAGCACAAGGCCTTCTA | |
| SCN1A | SCN1A-fw | GCTGTATACATCCGTGTGCAG |
| SCN1A-rv | GCGTCTTTCAATAGCCGCAA | |
| SCN1A-dsAS | SCN1A-dsAS-fw | CCTTGTACACCAGGGCATTC |
| SCN1A-dsAS-rv | GTGGTATAGGAACTGGCAGCA | |
| SCN1A-usAS | SCN1A-usAS-fw | CCAGGAAACAGGAATTCAGGC |
| SCN1A-usAS-rv | CGAGTGATCCGTCTTGCCA |
2.7. Statistical analysis
The correlation of SCN1A and its antisense transcripts were evaluated with R 3.6 using the Pearson correlation method to calculate R 2 and p-value. The student’s t-test for unpaired samples was applied to evaluate intergroup differences. The level of significance level was set to 0.05 (two-tailed). In case of multiple testing, the significance level was adjusted applying the correction method of Bonferroni [14].
2.8. ASOs directed against SCN1A-dsAS
ASO specifications are depicted in Table 2.
Table 2.
Detailed specifications of ASO. ASO2 is not shown as it was not ordered due to predicted inappropriate binding properties
| ASO | Modified seg: *phosphorothioates, 2MoEr: 2'methodxyethoxy moiety | Scale (nmol) |
|---|---|---|
| scr-ctrl | /52MOErC/*/i2MOErT/*/i2MOErA/*/i2MOErA/*/i2MOErG/*G*T*T*A*A*G*T*C*G*C*/i2MOErC/*/i2MOErC/*/i2MOErT/*/i2MOErC/*/32MOErG/ | 100 |
| ASO1 (nr110598-1) | /52MOErC/*/i2MOErA/*/i2MOErC/*/i2MOErG/*/i2MOErG/*A*A*G*A*C*T*T*T*A*G*/i2MOErT/*/i2MOErA/*/i2MOErG/*/i2MOErT/*/32MOErG/ | 100 |
| ASO3 (nr110598-3 | /52MOErG/*/i2MOErG/*/i2MOErT/*/i2MOErA/*/i2MOErT/*A*G*G*A*A*C*T*G*G*C*/i2MOErA/*/i2MOErG/*/i2MOErC/*/i2MOErA/*/32MOErG/ | 100 |
2.9. Cell culture and transfection of cell lines
SK-N-AS cells (human neuroblastoma derived cell line) were cultured for 48 h (5% CO2) at 37°C. 40pmol of ASO were transfected with Lipofectamin2000 (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufactures’ protocol. RT-PCR of SCN1A and SCN1A-dsAS was performed 48 h after transfection as described in Section 2.6.
Ethical approval: The research related to human use has been complied with all the relevant national regulations, institutional policies and in accordance with the tenets of the Helsinki Declaration, and has been approved by the authors’ institutional review board or equivalent committee. The responsible ethical boards (Munich: ethical board of the Medical Faculty of the University of Munich, Vogtareuth: ethical board of the Bavarian professional association of physicians) approved the study (# 18-331 and # 18042, respectively). Patient data after study entry were pseudonymized.
Informed consent: Informed consent has been obtained from all individuals included in this study.
3. Results
3.1. Demographic data
We investigated the brain specimens of a total of 18 patients (for summary of demographic data, see Table 3, for a detailed individual case description, see Table 4) of whom 14 were females (77.8%). The mean age at the time of surgery was 9.1 years (±5.1, range 0.8–18.7) and the mean duration of epilepsy was 6.5 years (±4.5, range 0.8–15.7). Most of the patients suffered from daily seizures at the time of surgery (n = 12, 66.7%). Only a minority of the patients had seizures on a weekly or monthly basis (n = 3, 16.7% and n = 3, 16.6%). The regional distribution of specimen origin was as follows: frontal lobe 50% (n = 10), temporal lobe 40% (n = 8), parietal lobe 5% (n = 1), and insula 5% (n = 1).
Table 3.
Summary of demographic patient data
| Total (n =) | 18 |
| Sex (f/m) | 14/4 |
| Mean age at surgery (years) | 9.1 (±5.1, range 0.8–18.7) |
| Mean age at seizure manifestation (years) | 2.6 (±2.5, range 0.1–8.0) |
| Mean epilepsy duration (years) | 6.5 (±4.5, range 0.8–15.7) |
| Seizure frequency ( n =) | |
| Daily | 12 |
| Weekly | 3 |
| Monthly | 3 |
| Number of previous ASMs (mean value) | 5 (±3, range 1–12) |
| Previous KD (n =) | 2 |
| Number of ASMs at surgery (mean value) | 2 (±1, range 1–4) |
| Resected tissue area ( n =) | |
| Frontal | 10 |
| Temporal | 8 |
| Parietal | 1 |
| Insular | 1 |
| Pathology ( n =) | |
| MCD | 13 |
| HS | 3 |
| LGT | 2 |
| Gliosis | 3 |
ASMs = antiseizure medications, KD = ketogenic diet, MCD = malformation of cortical development, HS = hippocampal sclerosis, LGT = low grade tumor.
Table 4.
Detailed summary of individual patient specifications
| Patient ID | Sex | Age at surgery (years) | Seizure manifestation (years) | Epilepsy duration (years) | Seizure frequency | Number of previous ASMs | ASMs at surgery | Type of surgery/resection | Investigated tissue | Pathology |
|---|---|---|---|---|---|---|---|---|---|---|
| SKV-13 | M | 13.3 | 0.3 | 12.9 | Daily | 2 | 1 | Temporal lobe resection, amygdalohippocampectomy, anterior insular, frontal opercular, and basal cortex | Anterior temporal lobe | HS, FCD IIIa |
| SKV-04 | W | 4.2 | 0.2 | 4.0 | Daily | 12 | 2 | Subtotal frontal and anterior insular resection | Frontal lobe | MOGHE |
| SKV-03 | M | 4.8 | 1.4 | 3.4 | Daily | 10 | 2 | Subtotal frontal and anterior insular and anterior cingular resection | Frontal lobe | MOGHE |
| SKV-05 | M | 11.5 | 8.0 | 3.5 | Daily | 1 | 1 | Temporal lobe resection | Anterior temporal lobe | MVNT |
| SKV-08 | W | 8.5 | 5.0 | 3.5 | Weekly | 2 | 1 | Temporal lobe resection | Anterior temporal lobe | GG |
| LMU-16 | W | 6.5 | 5.0 | 1.5 | Weekly | 3 | 1 | Parietal lesionectomy | Parietal lobe | FCD Ia |
| LMU-17 | W | 0.8 | 0.1 | 0.8 | Daily | 4 | 4 | Hemispherotomy | Frontal and parietal lobe, insula | FCD IIb, PMG |
| SKV-01 | W | 1.3 | 0.3 | 1.0 | Weekly | 7 | 4 | Hemispherotomy | Frontal lobe | FCD IIa |
| SKV-06 | W | 7.3 | 2.0 | 5.3 | Daily | 5 | 2 | Hemispherotomy | Frontal lobe | Gliosis |
| SKV-07 | M | 13.2 | 0.5 | 12.7 | Daily | 4 | 2 | Hemispherotomy | Frontal lobe | Gliosis |
| SKV-10 | W | 7.3 | 1.4 | 5.9 | Daily | 4 | 4 | Subtotal frontal lobe resection | Frontal lobe | MOGHE |
| SKV-14 | W | 7.7 | 2.0 | 5.7 | Monthly | 4 | 2 | Temporal lobe resection | Anterior temporal lobe | HS |
| SKV-15 | W | 18.7 | 3.0 | 15.7 | Daily | 3 | 2 | Temporal lobe resection | Anterior temporal lobe | HS |
| SKV-12 | W | 12.1 | 3.5 | 8.6 | Daily | 4 | 2 | Hemispherotomy | Frontal lobe | PMG |
| SKV-02 | W | 12.2 | 0.6 | 11.6 | Daily | 10 | 4 | Hemispherotomy | Frontal lobe | FCD IIId, gliosis |
| SKV-09 | W | 13.9 | 7.2 | 6.8 | Monthly | 5 | 2 | Temporal lobe resection | Temporal lobe | mMCD |
| SVK-11 | W | 16.1 | 5.5 | 10.6 | Month | 3 | 2 | Temporal lobe resection | Anterior temporal lobe | FCD IIa |
| LMU-18 | W | 4.0 | 0.6 | 3.4 | Daily | 6 | 2 | Hemispherotomy | Frontal lobe | MOGHE |
Note: Patients SVK-03 and 13 received ketogenic diet as a presurgical antiseizure treatment. Abbreviations: HS = hippocampal sclerosis, FCD = focal cortical dysplasia, GG = ganglioglioma, MOGHE = mild malformation of cortical development with oligodendroglial hyperplasia, MVNT = multinodular and vacuolating neuronal tumor, mMCD = mild malformation of cortical development with heterotopic neurons in the white matter, PMG = polymicrogyria, ASMs = antiseizure medications.
3.2. SCN1A and SCN1A-lnc expression and correlation
In the first extraction step, 51.8 ± 24.9 µg total RNA was purified from 100 mg tissue. After the second column purification step, we obtained 63.26 ± 37.9% of the used RNA with an RNA-IQ value of 8.2 ± 0.4, showing that the purification procedure yielded RNA with high quality. Next we measured the level of SCN1A, SCN1A-dsAS, and SCN1A-usAS with quantitative RT-PCR using TBP as a housekeeping gene [15].
SCN1A was the most abundant transcript (Figure 2a). Among the regulatory antisense RNAs, SCN1A-dsAS was the least abundant antisense transcript (Figure 2b), and SCN1A-usAS exhibited a moderate expression (Figure 2c). The ratio between the individual expression of SCN1A, SCN1A-dsAS, and SCN1A-usAS was comparable among patients (Figure 2g).
Figure 2.

SCN1A expression correlates with the expression of SCN1A-dsAS and SCN1A-usAS. (a–c) RNA expression levels of SNC1A (a), SCN1A-dsAS (b), and SCN1A-usAS (c) determined by quantitative RT-PCR in biopsy specimens dissected from patients who underwent surgery for drug resistant epilepsy. Values are normalized to TBP. Expression values were measured in 2–4 biopsy specimens per patient. (d–f) Correlation analysis of the expression of SCN1A and its antisense transcripts. The correlation coefficient is provided as R 2-value in the plot. (d) Correlation of SCN1A expression levels with SCN1A-dsAS expression levels (log scale). (e) Correlation of SCN1A expression levels with SCN1A-usAS expression levels. (f) Correlation of SCN1A expression levels with the ratio of SCN1A-usAS and SCN1A-dsAS expression levels. (g) Boxplot analysis showing distribution of: SCN1A-dsAS/SCN1A ratio (red), SCN1A-usAS/SCN1A ratio (green), and SCN1A-usAS/SCN1A-dsAS/SCN1A ratio (blue, log scale).
Next we completed a correlation analysis to assess a putative regulatory impact of the antisense transcripts on SCN1A expression by correlation analysis. First, we analyzed a potential correlation of SCN1A’s with SCN1A-dsAS’s expression. This analysis demonstrated a significant negative correlation (R 2 = 0.5, p < 0.0001, Figure 2d). In contrast, the respective analysis for SCN1A and SCNA1-usAS’s expression revealed a positive correlation (R 2 = 0.52, p < 0.0001, Figure 2e).
To test whether both transcripts co-regulate SCN1A’s expression, we additionally investigated the correlation of SCN1A expression with the expression ratio of both antisense transcripts. A strong correlation became evident between SCN1A expression and the expression ratio of the antisense transcripts (R 2 = 0.87, p < 0.0001, Figure 2f). An assessment of a potential direct correlation between the regulatory RNAs did not confirm a significant correlation (R 2-value = 0.0625; p = 0.32, no plot shown).
We detected no difference in the expression patterns of SCN1A, SCN1A-dsAS, and SCN1A-usAS with respect to lobar location (Figure 3a–c) and sex (Figure 3d–f). Furthermore, there was no correlation of either transcript expression for age at surgery (Figure 4a–c), age of epilepsy onset (Figure 4d–f) or epilepsy duration (Figure 4g–i).
Figure 3.

SCN1A, SCN1A-dsAS, and SCN1A-usAS are equally expressed in frontal and temporal lobes and in both sexes. (a–c) Boxplots show the expression of SCN1A (a), SCN1A-dsAS (b), and SCN1A-usAS (c) in the frontal lobe and temporal lobe. (d–f) Boxplots show the expression of SCN1A (d), SCN1A-dsAS (e), and SCN1A-usAS (f) in the male and female patients.
Figure 4.

Age at surgery, age of disease onset, and duration of epilepsy do not correlate with SCN1A, SCN1A-dsAS, and SCN1A-usAS expression. (a–c) Correlation of SCN1A expression (a), SCN1A-dsAS (b), and SCN1A-usAS (c) for age at surgery. (d–f) Correlation of SCN1A expression (d), SCN1A-dsAS (e), and SCN1A-usAS (f) for age of onset. (g–i) Correlation of SCN1A expression (g), SCN1A-dsAS (h), and SCN1A-usAS (i) for duration of epilepsy.
3.3. Treatment of SK-N-AS cells with ASOs directed against SCN1A-dsAS
Three ASOs targeted against SCN1A-dsAS were designed. Treatment with ASO3 revealed a significant increase (p < 0.001) in SCN1A mRNA expression by about 15-fold compared to control or mock transfected cells (Figure 5a). Furthermore, ASO3 treatment was associated with a reduction in SCN1A-dsAS expression by about 40% (p < 0.05) compared to control and mock transfected cells (Figure 5b). ASO1 did not alter the expression of either SCN1A mRNA or SCN1A-dsAS transcript (not shown).
Figure 5.
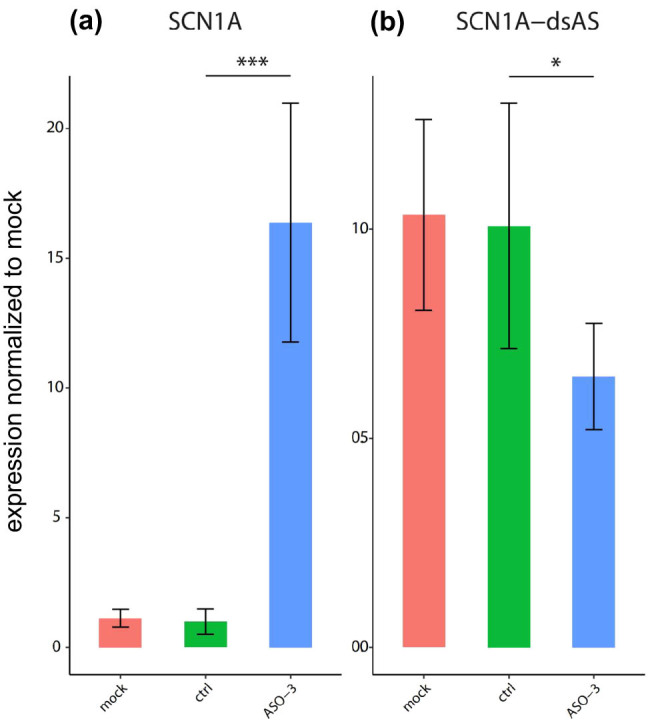
Transfection of SK-N-AS cells with ASOs directed against SCN1A-dsAS. Treatment with ASO3 showed a significant increase in SCN1A mRNA expression by about 15-fold (a, ***p < 0.001) and reduction in SCN1A-dsAS expression by about 40% compared to control and mock transfected cells (b, *p < 0.05).
4. Discussion
In the present study, we analyzed RNA-expression of SCN1A and two SCN1A antisense RNAs (SCN1A-dsAS and SCN1A-usAS) in brain specimens of children and adolescents undergoing surgery for medically refractory epilepsy. SCN1A-dsAS correlated negatively and SCN1A-usAS positively with SCN1A expression suggesting a contrary role in SCN1A gene regulation for both antisense RNAs.
SCN1A related DS is a devastating DEE. There are recent developments of new antiseizure medications as fenfluramine and cannabidiol. These drugs can significantly reduce seizure frequency compared to placebo though seizure freedom is only rarely achieved [16,17]. However, the impact of “newer” and “older” antiseizure medications on further clinically relevant symptoms in DS patients including cognitive decline, behavioral disturbances, and atactic gait disorder seems to be rather limited. Thus, there is an urgent need for disease-modifying agents overcoming NaV1.1 haploinsufficiency. This need is underlined by both clinical and experimental findings that seizure activity alone is only a poor contributor to cognitive deterioration [9,18]. In the sense of precision medicine, various options are currently under investigation. Among others, these include the application of selective Nav1.1 activators, CRISPRa/dCAS technologies targeting transcription, of ASOs to target either mRNA processing or, as antagonists of cis-acting antisense RNAs [19–22]. These approaches aim to either ameliorate the haploinsufficiency of Nav1.1 by enhancing the activity or by upregulating the expression of residual Nav1.1. The inhibition of antisense RNAs on SCN1A has been recently extensively investigated in vitro and in vivo [12]. The latter study by Hsiao and colleagues revealed that SCN1A expression can be enhanced by application of ASOs against SCN1A related antisense RNAs in several cell lines, fibroblasts of patients with DS, and brain tissue of both mice and monkeys [12]. Moreover, seizure activity was ameliorated by this approach in mice carrying a pathogenic heterozygous SCN1A mutation.
4.1. Expression of SCN1A and SCN1A-dsAS/SCN1A-usAS in human brain tissue
SCN1A and SCN1A-dsAS/SCN1A-usAS proved to be expressed in the investigated brain regions primarily comprising the frontal and temporal lobe. According to the human protein atlas (brain-map.org) and previous reports, SCN1A is known to be widely expressed in these areas [23,24]. The expression in these brain regions is thought to be connected to the key clinical features in DS as cognitive and behavioral disturbances (frontal lobe) and seizures (temporal lobe, hippocampus) [18,25]. We noticed that the expression of all three transcripts varied significantly between the patients. However, the ratio of the individual expression of SCN1A, SCN1A-dsAS, and SCN1A-usAS was comparable among patients. These results suggest similar regulatory mechanisms between the transcripts despite interindividual differences in quantitative expression levels. There was no correlation between transcript and the age of investigation (= age at surgery). These findings support that potential targets for treatment, i.e., with ASOs against SCN1A-dsAS are detectable within the time span of the investigated patients from 1 to 19 years of age. This finding is of particular relevance as any treatment option aiming to modify the disease should be most likely implemented as early as possible in order to overcome long term sequelae of concurrent Nav1.1 dysfunction and ongoing seizure activity on the developing brain [26–28].
Epilepsy duration had no impact on regulation of SCN1A, SCN1A-dsAS, and SCN1A-usAS (Figure 4g–i). As epilepsy duration also reflects the total load of seizures until surgery, the findings may suggest that seizure activity does not affect SCN1A, SCN1A-dsAS, and SCN1A-usAS expression in brain tissues derived from non-Dravet patients. Our data on expression of SCN1A-dsAS in human brain tissues complement findings of previous studies reporting SCN1A antisense RNAs expression in mouse and monkey brain tissue [12]. In the latter study, the researchers also detected SCN1A antisense RNAs expression in brain tissue derived from a human tissue panel.
4.2. Correlation of expression levels of SCN1A and SCN1A-dsAS/SCN1A-usAS in human brain tissue
SCN1A expression correlated in an opposite manner with the two SCN1A related antisense RNAs: SCN1A-dsAS correlated negatively and SCN1A-usAS positively with SCN1A expression on mRNA level. The data suggest that both antisense transcripts co-modulate SCN1A’s expression without affecting the expression of the other antisense transcript. While SCN1A-dsAS most likely suppresses SCN1A expression, SCN1A-usAS may enhance SCN1A expression. Thus, these results support that inhibition of SCN1A-dsAS, i.e., by ASOs might help to increase SCN1A gene expression. As mentioned above, functional evidence has been reported that SCN1A gene expression on mRNA and protein level in vitro is enhanced by ASOs against SCN1A-dsAS RNA and improves in vivo seizure activity in a Dravet mouse model [12]. Contrary to our results in human tissue, the latter study reported a positive correlation of the downstream antisense RNA and SCN1A in primate (green monkey) tissue samples. They interpreted these findings as an increased turnover of antisense RNAs in more “active” loci. However, the results are difficult to compare with our findings as they used mixed tissue samples consisting of more than only brain tissue. Furthermore, potential species differences might be applicable in this context.
As DS is commonly caused by haploinsufficiency of truncating or less commonly missense mutations of the SCN1A gene, there is no dominant negative effect of mutations [29]. Thus, it is rather unlikely that upregulation of SCN1A will exert detrimental effects in the affected neurons. The unlikelihood of dominant negative effects further underlines the potential value of ASOs against SCN1A-dsAS as a precise and disease modifying therapeutic concept for patients with DS. This is further underlined by current approaches and ongoing phase 1/2 studies to enhance SCN1A expression by overcoming introduction of the poison exon 20 N insertion within the SCN1A transcript [20,30]. Insertion of exon 20 N into a SCN1A transcript is known to reduce functioning Nav1.1 channel expression. Nevertheless, recent findings have reported on patients with SCN1A gain of function mutations [31]. In these patients, enhancement of SCN1A mRNA expression is not warranted as worsening of the condition is very likely.
In addition to SCN1A-dsAS, SCN1A-usAS, whose expression positively correlates with SCN1A expression, could also be a suitable target for increasing SCN1A expression. Similar to the reported cases in which lncRNAs such as PCNA-AS enhance the expression of nearby protein-coding genes by repressing miRNAs targeting these genes, there is a possibility that a similar regulatory network exists between SCN1A-usAS and SCN1A [32]. If this regulatory link is confirmed, exogenous expression of SCN1A-usAS to enhance SCN1A expression could be exploited using recombinant adeno-associated viruses that do not deliver 13kbp targets like SCN1A [33].
4.3. Treatment of SK-N-AS cells with ASOs directed against SCN1A-dsAS
Treatment with ASO3 targeted against SCN1A-dsAS revealed a significant increase in SCN1A mRNA expression and a reduced abundance of SCN1A-dsAS transcript in SK-N-AS cells. In our study, ASO3 demonstrated superior efficiency when compared to a previously published ASO (Cur-1740, 11), despite both targeting similar sites within the SCN1A-dsAS sequence. This enhanced efficacy could be attributed to distinct chemical modifications employed in our ASO design, specifically the incorporation of 2′O-methoxy-ethyl as opposed to alternative 2′O modification methods. This choice of chemical modification is known to elevate the melting temperature of the ASO and enhance its overall affinity towards the target gene [34]. The increased stability conferred by the 2′O-methoxy-ethyl modification potentially contributes to a more robust and effective binding of the ASO3 towards SCN1A-dsAS, resulting in a higher overall efficiency in upregulation of SCN1A
These data further underline the role of SCN1A-dsAS in regulation and especially in inhibition of SCN1A mRNA expression as investigated previously [12]. Reduced SCN1A-dsAS transcripts in ASO3 transfected cells further suggest that the enhancement of SCN1A mRNA Expression is in part negatively correlated with the amount SCN1A-dsAS lncRNA.
4.4. Limitations
There are several limitations, which should be acknowledged when interpreting the results of the present study. Measuring gene expression is a descriptive rather than a functional approach. In addition, due to posttranscriptional and posttranslational regulation mRNA expression does not always correlate with the expression of a fully functional protein [35). Nevertheless, a correlation of SCN1A mRNA expression and protein function was demonstrated in recent studies [12,20). In addition, concern on interpreting only quantitative RNA levels does not apply to regulatory RNA levels including lncRNAs.
One needs further to consider the heterogeneity of the samples (i.e., different pathological findings) and of the patient specifications for interpretation of the findings. However, the aim of this study was to determine whether the regulatory antisense RNAs are expressed at a relevant level in the epileptic brain at different age levels. Therefore, the aim implies a respective heterogeneity. In this context, it also should be underlined that we did not investigate brain tissues from SCN1A positive patients as these do not qualify for epilepsy surgery as investigated previously [36,37]. Thus, investigating brain specimen from SCN1A patients appears to be more than challenging. Nevertheless, caution is warranted when comparing results from non-Dravet to DS patients. We cannot exclude that SCN1A antisense transcripts are differently regulated in the brains of SCN1A-haploinsufficient patients. However this is at least not true for mouse models [12].
Furthermore, the use of control brain tissue could be suggested. However, taking intact brain specimen of children for a scientific study is questionable from an ethical point of view. Thus, we decided against using normal tissue in this study.
It is also emphasized that the sample size did not allow a meaningful subgroup comparison. While the data did not point to differences related to lobar location and sex or to a correlation of expression data with age of onset, age at surgery, and epilepsy duration, we cannot exclude that a larger sample size would have revealed differences or an association. There were no significant changes in the expression of SCN1A and both of its antisense transcripts with respect to lobar location and sex. Furthermore, age of onset, age at surgery, and epilepsy duration did not correlate with expression levels of either transcript. These findings might not only suggest a rather homogenous sample size but also could be interpreted as a result of the small sample size.
4.5. Conclusion
The findings of the present study revealed that SCN1A antisense RNAs are widely expressed across different human brain regions and age range. Correlation analysis suggests inhibitory effects of SCN1A-dsAS and enhancing effects of SCN1A-usAS on SCN1A expression. The data support SCN1A-dsAS as an interesting target for development of precision medicine therapeutic approaches for SCN1A related DS aiming to enhance expression of functional Nav1.1 and to overcome haploinsufficiency.
Acknowledgements
We thank Andreas Kutschka for sample transfer.
Footnotes
Funding information: This study was funded by Roche Ltd, Basel, Switzerland.
Conflict of interest: M.F.S., J.S., V.M., M.V., A.H.L.F., H.P., and I.Bor. received funding from Roche Ltd, Basel, Switzerland. T.K. and S.L. are employee at F. Hoffmann-La Roche Ltd, Basel, Switzerland. F.M. is a former employee of F. Hoffmann-La Roche Ltd, Basel, Switzerland. This does not alter our adherence to policies on sharing data and materials. All other authors state no conflict of interest.
Data availability statement: The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- [1].Wu YW, Sullivan J, McDaniel SS, Meisler MH, Walsh EM, Li SX, et al. Incidence of Dravet syndrome in a US population. Pediatrics. 2015;136:e1310. [DOI] [PMC free article] [PubMed]
- [2].Volkers L, Kahlig KM, Verbeek NE, Das J, van Kempen M, Stroink H, et al. Nav 1.1 dysfunction in genetic epilepsy with febrile seizures-plus or Dravet syndrome. Eur J Neurosci. 2011;34:1268–75. [DOI] [PMC free article] [PubMed]
- [3].Jensen HS, Grunnet M, Bastlund JF. Therapeutic potential of Na(V)1.1 activators. Trends Pharmacol Sci. 2014;35:113. [DOI] [PubMed]
- [4].Dutton SB, Makinson CD, Papale LA, Shankar A, Balakrishnan B, Nakazawa K, et al. Preferential inactivation of SCN1A in parvalbumin interneurons increases seizure susceptibility. Neurobiol Dis. 2013;49:211. [DOI] [PMC free article] [PubMed]
- [5].Dravet C. The core Dravet syndrome phenotype. Epilepsia. 2011;52(Suppl 2):3. [DOI] [PubMed]
- [6].Kalume F, Westenbroek RE, Cheah CS, Yu FH, Oakley JC, Scheuer T, et al. Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest. 2013;123:1798–808. [DOI] [PMC free article] [PubMed]
- [7].Kearney J. Sudden unexpected death in Dravet syndrome. Epilepsy Curr. 2013;13:264–5. [DOI] [PMC free article] [PubMed]
- [8].Gataullina S, Dulac O. Is epilepsy the cause of comorbidities in Dravet syndrome? Dev Med Child Neurol. 2018;60:8. [DOI] [PubMed]
- [9].Nabbout R, Chemaly N, Chipaux M, Barcia G, Bouis C, Dubouch C, et al. Encephalopathy in children with Dravet syndrome is not a pure consequence of epilepsy. Orphanet J Rare Dis. 2013;8:176. [DOI] [PMC free article] [PubMed]
- [10].Gil N, Ulitsky I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat Rev Genet. 2020;21:102–17. [DOI] [PubMed]
- [11].Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24. [DOI] [PMC free article] [PubMed]
- [12].Hsiao J, Yuan TY, Tsai MS, Lu CY, Lin YC, Lee ML, et al. Upregulation of haploinsufficient gene expression in the brain by targeting a long non-coding RNA improves seizure phenotype in a model of Dravet syndrome. EBioMedicine. 2016;9:257–77. [DOI] [PMC free article] [PubMed]
- [13].Noachtar S, Borggraefe I. Epilepsy surgery: A critical review. Epilepsy Behav. 2009;15:66–72. [DOI] [PubMed]
- [14].Bland JM, Altman DG. Multiple significance tests: The Bonferroni method. BMJ. 1995;310:170. [DOI] [PMC free article] [PubMed]
- [15].Zhang Q, Zhang H, Liu F, Yang Q, Chen K, Liu P, et al. Comparison of reference genes for transcriptional studies in postmortem human brain tissue under different conditions. Neurosci Bull. 2019;35:225–8. [DOI] [PMC free article] [PubMed]
- [16].Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R, et al. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N Engl J Med. 2017;376:2011–20. [DOI] [PubMed]
- [17].Lagae L, Sullivan J, Knupp K, Laux L, Polster T, Nikanorova M, et al. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: a randomised, double-blind, placebo-controlled trial. Lancet. 2019;394:2243–54. [DOI] [PubMed]
- [18].Bender AC, Natola H, Ndong C, Holmes GL, Scott RC, Lenck-Santini PP. Focal SCN1A knockdown induces cognitive impairment without seizures. Neurobiol Dis. 2013;54:297–307. [DOI] [PMC free article] [PubMed]
- [19].Johannessen Landmark C, Potschka H, Auvin S, Wilmshurst JM, Johannessen SI, Kasteleijn-Nolst Trenité D, et al. The role of new medical treatments for the management of developmental and epileptic encephalopathies: Novel concepts and results. Epilepsia. 2021;62:857. 10.1111/epi.16849. [DOI] [PubMed]
- [20].Han Z, Chen C, Christiansen A, Ji S, Lin Q, Anumonwo C, et al. Antisense oligonucleotides increase SCN1A expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci Transl Med. 2020;12. 10.1126/scitranslmed.aaz6100. [DOI] [PubMed]
- [21].Chow CY, Chin Y, Ma L, Undheim E, Herzig V, King GF. A selective NaV1.1 activator with potential for treatment of Dravet syndrome epilepsy. Biochem Pharmacol. 2020;181:113991. [DOI] [PubMed]
- [22].Yamagata T, Raveau M, Kobayashi K, Miyamoto H, Tatsukawa T, Ogiwara I, et al. CRISPR/dCas9-based SCN1A gene activation in inhibitory neurons ameliorates epileptic and behavioral phenotypes of Dravet syndrome model mice. Neurobiol Dis. 2020;141:104954. [DOI] [PubMed]
- [23].Bilginer B, Yalnizoglu D, Soylemezoglu F, Turanli G, Cila A, Topçu M, et al. Surgery for epilepsy in children with dysembryoplastic neuroepithelial tumor: clinical spectrum, seizure outcome, neuroradiology, and pathology. Childs Nerv Syst. 2009;25:485. [DOI] [PubMed]
- [24].Wang W, Takashima S, Segawa Y, Itoh M, Shi X, Hwang SK, et al. The developmental changes of Na(v)1.1 and Na(v)1.2 expression in the human hippocampus and temporal lobe. Brain Res. 2011;1389:61–70. [DOI] [PubMed]
- [25].Ohno Y, Ishihara S, Mashimo T, Sofue N, Shimizu S, Imaoku T, et al. SCN1A missense mutation causes limbic hyperexcitability and vulnerability to experimental febrile seizures. Neurobiol Dis. 2011;41:261. [DOI] [PubMed]
- [26].Lopez-Santiago L, Isom LL. Dravet Syndrome: A developmental and epileptic encephalopathy. Epilepsy Curr. 2019;19:51–3. [DOI] [PMC free article] [PubMed]
- [27].Favero M, Sotuyo NP, Lopez E, Kearney JA, Goldberg EM. A transient developmental window of fast-spiking interneuron dysfunction in a mouse model of Dravet syndrome. J Neurosci. 2018;38:7912–27. [DOI] [PMC free article] [PubMed]
- [28].Richards KL, Milligan CJ, Richardson RJ, Jancovski N, Grunnet M, Jacobson LH, et al. Selective NaV1.1 activation rescues Dravet syndrome mice from seizures and premature death. Proc Natl Acad Sci U S A. 2018;115:E8077. [DOI] [PMC free article] [PubMed]
- [29].Bechi G, Scalmani P, Schiavon E, Rusconi R, Franceschetti S, Mantegazza M. Pure haploinsufficiency for Dravet syndrome Na(V)1.1 (SCN1A) sodium channel truncating mutations. Epilepsia. 2012;53:87–100. [DOI] [PubMed]
- [30].Voskobiynyk Y, Battu G, Felker SA, Cochran JN, Newton MP, Lambert LJ, et al. Aberrant regulation of a poison exon caused by a non-coding variant in a mouse model of SCN1A-associated epileptic encephalopathy. PLoS Genet. 2021;17:e1009195. [DOI] [PMC free article] [PubMed]
- [31].Beck VC, Hull JM, Isom LL. SCN1A gain of function in early infantile encephalopathy. Ann Neurol. 2019;85:514–25. [DOI] [PubMed]
- [32].Hu T, Niu Y, Fu J, Dong Z, He D, Liu J. Antisense lncRNA PCNA-AS1 promotes esophageal squamous cell carcinoma progression through the miR-2467-3p/PCNA axis. Open Med (Wars). 2022;17:1483–94. [DOI] [PMC free article] [PubMed]
- [33].Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol Ther. 2010;18:80. [DOI] [PMC free article] [PubMed]
- [34].Deleavey GF, Damha MJ. Designing chemically modified oligonucleotides for targeted gene silencing. Chem Biol. 2012;19:937. [DOI] [PubMed]
- [35].Koussounadis A, Langdon SP, Um IH, Harrison DJ, Smith VA. Relationship between differentially expressed mRNA and mRNA-protein correlations in a xenograft model system. Sci Rep. 2015;5:10775. [DOI] [PMC free article] [PubMed]
- [36].Skjei KL, Church EW, Harding BN, Santi M, Holland-Bouley KD, Clancy RR, et al. Clinical and histopathological outcomes in patients with SCN1A mutations undergoing surgery for epilepsy. J Neurosurg Pediatr. 2015;16:668. [DOI] [PubMed]
- [37].Vezyroglou A, Varadkar S, Bast T, Hirsch E, Strobl K, Harvey AS, et al. Focal epilepsy in SCN1A-mutation carrying patients: Is there a role for epilepsy surgery? Dev Med Child Neurol. 2020;62:1331–5. [DOI] [PubMed]