

Abstract
Coastal shelf sediments are hot spots of organic matter mineralization. They receive up to 50% of primary production, which, in higher latitudes, is strongly seasonal. Polar and temperate benthic bacterial communities, however, show a stable composition based on comparative 16S rRNA gene sequencing despite different microbial activity levels. Here, we aimed to resolve this contradiction by identifying seasonal changes at the functional level, in particular with respect to algal polysaccharide degradation genes, by combining metagenomics, metatranscriptomics, and glycan analysis in sandy surface sediments from Isfjorden, Svalbard. Gene expressions of diverse carbohydrate-active enzymes changed between winter and spring. For example, β-1,3-glucosidases (e.g. GH30, GH17, GH16) degrading laminarin, an energy storage molecule of algae, were elevated in spring, while enzymes related to α-glucan degradation were expressed in both seasons with maxima in winter (e.g. GH63, GH13_18, and GH15). Also, the expression of GH23 involved in peptidoglycan degradation was prevalent, which is in line with recycling of bacterial biomass. Sugar extractions from bulk sediments were low in concentrations during winter but higher in spring samples, with glucose constituting the largest fraction of measured monosaccharides (84% ± 14%). In porewater, glycan concentrations were ~18-fold higher than in overlying seawater (1107 ± 484 vs. 62 ± 101 μg C l−1) and were depleted in glucose. Our data indicate that microbial communities in sandy sediments digest and transform labile parts of photosynthesis-derived particulate organic matter and likely release more stable, glucose-depleted residual glycans of unknown structures, quantities, and residence times into the ocean, thus modulating the glycan composition of marine coastal waters.
Keywords: marine sediment, glycan degradation, microbial diversity, metatranscriptomics, rRNA, Colwellia, Svalbard, Isfjorden
Introduction
Continental shelf ecosystems contribute 15%–21% of global primary production [1] of which up to 50% reaches the shallow seafloor. About half of the continental shelf area is covered by sandy sediments [2, 3]. Their high permeability enhances the advective flow of bottom water with organic matter (OM) [4, 5]. Heterotrophic benthic bacteria remineralize this imported OM as well as OM derived from benthic primary production [2, 4, 6].
A major fraction of the OM consists of polysaccharides that phytoplankton produces for energy storage, for cell wall building blocks or as exudates. Glycans constitute up to 80% of the algae dry weight, depending on the species and growth phase [7]. They are structurally complex, in terms of linkage, configuration, and diversity of monosaccharide building blocks [8]. This structural diversity makes marine glycans an important theme of ongoing research; reviewed in, e.g. [9]. For the utilization of these carbohydrates, heterotrophic bacteria use a diverse set of carbohydrate-active enzymes (CAZymes). They include glycoside hydrolases (GH), polysaccharide lyases (PL), carbohydrate esterases (CE), and accessory proteins, such as proteins carrying carbohydrate-binding modules; CBMs [10]. The number of CAZymes required for the degradation of a glycan scales linearly with its structural complexity [11]. For example, for the digestion of complex, branched, and highly sulfated fucoidan consisting of multiple monosaccharides, including methylpentose (fucose) from brown algae, many enzymes are required [12]. By contrast, for the degradation of laminarin, which is the most abundant marine glycan and contributes 26% ± 17% to the particulate organic carbon pool [13], two or three enzymes are—at least ex situ—sufficient to degrade laminarin into glucose [14]. Due to the structural diversity, direct quantification of specific polysaccharides in the environment remains technologically challenging [15]. Inventories of bacterial CAZymes, therefore, offer an alternative approach for studying bacterial glycan utilization [16, 17].
While the bacterial glycan degradation in temperate surface waters was shown to be highly dynamic, e.g. [17, 18], benthic bacterial communities have limited seasonality [19, 20]. Polar regions with their prolonged periods of complete darkness in winter and 24 h of sunlight in spring and summer are ideal environments to study the seasonality of bacterial glycan degradation. Arctic fjords of Svalbard (74–81°N) have strong peaks of primary production in spring and summer, fueling the entire coastal ecosystem, including its sediments [21]. Of particular importance is benthic photosynthesis, which adds a significant amount of fresh, labile OM to the seafloor in shallow coastal regions. For example, in Kongsfjorden, microphytobenthos primary production is comparable to those from pelagic production [22]. In addition, there is a terrestrial input from the glacial run-off and a contribution of ice algae, which are also both sources of OM, which vary with season. Large datasets from Svalbard fjords have repeatedly underscored seasonal changes in respiration, sulfate reduction, and mineralization in these permanently cold sediments (−1°C to +4°C); for review, see [23]. Respiration is mostly driven by the input of fresh OM and it is plausible to assume that fresh OM will also drive seasonal succession of heterotrophic bacteria in sandy surface sediments. However, benthic and pelagic microbial communities differ fundamentally in their ability to access high molecular weight substrates such as polysaccharides in arctic sediments [24]. Furthermore, rRNA gene-based studies showed a stable community composition over 2 years in coastal sands from Isfjorden [19]. Since studies based on the comparative sequence analysis of rRNA genes are limited in taxonomic resolution, we revisited Isfjorden sediments and studied them by a combination of metagenomics and metatranscriptomics. Thereby, we expected to detect subtle differences in the gene repertoire of bacteria and in gene expression. In this study, we tested the following three hypotheses: (i) metagenomic (and metatranscriptomics) data reveal seasonal differences in the genes encoding glycan utilization. Furthermore, benthic bacterial communities respond to the seasonally changing input of fresh OM by changing the regulation of genes encoding CAZymes. (ii) The utilization of continuously available, less labile substrates explains the high overall stability in the benthic bacterial community composition. (iii) The main glycans used by heterotrophic benthic bacteria change between seasons and can be predicted based on gene expressions. For addressing the third hypothesis, we additionally performed glycan analyses.
Materials and methods
Sampling
Sediment samples (fine sand) were taken from Isfjorden, Svalbard (Fig. 1), using a van Veen grab. Surface layers (0–2 cm depth) were sampled at Station 5 (78.11°N/14.35°E) and at Station 7 (78.10°N/14.38°E) in 2017 (20 December), 2018 (6 February, 1 May, and 17 December), and 2019 (25 April). Sediment temperatures ranged between −0.6°C and 2.2°C, water depth was between 2.7 and 8.8 m. Chlorophyll a in seawater was 6.3–8 μg l−1 in spring and 0.4 μg l−1 in winter. In sediments, Chlorophyll a was 0.4–1.0 μg ml−1 in spring and was 0.2–0.3 μg ml−1 in winter. Contextual data have been reported previously [19]. In addition, sediments, porewater, and bottom water (sampled above the sediment surface; hereafter, referred to as “overlying seawater,” OSW) were sampled for glycan analysis using a Shipek-type grab at Station 5 in 2022 (29 April and 2 May). All samples were immediately frozen in dry ice.
Figure 1.

Sampling area in Isfjorden, Svalbard; surface sediments were retrieved from two shallow sites (78°N, 4–8-m water depth) close to Kapp Dresselhuys.
DNA and RNA extraction
DNA for metagenome analysis was extracted from the 0–2-cm depth horizon of selected sediment samples of December 2017, February 2018, May 2018, December 2018, and April 2019 after Zhou et al. [25], including three additional freeze-thawing steps. RNA was extracted from December 2017, February 2018, and May 2018 samples using RNeasy PowerSoil Total RNA Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s recommendation with minor modifications. For an overview of samples and details, see Supplementary Table S1 and Supplementary Information.
Library preparation, sequencing, assembly, and binning
Illumina-compatible libraries were prepared from genomic DNA with NEBNext Ultra™ DNA v2 Library Prep Kit for Illumina (New England Biolabs, Frankfurt, Germany), starting with initial DNA fragmentation using a Covaris S2 ultrasonicator (Covaris, Woburn, MA, USA). Illumina-compatible RNAseq libraries were prepared from total RNA with NEBNext UltraII Directional RNA Library Prep Kit for Illumina (New England Biolabs). In addition, three Illumina-compatible RNAseq libraries from Station 5 were prepared from bacterial rRNA-depleted RNA using the Illumina Ribo-Zero rRNA depletion kit (“Bacteria”). Metagenome and metatranscriptome sequencing were done on a HiSeq 2500 System (Illumina, San Diego, CA, USA, 2 × 250 bases) at the Max Planck-Genome Center in Cologne (Germany). Detailed settings of the programs used for sequence analysis are given in the Supplementary Information. In short, sequences were quality-controlled using BBTools v37.62 (quality < 20, minimum length 140 nt). Coverage of sequence diversity was analyzed using nonpareil v3.303 [26]. Assembly of reads was done with SPAdes v3.13.1 [27] (meta option) and the quality was evaluated using QUAST v4.5 [28]. Contigs < 1 kb length were excluded from further analyses.
For each dataset, binning was done using MaxBin v2.2.7 [29] and MetaBAT v2:2.15 [30]. Bin refinement was performed using DAS_Tool v1.1.2 [31]. Mapping for differential coverage binning was done using bbmap v38.70 [32] at default settings and a minid = 0.99. Dereplication was performed with dRep v3.1.1 [33] (−comp 50, −con 15) and classification using the Genome Taxonomy Database (GTDB)-Tk v2.1.1 and the GTDB release r214 [34]. Completeness and contamination were assessed in checkM v1.0.7 [35].
Gene annotation and analyses
Gene predictions and annotations from bins were done using Prokka v1.14.6 [36], dbCAN (run_dbCAN v2.0.11 workflow; https://github.com/linnabrown/run_dbcan) [37], Swiss-Prot release 2021_04 [38], SulfAtlas v1.0 [39], and transporterDB (download Oct 2021) [40]. The latter three databases were searched using DIAMOND blastp (v2.0.15.153) [41]. Results were filtered for the best hit using the enveomics script BlastTab.best_hit_sorted [42] (>60% identity, query coverage > 70%).
CAZyme annotations obtained from dbCAN were accepted when two of the three integrated annotation methods (HMMER v3.3.2, diamond v2.0.9.147, Hotpep version included in run_dbCAN workflow) matched [37].
Transcriptomic analyses
Quality-controlled RNA reads were sorted using SortMeRNA 4.0.4 [43]. Reads identified as rRNA were taxonomically classified by using the SILVAngs pipeline (https://ngs.arb-silva.de/silvangs/, release 138.1) [44]. All reads that were not classified as rRNA or tRNA were considered as mRNA.
Annotation of transcripts was done by mapping mRNA to predicted genes from metagenomics contigs and bins using DIAMOND blastx (v2.0.15.153) [41]. Results were filtered for the best hit using the enveomics script BlastTab.best_hit_sorted [42] (>60% identity, query coverage > 70%). Values of transcripts per million (TPM) mapped reads were calculated after normalization by gene length.
Calculations of predicted monosaccharide patterns were done based on the expression of GH genes (TPM) by referring enzyme activities given in the CAZy database to one or several monosaccharide types released/degraded. Details are provided in the Supplementary Information; the script was deposited on Gitlab (https://gitlab.mpi-bremen.de/smiksch/gh_family_to_monosaccharides). Data transformation and plotting were done using R and the tidyverse packages [45].
Monosaccharide analysis
Polysaccharides extracted from sediment, porewater, and OSW were acid hydrolyzed and the resulting monosaccharides were analyzed using high-performance anion exchange chromatography (HPAEC) with pulsed amperometric detection (PAD) according to Vidal-Melgosa et al. [46]. For details, see Supplementary Information. Values measured for calibration standards having high monosaccharide concentrations were consistent between injections during the chromatographic run. Low-concentrated calibration standards, however, showed much lower values at the second injection. To account for this decrease in detector sensitivity with time, a threshold concentration for each monosaccharide was set to the value at which the variation between two injections was ±20%. Values lower than the threshold concentrations defined for each monosaccharide were rejected.
Results
Bacterial community composition as revealed by rRNA read frequencies
As a proxy for activity of a population, we used rRNA read frequencies from 11 metatranscriptomes recovered from Isfjorden sediments (December 2017, February 2018, May 2018; Fig. 2A). The rRNA read frequencies of the majority of clades were not remarkably different between seasons. Notable exceptions were rRNA reads affiliated with the genera Colwellia and Polaribacter that showed increased relative abundance from winter (average of 0.6% and 0.1% of total 16S rRNA reads, respectively) to spring (average of 3.3% and 0.6%, respectively).
Figure 2.
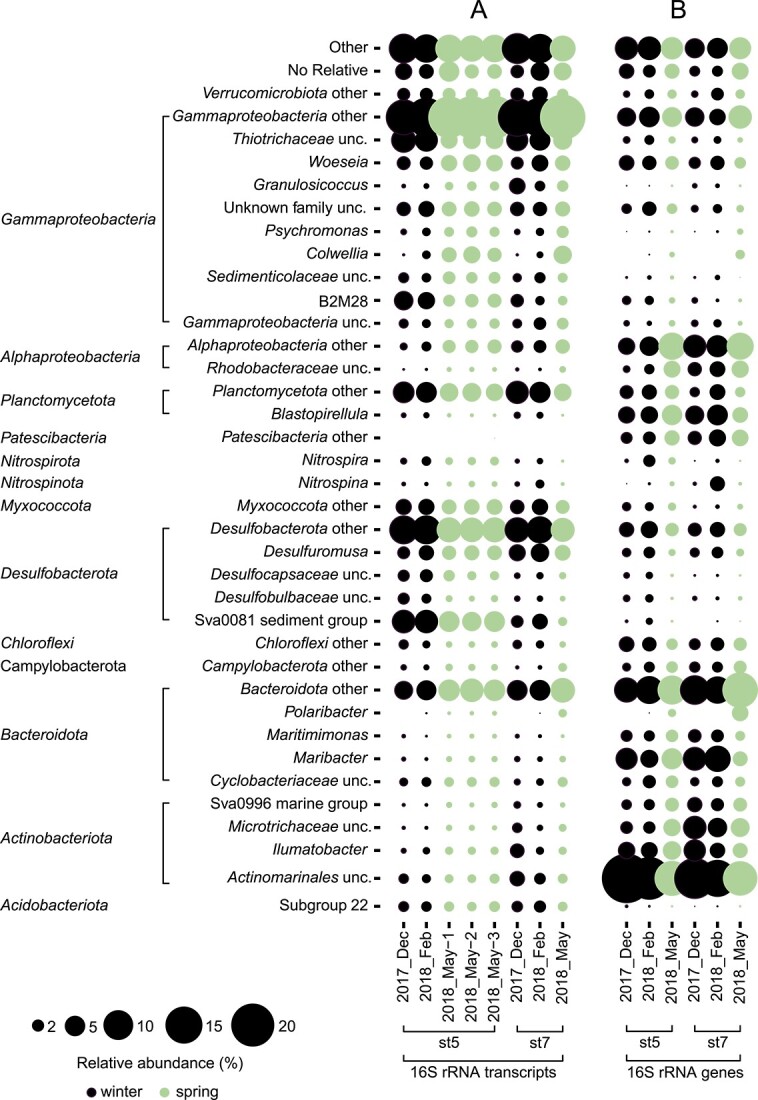
Major taxa of bacterial communities in Svalbard surface sediments are seasonally stable; comparison of the benthic community composition as revealed by (A) rRNA read frequencies in metatranscriptomes and (B) 16S rRNA genes frequencies from amplicon tag sequencing; extracted from [19]; for classification, 20 000 reads were randomly subsampled from each dataset and were submitted to SILVAngs (https://ngs.arb-silva.de/silvangs/, release 138.1) [44]; only taxa with a read frequency of >2% are shown, minor taxa are summarized as “other”; both rRNA and rRNA genes revealed a stable bacterial community in winter and spring; only minor taxa such as Colwellia and Polaribacter spp. showed a clear seasonal variation in abundance.
Although no clear differences in the community composition between seasons were detected, relative abundance in metatranscriptomic 16S rRNA versus amplicon 16S rRNA genes [19] differed for several taxa (Fig. 2B). A greater relative abundance in metatranscriptomic 16S rRNA versus amplicon 16S rRNA genes was determined for Verrucomicrobiota (2.3 ± 0.4% rRNA vs. 1.3 ± 0.5% amplicon rRNA genes), Planctomycetota (4.8 ± 0.9% vs. 2.4 ± 0.7%), Desulfobacterota (7.9 ± 1.1% vs. 2.8 ± 0.5%), Thiotrichaceae (5.0 ± 1.3% vs. 0.9 ± 0.3.), and Myxococcota (3.0 ± 0.3% vs. 0.8 ± 0.4%). By contrast, a lower relative abundance in rRNA read frequencies in metatranscriptomic 16S rRNA versus amplicon 16S rRNA genes was found for Blastopirellula (0.6 ± 0.2% vs. 4.2 ± 0.8%), Bacteroidota (Maribacter 0.6 ± 0.1% vs. 5.4 ± 1.8%; Maritimimonas 0.4 ± 0.1 vs. 2.0 ± 0.4%), and Actinomarinales unc. (1.6 ± 0.6% vs. 17.8 ± 5.3%). The metatranscriptomes comprised only few archaeal 16S rRNA sequences (<1%).
Addressing changes in functional potential of benthic bacteria by omics
Metagenomes and metatranscriptomes from Svalbard sediments were used to study possible changes in the functional potential of the bacterial community and to detect differences in the genomic repertoire between species of the same genus and in gene expression of CAZymes. Three metagenomes were obtained from Station 5 samples in winter (December 2017, February 2018, December 2018; hereafter, referred to as “winter”) and three metagenomes in spring (Station 5: May 2018, April 2019; Station 7: April 2019; hereafter, referred to as “spring”). Nonpareil, a redundancy-based approach to assess the level of coverage, ranged between 0.46 and 0.5 for all metagenomes, indicating that about half of the total diversity was covered (Supplementary Table S2). A total of 9 207 104 genes were predicted of which about one-third remained hypotheticals after annotation. A total of 183 bins (16–42 bins per sample) were recovered of which 36 bins (Supplementary Table S3) were selected based on the diversity and quality for further analysis. The bins represented all major taxa previously found in sandy surface sediments [19], including Acidimicrobiia, Bacteroidia, Desulfobacteria, Planctomycetota, and Gammaproteobacteria.
Seasonal expression of bins
As a proxy for activity, 11 metatranscriptomes (December 2017, February 2018, May 2018) were mapped on the 36 bins. A bin was considered being upregulated in spring when the ratio (average spring TPM mapped reads/average winter TPM mapped reads) was ≥2 (=log2-fold change of >1; green bars, Fig. 3A) and being upregulated in winter when the ratio (average spring TPM mapped reads/average winter TPM mapped reads) was <0.5 (=log2-fold change of <−1; black bars). Bins with log2-fold changes −1 ≤ x ≤ 1 were considered as constantly expressed and therefore unregulated (gray bars). According to this definition, 10 of 36 bins were upregulated in spring. Of this group, seven belonged to Bacteroidia and three belonged to Gammaproteobacteria. Among the most upregulated bins were Flavobacteraceae-bin Sval_st7_May.bin.40 and Colwellia-bin Sval_st7_May.bin.39 with a 33-fold and 19-fold higher TPM in spring versus winter, respectively. In winter, six bins showed an increased expression (Fig. 3A, black bars) affiliated with Gammaproteobacteria, Desulfobacteria, Acidimicrobiia, and Planctomycetota. The majority of unregulated bins (9 of 20 bins, Fig. 3A, gray bars) were Gammaproteobacteria. Many of these bins had high TPM values (Fig. 3B).
Figure 3.

Expression of bins from Svalbard sediment metagenomes; (A) changes of bin expression given as a ratio of TPM mapped reads from spring metatranscriptomes divided by TPM mapped reads from winter metatranscriptomes; values are plotted as log2-fold change; a bin was defined being upregulated in spring for log2-fold changes of >1 (corresponding to a ratio TPM spring/TPM winter of >2; green bars) and being upregulated in winter for log2-fold changes of <−1 (corresponding to a ratio TPM spring/TPM winter of <0.5; black bars); gray bars show less regulated bins not matching these thresholds; (B) expression of bins in TPM given as an average of all sampling time points and metatranscriptomes; some bins of Bacteroidia and Gammaproteobacteria were upregulated on mRNA level in spring, while clade UBA9214 (Bins 80 and 103), Acidimicrobiia (Bin 66), and Desulfobacteria bins (Bins 47 and 161) were more expressed in winter; most highly expressed Bins 207 and 11 were less regulated.
Carbohydrate-active enzymes and polysaccharide utilization loci
We focused our analysis on CAZymes, in particular on GH families, as they can be used as a proxy for polysaccharide degradation. Overall, GH23 (peptidoglycan lyase) was the most abundant family in the metagenomes (Supplementary Fig. S1) and was not remarkably changing between seasons. Most members of the GH23 family have peptidoglycan lyase activity and are widely distributed among many phyla such as Proteobacteria and Firmicutes [10]. In winter metagenomes, e.g. GH29 (fucosidase), GH106 (rhamnosidase), and GH165 (galactosidase) were more abundant than in summer with a log2-fold increase of −1.2 to −2.2, yet with lower frequencies than the less regulated representatives GH16, GH17, GH23, and GH103. Spring-induced GH were less frequent (below threshold of >0.1% metagenomics abundance).
The number of GH in the bins varied between 3 and 16 GH Mbp−1 (Supplementary Fig. S2, Supplementary Table S4) and the number of total CAZymes (GH, CE, and PL) varied between 5 and 22 Mbp−1. Three of the bins showed a high density of peptidases with 8–11 Mbp−1 but comprised only 6–9 CAZymes Mbp−1. Major substrates expected to be consumed by these Bacteroidia were laminarin or other β-glucans (GH16_3, GH2, GH3, GH149, GH17, and GH30_1), α-glucans such as glycogen (GH13, GH13_19, GH31, and GH65), mannans (GH92), xylans (GH3), and alginates (PL7 and PL17).
Polysaccharide utilization loci (PUL) are structured genomic regions that are used to predict the substrate of heterotrophic bacteria and are common in Bacteroidia [17, 47]. Canonical bacteroidetal PUL include a pair of susCD-like transporter genes and ≥2 CAZyme genes, like GH, PL, CE, or CBM, within a 10-genes-sliding window [48]. Automated prediction of canonical PUL and PUL-like structures (defined as susCD pair or a single susC and ≥ 1 CAZyme) identified 16 loci (Supplementary Table S4) in the 7 Bacteroidia bins that were upregulated in spring. Another 11 loci were identified with multiple CAZymes, but no susCD. The two seasonally unregulated bins, Sval1819_Apr.bin.26 and Sval_Feb.bin.86 (Fig. 3, gray bars) did not comprise contigs with canonical PUL or PUL-like structures, but four and eight single CAZymes Mbp−1 (Supplementary Fig. S2).
Seasonal changes in gene expression
To analyze the changes in gene expression, the average relative frequency of transcripts was calculated for winter and spring (Supplementary Fig. S3, Supplementary Table S5). The 10 most expressed genes comprised only hypothetical proteins. Expressions of most of these unknowns did not differ between seasons, but those contribute evenly to the gene expression by the sediment community. Genes related to photosynthesis were highly upregulated in spring: besides photosystem I- and II-related genes, other genes of presumably photosynthetic organisms, e.g. ribulose bisphosphate carboxylase, had up to 14-fold higher TPM values in spring than in winter. Furthermore, ammonia channel proteins/transporters and cytochromes were also upregulated in spring. By contrast, genes involved in nitrogen and sulfur cycling were upregulated in winter (Supplementary Fig. S4). These are, in particular genes for respiration, e.g. nitrite reductase and nitrate reductase (1.5 and 1.3 log2-fold change TPM winter vs. spring, respectively), as well as dissimilatory sulfate reductase (log2-fold change TPM winter vs. spring of 1.0) and adenylylsufate reductase (log2-fold change TPM winter vs. spring of 1.1).
Most prominent GH families upregulated in spring were GH30_1, GH17, GH16_3, and GH149 (Fig. 4). Enzymes of these families comprised β-glucanases and are likely degrading laminarin. GH149 also acts on β-1,3-linked glucan as phosphorylase. GH families that were downregulated in spring included GH130, GH63, GH13_18, GH15, GH23, and GH57. Enzymes characterized within these families showed a diverse range of activities such as mannoside phosphorylases (GH130), α-glucosidases (GH63), α-glycoside phosphorylases (GH13_18), glucan-1,4-α-glucosidase (e.g. glucoamylase, trehalase; GH15), peptidoglycan lyases (GH23), and α-glucanases (GH57).
Figure 4.
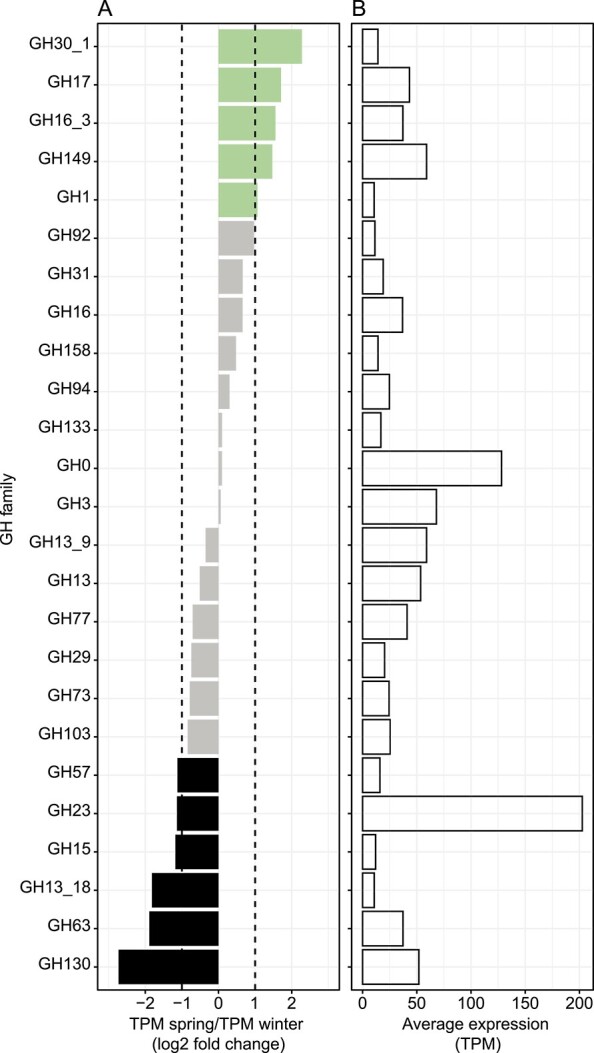
Expression of GH families in spring and winter; β-glucan utilization is upregulated in spring (GH17, GH16_3, and GH149), while α-glucan utilization is more prominent in winter (GH63, GH15, and GH57); (A) changes of GH expression are given as a ratio of TPM in spring metatranscriptomes divided by TPM in winter metatranscriptomes; values are plotted as log2-fold change; a GH family was defined as being upregulated in spring when log2-fold changes were >1 (TPM spring/TPM winter >2, green bars) and being upregulated in winter when log2-fold changes were <−1 (TPM spring/TPM winter < 0.5, black bars); gray bars show unregulated GH families not matching these thresholds; (B) average expression of GH families in TPM calculated from metatranscriptomes from all sampling time points; GH families shown are expressed in spring and winter (no infinite fold change) with TPM values > 10.
Monosaccharide concentrations in sediments
To link the gene expression of CAZymes with glycan concentrations in the sediment, we extracted glycans from the sediment with MilliQ water and quantified their monosaccharides after acid hydrolysis. The monosaccharide composition of the water extracts was dominated by glucose, accounting for 50%–80% of total monosaccharides (Fig. 5). In spring 2019, total monosaccharide content (sum of concentrations of all different monosaccharides) was on average lower than in spring 2018 (Station 5: ~3 μg C gdw−1 sediment vs. ~8.5 μg C gdw−1 and Station 7: 2.5 vs. 7.5 μg C gdw−1 in 2019 and 2018, respectively). Samples from station 7 (December 2017 and February 2018) contained only glucose in measurable amounts, while other monosaccharides were below the detection limit. Other abundant monosaccharides in our samples were mannose and galactose. In winter, the concentration of mannose increased by a factor of ~2.2 from 0.08 μg C gdw−1 sediment in spring to 0.17 μg C gdw−1 (average for station 5 and 7). By contrast, spring samples had a 7.9-fold higher concentration of galactose (winter: 0.05 μg C gdw−1; spring: 0.41 μg C gdw−1 sediment) and a 4.3-fold higher concentration of fucose than in winter (winter: 0.04 μg C gdw−1; spring: 0.17 μg C gdw−1 sediment).
Figure 5.

Monosaccharide concentrations measured and predicted based on expression patterns of GH genes in Svalbard sediments; glycans from the water extracts of sediment samples were acid hydrolyzed and the resulting monosaccharides were measured by HPAEC-PAD analysis; (A) concentrations and (B) relative fractions of total measured monosaccharides; (C) monosaccharide utilization deduced from predicted functions of expression patterns of GH genes; star, data from rRNA-depleted metatranscriptome; square, data from “full” metatranscriptome; all samples were dominated by glucose; in particular, in spring 2018, glucose and galactose concentrations strongly increased, while mannose was more prominent in winter samples; the measured monosaccharide composition is in line with the predicted trends of monosaccharide utilization patterns based on GHs’ expression.
In an additional sampling campaign at Station 5 in April 2022, we collected porewater and OSW along with sediment samples. Total concentrations of monosaccharides were similar to those measured in spring 2019 with on average 2.9 ± 2.6 μg C gdw−1 sediment for four replicate grabs (data not shown). Total concentrations in porewater were high, with 1107 ± 484 μg C l−1 being 18-fold higher than those measured for OSW (62 ± 101 μg C l−1, Supplementary Fig. S5, Supplementary Table S6). The monosaccharide composition differed between sediments and porewater: the porewater monosaccharide spectrum was not dominated by glucose, but it mostly had an even contribution of glucose, arabinose, fucose, galactose, glucosamine, and xylose (Supplementary Table S6).
Predicting monosaccharide utilization based on GH expression data
The frequency of mRNA reads annotated as GH was used to predict monosaccharide utilization in Svalbard sediments and to test if the predicted patterns correlate with the measured monosaccharide concentrations. We assigned one or more monosaccharides to each detected GH family based on information given in the CAZy database (matrix available as Supplementary Table S7). The monosaccharide utilization pattern predicted based on the transcriptomic data (Fig. 5C) was similar to the pattern of measured monosaccharides at Station 5 (Fig. 5B): it indicated a dominance of glucose utilization, accounting for >60% of the total used monosaccharides in sediments in spring. Analog to measured monosaccharide concentrations, transcripts mapping to mannose-related GH families were more prominent in winter, while galactose-related GH families were more abundant in spring predictions. In line with monosaccharide measurements, fucose, rhamnose, arabinose, and xylose utilizations were detected, though in a less seasonally consistent manner.
Concentrations of most monosaccharides were below detection threshold at Station 7; thus, a comparison of measured and predicted monosaccharide composition is not meaningful.
Discussion
Bacterial communities in temperate and polar sediments were reported to be seasonally stable based on 16S rRNA gene frequencies [19]. In this study, we showed that also the ribosomal RNA expression of most taxa did not remarkably change between winter and spring (Fig. 2), supporting previous findings. Although the rRNA concentrations of diverse natural bacterial communities cannot be metrically linked to real-time activities due to the differences in life histories, life strategies, and nongrowth activities [49], rRNA frequencies have been used as a proxy for growth potential and activity of a population due to the relationship between cellular ribosome content and the ability to synthesize proteins [50]. Our data show that abundant taxa (>5% of total rRNA reads) such as Actinobacteria, Bacteroidia (except Polaribacter), Desulfobacterota, Myxococcota, and Woesiaceae, were seasonally stable. The detected stability gives a first hint that a major part of the bacterial community is thriving on constantly available substrates rather than seasonally fluctuating substrates like laminarin.
Colwellia and Polaribacter are prominent in spring
Despite the stability of major phyla, two genera, Polaribacter (Bacteroidia) and Colwellia (Gammaproteobacteria) showed a strong increase in spring, with average relative abundances rising from <0.1% to 1.1% and from 0.3% to 4.1% of total rRNA reads, respectively. Both genera are known for the degradation of various algal polysaccharides and are tightly associated with phytoplankton blooms; e.g. [16, 18, 51-53]. In particular, members of the genus Colwellia have been reported to be seasonally abundant in Arctic and Antarctic waters and sea ice [53-55]. The increase of Colwellia rRNA frequency in spring versus winter went along with a 19- to 36-fold higher expression of mRNA that mapped on the Colwellia bin Sval_st7_May.bin.39 (Fig. 3). Two contigs contained PUL-like loci (no susCD; Supplementary Fig. S6). Of the genes in these two loci, all GH and CBM genes were expressed in the spring metatranscriptomes, while they were not detected in winter metatranscriptomes. In particular, GH17, GH16_3, and GH149, indicative of the degradation of laminarin as the main storage glycan of diatoms [13] and brown algae were upregulated. The most likely explanation for their absence in our winter metatranscriptomes is a low expression combined with insufficient depth of sequencing. Only GH73 and GH103 (both genes outside the PUL-like loci) were slightly expressed in winter metatranscriptomes (TPM ~ 0.2). GH73 (peptidoglycan hydrolases) was higher expressed in winter than in spring, which is reasonable as the recycling of bacterial cell compounds becomes relatively more important. This Colwellia bin with a genome size of 3.65 Mbp (95% completeness; 1.9% contamination) represents a novel species according to the ANI-based genomic similarity criteria for delineating species used by the GTDB [56]. Its closest phylogenetic relative is an uncultured Colwellia sp. from marine water (ANI 85.9%; bioproject PRJEB37807, BioSample SAMEA9694887) with a genome size of 4.9 Mbp. We conclude that the high abundance and pronounced seasonality of Colwellia ask for future studies on the ecology of this gammaproteobacterial genus in polar systems.
Besides Colwellia, seven Bacteroidia bins showed a strong upregulation in spring (Fig. 3). These bins are on average of a size of 2.86 ± 0.35 Mbps and thereby significantly larger than the 2.0 Mbp bins of Bacteroidia obtained from temperate surface waters [17]. Surface water Bacteroidia are characterized by a wealth of PUL that encode CAZymes, carbohydrate-binding proteins, and SusCD-like transporters; for review, see [57]. Like planktonic Bacteroidia, the benthic bins also affiliated with the family Flavobacteriaceae and show a similar genomic organization regarding polysaccharide degradation. The variability of GH in our Bacteroidia bins was high (3–16 GH Mbp−1; Supplementary Fig. S2) and was only slightly lower than that found for pelagic Polaribacter spp.; e.g. [16-18]. Ratios of annotated degradative CAZymes versus peptidases suggest a niche separation of Bacteroidia into carbohydrate (four/seven bins) and protein degradation (three/seven bins; Supplementary Fig. S2).
Desulfobacteria is an abundant taxon slightly elevated in winter
Bins of sulfate-reducing Desulfobacteria (Fig. 3) showed an elevated relative expression in winter. The same was observed for key genes involved in sulfur cycling (Supplementary Fig. S4), both indicating a more prominent role of sulfur cycling in winter samples, potentially linked to more anoxia in the absence of benthic photosynthesis. Most sulfate-reducing bacteria rely on low molecular products, such as fatty acids and hydrogen [58], which are available throughout the year, while fresh, complex organic material gets limited in winter. Canonical denitrification as indicated by genes for nitrate and nitrite reductases (nir and nar) was more prominent in winter (Supplementary Fig. S4), which supported extended phases of anoxia.
Relative utilization of β-glucans increases in spring
In spring, the use of algae-derived β-glucans was most prominent by an elevated expression of mRNA of GH families GH30_1, GH17, GH16_3, and GH149, together indicating an increased degradation of laminarin (Fig. 4). Several GH families with galactosidase activities (according to CAZy database) [10] were also upregulated in spring such as GH1, GH4, or GH42 (Supplementary Table S5). Galactose has been described to be a main building block of several marine algal polysaccharides, like agar and carrageenan, which are important components of macroalgae cell walls [59]. Hydrolysis of such labile algal polysaccharides would be plausible to be induced in spring when algal biomass is increasing. The importance of laminarin as carbon source for benthic microbes is supported by short-term incubations of intact sediment cores from different fjords at Svalbard with fluorescently labeled polysaccharides, which showed a rapid hydrolysis of laminarin in surface sediments [24]. Sources of laminarin are micro- and macroalgae that extensively colonize coastal habitats in Arctic fjords such as Isfjorden and Kongsfjorden [60-62]. While Arctic kelp can grow even during polar night using stored carbohydrates and sugar alcohols derived from summer/autumn photosynthetic periods [63, 64], microalgae such as diatoms show a strong seasonality [60]. At the time point of spring sampling, chlorophyll a concentrations in seawater and sediments, as well as 16S rRNA amplicon frequencies from chloroplasts, were clearly higher than in winter [19], supporting the presence of a current or recent phytoplankton bloom.
Relative utilization of α-glucans increases in winter
Transcript levels for the degradation of α-glucans like glycogen (GH63, GH15, and GH57) increased in winter (between log2-fold change −1.1 and −1.9) but were detected in spring, too. α-Glucans are the intracellular storage products of not only many heterotrophic bacteria [65], but they are also the intracellular storage products of animals and protists as well as some fungi [66, 67]. Therefore, glycogen is continuously available, either in intracellular pools or recycled from bacterial and animal biomass. We assume that, in spring, benthic bacteria use large amounts of available glycans and transform part of them into glycogen, thus making it available later during the year. Thus, unlike laminarin, glycogen is a constantly available carbon source contributing to the high stability in the bacterial community composition. GH23 transcripts were also upregulated in winter. They likely encode hydrolysis of peptidoglycan [68]. This could be a result of starvation since bacteria are known to reduce their size and use cell wall compounds as energy source [69].
Utilization of constantly available substrates
Besides glycogen, we found multiple indications that other substrates are continuously used, likely contributing to the high stability of benthic bacterial communities. The transcription of most of the abundant GH families was independent of polar day and night and was not remarkably regulated across seasons (Fig. 4). Among these GH families, there were mannosidases (GH92), α-glucanases (GH31, GH133, GH13_9, and GH77), β-glucanases (GH16, GH158), fucosidases (GH29), peptidoglycan lyases (GH73 and GH103), and families without a clear substrate affiliation (GH94, GH0, GH3, and GH13).
Further constant carbon sources are chitin and mucin, which are both mostly of animal origin. Benthic meiofaunal and macrofaunal density and diversity in the close by Kongsfjorden have been shown to be stable throughout the year [70]. Chitin is the most abundant polysaccharide in surface marine sediments [71], yet we could not identify high transcription levels of known chitinases; e.g. GH18 [72], in our samples. However, in the Bins Sval_st7_May.bin.207 and Sval_Feb_bin.32, we did find expressed chitosan disaccharide transporters as well as key genes for further breakdown catalyzing deacetylation (chitooligosaccharide deacetylase) and hexosaminidases (GH3). Both bins were classified as Acidimicrobiia and were highly expressed in winter and spring (Fig. 3).
Mucins are glycoproteins copiously secreted by marine fauna, in particular by invertebrates [73]. They constitute a complex class of energy-rich substrates containing a protein backbone with side chains of oligosaccharides, which can be very diverse in nature, covering glycans composed of different monosaccharide building blocks [74]. Mucus is a potent substrate for marine microbes. Hannides and colleagues [75] showed a strong priming effect of gastropod mucus on benthic OM remineralization. Key enzymes for mucin degradation have been identified for gut bacteria comprising sialidases (GH33), fucosidases (GH29 and GH95), N-acetylgalactosaminidases (GH101 and 129), N -acetylglucosaminidases (GH84 and GH85), galactosidases (GH2, GH20, and GH42), and proteases [76, 77]. These were all present in our metagenomes and were expressed either all year or preferentially in winter metatranscriptomes. This corroborates that mucins are important substrates for benthic bacteria.
Monosaccharide measurements are consistent with carbohydrate-active enzyme expression
Glucose concentrations were seasonal with clear maxima in spring. Their up to 4-fold increase was consistent with higher transcription levels of β-1,3-glucan degradation genes, indicating substrate-related induction. Also in winter, glucose remained an important substrate, likely because the α-glucan storage products of animals and bacteria were recycled. Another direct relationship between the abundance of monosaccharides and transcript frequency of degradative enzymes was observed for mannose-containing substrates whose concentrations were higher in winter (0.38 ± 0.16 μg C g−1 sediment) compared to spring (0.16 ± 0.10 μg C g−1 sediment). Correspondingly, genes belonging to GH130 family (including activities like β-1,4-mannosylglucose phosphorylase, β-1,4-mannooligosaccharide phosphorylase, and β-1,4-mannosyl-N-acetyl-glucosamine phosphorylase) as well as genes encoding GH63 (α-glucosidase and α-mannosidase) and GH113 (β-mannanase and β-mannosidase) were upregulated toward winter. The α- and β-mannans are known to be important compounds of diatom cell walls [78, 79]. These cell walls are considered to be semi-labile OM and therefore are relatively more important in winter when labile glucans such as laminarin are long gone.
Overall, this study suggests that the transcription frequency of GH families is linked to monosaccharide concentrations in the natural environment. This comparison between detected monosaccharides and expressed GH should ideally be extended to the glycan level, so types and substrate classes are also considered. Few enzyme-based methods that allow quantification of specific glycan structures, such as laminarin and α-glucans, in marine samples have been recently developed [13, 80]. However, due to the glycans’ structural complexity and diversity, the quantification of individual glycan types remains technologically challenging.
Sandy sediments mineralize labile parts of photosynthesis-derived particulate organic matter and release more stable, glucose-depleted residual glycans
Marine dissolved organic matter (DOM) is the largest ocean reservoir of reduced carbon with ~662 Pg C [81]. Much of the porewater DOM originates from the deposited POM produced by primary production in surface waters [82]. While Svalbard sediments were rich in glucose (84% ± 14% of total glycans measured) and were similar to POM from other sites [83, 84], porewater showed a lower contribution of glucose (~15–25%; Supplementary Table S6), resulting in a more even distribution of the different monosaccharides. This is in line with previous findings for DOM composition in seawater (15% glucose) [85, 86] and porewater (average 28% glucose) [82, 87]. Together with our findings that the concentrations of monosaccharides in porewater were about one order of magnitude higher than in bottom water, these data suggest that benthic microbial communities transform OM, utilizing mostly glucose. Glucose-depleted DOM which is more stable against bacterial degradation is released into the water column by tidal pumping. Overall, we show that benthic microbiomes in sandy shelf sediments are major modulators of DOM composition, extending early findings by Burdige [87, 88] and Huettel and colleagues [89] who suggested that the sediments present a net source of dissolved organic carbon.
Conclusion and outlook
Our data show that the majority of the benthic bacterial community in Svalbard is present and active in two contrasting seasons despite the strong seasonality in polar regions. These findings highlight that the bacterial communities of the water column and of underlying sediments respond differently to fresh OM input from algae blooms. Nevertheless, we found some seasonality, such as degradation of β-glucans by Bacteroidia and Gammaproteobacteria in spring, supporting our hypothesis that benthic bacterial communities respond to the seasonally changing input of fresh OM. Similar to what occurs in the water column, laminarin degradation is a major process in sediments during spring, while utilization of α-glucans, in particular glycogen, occurs throughout the year. The stable expression of genes for the degradation of other constantly available substrates, such as mucin and chitin, is consistent with our hypothesis that the continuous utilization of less labile, permanently available substrates stabilizes benthic bacterial communities.
Future studies could aim at the autecology of taxa degrading these often complex, permanently available substrates, for example, by enrichment and isolation of pure cultures using mucin and chitin. Yet, we hypothesize that it is the tremendous diversity on various trophic levels, the multiple niches, the complexity of substrates, and the highly dynamic conditions of coastal sandy sediments with currents and storms that make the benthic microbiome so robust and stable, both with respect to taxonomy and function. Benthic microbiomes thereby will remain an ultimate challenge for ecologists.
Supplementary Material
Acknowledgements
We acknowledge the captain and crew of R/V Farm for great support. We are grateful to Kathrin Büttner, Mirja Meiners, Jörg Wulf, and Andreas Ellrott for excellent technical assistance and Alek Bolte for help with sugar analysis. Chyrene Moncada, Kyoko Kubo, Max Holthuis, Meike Knittel, Erich Nordmann, and David Probandt are acknowledged for help with field work, Bruno Hüttel and the Max Planck Genome Center in Cologne are acknowledged for excellent sequencing service, and Dirk de Beer and Tim Ferdelman are acknowledged for fruitful discussions.
Contributor Information
Sebastian Miksch, Department of Molecular Ecology, Max Planck Institute for Marine Microbiology, 28359 Bremen, Germany.
Luis H Orellana, Department of Molecular Ecology, Max Planck Institute for Marine Microbiology, 28359 Bremen, Germany.
Monike Oggerin de Orube, Department of Molecular Ecology, Max Planck Institute for Marine Microbiology, 28359 Bremen, Germany.
Silvia Vidal-Melgosa, Department of Molecular Ecology, Max Planck Institute for Marine Microbiology, 28359 Bremen, Germany; MARUM MPG Bridge Group Marine Glycobiology, Center for Marine Environmental Sciences, University of Bremen, 28359 Bremen, Germany.
Vipul Solanki, Department of Molecular Ecology, Max Planck Institute for Marine Microbiology, 28359 Bremen, Germany.
Jan-Hendrik Hehemann, Department of Molecular Ecology, Max Planck Institute for Marine Microbiology, 28359 Bremen, Germany; MARUM MPG Bridge Group Marine Glycobiology, Center for Marine Environmental Sciences, University of Bremen, 28359 Bremen, Germany.
Rudolf Amann, Department of Molecular Ecology, Max Planck Institute for Marine Microbiology, 28359 Bremen, Germany.
Katrin Knittel, Department of Molecular Ecology, Max Planck Institute for Marine Microbiology, 28359 Bremen, Germany.
Conflicts of interest
None declared.
Funding
S.V.-M. and J.-H.H. received funding from the Deutsche Forschungsgemeinschaft Emmy Noether program (HE 7217/1-1), Deutsche Forschungsgemeinschaft Heisenberg program (HE 7217/5-1), and Deutsche Forschungsgemeinschaft Exzellenzcluster (2077), and K.K. received funding from the Andreas Rühl foundation. Further funding came from MARUM Center for Marine Environmental Science and the Max Planck Society.
Data availability
Sequence data are available at ENA under the project accession PRJEB53193.
References
- 1. Jahnke RA. Global Synthesis1. In: Liu KK, Atkinson L, Quiñones Ret al. (eds), Carbon and Nutrient Fluxes in Continental Margins. Global Change. The IGBP Series. Berlin, Heidelberg: Springer, 2010, 597–615. 10.1007/978-3-540-92735-8_16. [DOI] [Google Scholar]
- 2. Boudreau BP, Huettel M, Forster Set al. Permeable marine sediments: overturning an old paradigm. Eos Trans AGU 2001;82:133–6. 10.1029/EO082i011p00133-01. [DOI] [Google Scholar]
- 3. Hall SJ. The continental shelf benthic ecosystem: current status, agents for change and future prospects. Environ Conserv 2002;29:350–74. 10.1017/S0376892902000243. [DOI] [Google Scholar]
- 4. Huettel M, Berg P, Kostka JE. Benthic exchange and biogeochemical cycling in permeable sediments. Annu Rev Mar Sci 2014;6:23–51. 10.1146/annurev-marine-051413-012706. [DOI] [PubMed] [Google Scholar]
- 5. Ahmerkamp S, Winter C, Krämer Ket al. Regulation of benthic oxygen fluxes in permeable sediments of the coastal ocean. Limnol Oceanogr 2017;62:1935–54. 10.1002/lno.10544. [DOI] [Google Scholar]
- 6. Middelburg JJ, Barranguet C, Boschker HTSet al. The fate of intertidal microphytobenthos carbon: an in situ 13C-labeling study. Limnol Oceanogr 2000;45:1224–34. 10.4319/lo.2000.45.6.1224. [DOI] [Google Scholar]
- 7. Myklestad S. Production of carbohydrates by marine planktonic diatoms. I. Comparison of nine different species in culture. J Exp Mar Biol Ecol 1974;15:261–74. 10.1016/0022-0981(74)90049-5. [DOI] [Google Scholar]
- 8. Laine RA. A calculation of all possible oligosaccharide isomers both branched and linear yields 1.05 x 1012 structures for a reducing hexasaccharide: the isomer barrier to development of single-method saccharide sequencing or synthesis systems. Glycobiology 1994;4:759–67. 10.1093/glycob/4.6.759. [DOI] [PubMed] [Google Scholar]
- 9. Arnosti C, Wietz M, Brinkhoff Tet al. The biogeochemistry of marine polysaccharides: sources, inventories, and bacterial drivers of the carbohydrate cycle. Annu Rev Mar Sci 2021;13:81–108. 10.1146/annurev-marine-032020-012810. [DOI] [PubMed] [Google Scholar]
- 10. Drula E, Garron M, Dogan Set al. The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res 2021;50:D571–7. 10.1093/nar/gkab1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bligh M, Nguyen N, Buck-Wiese Het al. Structures and functions of algal glycans shape their capacity to sequester carbon in the ocean. Curr Opin Chem Biol 2022;71:102204. 10.1016/j.cbpa.2022.102204. [DOI] [PubMed] [Google Scholar]
- 12. Sichert A, Corzett CH, Schechter MSet al. Verrucomicrobia use hundreds of enzymes to digest the algal polysaccharide fucoidan. Nat Microbiol 2020;5:1026–39. 10.1038/s41564-020-0720-2. [DOI] [PubMed] [Google Scholar]
- 13. Becker S, Tebben J, Coffinet Set al. Laminarin is a major molecule in the marine carbon cycle. Proc Natl Acad Sci U S A 2020;117:6599–607. 10.1073/pnas.1917001117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Becker S, Scheffel A, Polz MFet al. Accurate quantification of laminarin in marine organic matter with enzymes from marine microbes. Appl Environ Microbiol 2017;83:e03389–16. 10.1128/AEM.03389-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vidal-Melgosa S, Pedersen HL, Schückel Jet al. A new versatile microarray-based method for high throughput screening of carbohydrate-active enzymes. J Biol Chem 2015;290:9020–36. 10.1074/jbc.M114.630673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Avci B, Krüger K, Fuchs BMet al. Polysaccharide niche partitioning of distinct Polaribacter clades during North Sea spring algal blooms. ISME J 2020;14:1369–83. 10.1038/s41396-020-0601-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Krüger K, Chafee M, Ben Francis Tet al. In marine Bacteroidetes the bulk of glycan degradation during algae blooms is mediated by few clades using a restricted set of genes. ISME J 2019;13:2800–16. 10.1038/s41396-019-0476-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Teeling H, Fuchs BM, Bennke CMet al. Recurring patterns in bacterioplankton dynamics during coastal spring algaep blooms. eLife 2016;5:e11888. 10.7554%2FeLife.11888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miksch S, Meiners M, Meyerdierks Aet al. Bacterial communities in temperate and polar coastal sands are seasonally stable. ISME Commun 2021;1:29. 10.1038/s43705-021-00028-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Probandt D, Knittel K, Tegetmeyer HEet al. Permeability shapes bacterial communities in sublittoral surface sediments. Environ Microbiol 2017;19:1584–99. 10.1111/1462-2920.13676. [DOI] [PubMed] [Google Scholar]
- 21. Hodal H, Falk-Petersen S, Hop Het al. Spring bloom dynamics in Kongsfjorden, Svalbard: nutrients, phytoplankton, protozoans and primary production. Polar Biol 2012;35:191–203. 10.1007/s00300-011-1053-7. [DOI] [Google Scholar]
- 22. Woelfel J, Schumann R, Peine Fet al. Microphytobenthos of Arctic Kongsfjorden (Svalbard, Norway): biomass and potential primary production along the shore line. Polar Biol 2010;33:1239–53. 10.1007/s00300-010-0813-0. [DOI] [Google Scholar]
- 23. Jørgensen BB, Laufer K, Michaud ABet al. Biogeochemistry and microbiology of high Arctic marine sediment ecosystems—case study of Svalbard fjords. Limnol Oceanogr 2020;66:S273–92. 10.1002/lno.11551. [DOI] [Google Scholar]
- 24. Arnosti C. Functional differences between Arctic seawater and sedimentary microbial communities: contrasts in microbial hydrolysis of complex substrates. FEMS Microbiol Ecol 2008;66:343–51. 10.1111/j.1574-6941.2008.00587.x. [DOI] [PubMed] [Google Scholar]
- 25. Zhou J, Bruns MA, Tiedje JM. DNA recovery from soils of diverse composition. Appl Environ Microbiol 1996;62:316–22. 10.1128/aem.62.2.316-322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rodriguez-R LM, Konstantinidis KT. Nonpareil: a redundancy-based approach to assess the level of coverage in metagenomic datasets. Bioinformatics 2013;30:629–35. 10.1093/bioinformatics/btt584. [DOI] [PubMed] [Google Scholar]
- 27. Bankevich A, Nurk S, Antipov Det al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 2012;19:455–77. 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gurevich A, Saveliev V, Vyahhi Net al. QUAST: quality assessment tool for genome assemblies. Bioinformatics 2013;29:1072–5. 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu YW, Tang YH, Tringe SGet al. MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome 2014;2:26. 10.1186/2049-2618-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kang DD, Li F, Kirton Eet al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019;7:e7359–9. 10.7717/peerj.7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sieber CMK, Probst AJ, Sharrar Aet al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat Microbiol 2018;3:836–43. 10.1038/s41564-018-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bushnell B, Rood J, Singer E. BBMerge - accurate paired shotgun read merging via overlap. PLoS One 2017;12:e0185056. 10.1371/journal.pone.0185056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Olm MR, Brown CT, Brooks Bet al. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J 2017;11:2864–8. 10.1038/ismej.2017.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chaumeil P-A, Mussig AJ, Hugenholtz Pet al. GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 2019;36:1925–7. 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Parks DH, Imelfort M, Skennerton CTet al. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 2015;25:1043–55. 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 2014;30:2068–9. 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 37. Huang L, Zhang H, Wu Pet al. dbCAN-seq: a database of carbohydrate-active enzyme (CAZyme) sequence and annotation. Nucleic Acids Res 2018;46:D516–21. 10.1093/nar/gkx894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Consortium TU . UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 2020;49:D480–9. 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Barbeyron T, Brillet-Guéguen L, Carré Wet al. Matching the diversity of sulfated biomolecules: creation of a classification database for sulfatases reflecting their substrate specificity. PLoS One 2016;11:e0164846. 10.1371/journal.pone.0164846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Elbourne LDH, Tetu SG, Hassan KAet al. TransportDB 2.0: a database for exploring membrane transporters in sequenced genomes from all domains of life. Nucleic Acids Res 2016;45:D320–4. 10.1093/nar/gkw1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Buchfink B, Reuter K, Drost H-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods 2021;18:366–8. 10.1038/s41592-021-01101-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rodriguez-R LM, Konstantinidis KT. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Preprints 2016;4:e1900v1. 10.7287/peerj.preprints.1900v1. [DOI] [Google Scholar]
- 43. Kopylova E, Noé L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012;28:3211–7. 10.1093/bioinformatics/bts611. [DOI] [PubMed] [Google Scholar]
- 44. Quast C, Pruesse E, Yilmaz Pet al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 2012;41:D590–6. 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wickham H, Averick M, Bryan Jet al. Welcome to the tidyverse. J Open Source Softw 2019;4:1686. 10.21105/joss.01686. [DOI] [Google Scholar]
- 46. Vidal-Melgosa S, Sichert A, Francis TBet al. Diatom fucan polysaccharide precipitates carbon during algal blooms. Nat Commun 2021;12:1150. 10.1038/s41467-021-21009-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kappelmann L, Krüger K, Hehemann J-Het al. Polysaccharide utilization loci of North Sea Flavobacteriia as basis for using SusC/D-protein expression for predicting major phytoplankton glycans. ISME J 2019;13:76–91. 10.1038/s41396-018-0242-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lu D-C, Wang F-Q, Amann RIet al. Epiphytic common core bacteria in the microbiomes of co-located green (Ulva), brown (Saccharina) and red (Grateloupia, Gelidium) macroalgae. Microbiome 2023;11:126. 10.1186/s40168-023-01559-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Blazewicz SJ, Barnard RL, Daly RAet al. Evaluating rRNA as an indicator of microbial activity in environmental communities: limitations and uses. ISME J 2013;7:2061–8. 10.1038/ismej.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Poulsen LK, G B, Stahl DA. Use of rRNA fluorescence in situ hybridization for measuring the activity of single cells in young and established biofilms. Appl Environ Microbiol 1993;59:1354–60. 10.1128/aem.59.5.1354-1360.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zorz J, Willis C, Comeau AMet al. Drivers of regional bacterial community structure and diversity in the Northwest Atlantic Ocean. Front Microbiol 2019;10:281. 10.3389/fmicb.2019.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. El-Swais H, Dunn KA, Bielawski JPet al. Seasonal assemblages and short-lived blooms in coastal north-west Atlantic Ocean bacterioplankton. Environ Microbiol 2015;17:3642–61. 10.1111/1462-2920.12629. [DOI] [PubMed] [Google Scholar]
- 53. Thiele S, Storesund JE, Fernández-Méndez Met al. A winter-to-summer transition of bacterial and archaeal communities in arctic sea ice. Microorganisms 2022;10:1618. 10.3390/microorganisms10081618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jain A, Krishnan KP, Begum Net al. Response of bacterial communities from Kongsfjorden (Svalbard, Arctic Ocean) to macroalgal polysaccharide amendments. Mar Environ Res 2020;155:104874. 10.1016/j.marenvres.2020.104874. [DOI] [PubMed] [Google Scholar]
- 55. Wietz M, Bienhold C, Metfies Ket al. The polar night shift: seasonal dynamics and drivers of Arctic Ocean microbiomes revealed by autonomous sampling. ISME Commun 2021;1:76. 10.1038/s43705-021-00074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Parks DH, Chuvochina M, Rinke Cet al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res 2022;50:D785–94. 10.1093/nar/gkab776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Grondin JM, Tamura K, Déjean Get al. Polysaccharide utilization loci: fuelling microbial communities. J Bacteriol 2017;199:e00860–16. 10.1128/jb.00860-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Widdel F, Bak F. Gram-negative mesophilic sulfate-reducing bacteria. In: Balows A, Truper HG, Dworkin Met al. (eds), The Prokaryotes. 2nd ednNew York: Springer, 1992, 3352–78. 10.1007/978-1-4757-2191-1_21. [DOI] [Google Scholar]
- 59. Bäumgen M, Dutschei T, Bornscheuer UT. Marine polysaccharides: occurrence, enzymatic degradation and utilization. Chembiochem 2021;22:2247–56. 10.1002/cbic.202100078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Marquardt M, Vader A, Stübner EIet al. Strong seasonality of marine microbial eukaryotes in a high-arctic fjord (Isfjorden, in West Spitsbergen, Norway). Appl Environ Microbiol 2016;82:1868–80. 10.1128/aem.03208-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fredriksen S, Kile MR. The algal vegetation in the outer part of Isfjorden, Spitsbergen: revisiting per Svendsen’s sites 50 years later. Polar Res 2012;31:17538. 10.3402/polar.v31i0.17538. [DOI] [Google Scholar]
- 62. Hop H, Kovaltchouk NA, Wiencke C. Distribution of macroalgae in Kongsfjorden. Svalbard Polar Biol 2016;39:2037–51. 10.1007/s00300-016-2048-1. [DOI] [Google Scholar]
- 63. Berge J, Renaud PE, Darnis Get al. In the dark: a review of ecosystem processes during the Arctic polar night. Prog Oceanogr 2015;139:258–71. 10.1016/j.pocean.2015.08.005. [DOI] [Google Scholar]
- 64. Chapman ARO, Lindley JE. Seasonal growth of Laminaria solidungula in the Canadian high Arctic in relation to irradiance and dissolved nutrient concentrations. Mar Biol 1980;57:1–5. 10.1007/BF00420961. [DOI] [Google Scholar]
- 65. Wang L, Wang M, Wise MJet al. Recent progress in the structure of glycogen serving as a durable energy reserve in bacteria. World J Microbiol Biotechnol 2020;36:14. 10.1007/s11274-019-2795-6. [DOI] [PubMed] [Google Scholar]
- 66. Synytsya A, Novak M. Structural analysis of glucans. Ann Transl Med 2014;2:17. 10.3978/j.issn.2305-5839.2014.02.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wu G, Müller M. Glycogen phosphorylase sequences from the Amitochondriate protists, Trichomonas vaginalis, Mastigamoeba balamuthi, Entamoeba histolytica and Giardia intestinalis. J Eukaryot Microbiol 2003;50:366–72. 10.1111/j.1550-7408.2003.tb00151.x. [DOI] [PubMed] [Google Scholar]
- 68. Lapébie P, Lombard V, Drula Eet al. Bacteroidetes use thousands of enzyme combinations to break down glycans. Nat Commun 2019;10:2043. 10.1038/s41467-019-10068-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mohiuddin SG, Ghosh SA-O, Ngo HA-Oet al. Cellular self-digestion and persistence in bacteria. Microorganisms 2021;9:2269. 10.3390/microorganisms9112269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Włodarska-Kowalczuk M, Górska B, Deja Ket al. Do benthic meiofaunal and macrofaunal communities respond to seasonality in pelagial processes in an Arctic fjord (Kongsfjorden, Spitsbergen)? Polar Biol 2016;39:2115–29. 10.1007/s00300-016-1982-2. [DOI] [Google Scholar]
- 71. Souza CP, Almeida BC, Colwell RRet al. The importance of chitin in the marine environment. Mar Biotechnol 2011;13:823–30. 10.1007/s10126-011-9388-1. [DOI] [PubMed] [Google Scholar]
- 72. Huang Q-S, Xie X-L, Liang Get al. The GH18 family of chitinases: their domain architectures, functions and evolutions. Glycobiology 2011;22:23–34. 10.1093/glycob/cwr092. [DOI] [PubMed] [Google Scholar]
- 73. Decho A. Microbial exopolymer secretions in ocean environments: their role(s) in food webs and marine processes. Oceanogr Mar Biol Annu Rev 1990;28:73–154. [Google Scholar]
- 74. Hansson GC. Mucins and the microbiome. Annu Rev Biochem 2020;89:769–93. 10.1146/annurev-biochem-011520-105053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hannides AK, Aller RC. Priming effect of benthic gastropod mucus on sedimentary organic matter remineralization. Limnol Oceanogr 2016;61:1640–50. https://doi.org/10.1002%2Flno.10325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Crost EH, Tailford LE, Monestier Met al. The mucin-degradation strategy of Ruminococcus gnavus: the importance of intramolecular trans-sialidases. Gut Microbes 2016;7:302–12. 10.1080/19490976.2016.1186334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Derrien M, Collado MC, Ben-Amor Ket al. The mucin degrader Akkermansia muciniphila is an abundant resident of the human intestinal tract. Appl Environ Microbiol 2008;74:1646–8. https://doi.org/10.1128%2FAEM.01226-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Beidler I, Robb CS, Vidal-Melgosa Set al. Marine bacteroidetes use a conserved enzymatic cascade to digest diatom β-mannan. ISME J 2023;17:276–85. 10.1038/s41396-022-01342-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Le Costaouëc T, Unamunzaga C, Mantecon Let al. New structural insights into the cell-wall polysaccharide of the diatom Phaeodactylum tricornutum. Algal Res 2017;26:172–9. 10.1016/j.algal.2017.07.021. [DOI] [Google Scholar]
- 80. Steinke N, Vidal-Melgosa S, Schultz-Johansen Met al. Biocatalytic quantification of α-glucan in marine particulate organic matter. MicrobiologyOpen 2022;11:e1289. 10.1002/mbo3.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hansell DA, Carlson CA, Repeta DJet al. Dissolved organic matter in the ocean: a controversy stimulates new insights. Oceanography 2009;22:202–11. 10.5670/oceanog.2009.109. [DOI] [Google Scholar]
- 82. Burdige DJ, Komada T. Chapter 12- sediment pore waters. In: Hansell DA, Carlson CA (eds), Biogeochemistry of Marine Dissolved Organic Matter. Boston: Academic Press, 2015, 535–77. 10.1016/B978-012323841-2/50015-4. [DOI] [Google Scholar]
- 83. Panagiotopoulos C, Sempéré R. Analytical methods for the determination of sugars in marine samples: a historical perspective and future directions. Limnol Oceanogr-Meth 2005;3:419–54. 10.4319/lom.2005.3.419. [DOI] [Google Scholar]
- 84. Tanoue E, Handa N. Monosaccharide composition of marine particles and sediments from the Bering Sea and northern North Pacific. Oceanol Acta 1987;10:91–9https://archimer.ifremer.fr/doc/00110/22086/. [Google Scholar]
- 85. Aluwihare LI, Repeta DJ, Chen RF. Chemical composition and cycling of dissolved organic matter in the Mid-Atlantic Bight. Deep Sea Res (II Top Stud Oceanogr) 2002;49:4421–37. 10.1016/S0967-0645(02)00124-8. [DOI] [Google Scholar]
- 86. Aluwihare LI, Repeta DJ, Chen RF. A major biopolymeric component to dissolved organic carbon in surface sea water. Nature 1997;387:166–9. 10.1038/387166a0. [DOI] [Google Scholar]
- 87. Burdige DJ, Skoog A, Gardner K. Dissolved and particulate carbohydrates in contrasting marine sediments. Geochim Cosmochim Acta 2000;64:1029–41. 10.1016/S0016-7037(99)00361-0. [DOI] [Google Scholar]
- 88. Burdige DJ, Alperin MJ, Homstead Jet al. The role of benthic fluxes of dissolved organic carbon in oceanic and sedimentary carbon cycling. Geophys Res Lett 1992;19:1851–4. 10.1029/92GL02159. [DOI] [Google Scholar]
- 89. Huettel M, Rusch A. Advective particle transport into permeable sediments—evidence from experiments in an intertidal sandflat. Limnol Oceanogr 2000;45:525–33. 10.4319/lo.2000.45.3.0525. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data are available at ENA under the project accession PRJEB53193.