

Abstract
Glioblastoma (GBM) is the most prevalent and aggressive type of adult brain tumors with low 5-year overall survival rates. Epidemiologic data suggest that estrogen may decrease brain tumor growth, and estrogen receptor beta (ERβ) has been demonstrated to exert anti-tumor functions in GBM. The lack of potent, selective and brain permeable ERβ agonist to promote its anti-tumor action is limiting the therapeutic promise of ERβ. In this study, we discovered that Indanone and tetralone-keto or hydroxyl oximes are a new class of ER agonists. Due to its high activity in ERβ reporter assays, specific binding to ERβ in polar screen assays, and potent growth inhibitory activity in GBM cells, CIDD-0149897 was discovered as a possible hit by screening a library of compounds. CIDD-0149897 is more selective for ERβ than ERα (40-fold). Treatment with CIDD-0149897 markedly reduced GBM cell viability with an IC50 of ~7-15 μM, while having little to no effect on ERβ-KO cells and normal human astrocytes. Further, CIDD-0149897 treatment enhanced expression of known ERβ target genes and promoted apoptosis in established and patient-derived GSC models. Pharmacokinetic studies confirmed that CIDD-0149897 has systemic exposure, and good bioavailability in the brain. Mice tolerated daily intraperitoneal treatment of CIDD-0149897 (50 mg/kg) with a 7-day repeat dosage with no toxicity. Additionally, CIDD-0149897 treatment significantly decreased tumor growth in U251 xenograft model and extended the survival of orthotopic GBM tumor-bearing mice. Collectively, these findings pointed to CIDD-0149897 as a new class of ERβ agonist, offering GBM patients a potential means of improving survival.
Keywords: estrogen receptor beta, glioblastoma, ERβ agonist
Introduction
Glioblastoma (GBM) is among the most aggressive and often diagnosed primary malignant brain tumors in adults (1,2). GBM is a deadly neoplasm associated with the worst 5-year overall survival (OS) rates amid all human cancers (3) with 1 year survival at 65% and the 5-year survival at 12% (4). GBMs affect ~13,000 patients per year in United States (2,5). The molecular factors that contribute to GBM development are incompletely understood. Female sex hormone estrogen may prevent the progression of GBM, according to research that has been published (6–9).
Two estrogen receptor (ER) subtypes, ER-alpha (ERα, which acts as a tumor promoter) and ER-beta (ERβ, which acts as a tumor suppressor), primarily mediate the effects of estrogen (6,7). Earlier studies confirmed expression of ERβ in GBM cells and glioma stem cells (GSCs) (10–13). The potential for ERβ to limit tumor growth in GBM was recently verified using CRISPR/Cas9 ERβ knockout (ERβ-KO) in human GBM models (10). Additionally, ERβ is necessary for the optimal chemotherapy-induced DNA damage response, to promote GBM cell death, and to minimize GSC stemness (13,14). These findings imply that ERβ activation may be a promising treatment approach to reduce the progression of GBM. Due to the lack of specific ERβ agonists that could enhance its anti-tumor action in the brain, the therapeutic promise of ERβ has not been fully realized.
While ERα and ERβ are structurally similar, their ligand-binding domains differ enough to be selective for unique ligands. Various selective ERβ agonists are being investigated for therapeutic use (15,16). Several plant-based selective ERβ agonists (such as liquiritigenin, and S-equol) have been identified, and these ERβ agonists have demonstrated efficacy in preclinical models (17,18). The utility of natural phytoestrogens in the clinic is limited primarily due to difficulty in the synthesis, the lack of selectivity for ERβ, and when administered at high doses leads to activation of ERα. For example, S-equol exhibits an IC50 of 100-200 μM in GBM cells and lacks brain penetration. Several synthetic ERβ agonists (LY500307 and ERB041) have been identified as more potent than natural ERβ agonists, proven to be safe, and are able to penetrate the blood brain barrier (BBB). ERβ agonist LY500307 shows the selectivity for ERβ by 14-fold compared to ERα (19). However, these are no longer in clinical development by industry due to failure to meet clinical endpoints in non-oncological clinical studies.
In this study, we present the discovery of CIDD-0149897, a new ERβ agonist that preferentially binds ERβ and supports its tumor suppressor activities in GBM. Using molecular modeling, in vitro reporter, and binding assays, we confirmed that CIDD-0149897 binds ERβ. CIDD-0149897 reduces GBM cell viability while decreasing cell invasion and promoting apoptosis. Further pharmacokinetics and biodistribution studies showed that CIDD-0149897 is brain permeable and significantly slowed the progression of the GBM tumor in xenograft while increasing the survival in orthotopic mice models.
Methods
Cell lines and reagents
GBM cell lines U87 and U251 were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and were maintained as per ATCC guidelines. Cell identity was confirmed with Short Tandem Repeat (STR) polymorphism analysis. Human astrocytes were purchased from ScienCell Research Laboratories (Carlsbad, CA). Patient derived primary glioma stem cells (GSC-090909, GSC-031417, GSC-040815, GSC-082209, GSC-101310, GSC-111010, and GSC-012015), developed and characterized in earlier publications (12,20–22). All studies were carried out in compliance with the Helsinki Declaration and the guidelines established by the UT Health San Antonio Institutional Review Board. Using the Mycoplasma PCR Detection Kit, we confirmed that none of the model cells used were contaminated with mycoplasma (Sigma, St. Louis, MO, USA). Primary GBM cells, were cultured as neurospheres in neurobasal medium supplemented with B27 serum-free supplement, EGF (20 ng/ml), bFGF (20 ng/ml), LIF (10 ng/ml) and heparin (5 μg/ml). ERβ agonist LY500307 was purchased from Cayman Chemical (Ann Arbor, Michigan, USA). CellTiter-Glo® Luminescent Cell Viability Assay kit and Luciferase Assay system were obtained from Promega (Madison, WI, USA). ERβ-FLAG overexpressed model cells were generated by infecting them with pLenti6/V5-D-FLAG ERβ and empty control vectors and stable clones were selected with blasticidin (5 μg/ml). ERβ-KO cells were generated as described previously (10). Annexin V/PI kit was purchased from BioLegend (San Diego, CA, USA). ERβ antibody (Cat # GTX70174, WB-1:200, ICC 1:100) was purchased from GeneTex (Irvine, CA, USA). SOX2 (Cat # 3579, 1:1000), Oct4 (Cat # 2890, 1:1000) antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA) and β-actin (Sigma, Cat # A2066, WB-1:1000) antibody was purchased from Sigma (Sigma, St. Louis, MO, USA). Ki67 antibody (Cat # ab16667, IHC-1:50) was purchased from Abcam (Waltham, MA).
Synthesis of CIDD ERβ agonists
In order to quickly assess the desired indanone analogs for ERβ activity, we developed a short, two-step synthesis from commercially available starting materials. The representative synthesis shown in Scheme 1 (Figure. S1) supported our desired 3-point ERβ structure-activity relationship studies, as well as providing larger amounts of CIDD-0149897 for in vivo and PK studies. Starting from the commercially available bromo-indanone 1, Suzuki coupling utilizing a palladium-RuPhos catalyst system in the presence of boronic acid 2 and K3PO4 provided the desired biaryl compound 3 in good yield. Reaction of bromo-ketone 3 with isopentyl nitrite in the presence of HCl produced the corresponding keto-oxime CIDD-0149897 in moderate yield.
Cell viability, clonogenic, invasion, and apoptosis
MTT and the Cell Titer-Glo Luminescent Cell Viability Assays were used to determine the cell viability rates of GBM cells treated with CIDD-ERβ agonists as described (20). Clonogenic experiments were performed with U251 and U87 cells in 6-well plates as described (20). Briefly, cells were treated with CIDD-0149897 for 5 days, fixed with ice-cold methanol after 14 days, and stained with 0.5% crystal violet solution. In the analysis, colonies were counted using NIH Image J software. CIDD-149897-treated GBM cells’ capacity to invade Matrigel was assessed utilizing the Corning® BioCoat™ Growth Factor Reduced Matrigel Invasion Chamber test as previously mentioned (20). The effect of CIDD-0149897 on apoptosis of GBM cells was measured using annexin V/PI assay as previously described (22). Western blotting, immunohistochemistry (IHC) and Immunocytochemistry (ICC) experiments were performed as described earlier (20).
RNA sequencing, RT-qPCR, Western, ICC, and IHC studies
GSC-040815 cells were treated with vehicle or CIDD-0149897 for 48 hours and RNA was isolated using RNeasy mini kit (Qiagen, Hilden, Germany). The Genome Sequencing Facility (UT Health SA) established protocol was used to perform RNA-Seq (23). Genes with a fold change > 1.5 and an adjusted p-value 0.05 were classified as differentially expressed genes in a DEseq2 study of differential gene expression (24). Data from RNA-Seq was uploaded to the GEO database and given the accession number (GSE226765). Real time quantitative PCR (RT-qPCR) was used to verify the status of the ERβ target genes. Data were normalized to GAPDH or β-actin, and the delta-delta-CT method was used to determine the difference in fold change. Earlier publication described Western blotting, immunohistochemistry (IHC), Immunocytochemistry (ICC) experiment conditions and primer sequences (20).
Polarization studies
We utilized PolarScreen™ ERα (catalogue#A15883) or ERβ (catalogue#A15890) Competitor Assay kits Green (Thermo Fisher Scientific), to evaluate the specificity of ERα versus ERβ. This binding test was carried out in accordance with the manufacturer’s protocol to determine the EC50 values of ERβ agonists. The complex formed when the ER binds to the Fluormone™ ligand has a high polarization value. The polarization value is decreased if the Fluormone ligand is displaced by the ERβ agonist from the ER complex. The change in polarization values can be used to assess the relative affinity of an ERβ agonist precisely and simply for the ERα or ERβ because this only happens in the presence of a test substance. The 3.5 nM ERα or ERβ, 10 nM Fluormone™ ES2 Green, and ERβ agonists were evaluated in the range of 0.00006-10000 nM using repeated dilutions in the final assay buffer composition. The plate was incubated at room temperature for 2 hours, and the fluorescence polarization value (millipolarization, mP) of each well was measured using a PHERAstar microplate reader set to the following settings: 535 nm for emission and 485 nm for excitation. Analyses and plots of the emission ratio (535:485) were made. GraphPad Prism™ 9.0 software was used to create curves using a sigmoidal dose response equation (varying slope).
Molecular modeling studies
Crystal structure 1X7B was obtained from the PDB and used for all molecular modeling studies. Chain B was used for all docking calculations. The protein structure was prepared using Schrodinger’s automated protein preparation workflow. Docking was performed using Glide within the Schrodinger suite at the SP level with the grid centered on the crystallographic ligand-binding region. Induced fit docking was also performed using Schrodinger’s automated workflow. Docking poses were visualized using Maestro.
Reporter gene assays
Reporter gene assays were done as previously described(20). HEK-293T or U251 model cells stably transfected with either ERα or ERβ along with an ERE reporter were used in these assays. Model cells were then plated in 24 well plates and CIDD-ERβ agonists were treated to cells for a 24-hour period. The luciferase activity was determined after the cells had been lysed in luciferase lysis buffer and by using a dual luciferase assay kit (Promega, Madison, WI, USA).
Neurosphere formation Assay
For neurosphere formation assay, single cell suspension of GSCs were seeded in low attachment 96 well plate (5 cells/well) and treated with CIDD-0149897 (20 μM) for 8-10 days. After 8-10 days, neurospheres were imaged and neutrosphere diameter was calculated using ImageJ software.
Pharmacokinetic and brain biodistribution studies
A pharmacokinetic (PK) study of CIDD-0149897 was conducted in C57BL/6 mice following intraperitoneal (i. p.) administration of the compound. Since the C57BL/6 strain of mice has an intact immune system, we have used it for the maximum tolerable dose (MTD) and PK studies. Briefly, i.p., formulation (20 mg/kg) was prepared at the required volume of 0.1 mL by dissolving CIDD-0149897 in 0.3% Hydroxypropyl cellulose. The formulation was administered to a group of mice (n = 3) via i.p. and blood samples aliquots (10 μL) were collected via mouse tail vein at 15 minutes, 1 hr, 2 hr, 4 hr, 8hr and 24 hr after administration. To examine the availability of CIDD-149897 in the brain, i.p. formulation (50 mg/kg) was administered to another group of female mice (n = 3). Whole brain samples were collected at 15 minutes, 30 minutes, and 60 minutes after administration, respectively. CIDD-0149897 concentrations in mice blood and brain homogenates were quantitated using validated LC-MS/MS bioanalytical methods developed by GCC Center for Comprehensive PK/PD & Formulation at Texas Southern University (Houston, TX). Briefly, bio-samples were extracted by protein precipitation using 5X volume of acetonitrile containing an internal standard (CIDD-0149897 as IS, 20 ng/mL). After mixing, the samples were vortexed for 2 minutes followed by 20 minutes of centrifugation at 14,000 rpm, 4°C. An aliquot of the supernatant was injected onto the LC-MS/MS for analysis. CIDD-0149897 was separated in an ACE Excel 2 Super C18 column (50 x 2.1 mm, 2.0 μm) using a gradient mobile phase consisting of solvent A (0.05% formic acid in water) and solvent B (0.05% formic acid in acetonitrile) in Exion LC™ system. The drug concentrations were determined in a SCIEX TRIPLE QUAD™ 6500+ Mass Spectrometry using ESI source monitored in positive ion mode under multiple reaction monitoring (MRM). Mass transitions were m/z 272.034 152 for CIDD-0149897 and 280.025 244.2 for IS. The assay was linear in mouse blood from 0.25 ng/mL to 500 ng/mL and in mouse brain homogenate from 0.1 ng/g brain tissue to 300 ng/g brain tissue.
In vivo xenograft studies
All animal studies were carried out using IACUC approval from UT Health San Antonio. SCID mice (8 to 10 weeks old) were purchased from Charles River (Wilmington, MO, USA). U251 (2 x 106) cells were injected into the flank of SCID mice (male mice, n = 8 tumors) for xenograft investigations. The mice were divided into the vehicle or CIDD-0149897 treatment groups when the tumor reached quantifiable size. At intervals of three to four days, the tumor’s volume was measured using a caliper, and its volume was computed using a modified ellipsoidal formula: tumor volume = 1/2(L x W2), where L stands for the tumor’s longitudinal diameter and W for its transverse diameter. At the conclusion of the study, all mice were euthanized, and tumors were removed from the mice, and processed for histological analysis. For orthotopic xenograft investigations, GFP-Luciferase reporter-labeled U251 (1 x 106) or GFP-Luciferase reporter-labeled GSC-040815 (1 x 104) cells were orthotopically injected into the female mouse’s right cerebrum (female mice, n=5) using an established protocol. Dose was selected based on pilot MTD study of 10, 20, 50 mg/kg of CIDD-149897 for 7 days using C57BL/6 mice. The mice were monitored daily for adverse toxic effects. Immunohistochemical investigations were carried out as previously described(20). Five microscopic fields were randomly chosen and were used to calculate the percentage of Ki-67 positive proliferating cells.
Statistical analysis
GraphPad Prism 9 software was used for all statistical analyses (GraphPad Software, San Diego, CA). Statistics were compared between the control and CIDD-ERβ agonist treated groups using a student’s t-test and ANOVA. All the data in bar graphs are displayed as mean ± SE. Significant results were defined as a p-value less than 0.05. For the longitudinal examination of tumor growth on log-pre-processed tumor surface, linear mixed effect modeling was employed. By comparing the tumor growth intercepts and slopes (on a log scale) for each treatment group of interest, p values were determined. Mouse survival was calculated using Kaplan-Meier survival curves and the log-rank test.
Data and materials availability:
All data supporting the conclusions is included in the paper and/or in the Supplementary Materials.
Results
Identification of CIDD-0149897 as a novel ERβ agonist
To identify structurally novel ERβ agonists that are both potent and selective, and possess favorable CNS properties for BBB penetration, we carried out a structure-based drug design approach based on existing X-ray structural data across multiple chemotypes on the ERβ-LBD binding pocket (25). Within this design approach, we also analyzed key physicochemical properties (PCP) such as MW, LogD, tPSA and central nervous system multiparameter optimization (CNS-MPO) score to optimize properties for favorable BBB-penetration. This resulted in the identification of biphenyl Indanone and tetralone-based keto-oximes compounds, as represented by CIDD-0149897 (Figure 1A).
Figure 1.
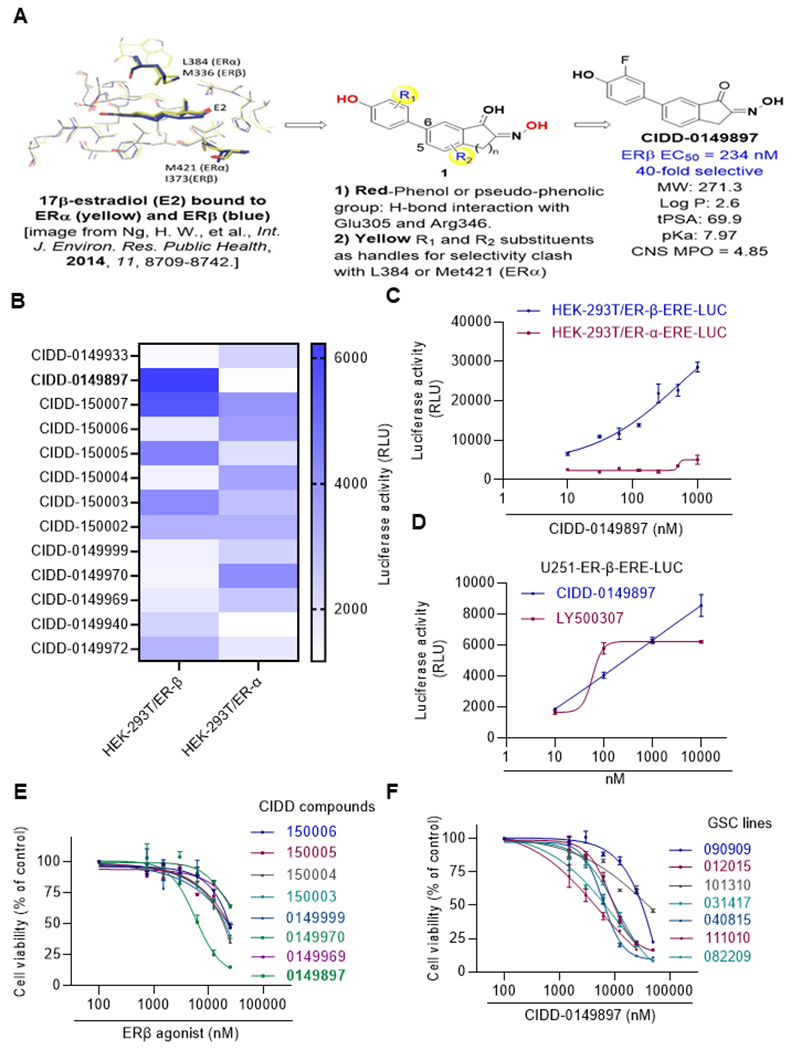
Identification of CIDD-0149897 as novel ERβ agonist. A, Schematic representation of ERβ ligand design strategy and the structure of CIDD-0149897. B HEK293T model cells stably expressing ERβ or ERα along with ERE-Luc reporter was used for initial screening of compounds testing for ER activity (n=3). Heat map showing ERβ and ERα reporter activity of selective compounds (n=3). C, HEK293Tcells expressing ERβ or ERα were treated with various doses of CIDD-0149897 and ERE-Luc reporter activity was measured after 24 hours (n=2). D, U251 GBM cells expressing ERβ were treated with indicated doses of CIDD-0149897 or LY500307 and after 24 h, ERE-Luc reporter activity was measured (n=3). E. Effect of increasing doses of selected ERβ agonists on the cell viability of U251 GBM model cells expressing ERβ was measured using the MTT cell viability assay (n=3). F, Effect of increasing doses of CIDD-0149897 on the cell viability of indicated patients derived GSCs was measured using CellTiter Glo assay (n=3). Data are presented as mean ± SEM.
Our design hypothesis (compound 1) based on the X-ray crystal structure of the unselective E2 compound, focused on the required diphenol moieties (red) for interactions with Glu305 and Arg346 in ERβ, substituents R1 and R2 as selectivity handles based on the known crystal structures, and keto-oximes or hydroxy-oximes to introduce steric clashes and modulate pKa of the pseudophenol. Thus, we envisioned a design strategy that would allow us to target the synthesis of the C5 or C6 substituted diphenyl-indanone (n=1) or diphenyl-tetralone (n=2) cores, with various substituents (R1 and R2), while incorporating oximes, keto-oximes, or hydroxyl-oximes.
We designed 27 compounds and tested those using HEK-293T reporter cells that uniquely express either ERα or ERβ. The results identified CIDD-0149897 as a potent and selective ERβ agonist (Figure 1B). The compound possesses low MW (<300), a suitable logP, tPSA and pKa profile for a CNS compound, and a very favorable CNS-MPO score of 4.85 (26). CIDD-0149897 enhanced ERβ reporter activity in a dose dependent manner and with no activation of ERα reporter (Figure 1C). CIDD-0149897 also enhanced ERβ reporter activity in a dose dependent manner in U251 GBM cells (Figure 1D). For comparison, currently available ERβ agonist LY500307 was used as a positive control. Among a library of compounds, CIDD-0149897 showed potent growth inhibitory activity in the GBM cells (IC50 =7.4 μM) (Figure 1E). In cell viability assays, CIDD-0149897 also showed potent cytotoxic activity against multiple primary GSCs (Figure 1F).
Target specificity of CIDD-0149897
To assess specificity of ERβ versus ERα, we established a polar screen nuclear receptor (NR) competitive binding assay (Thermo Fisher Scientific). In this assay, the complex formed when the ER binds to the fluormone ligand has a high polarization value. The polarization value is decreased if the ERβ agonist removes the fluormone ligand from the complex (Figure 2A). The change in polarization values is a simple and accurate method for determining the relative affinity of a test compound for the ER because it only happens in the presence of a test compound. We have used separate assays for ERα and ERβ. These results showed that CIDD-0149897 was 40-fold more selective for ERβ than ERα based on the EC50 value calculated using polarization values (Figure 2B). Docking studies (Figure 2C) suggest binding to key residues in ERβ including R346, E305, and F356, and H475. We then examined whether CIDD-0149897 treatment induces expression of known several ERβ target genes(27–29).As expected, RT-qPCR results showed activation of ERβ target genes in patient derived GSCs models (Figure 2D). Further, CIDD-0149897 also induced known ERβ target genes involved in DNA damage response and apoptosis such as p21 and GADD45A (Figure 2D, Supplementary Fig. S2A.). We also confirmed the specificity using ERβ KO cells (Supplementary Fig. S2C). Importantly, treatment of CIDD-014987 significantly decreased U251 cell viability with no/limited effect on normal human astrocytes and U251-ERβ-KO cells (Figure 2E).
Figure 2.
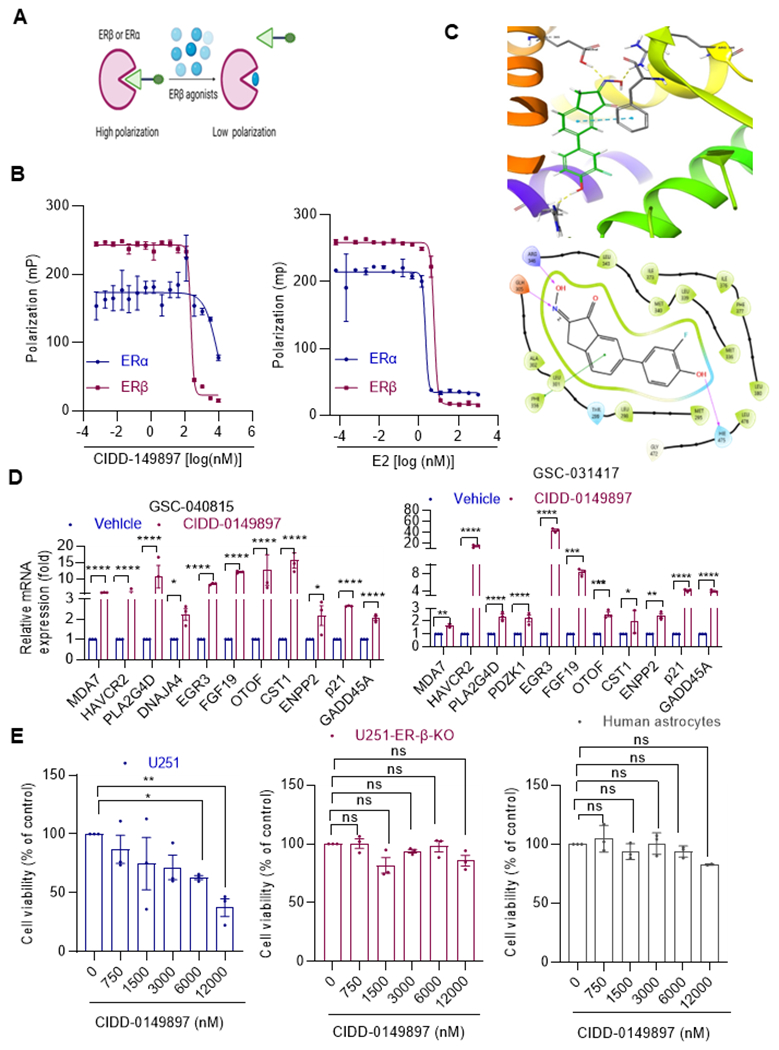
Target specificity of CIDD-0149897. A, Schematic of polar screen ER binding assay. B, Specificity of E2 (left panel) and CIDD-0149897 (right panel) to ERα and ERβ determined using polar screen ER competitive binding assays. Fluorescence polarization is measured and shown as millipolarization (mP) (n=2) C, Induced fit docking pose of CIDD-0149897 in the ligand binding site of PDB structure 1X7B. (Top panel) 3D rendering of docking pose indicating hydrogen bonding (yellow) and pi-stacking (cyan) interactions. (Bottom panel) 2D representation of ligand interactions. D, Effect of CIDD-0149897 treatment (10 μM, 72h) on ERβ targeted genes was measured using RT-qPCR analysis in GSC-040815, GSC-031417cells (n=3). E, Effect of CIDD-0149897 on normal human astrocytes as well as on WT and ERβ-KO U251 GBM cells was measured using the MTT cell viability assay (n=3). Data are presented as mean ± SEM * p<0.05, ** p<0.01, *** p<0.001, ****p<0.0001. In D, p-values were calculated using two-way ANOVA. In E, p-values were calculated using one-way ANOVA.
Biological activity of CIDD-0149897
Earlier studies showed that ERβ agonists reduce oncogenic properties of GBM cells. We therefore tested the activity of CIDD-0149897 using invasion, colony formation, neurosphere formation and apoptosis assays. Treatment of CIDD-0149897 significantly reduced GBM cells invasion (Figure 3A), long-term colony formation (Figure 3B). The observed effects on invasion of GBM cells is not due to CIDD-0149897 effect on cell viability, as we did not observe a significant reduction in cell viability during the short period of 24 hours of treatment (Supplementary Fig. S2D). Similarly, CIDD-0149897 treatment has no significant effect on the invasion and colony formation of U251- ERβ KO cells confirming that the effects seen in invasion and colony formation by CIDD-0149897 require the presence of ERβ (Supplementary Fig. S2E, 2F). Further, treatment with CIDD-0149897 significantly reduced the neurosphere growth of three different GSC model cells (Figure 3C), reduced the expression of stemness markers such as SOX2, and OCT4(Figure 3D). Additionally, treatment with CIDD-0149897 significantly promoted apoptosis of GBM cells as evident by Annexin V assays (Figure 3E). Earlier studies showed that activation of ERβ enhances chemotherapy response (14). We then examined whether CIDD-0149897 enhanced the efficacy of TMZ. The results showed that combination of CIDD-0149897 +TMZ has potent growth inhibition compared to monotherapy (Supplementary Fig. S3). Collectively these results confirm that CIDD-0149897 exerted anti-tumor activity in GBM.
Figure 3.
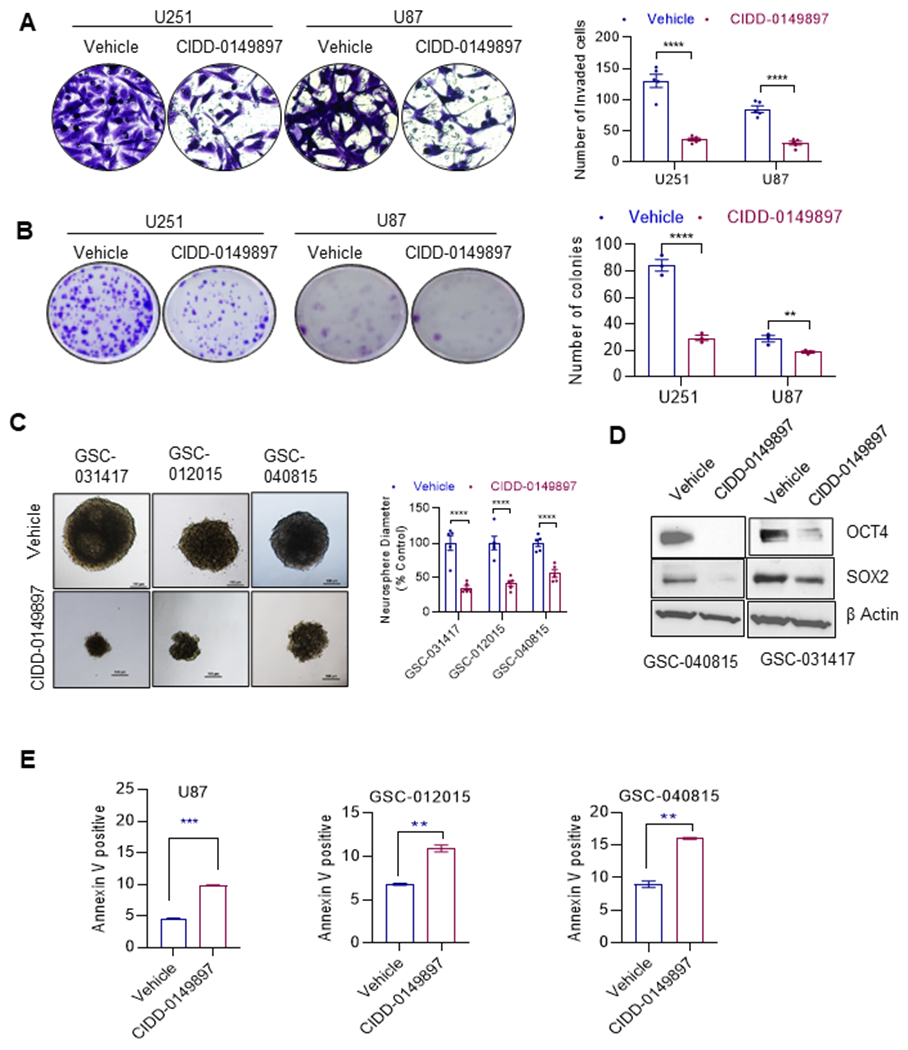
CIDD-0149897 decreases invasion, colony formation, neurosphere formation, stemness and promotes apoptosis of GBM cells. A, Effect of CIDD-0149897 (10μM, 22h) on cell invasion of U251 and U87 GBM model cells was determined using Matrigel invasion chamber assays (n=3). The number of invaded cells in five randomly selected fields was quantitated, and representative photos of invading cells are displayed. B, Effect of CIDD-0149897 (10 μM) on cell survival was measured using colony formation assays. C, Effect of CIDD-0149897 (20 μM) on neurosphere formation was measured in GSC-031417, GSC-012015, and GSC-040815 cells (n=3). Single cell suspension of GSCs were seeded in 24 well plates (100 cells/well) and treated with vehicle or CIDD-0149897 and after 7 days neurosphere were photographed and quantitated. GSCs were treated with vehicle or CIDD-0149897 and the expression levels of stemness genes were determined using western blotting (D). E, Effect of CIDD-0149897 (20 μM) on apoptosis was measured by Annexin V staining in GBM model cells. Data are presented as mean ± SEM. ** p<0.01, *** p<0.001, ****p<0.0001. In A, B and C, p-values were calculated using two-way ANOVA. In E, p-values were calculated using the t test.
CIDD-0149897 treatment modulated known ERβ regulated pathways
To understand the mechanistic insights of CIDD-0149897 activity, we conducted a global RNA-Seq study to identify the changes in gene expression. Total RNA was extracted from GSCs after a 48-hour treatment with vehicle or CIDD-0149897 and subjected to RNA-seq studies. Overall, 1054 genes (fold change >1.5 fold, adjusted p<0.05) showed differential expression in GSCs treated with CIDD-0149897; 494 genes showed upregulation and 560 genes showed downregulation. In Fig. 4A, a heat map of DEGs was displayed. GSEA-Hallmark pathway analyses of genes regulated by CIDD-0149897 treatment demonstrated modulation of several known ERβ-regulated pathways, including activation of the apoptosis, UPR, interferon γ response, UV response, and p53 pathways and downregulation of EMT, Myc, and E2F, G2/M checkpoint, and interferon alpha pathways (Fig. 4B). Representative GSEA plots of positive correlation with the p53 and apoptosis pathway gene sets and a negative correlation with the G2M check point and E2F targets for CIDD-0149897 regulated genes were shown (Figure 4C, D). Collectively these results confirm that CIDD-0149897 promotes ERβ mediated transcriptional changes.
Figure 4.
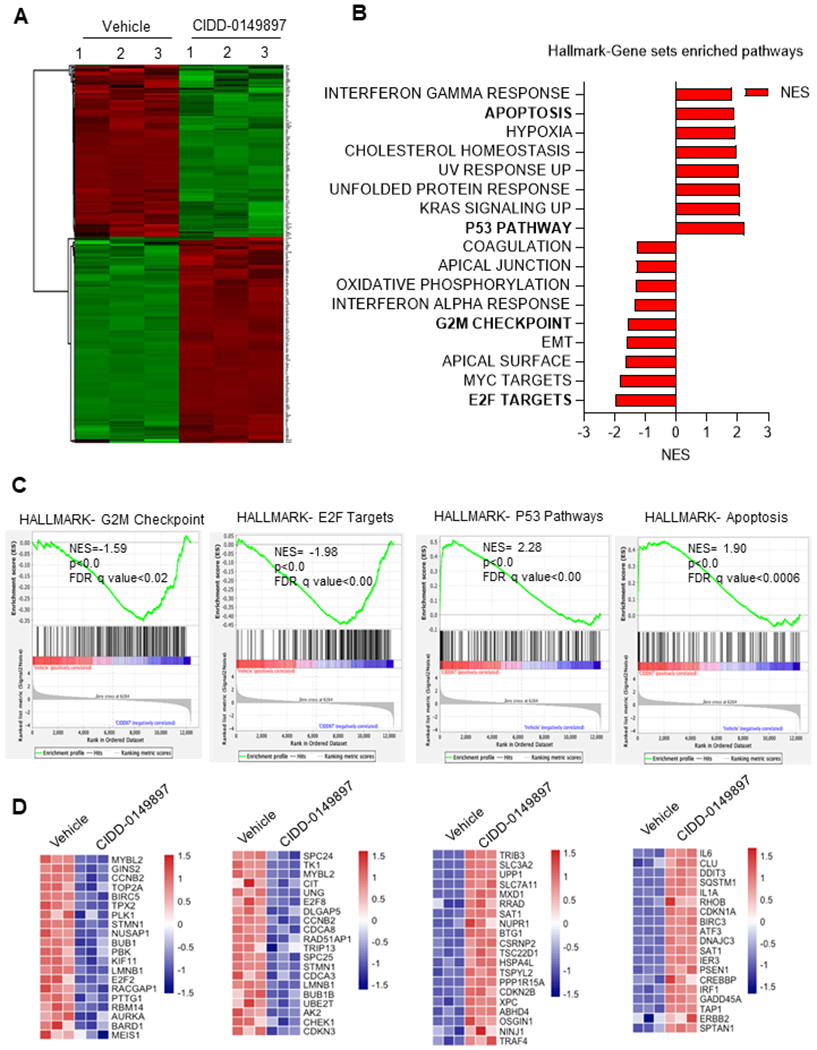
Global analysis of transcriptional changes altered by CIDD-0149897 treatment. A, DEGs identified from RNA-seq data are displayed in Heatmap. B, CIDD-0149897 regulated genes were subjected to GSEA and top positively or negatively enriched gene sets from Hallmark pathways were shown. C, GSEA enrichment plots of the apoptosis, p53, E2F, and G2/M checkpoint pathways were shown. D, heatmap of the selected genes related to the above indicated pathways are shown.
CIDD-0149897 has favorable PK and is safe for in vivo use
We then conducted PK analysis of CIDD-0149897 in C57BL/6 mice after intraperitoneal administration of 20 mg/kg of CIDD-0149897 (Figure 5A). A typical blood CIDD-0149897 concentration versus time profile was shown in Figure 5B. CIDD-149897 absorbed rapidly following intraperitoneal administration that resembled an intravenous bolus drug administration. PK model analysis showed negligible absorption phase and reached maximum CIDD-0149897 blood concentration in less than 15 min after dosing. Estimated maximum mean blood concentrations were 2,976 ng/mL (10,900 nM). CIDD-149897 distributed quickly in the central and peripheral compartment followed by a slower terminal elimination phase with a mean half-life of 9.59 hours. There were measurable drug concentrations (>1.5 ng/mL) in the systemic blood circulation by 8 hours post dose. CIDD-0149897 PK characteristics could be best described as a two-compartmental model as: Ct = 2,972 e−3.45t + 4.19 e−0.08t, where C (ng/mL) is blood concentration at time t (hours). (Figure 5B, C). We also conducted a toxicity study using C57BL/6 mice. Daily intraperitoneal administration of CIDD-0149897 (50 mg/kg) with 7-Day repeat dose was well tolerated by mice (n=3), with no changes in body weight and no organ toxicity was observed in H&E histology evaluation (n=3) (Figure 5D, E).
Figure 5.
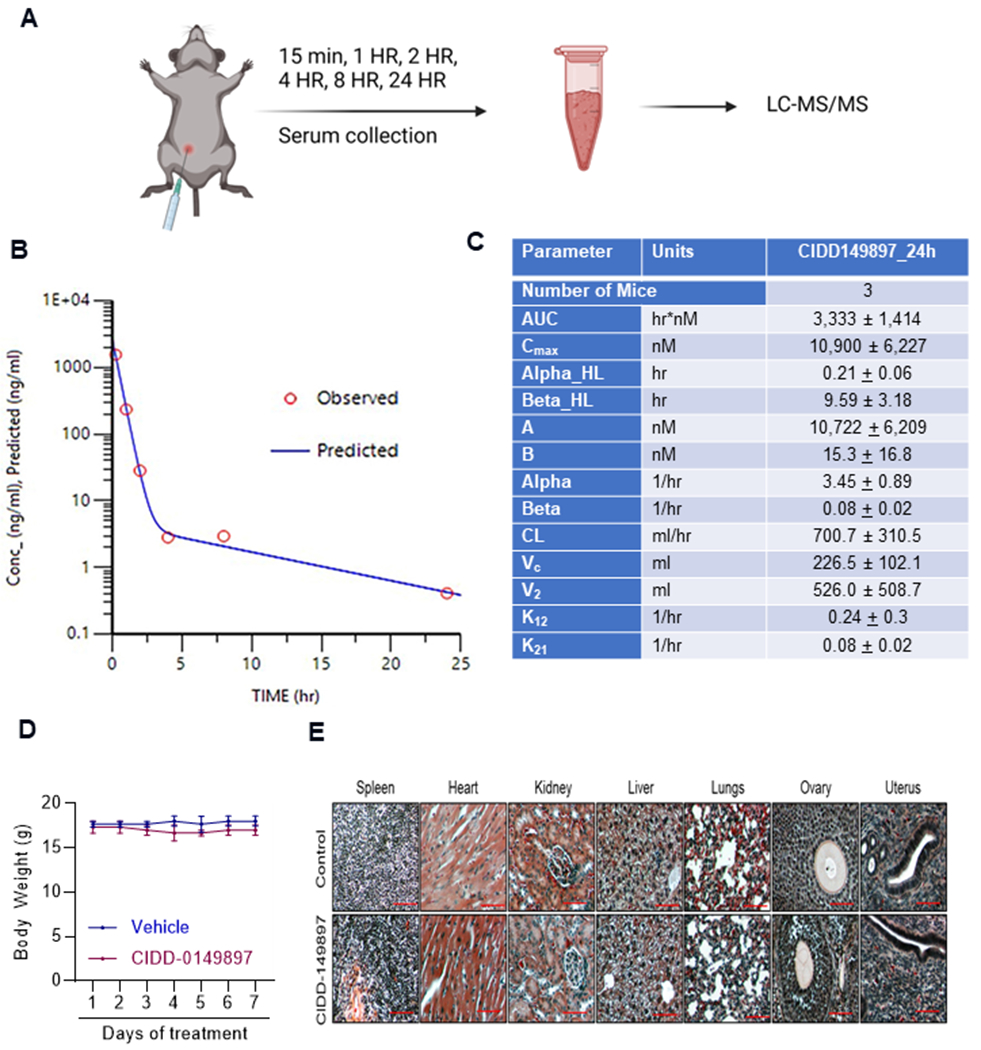
PK and toxicity analyses of CIDD-0149897. A, schematic of PK study design. B, C57/BL6 female mice (n=3) were administered with CIDD-0149897 (20mg/kg/single dose) intraperitoneally, and PK of the compounds at different time points was analyzed in blood. C, Summary of PK data for CIDD-0149897. D, C57BL6 mice were treated with CIDD-0149897 (50 mg/kg/i.p./day) or vehicle treatment for seven days and body weights were recorded. E, Histologic architecture of multiple organs in C57BL/6 mice were evaluated using H&E staining. Data are presented as mean ± SEM.
CIDD-0149897 reduced GBM xenograft tumor growth in vivo
To test the efficacy of CIDD-0149897 on in vivo tumor progression, we initially conducted in vivo studies using U251 subcutaneous xenograft tumor model in NOD/SCID mice Tumor bearing mice were randomized to receive vehicle or CIDD-0149897 (20 mg/kg/day/i. p.) 5 days/week. CIDD-0149897 treatment significantly reduced the tumor progression compared to vehicle (Figure 6A). Further, tumor weights from the CIDD-0149897 treated group (after 35 days of treatment) were significantly smaller compared to vehicle (Figure 6B). Mice body weights in the vehicle and CIDD-0149897 treated groups were not significantly altered (Figure 6C) confirming the low toxicity of CIDD-0149897. IHC analysis showed that CIDD-0149897 treated tumors exhibited fewer proliferating cells (Ki-67 positive cells) compared to vehicle treated tumors (Figure 6D). Further, RT-qPCR analysis of tumor tissues confirmed significant increase in the ERβ target genes in CIDD-0149897 treated tumors compared to vehicle (Figure 6E). Collectively, these results suggest that CIDD-0149897 has potent anti-tumor and on-target activity on subcutaneous GBM xenografts.
Figure 6.
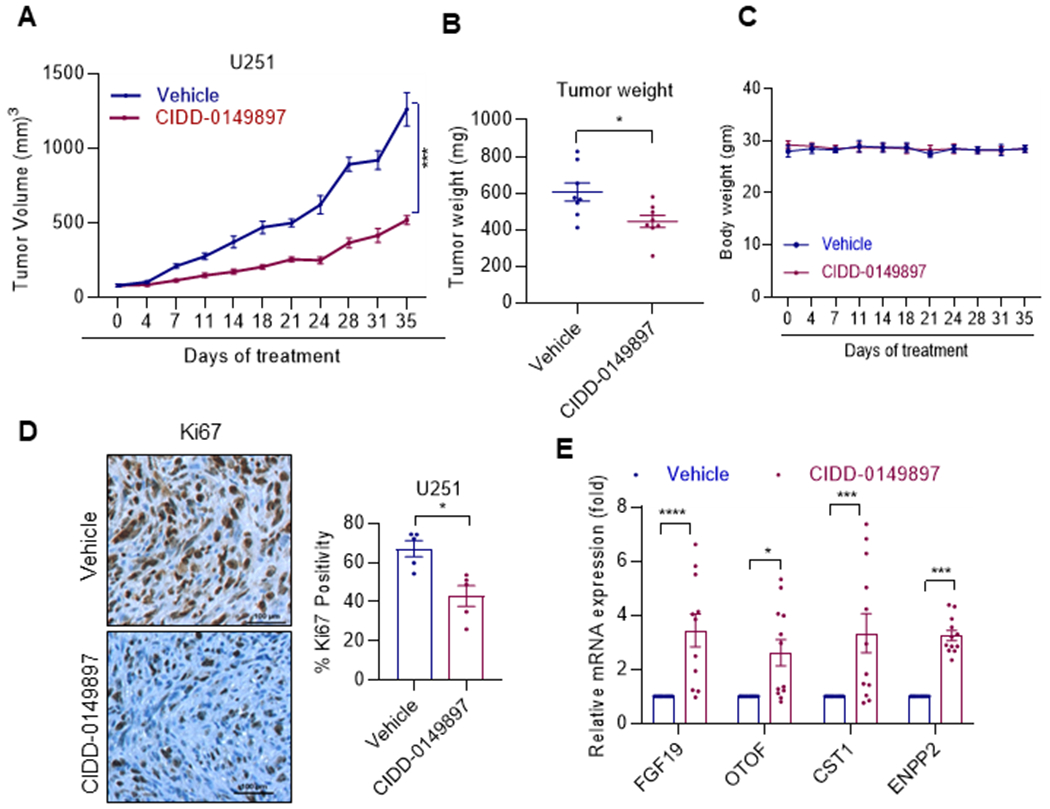
CIDD-0149897 inhibits the growth of GBM xenograft tumors. A, U251 xenografts (n=4) were treated with vehicle or CIDD-014897 (20mg/kg/i. p./5 days/week). B, Tumor volumes are shown in the graph. C, Body weights of vehicle and CIDD-014897 treated mice are shown. D, Ki-67 expression as a marker of proliferation was analyzed by IHC and quantitated. E, Status of ERβ induced target genes were measured by using RT-qPCR analysis (n=3). * p<0.05, *** p<0.001, ****p<0.0001. Data are presented as mean ± SEM. In A, p-values were calculated using linear mixed effect model. In B and D, p-values were calculated using the t test and in E, p value were calculated using two-way ANOVA.
CIDD-0149897 has brain permeability and therapeutic effect on orthotopic xenografts of GBM
We then examined whether CIDD-0149897 is capable of brain penetration. Brains from C57BL/6 mice were collected 15, 30, 60 min after administration of CIDD-0149897 (50 mg/kg/i.p.). The biodistribution study confirmed that CIDD-0149897 was detected in the brain, with peak detectable levels of 782 ng/g (2860 nM) (CIDD-0149897) at 15 min after i. p. administration, indicating that CIDD-0149897 crosses the BBB (Figure 7A, B). Importantly, treatment of CIDD-0149897 (50 mg/kg/i.p.) significantly increased the survival of orthotopic U251 tumor bearing mice (Fig 7C). IHC analysis of tumor sections revealed that CIDD-0149897 treated tumors have fewer proliferating cells (Ki-67 positive cells) in comparison to the vehicle treated group (Figure 7D). We also examined the activity of CIDD-0149897 in orthotopic xenograft model using patient-derived GSCs. After the tumors were established, mice were treated with vehicle or CIDD-0149897 (50 mg/kg/i. p.). Results showed that CIDD-0149897 significantly enhanced the survival of GSC tumor-bearing mice and IHC results showed significantly decreased number of Ki-67 positive cells in CIDD-0149897 tumors compared to vehicle (Figure 7E, F). Collectively, these results suggest that CIDD-0149897 has good blood-brain barrier permeability and increases the survival of tumor bearing mice.
Figure 7.
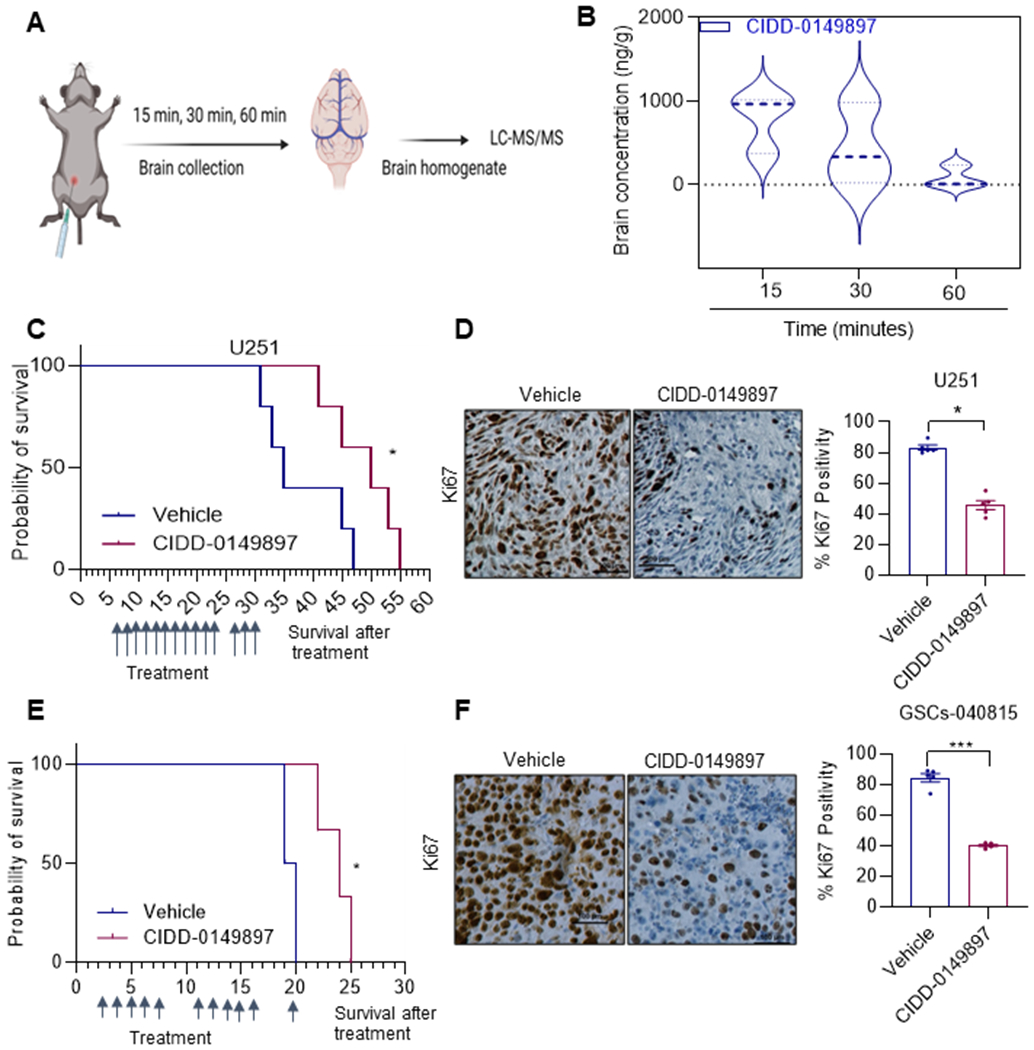
CIDD-0149897 has BBB permeability and therapeutic effects on orthotopic xenografts of GBM. A, Schematic of design used for studying brain tissue concentration of CIDD-0149897. B, C57BL/6 mice (female, n=3) were treated with a single dose of CIDD-0149897 (50 mg/kg/i.p./day) and the brain tissue concentrations were determined at indicated times. C, Mice were implanted with U251 cells orthotopically in the right cerebrum. After the tumor establishment, mice (n=5) were treated with vehicle (control) or CIDD-0149897 (50 mg/kg/i.p./day) and the survival of the mice was plotted using Kaplan-Meier curve. E, Patient derived GSC-040815- cells were implanted orthotopically in SCID mice, treated with vehicle (control) or CIDD-014987 (50 mg/kg/i.p.) for 11 days as indicated and the survival of the mice was plotted using Kaplan-Meier curve (n = 5). D, F, Ki-67 expression as a marker of proliferation was analyzed by IHC and quantitated. * p<0.05, *** p<0.001. Data are presented as mean ± SEM. In D and E, p-values were calculated using the t test. In C and E, p-values were calculated using Log-rank (Mantel-Cox) test.
Discussion
In this study to improve the specificity and potency of ERβ agonists, we have modeled, synthesized, and tested Indanone and tetralone-keto or hydroxyl oximes as novel ERβ agonists. These molecules are designed to better fit the ERβ-LBD binding pocket. Evaluation of these compounds using ERα and ERβ specific reporter assays and binding assays have identified several compounds with higher selectivity to ERβ. Of these compounds, we identified compound CIDD-0149897 as a potential lead due to its specific binding to ERβ (EC50 ~ 234 nM), potent inhibitory activity (IC50 ~7.4 μM) against ERβ expressing GBM cells and highly selective for ERβ over ERα (∼40-fold). Using multiple established GBM cells and patient-derived GSCs, we demonstrated that CIDD-0149897 exerts anti-tumor functions.
The biological effects of 17β-estradiol (E2) are mediated through ERα and ERβ. Despite extensive sequence similarities, these ER subtypes have distinctly unique biological functions. For example, ERβ exhibits antitumor activity, unlike ERα (15). Overexpression of ERβ reduces cell proliferation, whereas knockdown of ERβ enhances cell proliferation in cancer cells (30,31). As transcription factors, ERα and ERβ share many target genes; however, ERβ activates a unique set of genes (32,33) via its direct DNA binding or its interactions with other transcription factors (32,34). Supporting tumor suppressor functions of ERβ, our results showed that CIDD-0149897 treatment substantially enhanced the expression of ERβ target genes in GBM model cells.
The currently available synthetic ERβ agonists (such as LY500307 and ERB041) are superior to natural ERβ agonists and have been shown to be safe for human use (19). However, due to failure to meet clinical endpoints in non-oncological clinical studies, the industry has stopped developing these drugs for use in humans. ER cross-reactivity is a potential drawback with synthetic ERβ agonists. The discovery of new ERβ agonists with improved selectivity and efficacy increases the translational potential of ERβ as a therapeutic target. In this study we developed CIDD-0149897 as a novel brain penetrant ERβ agonist. We confirmed the specificity of ERβ versus ERα, using Polar Screen Nuclear Receptor Competitive Binding Assay. Our results showed higher selectivity of new CIDD-0149897 agonist to ERβ by 40-fold compared to ERα (35).
Published studies reported that ERβ functions as a tumor suppressor in GBM. In our studies using GBM xenograft model, we found that CIDD-0149897 treatment significantly reduced tumor progression in vivo. Using patient-derived xenograft (PDX) models, we also demonstrated that CIDD-0149897 enhances the survival of mice bearing orthotopic tumors. However, our studies are conducted using SCID mice models which lack intact immune system, therefore, the tumor extrinsic effect of CIDD-0149897 cannot be studied in our assays. Previous studies using ERβ knockdown, or overexpression suggested that ERβ activation enhances chemotherapy response (11,14). Our in vitro results showed that combination therapy of temozolomide with CIDD-0149897 has higher growth inhibitory effort compared to monotherapy which is in agreement with the published studies. However, future studies are needed to establish the utility of CIDD-0149897 in attenuating the therapy resistance of GBM as well as identifying the potential mechanism of combination effect.
In conclusion, we developed novel CIDD-ERβ agonists by designing, synthesizing, and testing keto-oximes based on biphenyl Indanone and tetralone. The lead ERβ agonist CIDD-0149897 that we discovered is more ERβ selective, and effective at reducing the growth of GBM in both in vitro and in vivo assays. Since CIDD-0149897 is a small molecule with good PK properties, it can be used as a monotherapy or in combination with the current standard of care for treating GBM.
Supplementary Material
Funding:
This study was supported by the NCI grant 1R01CA269866-01A1 (R.K.V., S.M., A.B.); R01 NS106173-01A1 (G.R.S,) and R50 CA265339-01A1 (Z.L.); and NCI Cancer Center Support Grant P30CA054174-17; Data generated in the Genome Sequencing Facility was supported by NIH Shared Instrument grant RP 1S10OD030311-01 (S10 grant), CPRIT Core Facility Award (RP220662), and CPRIT Core Facility Award (RP180748).
Footnotes
Competing interests: UT System filed a patent application on ER beta agonists
References
- 1.Ohgaki H, Kleihues P. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci 2009;100(12):2235–41 doi 10.1111/j.1349-7006.2009.01308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA 2013;310(17):1842–50 doi 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- 3.Tykocki T, Eltayeb M. Ten-year survival in glioblastoma. A systematic review. J Clin Neurosci 2018;54:7–13 doi 10.1016/j.jocn.2018.05.002. [DOI] [PubMed] [Google Scholar]
- 4.Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017;318(23):2306–16 doi 10.1001/jama.2017.18718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson DR, O’Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. Journal of neuro-oncology 2012;107(2):359–64 doi 10.1007/s11060-011-0749-4. [DOI] [PubMed] [Google Scholar]
- 6.Kabat GC, Etgen AM, Rohan TE. Do steroid hormones play a role in the etiology of glioma? Cancer Epidemiol Biomarkers Prev 2010;19(10):2421–7 doi 10.1158/1055-9965.EPI-10-0658. [DOI] [PubMed] [Google Scholar]
- 7.Hatch EE, Linet MS, Zhang J, Fine HA, Shapiro WR, Selker RG, et al. Reproductive and hormonal factors and risk of brain tumors in adult females. Int J Cancer 2005;114(5):797–805 doi 10.1002/ijc.20776. [DOI] [PubMed] [Google Scholar]
- 8.Carroll RS, Zhang J, Dashner K, Sar M, Black PM. Steroid hormone receptors in astrocytic neoplasms. Neurosurgery 1995;37(3):496–503; discussion −4. [DOI] [PubMed] [Google Scholar]
- 9.Michaud DS, Gallo V, Schlehofer B, Tjonneland A, Olsen A, Overvad K, et al. Reproductive factors and exogenous hormone use in relation to risk of glioma and meningioma in a large European cohort study. Cancer Epidemiol Biomarkers Prev 2010;19(10):2562–9 doi 10.1158/1055-9965.EPI-10-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J, Sareddy GR, Zhou M, Viswanadhapalli S, Li X, Lai Z, et al. Differential Effects of Estrogen Receptor beta Isoforms on Glioblastoma Progression. Cancer Res 2018;78(12):3176–89 doi 10.1158/0008-5472.CAN-17-3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sareddy GR, Li X, Liu J, Viswanadhapalli S, Garcia L, Gruslova A, et al. Selective Estrogen Receptor beta Agonist LY500307 as a Novel Therapeutic Agent for Glioblastoma. Sci Rep 2016;6:24185 doi 10.1038/srep24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sareddy GR, Nair BC, Gonugunta VK, Zhang QG, Brenner A, Brann DW, et al. Therapeutic significance of estrogen receptor beta agonists in gliomas. Mol Cancer Ther 2012;11(5):1174–82 doi 10.1158/1535-7163.MCT-11-0960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sareddy GR, Pratap UP, Venkata PP, Zhou M, Alejo S, Viswanadhapalli S, et al. Activation of estrogen receptor beta signaling reduces stemness of glioma stem cells. Stem Cells 2021;39(5):536–50 doi 10.1002/stem.3337. [DOI] [PubMed] [Google Scholar]
- 14.Zhou M, Sareddy GR, Li M, Liu J, Luo Y, Venkata PP, et al. Estrogen receptor beta enhances chemotherapy response of GBM cells by down regulating DNA damage response pathways. Sci Rep 2019;9(1):6124 doi 10.1038/s41598-019-42313-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nilsson S, Gustafsson JA. Estrogen receptors: therapies targeted to receptor subtypes. Clin Pharmacol Ther 2011;89(1):44–55 doi 10.1038/clpt.2010.226. [DOI] [PubMed] [Google Scholar]
- 16.Lo R, Matthews J. A new class of estrogen receptor beta-selective activators. Mol Interv 2010;10(3):133–6 doi 10.1124/mi.10.3.3. [DOI] [PubMed] [Google Scholar]
- 17.Mersereau JE, Levy N, Staub RE, Baggett S, Zogovic T, Chow S, et al. Liquiritigenin is a plant-derived highly selective estrogen receptor beta agonist. Mol Cell Endocrinol 2008;283(1–2):49–57 doi 10.1016/j.mce.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hernandez G, Zhao L, Franke AA, Chen YL, Mack WJ, Brinton RD, et al. Pharmacokinetics and safety profile of single-dose administration of an estrogen receptor beta-selective phytoestrogenic (phytoSERM) formulation in perimenopausal and postmenopausal women. Menopause 2018;25(2):191–6 doi 10.1097/GME.0000000000000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Norman BH, Dodge JA, Richardson TI, Borromeo PS, Lugar CW, Jones SA, et al. Benzopyrans are selective estrogen receptor beta agonists with novel activity in models of benign prostatic hyperplasia. J Med Chem 2006;49(21):6155–7 doi 10.1021/jm060491j. [DOI] [PubMed] [Google Scholar]
- 20.Pratap UP, Sareddy GR, Liu Z, Venkata PP, Liu J, Tang W, et al. Histone deacetylase inhibitors enhance estrogen receptor beta expression and augment agonist-mediated tumor suppression in glioblastoma. Neurooncol Adv 2021;3(1):vdab099 doi 10.1093/noajnl/vdab099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sareddy GR, Nair BC, Krishnan SK, Gonugunta VK, Zhang QG, Suzuki T, et al. KDM1 is a novel therapeutic target for the treatment of gliomas. Oncotarget 2013;4(1):18–28 doi 10.18632/oncotarget.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sareddy GR, Viswanadhapalli S, Surapaneni P, Suzuki T, Brenner A, Vadlamudi RK. Novel KDM1A inhibitors induce differentiation and apoptosis of glioma stem cells via unfolded protein response pathway. Oncogene 2017;36(17):2423–34 doi 10.1038/onc.2016.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015;31(2):166–9 doi 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ng HW, Perkins R, Tong W, Hong H. Versatility or promiscuity: the estrogen receptors, control of ligand selectivity and an update on subtype selective ligands. Int J Environ Res Public Health 2014;11(9):8709–42 doi 10.3390/ijerph110908709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wager TT, Hou X, Verhoest PR, Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem Neurosci 2010;1(6):435–49 doi 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shanle EK, Zhao Z, Hawse J, Wisinski K, Keles S, Yuan M, et al. Research resource: global identification of estrogen receptor beta target genes in triple negative breast cancer cells. Mol Endocrinol 2013;27(10):1762–75 doi 10.1210/me.2013-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vivar OI, Zhao X, Saunier EF, Griffin C, Mayba OS, Tagliaferri M, et al. Estrogen receptor beta binds to and regulates three distinct classes of target genes. J Biol Chem 2010;285(29):22059–66 doi 10.1074/jbc.M110.114116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan B, Cheng L, Chiang HC, Xu X, Han Y, Su H, et al. A phosphotyrosine switch determines the antitumor activity of ERbeta. J Clin Invest 2014;124(8):3378–90 doi 10.1172/JCI74085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strom A, Hartman J, Foster JS, Kietz S, Wimalasena J, Gustafsson JA. Estrogen receptor beta inhibits 17beta-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc Natl Acad Sci U S A 2004;101(6):1566–71 doi 10.1073/pnas.0308319100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hartman J, Edvardsson K, Lindberg K, Zhao C, Williams C, Strom A, et al. Tumor repressive functions of estrogen receptor beta in SW480 colon cancer cells. Cancer Res 2009;69(15):6100–6 doi 10.1158/0008-5472.CAN-09-0506. [DOI] [PubMed] [Google Scholar]
- 32.Charn TH, Liu ET, Chang EC, Lee YK, Katzenellenbogen JA, Katzenellenbogen BS. Genome-wide dynamics of chromatin binding of estrogen receptors alpha and beta: mutual restriction and competitive site selection. Mol Endocrinol 2010;24(1):47–59 doi 10.1210/me.2009-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grober OM, Mutarelli M, Giurato G, Ravo M, Cicatiello L, De Filippo MR, et al. Global analysis of estrogen receptor beta binding to breast cancer cell genome reveals an extensive interplay with estrogen receptor alpha for target gene regulation. BMC Genomics 2011;12:36 doi 10.1186/1471-2164-12-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakajima Y, Akaogi K, Suzuki T, Osakabe A, Yamaguchi C, Sunahara N, et al. Estrogen regulates tumor growth through a nonclassical pathway that includes the transcription factors ERbeta and KLF5. Sci Signal 2011;4(168):ra22 doi 10.1126/scisignal.2001551. [DOI] [PubMed] [Google Scholar]
- 35.Pratap U TM, Balinsa HU, Viswabadhpalli S, Sareddy G, McHardy S, Brenner A, Vadlamudi RK. Development of potent estrogen receptor beta agonists for treating glioblastoma. 2023 Proceedings of AACR annual meeting, Orlando, USA 2023(Abstract 1718). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.