

Abstract
PIK3CA is the second most mutated gene in cancer leading to aberrant PI3K/AKT/mTOR signaling and increased translation, proliferation, and survival. Some 4–25% of gastric cancers display activating PIK3CA mutations including 80% of EBV-associated GCs. Small molecules including pan-PI3K and dual PI3K/mTOR inhibitors have shown moderate success clinically, due to broad on-target/off-tissue effects. Thus, isoform specific and mutant selective inhibitors have been of significant interest. However, drug resistance is a problem and has affected success of new drugs. There has been a concerted effort to define mechanisms of resistance and identify potent combinations in many tumor types, though gastric cancer is comparatively understudied. In this study we identified modulators of the response to the PI3Kα-specific inhibitor, BYL719, in PIK3CA mutant GCs. We found that loss of NEDD9 or inhibition of BCL-XL conferred hyper-sensitivity to BYL719, through increased cell cycle arrest and cell death, respectively. Additionally, we discovered that loss of CBFB conferred resistance to BYL719. CBFB loss led to up-regulation of the protein kinase PIM1, which can phosphorylate and activate several overlapping downstream substrates as AKT thereby maintaining pathway activity in the presence of PI3Kα inhibition. The addition of a pan-PIM inhibitor re-sensitized resistant cells to BYL719. Our data provide clear mechanistic insights into PI3Kα inhibitor response in PIK3CA mutant gastric tumors and can inform future work as mutant selective inhibitors are in development for diverse tumor types.
Keywords: PIK3CA, gastric adenocarcinoma, drug resistance, CBFB, PIM1
INTRODUCTION
The phosphoinositide 3-kinase (PI3K/AKT/mTOR) signaling pathway regulates a number of crucial cellular functions such as proliferation, survival, differentiation, protein translation and glucose metabolism (1). The upstream component of this pathway, PI3K, has eight total isoforms in humans, split into three classes. The class I PI3Ks are responsible for directly activating signal transduction pathways and are heterodimers consisting of an 85kDa regulatory subunit that stabilizes a 110kDa catalytic subunit. There are four class I PI3K catalytic subunits, p110α, p110β, p110γ and p110δ, encoded by the PIK3CA, PIK3CB, PIK3CG and PIK3CD genes, respectively (2). Mutations in these genes are nearly ubiquitous in human cancers and specifically, PIK3CA is the second most commonly mutated gene in cancer. PIK3CA mutations are often activating and lead to dysregulation of normal cellular proliferation and survival signals thereby promoting aberrant cellular growth and tumor development (3–5). Since PIK3CA and other class I isoforms of PI3K are known common oncogenes, small molecules have been developed including pan-PI3K inhibitors, isoform-specific PI3K inhibitors, and dual PI3K/mTOR inhibitors (6). Many of these small molecules have been tested in clinical trials and some are approved for treatment of a variety of solid tumors (6,7). While these drugs have appeared promising in preclinical settings, this has not always translated into clinical success. This is due to a few factors including on-target, off-tissue effects, as well as both acquired and intrinsic resistance (6,8).
Gastric cancer is the fifth most common cancer and the fourth leading cause of cancer-associated mortality worldwide (9). PIK3CA mutation is frequent in gastric cancer and it is estimated that mutations occur in 4–25% of cases (10,11). Interestingly, in Epstein-Barr virus (EBV)-associated gastric cancers, 80% of patients display activating mutations in PIK3CA suggesting a crucial role for PI3K/AKT signaling in tumor initiation and oncogenesis of this subset (12). PI3K pathway inhibition has been of interest in the treatment of gastric cancer. For example, dual PI3K/mTOR inhibitors such as BEZ235, GSK1059615 and PI103, have been tested preclinically in gastric cancers as monotherapies and in combination with chemotherapeutic agents including paclitaxel, 5-fluorouracil and chloroquine (13–21).
Pan-PI3K and dual PI3K/mTOR inhibitors can effectively block pathway signaling, however because of the vast range of biological processes downstream of PI3K, these therapeutics are limited by tolerability and toxicity due to on-target/off-tissue effects (22). Given the prevalence of PIK3CA mutations in cancer, PI3Kα isoform-specific inhibitors are of significant interest and have been shown to have high efficacy in PIK3CA mutant tumors (23,24). Several drugs in preclinical development including STX-478, INK1117 and aminopyrazine compounds have so far shown high selectivity and efficacy against PI3Kα in comparison to other PI3K isoforms (25,26). However, PI3Kα specific inhibitors still suffer from toxicity and tolerability issues due to inhibition of wild type PI3Kα in non-tumor tissues (27). PI3Kα mutant selective inhibitors, including GDC-0077 and RLY-2608, are in development and have the potential to significantly alter the treatment landscape for patients with PIK3CA mutant tumors (NCT05216432) (28). The most advanced PI3Kα-specific inhibitor is BYL719 (Alpelisib), which was recently approved in combination with Fulvestrant for the treatment of HR+/HER2- advanced breast cancer and is being evaluated in many clinical trials for additional solid tumors (23,29,30). Furthermore, BYL719 is being evaluated in combination with an HSP90 inhibitor in patients with advanced and metastatic gastric cancer (NCT01613950) and in combination with paclitaxel in patients with PIK3CA-altered gastric cancer (NCT04526470).
Despite the success of BYL719 in pre-clinical and early phase clinical studies, acquired resistance has emerged as a problem and has prompted follow-up studies to further describe mechanisms of resistance in a variety of cancer cell lines including breast, ovarian, pancreatic, uveal melanoma and head and neck cancer (31–39). In vitro, BYL719 was shown to be effective in inhibiting the growth of PIK3CA mutant gastric cancer cell lines and was synergistic with the chemotherapeutic agent paclitaxel (40). However, given the emergence of resistance in other tumor types treated with BYL719 and other PI3Kα targeting monotherapies, we expect similar outcomes in studies in PIK3CA mutant gastric cancers. In this study, we aimed to define novel mechanisms of sensitivity and resistance to BYL719 specifically in PIK3CA mutant gastric cancers.
MATERIALS AND METHODS
Cell Culture
Gastric cancer cell lines SNU484 (RRID:CVCL_0100), SNU1750 (RRID:CVCL_8914), SNU1967 (RRID:CVCL_8915), MKN1 (RRID:CVCL_1415), NCC-24 (RRID:CVCL_8899) and SNU719 (RRID:CVCL_5086) were kindly provided by Patrick Tan at Duke National University Singapore, Singapore. These cell lines were maintained in Roswell Park Memorial Institute (RPMI) medium 1640 (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Corning), 100 U/mL non-essential amino acids, 100 U/mL penicillin and 100 μg/mL streptomycin (Invitrogen). Gastric cancer cell line AGS (RRID:CVCL_0139) was purchased from the Duke Cell Culture Facility, Duke University, Durham, NC and maintained in Dulbecco’s Modified Eagle Medium (DMEM)/Nutrient Mixture F-12 (Invitrogen) supplemented with 10% heat-inactivated FBS (Corning). The following cell lines were kindly provided by Kris Wood at Duke University, Durham, NC: laryngeal cancer cell line 584-A2 (RRID:CVCL_V278), esophageal squamous cancer cell line KYSE510 (RRID:CVCL_1354) and colorectal cancer cell line CRC119 were maintained in RPMI medium 1640 supplemented with 10% heat-inactivated FBS (Corning), 2mM L-Glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin (Invitrogen), breast cancer cell line CAL51 (RRID:CVCL_1110), maintained in Minimum Essential Medium (MEM) supplemented with 20% heat-inactivated FBS (Corning), 100 U/mL penicillin and 100 μg/mL streptomycin (Invitrogen), breast cancer cell lines MDA-MB-453 (RRID:CVCL_0418) and BT-549 (RRID:CVCL_1092), and colorectal cancer cell line WiDr (RRID:CVCL_2760) maintained in MEM medium supplemented with 10% heat-inactivated FBS (Corning), 100 U/mL non-essential amino acids, 100 U/mL penicillin and 100 μg/mL streptomycin (Invitrogen) and breast cancer cell line MCF7 (RRID:CVCL_0031), maintained in DMEM supplemented with 10% heat-inactivated FBS (Corning), 2mM L-Glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin (Invitrogen). Cell lines are regularly tested for mycoplasma using the LookOut Mycoplasma PCR Detection Kit (Sigma-Aldrich Cat# MP0035).
BYL719 resistant clones were grown by culturing the AGS cell line in increasing concentrations of drug over a 3-month period. Specifically, cells were cultured in 25 nM BYL719 for 3–4 days, and the concentration was increased 25 nM every 3–4 days until 100 nM at which point the concentration was increased by 100 nM approximately once per week. The resistant clones were selected as single cells from the bulk population and all resistant clones were maintained in 1 μM BYL719 without significant adverse effects on growth.
Growth Inhibition Assays (GI50)
Cells were seeded at 2,500 cells per well of 96-well plates 24 hours prior to treatments. To generate GI50 curves, cells were treated with vehicle (DMSO) or serial dilutions of each drug for 72 hours. Each treatment condition was completed in technical triplicate. Cell viability was read-out using CellTiter Glo (Promega G7573) on a BioTek Synergy2 plate reader. The total number of live cells for each drug dose were normalized to the vehicle control and GI50 drug curves were established using GraphPad Prism. For combinations, one drug was added at a constant concentration across all wells and total live cell counts were normalized to the secondary drug-only condition. For three-day growth curves, cells were seeded at 2,500 cells per well of 96-well plates 24 hours prior to treatment and treated with stable concentrations of each drug. Total live cell counts were measured using CellTiter Glo on each day, and relative luciferase units were plotted to represent cell growth over time. Drugs used include BYL719 (Selleckchem S2814) and A-1331852 (Chemietek 1430844–80-6), PIM447 (Selleckchem S7985) and Dasatinib (ApexBio A3017).
CRISPR library Lentivirus Production and Transduction
Lentivirus production was adapted from Joung et al., 2017 (41). HEK293FT (RRID:CVCL_6911) cells were grown to ~80% confluency in 10-cm or 6-well plates, for 10mL or 2mL final viral media harvest respectively and transfection reagents were scaled according to seeding area. For 10-cm plate, 3.5 – 4E6 cells were seeded and incubated for 24 hours (37°C, 5% CO2). Transfection reagents were prepared in Opti-MEM™ reduced serum medium (Gibco) and performed using 94.2 μL Lipofectamine 2000 (ThermoFisher), 103.6 μL PLUS™ reagent (ThermoFisher), 8.2 μg psPAX2 (RRID:Addgene_12260), 5.4 μg pMD2.G (RRID:Addgene_12259) and 10.7 μg construct DNA (lentiCRISPRv2, RRID:Addgene_52961). The mixture was incubated at room temperature for 5 minutes and gently added to the HEK293FT cells for 4-hour incubation (37°C, 5% CO2). The medium was then replaced with pre-warmed harvest media (DMEM 30% FBS). 48 hours after the start of the transfection, lentivirus supernatant was collected and syringed through a 0.45 μm filter. Transductions were conducted directly at the time of lentivirus harvest or freshly thawed from frozen aliquots. 0.5–1 mL of virus media and polybrene (1 μg/mL) were added to cells seeded in 6-well plate in 1–1.5 mL of growth media. Cells were spinfected at 2250RPM, 1 hour, room temperature (25°C) and incubated overnight (37°C, 5% CO2). 24 hours post-transduction, cells were selected by puromycin (2 μg/mL) for 48 hours.
Pooled Customized CRISPR Drug Sensitizer Screen and Analysis
A miniaturized CRISPR library representing 378 genes (five sgRNAs per gene with 50 non-targeting controls, 1940 sgRNAs total) was previously designed and validated by Winter and Anderson et al. (42). Lentivirus production of this library was scaled up and conducted as described above. A selection of PIK3CA WT, namely 584-A2 (laryngeal), and PIK3CA mutant cell lines, namely CRC119 (colorectal), AGS (gastric), MKN1 (gastric) and KYSE510 (esophageal squamous) were transduced with library virus as described above.
For each cell line, cells were seeded into 6-well plates at a density of ~0.35 – 0.5E6 cells per well and transduced at a MOI less than 0.2. A total of 10E6 cells were transduced in 6-well plates. 24 hours post-transduction, cells were selected by puromycin (2 μg/mL) for 48 hours. Puromycin-selected cells were collected and counted to confirm at least 1000X library coverage. Transduced cells were propagated in puromycin-containing media for a total of 7 days and subsequently split into vehicle control (DMSO) and BYL719 treatment conditions in duplicates. Specific doses of BYL719 were reported in Figure 3A. The screen was conducted over a total of 3 weeks, for approximately 15 cell doublings. Cells were counted and passaged with replenished drug every 3 days. Each treatment condition and replicate were represented by a minimum of 2E6 cells to maintain at least 1000X library coverage (>1000 cells per unique sgRNA) during each split throughout the screen. A total of 2E6 cells were collected at 48 hours post-puromycin exposure, screen initiation (t0) and at every passage until screen termination (tfinal). DNA was extracted from cell pellets (DNeasy Blood & Tissue Kit, Qiagen) and stored at −80°C until completion of all screens. Samples were further processed for sequencing as previously described (43). Screen libraries were sequenced on an Illumina NextSeq 500 sequencer (75-bp, single-end reads) at the Duke University Genome Sequencing Facility to achieve a depth of 5 million reads total per sample (~200 reads per guide).
Figure 3: Application of CRISPR/Cas9-based screening method to identify modulators of BYL719 response.
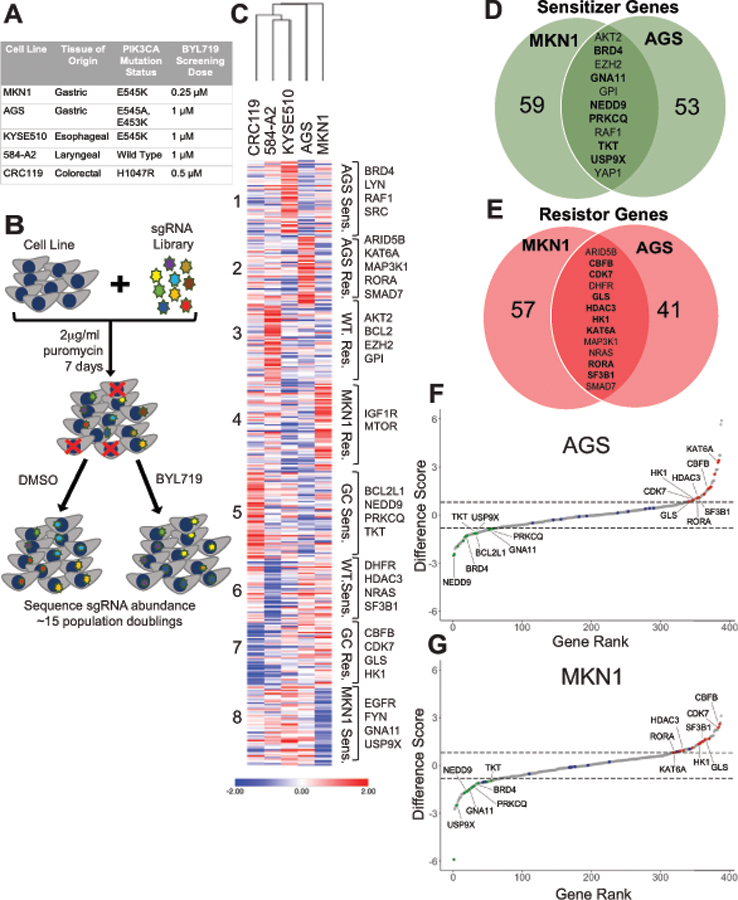
(A) Description of the cell lines used in the screen with tissue of origin, PIK3CA mutation status, and BYL719 screening dose used. (B) Schematic representation of CRISPR/cas9 screening approach. (C) Heat map of difference scores generated using MAGeCK analysis pipeline. Hierarchical clustering of cell lines and K-means clustering of genes was completed using Morpheus. (D & E) Venn diagram showing common sensitizer and resistor genes between AGS and MKN1 cell lines. Genes specific to gastric cancer cell lines in bold. (F & G) Ribbon plots generated using difference scores for AGS and MKN cell lines. Sensitizer genes highlighted in green, resistor genes highlighted in red and controls highlighted in blue. Genes specific to gastric cancer cell lines labeled.
Pooled samples were matched by barcoded reads and guide-level counts were computed using bcSeq (v1.12.0) Bioconductor package (44). Sequencing read counts for DMSO and BYL719-treated samples from each cell line were then processed for gene-level enrichment and depletion analysis relative to t0 samples to generate beta scores using the MAGeCK-MLE software analysis package under the default settings (45). Computed difference scores were subsequently normalized for each cell line by Z-transformation. Hierarchical and K-means clustering was completed using Z-transformed difference scores with Morpheus (https://software.broadinstitute.org/morpheus).
Generation of CRISPR Knockouts
Guide RNA sequences for CBFB, NEDD9, PIM1 and RUNX1 were designed with Synthego and sequences are listed in Table S6. Cells were plated 24 hours prior to transfection and guide RNAs were transfected as ribonucleoprotein complexes along with TrueCut Cas9 Protein V2 (ThermoFisher A36499) using the Lipofectamine CRISPRMAX Cas9 Transfection Reagent (ThermoFisher CMAX00008) following the manufacturer’s instructions. Single cell clones were developed from the bulk transfected populations and knockout was confirmed by western blot.
Immunoblotting
Cells were plated 24 hours prior to treatment. Following treatment with BYL719 or DMSO, cells were washed with ice cold 1X phosphate buffered saline (PBS) and lysed in ice-cold radio-immunoprecipitation assay lysis buffer (RIPA, Cell Signaling Technology 9806) supplemented with phosphatase and protease inhibitor cocktails (Roche 4906845001 and 11697498001). Cell lysates were kept on ice and sonicated with the QSonica three times each at 40 amps. Protein lysate was quantified by Bradford assay and 20 μg of protein was loaded into each lane. All protein lysates were run on NuPage 4 to 12% gradient gels (LifeTechnology) and transferred to polyvinylidene difluoride (PVDF) membrane (Bio-Rad). Membranes were first stained with Revert 700 Total Protein Stain (LI-COR 926–11021) and imaged. Membranes were then blocked in 5% milk in Tris-buffered saline-Tween 20 (TBST) for one hour, washed 3 times for 10 minutes each with TBST and stained with primary antibody overnight at +4°C, followed by 3 additional wash steps. Secondary antibody staining was done at room temperature for 1 hour with horseradish peroxidase (HRP)-conjugated antibodies. Primary antibodies against pAKT Ser473 (D9E; 4060), AKT (C67E7; 4691), pS6 Ser235/236 (91B2; 4857), pS6 Ser240/244 (D68F8; 5364), S6 (5G10; 2217), PIM1 (D8D7Y; 54523), BCL-XL (2762) and RUNX1/AML (4334) were purchased from Cell Signaling Technology. Antibody against CBF-beta (A303–547A) was purchased from Bethyl Laboratories. Antibody against NEDD9 (Cas-L 2G9; sc-33659) was purchased from Santa Cruz Biotechnology. Antibodies against pPRAS40 Thr246 and PRAS40 were a kind gift from Dr. Michael Brown, Duke University, Durham, NC. Secondary antibodies were purchased from Sigma. Antibody dilutions are listed in Table S8. Human Phospho-Kinase Array Kit (ARY003C) was purchased from R&D and used according to manufacturer’s instructions. Quantification was done with Image Studio software. Western blots signals were normalized first to total protein and phospho-proteome array signals were normalized first to the reference spots present on each membrane.
BrdU and Activated Caspase-3 Assays
Cells were plated 24 hours prior to treatment and treated with BYL719 or A-1331852 for either 24 hours or 48 hours. For cell cycle progression assays, cultures were pulsed with BrdU for 2 hours (BD Pharmingen 559619) and fixed and permeabilized. Cells were then treated with DNase to expose BrdU-bound epitopes and stained with a BrdU-fluorescent antibody and a total DNA marker 7-AAD. For activated caspase-3 assays, cells were fixed and permeabilized then stained with an activated caspase-3-fluorescent antibody (BD Pharmingen 550480). All fluorescence data was gathered on a flow cytometer (FACS Canto II) and analyzed with FlowJo.
RNA-Sequencing and Gene Expression Analysis
Total RNA was extracted using the Qiagen RNeasy Mini Prep Kit (74104) per the manufacturer’s instructions. Preparation of RNA library and transcriptome sequencing was conducted by Novogene Co., LTD (Beijing, China). Genes with adjusted p-value <0.05 and |log2(FoldChange)|>0 were considered differentially expressed. For gene expression analysis, RNA was reverse transcribed to gene complementary DNA (cDNA) using the High-Capacity cDNA kit (Invitrogen 4368814). qPCR was completed using Hi-Rox SYBR (Genessee 17–50608) in an Applied Biosystems Quant Studio S6 Pro instrument. QPCR primers are listed in Table S7.
Analysis of Clinical Data
RNA-sequencing data matrix from the TCGA stomach adenocarcinoma (STAD) dataset was downloaded from Broad GDAC Firehose (illuminahiseq_rnaseqv2-RSEM_genes_normalized). RNA-sequencing data was downloaded as RSEM (RNA-seq by Expectation Maximization) and converted to log2(RSEM+1) for analysis. Genomic data including PIK3CA mutation status and GC molecular subtypes were obtained from the cBioportal database. Sample type for each patient sample was determined using the TCGA barcode guideline (https://docs.gdc.cancer.gov/Encyclopedia/pages/TCGA_Barcode/). Tumor samples without available molecular subtype definition were not included in the sub-typing analysis but were included in analyses comparing PIK3CA mutant and wild type tumors. For all analyses, two-sided Wilcoxon statistical tests were used. All data analysis was completed using RStudio (source code available upon request).
Statistical Analysis
Unless otherwise specified, Student’s t tests or, for grouped analyses, two-way ANOVA with Tukey post-hoc correction was performed and P < 0.05 was considered significant (GraphPad Prism). Results are presented as means ± SEM except clinical data which is median ± interquartile range.
Data Availability:
Experimental data and results as well as CRISPR screen data and results are available in the manuscript and supplemental material. RNA-sequencing data was uploaded to the NCBI Gene Expression Omnibus (GEO) database and is available at accession GSE235631.
RESULTS
PIK3CA mutation predicts sensitivity to BYL719
We first assayed a panel of gastric, colorectal, breast, and head and neck cancer cell lines to assess whether PIK3CA mutant cell lines derived from diverse tissue types would be sensitive to single agent BYL719 treatment. Cells were treated with increasing concentrations of BYL719 for three days and live cells were measured to generate a GI50 value (growth inhibition dose of 50%) for each cell line (Fig. 1A). Cell lines with GI50 values below 1 μM were considered sensitive and cell lines with GI50 values ≥ 5 μM were considered resistant. These cutoffs were selected to remain consistent with prior studies using BYL719 (23,38,46). We found that the PIK3CA mutant gastric cancer cell lines AGS and MKN1 were sensitive to BYL719 with GI50 values below 1 μM. Conversely, we found that wild type gastric cancer cell lines were generally more resistant, in particular SNU484 and SNU1750 were resistant to BYL719 with GI50 values of 5 μM and 10 μM, respectively. Overall, we found that PIK3CA mutation status did predict sensitivity independent of tissue type to single agent BYL719 which is consistent with other studies (23,40,46–49). (Fig 1B).
Figure 1: PIK3CA mutation predicts sensitivity to BYL719.
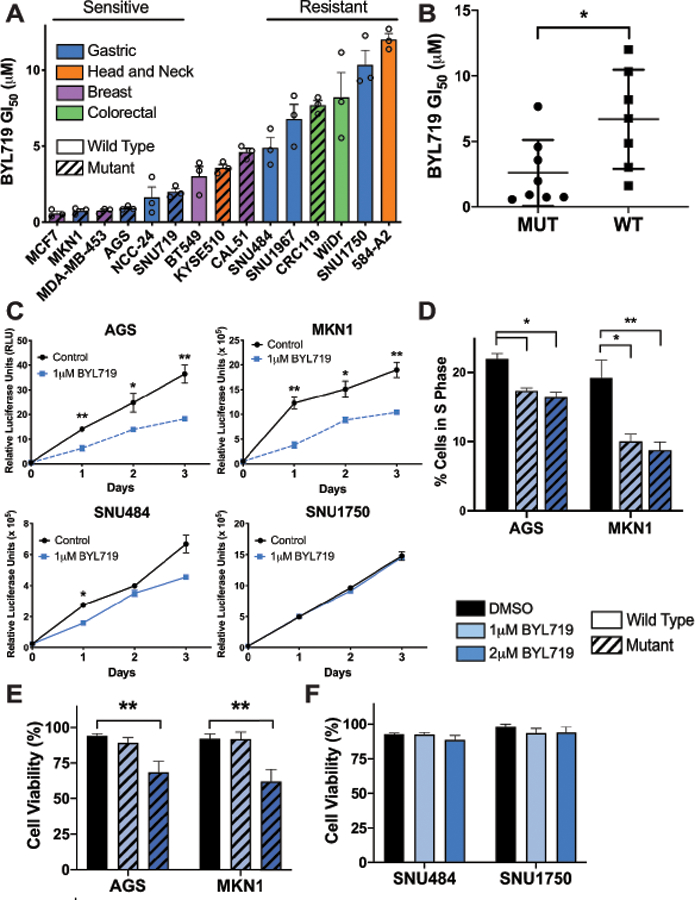
(A) Mean GI50 values generated for a panel of cancer cell lines derived from gastric (blue), head and neck (orange), breast (purple), and colorectal (green) tumors. Cells were treated for 3 days with PI3Kα inhibitor BYL719 and live cells were measured by CellTiter Glo and normalized to DMSO treated. Diagonal lines indicate cell lines with PIK3CA mutation. (B) Mean GI50 values separated by PIK3CA mutation, as calculated using data from panel A. (C) Growth measured by CellTiter Glo each day for 3 days in gastric cancer cell lines. Dashed blue line indicates PIK3CA mutant cell lines, AGS and MKN1, solid blue line indicates wild type cell lines, SNU484 and SNU1750. (D) Percentage of cells in S phase as measured by BrdU assay for cell cycle. Cells were treated with BYL719 for 48 hours and cell cycle was assessed by flow cytometry staining. (E and F) Viability of gastric cancer cell lines after 3 days treatment with DMSO or drug. Viability was determined by Trypan Blue exclusion. Diagonal lines indicate PIK3CA mutant cell lines. For all panels except B, error bars show data ± SEM, error bars in panel B show data ± SD. For experiments in panels A & C, n=3. For experiments in panels D-F, n=6. *p<0.05, **p<0.01.
We sought to further explore the observed differential sensitivity of the gastric cancer cell lines. PIK3CA mutant and wild type gastric cancer cell lines were grown in the presence of 1 μM BYL719 for three days. Growth of PIK3CA mutant cell lines AGS and MKN1 was significantly inhibited by BYL719 compared to untreated cells, whereas the PIK3CA wild type cell lines SNU484 and SNU1750 were largely unaffected (Fig. 1C). We found that the growth inhibitory phenotype seen in the PIK3CA mutant lines was due to cell cycle arrest as BYL719 treatment led to a significant decrease in the percentage of cells in S phase (Fig.1D). We next assessed whether BYL719 could induce cell death in PIK3CA mutant gastric cancer cells in addition to the growth inhibitory effects observed. Gastric cancer cells were treated with either DMSO, 1 μM or 2 μM BYL719 for three days and cell viability was assessed. The dosing of BYL719 was selected based on early phase clinical trial data that found that the maximum tolerated dose of BYL719 was either 400 mg once daily or 150 mg twice daily which led to mean plasma concentrations of approximately 2 μM two hours after drug administration. Therefore, we used 1 μM and 2 μM for experiments in vitro as these doses are equal to and under the maximum tolerated dose in vivo (30). There was significant cell death in PIK3CA mutant cell lines, AGS and MKN1, at 2 μM BYL719, but not at 1 μM BYL719 suggesting that growth inhibitory effects seen at 1 μM BYL719 are primarily due to cell cycle arrest rather than induction of cell death (Fig. 1E). We did not observe significant cell death in the PIK3CA wild type cell lines SNU484 and SNU1750 at either concentration of BYL719 thus confirming their resistance to the drug (Fig. 1F). Taken together, our data support prior preclinical studies showing that genotype-selective dependency extends to gastric cancer and that PIK3CA is a strong predictor of BYL719 response (40).
BYL719 effectively inhibits PI3K/AKT signaling in PIK3CA mutant cells
To confirm the efficacy and specificity of BYL719 at inhibiting the PI3K/AKT/mTOR signaling pathway, we examined phosphorylation of the proximal downstream effector, AKT (pAKT Ser473), as well as S6 (pS6 Ser235/6 and Ser240/4), which is downstream of mTORC1/S6K. First, cells were treated for 1 hour with increasing concentrations of BYL719. In PIK3CA mutant cell lines AGS and MKN1, phosphorylation of AKT and S6 were strongly inhibited at sub-micromolar concentrations of BYL719 (Fig. 2A). In the PIK3CA wild type cell lines SNU484 and SNU1750, AKT and S6 activity was maintained at low doses of BYL719 and inhibited at higher doses than in PIK3CA mutant lines. (Fig. 2B).
Figure 2: BYL719 effectively inhibits PI3K/AKT signaling in PIK3CA mutant cells.
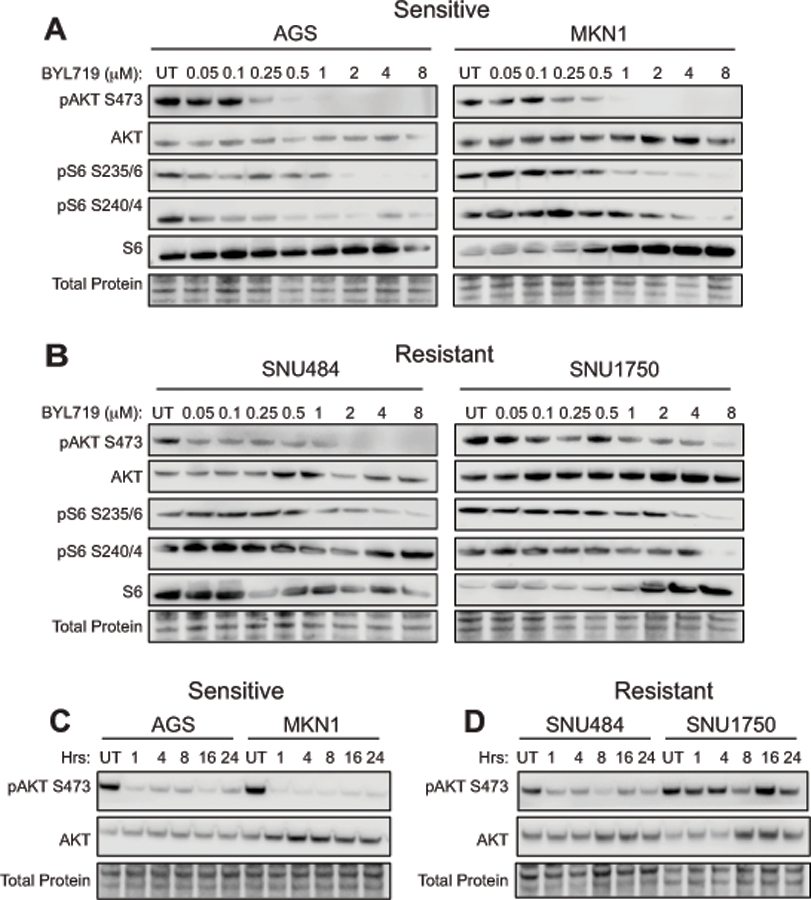
Western blots of PI3K pathway inhibition as measured by phosphorylation of AKT at Ser473 and phosphorylation of the downstream effector S6K at Ser235/236 and Ser240/244. (A) PIK3CA mutant gastric cancer cell lines AGS and MKN1 were treated for 1 hour with increasing doses of BYL719. (B) PIK3CA wild type gastric cancer cell lines SNU484 and SNU1750 were treated for 1 hour with increasing doses of BYL719. (C) AGS and MKN1 cell lines were treated with 1 μM BYL719 and protein lysates were harvested at the indicated time points. (D) SNU484 and SNU1750 cell lines were treated with 1 μM BYL719 and protein lysates were harvested at the indicated time points.
We next assessed pathway inhibition over a longer period of treatment time with BYL719, which is a key indicator of oncogene addiction in cancer cell lines. Cells were treated with 1 μM BYL719 and AKT phosphorylation was assayed after 1, 4, 8, 16 and 24 hours of drug treatment. We found that phosphorylation of AKT was inhibited over a sustained treatment time in the PIK3CA mutant cell lines (Fig. 2C). In the PIK3CA wild type cell lines, phosphorylation of AKT was moderately inhibited at later time points; however, we did not observe complete ablation of signal as in the mutant cell lines (Fig. 2D). Taken together with our earlier results, these data indicate that the PIK3CA mutant gastric cancer cell lines, AGS and MKN1, are dependent on PI3K/AKT/mTOR signaling for survival and signaling through this pathway is effectively inhibited by BYL719.
Application of CRISPR/Cas9-based screening method to identify modulators of BYL719 response
While we found that PIK3CA mutant gastric cancer cell lines were indeed sensitive to BYL719, clinically, resistance to monotherapies is common and thus there has been a push to identify co-dependencies that occur in cancer cells (50). This would allow for tumors to be targeted with multiple drugs at lower doses thereby decreasing toxicity as well as increasing efficacy and preventing resistance. We used a CRISPR/Cas9-based screening approach to query potential modulators of the response to BYL719 treatment. We used a targeted gRNA library made up of genes involved in cancer-associated survival pathways, key druggable targets, frequently mutated and amplified oncogenes, receptor tyrosine kinases (RTK), and metabolism genes (378 genes in library; 5 guides per gene) that had been previously designed and validated by the Wood lab (Table S1) (42). Five cancer cell lines were used in this study including two PIK3CA mutant gastric cancer lines (AGS and MKN1), one PIK3CA mutant esophageal squamous cancer line (KYSE510), one PIK3CA mutant colorectal cancer line (CRC119) and one PIK3CA wild type laryngeal cancer line (584-A2) (Fig. 3A). These were selected as a diverse panel of gastrointestinal-derived cell lines with differential sensitivity to BYL719 as defined by our GI50 data (Fig. 1A). Importantly, all of the tissue types represented cancers in clinical trials with PI3K inhibitors. Briefly, cell lines were transduced with the CRISPR gRNA library, selected for 7 days with puromycin and split into either BYL719 at each line’s IC30, or DMSO. Treated cells were grown for three weeks, or approximately 15 population doublings (Fig. 3B). We used the MAGeCK analysis pipeline to generate a difference score for each gene in the screen. The difference scores were used to identify sensitizer genes whose knockout conferred increased sensitivity to BYL719 and resistor genes whose knockout conferred increased resistance to BYL719 (Fig. S1, Tables S2, S3 & S4).
We used hierarchical and K-means clustering to identify distinct patterns of response based on tissue type as well as PIK3CA mutation status (Fig. 3C, Table S5). As expected, the two gastric cancer cell lines, AGS and MKN1, clustered together as did the esophageal squamous cancer cell line KYSE510 and laryngeal cancer cell line 584-A2. We identified 8 clusters that represent enriched functional phenotypes whose knockout rendered: 1) AGS cells hyper-sensitive to BYL719, 2) AGS cells resistant to BYL719, 3) PIK3CA wild type cells resistant to BYL719, 4) MKN1 cells resistant to BYL719, 5) gastric cancer cells hyper-sensitive to BYL719, 6) PIK3CA wild type cells sensitive to BYL719, 7) gastric cancer cells resistant to BYL719, and 8) MKN1 cells hyper-sensitive to BYL719 (Fig. 3C). There were no genes that were common sensitizers or resistors in all five cell lines, though AKT2 was a common sensitizer, and NRAS and DHFR were common resistors in the four PIK3CA mutant cell lines. Genes that scored as sensitizer genes in the PIK3CA wild type cell line 584-A2 and resistor genes in the four mutant lines, such as DHFR, HDAC3, NRAS and SF3B1, clustered together in cluster 6. Conversely, genes that scored as resistor genes in the PIK3CA wild type cell line 584-A2 while scoring as sensitizer genes in the four mutant lines, such as BCL-2, AKT2, EZH2 and GPI, clustered together in cluster 3 (Fig. 3C, Table S5).
We identified sensitizer genes that were common to the two PIK3CA mutant gastric cancer cell lines that clustered together in cluster 5 such as BCL2L1, NEDD9, PRKCQ, and TKT. We also identified genes that scored as common sensitizers but were particularly enriched in one of the cell lines and were present in clusters 1 and 8, including BRD4, RAF1, GNA11 and USP9X (Fig. 3C, Table S5). Resistor genes common to gastric cancer cell lines clustered together in cluster 7 including CBFB, CDK7, GLS and HK1. Similarly, we found genes that scored as common resistor genes but were enriched in the AGS cell line in cluster 2 including ARID5B, KAT6A, MAP3K1, RORA, and SMAD7 (Fig. 3C, Table S5).
We were primarily focused on identifying genetic vulnerabilities in the PIK3CA mutant gastric cancer cell lines and therefore we focused on the common sensitizer and resistor genes between the cell lines AGS and MKN1 (Fig. 3D & E). We found 11 common sensitizer genes in the gastric cancer cell lines, 6 of which were unique to the gastric cancer cell lines (indicated in bold) and 5 of which were also sensitizer genes in one or more of the non-gastric cancer cell lines (Fig. 3D). Conversely, we found 13 resistor genes common to the gastric cancer cell lines, 8 of which were unique to the gastric cancer cell lines (indicated in bold), and 5 of which were also resistor genes in one or more of the non-gastric cancer cell lines (Fig. 3E). We generated ribbon plots based on the difference scores for AGS and MKN1 to visualize the common sensitizer genes (highlighted in green), common resistor genes (highlighted in red) and control genes (highlighted in blue, Fig. 3F & G, Fig. S2). Ultimately, we selected NEDD9, a gastric cancer specific sensitizer and CBFB, a gastric cancer specific resistor for further validation. We additionally chose to follow up on BCL2L1, which encodes BCL-XL, in the AGS cell line as it scored as a sensitizer gene, and prior research suggests that PI3Kα and BCL-XL inhibition are synergistic (51,52).
Loss of NEDD9 or inhibition of BCL-XL renders PIK3CA mutant gastric cancer cells hyper-sensitive to BYL719 treatment by increasing cell cycle arrest and cell death, respectively
The data from the CRISPR screen identified NEDD9 as a strong sensitizer gene in both PIK3CA mutant gastric cancer cell lines. NEDD9 is a docking and scaffold protein involved in RTK and integrin signaling, as well as regulation of the cell cycle (53,54). In order to assess whether NEDD9 loss could confer increased sensitivity to BYL719, we generated NEDD9 knockout AGS cells (Fig. 4A). NEDD9 knockout cells were treated with BYL719 for three days to determine the GI50 and we found that the knockout cells were significantly more sensitive to BYL719 than the parental cells (Fig. 4B). NEDD9 plays a crucial role in regulating the cell cycle and depletion of NEDD9 has been shown to induce cell cycle arrest (54). Therefore, we hypothesized that the growth suppression phenotype in the NEDD9 knockout cells may be due to cell cycle arrest. Therefore, we assayed cell cycle status after treatment with BYL719 in the NEDD9 knockout cells and parental AGS cells (Fig. 4C). We found a significantly larger reduction in cells in S phase in the NEDD9 knockout cells compared to the parental cell line which correlated with an increase in cells arresting at G2/M phase (Fig. 4D & E).
Figure 4: Loss of NEDD9 or inhibition of BCL-XL renders PIK3CA mutant gastric cancer cells hyper-sensitive to BYL719 treatment by increasing cell cycle arrest and cell death, respectively.

(A) Western blot of NEDD9 expression in parental and NEDD9 knockout AGS cells. (B) GI50 curve of BYL719 in parental and NEDD9 knockout AGS cells measured by CellTiter Glo at 3 days. (C) Representative BrdU plots of AGS parental and NEDD9 knockout cells treated with either DMSO or 2 μM BYL719 for 24 hours. (D) Reduction in S phase in parental and NEDD9 knockout AGS cells after 24 hours of treatment with 2 μM BYL719. (E) Increase in cells in G0/G1 and G2/M phase after 24 hours of treatment with 2 μM BYL719. (F) GI50 curve of BYL719 only and the addition of 500 nM A-1331852 measured by CellTiter Glo at 3 days. (G) Cells were treated with 1 μM BYL719 or 500 nM A-1331852 alone and in combination. Cell viability was measured by Trypan Blue exclusion after 3 days. (H) Cells were treated with 1 μM BYL719 or 500 nM A-1331852 alone and in combination. After 24 hours, cells were harvested and stained for activated caspase-3 for flow cytometry. For all panels, error bars show mean ± SEM. For experiments in panel B, F, & H, n=3. For experiments in panels D & E, n=5. For experiments in panel G, n=6. *p<0.05, **p<0.01, ***p<0.005, ****p<0.001.
Interestingly, in our screen data we also found that both SRC and LYN scored as strong sensitizer genes in the AGS cell line. Src and Lyn kinases are crucial for NEDD9 activity as they extensively phosphorylate NEDD9 to enable downstream effector binding facilitating growth and migration (53–56). Consistent with our genetic findings, we found that the addition of the Src family kinase inhibitor, Dasatinib, significantly increased the sensitivity of AGS cells to BYL719 suggesting a Src/NEDD9/PI3K signaling axis (Fig. S3).
BCL2L1, which encodes the anti-apoptotic protein BCL-XL, scored in the screen as a strong sensitizer gene in the AGS cell line. We sought to orthogonally validate the genetic finding from our screen using A-1331852, a potent and selective inhibitor of BCL-XL (57). We first defined the GI50 value of A-1331852 alone in the AGS cell line as 2 μM and added 500 nM A-1331852 to assay whether the addition of the BCL-XL inhibitor would render AGS cells hyper-sensitive to BYL719 (Fig. S4A). We observed a significant decrease in the BYL719 GI50 with the addition of 500 nM A-1331852 (Fig. 4F). We additionally found that genetic knockout of BCL2L1 conferred hyper-sensitivity to BYL719 and led to two-fold decrease in GI50 value (Fig. S4D & E). Despite BCL2L1 scoring as inert in MKN1, we also found that the addition of A-1331852 conferred significantly increased sensitivity to BYL719 in MKN1 cells and that the combination of these two inhibitors was synergistic in both cell lines (Fig. S4B & F) (58). This result was not wholly unexpected as BCL-XL inhibition has been previously shown to sensitize cells to PI3K pathway inhibitors in a variety of tumor settings (51,52,59–62).
In addition to the growth inhibitory effect of PI3Kα and BCL-XL inhibition, we observed a significant loss in cell viability in cells treated with a combination of A-1331852 and BYL719 (Fig. 4G & Fig. S4C). We found that this effect was specific to BCL-XL inhibition as other BH3 mimetics or drugs that act on MCL-1 inhibited cell growth but were ultimately unable to induce significant cell death (Fig. S5). We additionally measured activated caspase-3 by flow cytometry and found that cells treated with the combination expressed significantly increased levels of activated caspase-3 (Fig. 4H). BCL-XL inhibition alone also significantly increased activated caspase-3 compared to the DMSO control.
Loss of CBFB confers resistance to BYL719
In addition to sensitizer genes, we also identified common resistor genes of PI3Kα inhibition in PIK3CA mutant gastric cancer cells. CBFB scored as a gastric specific resistor gene in the PIK3CA mutant cell lines. To determine whether loss of CBFB could confer resistance to BYL719, we generated CBFB knockout AGS cells (Fig. 5A). CBFB knockout cells were treated for three days with increasing concentrations of BYL719 and we found that the CBFB knockout lines were 5–10-fold more resistant to BYL719 relative to the parental cell line (Fig. 5B). CBFB heterodimerizes with RUNX family proteins RUNX1, RUNX2 and RUNX3 to form the core-binding factor complex which regulates transcription. We therefore additionally generated a RUNX1 knockout and found that RUNX1 knockout cells were approximately 5-fold more resistant to BYL719 than the parental cell line thereby phenocopying the resistance seen with CBFB knockout (Fig. S6A & B).
Figure 5: Loss of CBFB confers resistance to BYL719 in PIK3CA mutant gastric cancer cells.
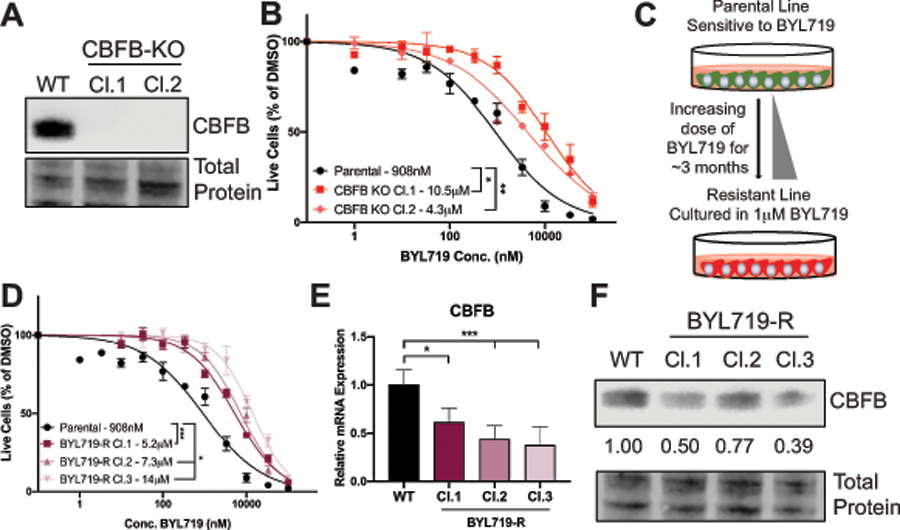
(A) Western blot of CBFB expression in parental and CBFB knockout AGS cells. (B) GI50 curve of BYL719 in WT and CBFB knockout AGS cells measured by CellTiter Glo at 3 days. (C) Schematic representation of generation of AGS cells with resistance to BYL719. (D) GI50 curve of BYL719 in parental and BYL719 resistant clones measured by CellTiter Glo at 3 days. (E) QPCR analysis of CBFB mRNA expression in parental and BYL719 resistant clones. (F) Western blot of CBFB expression in parental and BYL719 resistant clones. Quantification of BYL719 resistant clones relative to parental expression. For all panels, error bars show mean ± SEM. For experiments in panel B, D & E, n=3. *p<0.05, **p<0.01, ***p<0.005.
To further probe mechanisms of drug resistance, we generated single cell clones with resistance to BYL719. We cultured the sensitive parental AGS cell line in increasing doses of BYL719 for approximately 3 months until the cells were being cultured in 1 μM BYL719 without adverse effects on growth and developed single cell clonal populations (Fig. 5C). We confirmed that the clones were significantly more resistant to treatment with BYL719 than the parental AGS cell line (Fig. 5D). Further characterization of the BYL719 resistant clones revealed that CBFB was down-regulated at both the mRNA and protein level (Fig. 5E & F). Additionally, the BYL719 resistant clones also displayed down-regulation of RUNX1, the binding co-factor of CBFB, at both the RNA and the protein level (Fig. S6C-E). These data suggest that loss of CBFB function is a common mechanism of resistance to PI3Kα inhibition.
CBFB loss drives resistance to BYL719 through PIM1 kinase
To define the mechanism by which CBFB loss leads to resistance to BYL719, we queried the phosphorylation status of 37 unique substrates in the parental and CBFB KO cells. These included substrates involved in core intracellular signaling pathways such as PI3K/AKT, JAK/STAT, MAPK/ERK, and p53. We used this approach primarily to determine whether CBFB KO cells displayed up-regulation of compensatory signaling pathways that could confer PI3Kα inhibitor resistance. We observed significantly increased phosphorylation of five substrates in CBFB KO Cl.1, and seven substrates in CBFB KO Cl.2 relative to the parental cell line. Three of these substrates were common to both CBFB KO lines, including pPRAS40, a known target of both AKT and PIM kinase (Fig. 6A & B, Table S9). The PIM kinases are serine/threonine protein kinases that phosphorylate a number of targets involved in regulation of the cell cycle and apoptosis and provide pro-growth and survival signals in cells. The PIM kinase family is composed of three isoforms, PIM1, PIM2 and PIM3. PIM1 specifically is known to play an important role in cancer cell growth (63,64). Because CBFB is a transcription regulator, we additionally performed RNA-sequencing in the parental AGS cell line as well as the CBFB knockouts and resistant cell clones. We found that PIM1, but not PIM2 or PIM3, mRNA was expressed at a significantly higher level in the CBFB knockout cells and the BYL719 resistant clones compared to the parental cells (Fig. 6C). Furthermore, acute treatment with 1 μM BYL719 led to a more pronounced increase in PIM1 mRNA comparing CBFB knockout cells and the resistant clones to the parental (Fig. 6D). We also found increased basal expression of PIM1 protein in the CBFB KO and resistant clones compared to the parental (Fig. 6E).
Figure 6: CBFB loss drives resistance to BYL719 through PIM1.
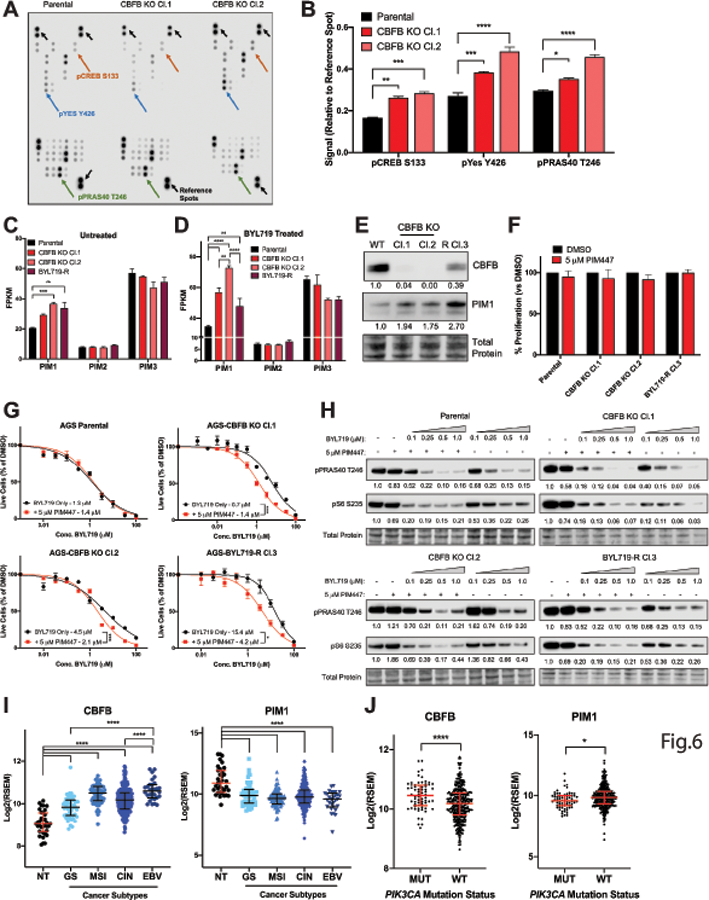
(A) Phospho-Proteome Array image with significantly up-regulated substrates and reference spots labeled. (B) Quantification of significantly up-regulated substrates from Phospho-Proteome Array. Signal was first normalized to the reference spots on each membrane and then to the parental cell lines. Each substrate was measure in technical duplicate. (C) RNA-sequencing data showing FPKM values of each of the PIM kinases in parental, CBFB knockout, and BYL719 resistant clones untreated and (D) treated with 1 μM BYL719 for 24 hours. CBFB knockout clones in duplicate, parental and BYL719 resistant clones in triplicate. (E) Western blot showing protein expression of CBFB and PIM1 in parental, CBFB KO and BYL719-R Cl.3. (F) Cells were treated with 5 μM PIM447 for 72 hours and total live cell number was measured by CellTiter Glo. (G) GI50 curves of BYL719 alone or in combination with 5 μM PIM447. (H) Western blot of pPRAS40 Thr246 and pS6 Ser235/6 in lysates from cells treated with increasing concentrations of BYL719 alone or in combination with 5 μM PIM447 for 1 hour. (I) RNA expression of CBFB and PIM1 in gastric tumors and normal tissue in TCGA dataset stratified by tumor subtype. NT=normal tissue, GS=genomically stable, MSI=microsatellite instable, CIN=chromosomal instable, EBV=Epstein-Barr Virus positive. (J) RNA expression of CBFB and PIM1 in gastric tumors in TCGA dataset stratified by PIK3CA mutation status. For experiments in panels, B-F, error bars show mean ± SEM. For clinical data in panels H & I, error bars show median ± interquartile range. *p<0.05, **p<0.01, ***p<0.005, ****p<0.001.
Visual Overview. (A) In PIK3CA mutant cells with mid-high CBFB expression, BYL719 effectively inhibits signaling via AKT-PRAS40-mTOR. (B) In PIK3CA mutant cells with low CBFB or CBFB knockout, PIM1 is over-expressed (C) and is further up-regulated in the presence of BYL719 leading to maintenance of signaling through PRAS40 and cell proliferation. (D) The addition of a pan-PIM inhibitor re-sensitizes the cells to BYL719 by inhibiting signaling through PRAS40 resulting in inhibition of cell growth.
Given the significant up-regulation of PIM1 in the CBFB KO and resistant clones, we next asked whether PIM kinase inhibition could reverse the BYL719 resistance phenotype. We first treated cells with 5 μM PIM447 alone and confirmed that inhibition of PIM kinases had no effect on cell growth (Fig. 6F). We then generated GI50 curves in the CBFB KO and resistant clones. We did not observe a difference in sensitivity in the parental cell line but found a significant decrease in the GI50 of BYL719 in the CBFB KO and resistant clones (Fig. 6G & Fig. S7A). Specifically, the addition of 5 μM PIM447 resulted in a 4.5-fold decrease and a 2.1-fold decrease in the GI50 value of CBFB KO Cl.1 and CBFB KO Cl.2, respectively, and a 3.7-fold decrease in BYL719-R Cl.3. We additionally found that the combination of 5 μM PIM447 and 1 μM BYL719 significantly inhibited cell growth of the CBFB KO clones over a longer period of time compared to 1 μM BYL719 alone (Fig. S7B). To confirm our hypothesis that PIM1 could phosphorylate PRAS40 in the presence of PI3Kα inhibition, we assessed pPRAS40 Thr246 expression by western blot. We found that pPRAS40 Thr246 was inhibited in a dose dependent manner when cells were treated with BYL719 alone, and that the inhibitory effect was enhanced with the addition of 5 μM PIM447 in the CBFB knockout clones, but not the parental cell line (Fig. 6H). Lastly, to complement our pharmacological approaches, we also generated PIM1 knockout clones and we found that genetic knockout of PIM1 significantly re-sensitized CBFB KO Cl.1 and BYL719-R Cl.3 to treatment with BYL719 (Fig. S7C & D).
Since we determined that low expression of CBFB, and high expression of PIM1 could confer resistance to BYL719, we were curious as to the basal expression of these two genes in gastric tumors. Using the TCGA dataset, we compared RNA expression of CBFB and PIM1 in the four unique gastric cancer subtypes to normal tissue. CBFB expression was significantly higher in all four subtypes compared to normal tissue and was the highest in the EBV-associated subtype, while PIM1 displayed the inverse phenotype (Fig. 6I). We also compared expression of CBFB and PIM1 in gastric cancers stratified by PIK3CA mutation status and found that CBFB expression was significantly higher in the mutant tumors while PIM1 again showed the inverse phenotype (Fig. 6J).
DISCUSSION
Small molecules targeting the PI3K pathway such as pan-PI3K and dual PI3K/mTOR inhibitors have shown promise in pre-clinical studies however this has not translated to clinical success. This is primarily due to broad on-target/off-tissue effects and high toxicity (6). Isoform-specific inhibitors of PI3K have garnered significant interest however, they have faced similar issues with toxicity and tolerability (27). Additionally, resistance, both intrinsic and acquired, still poses a major problem in the success of single agent inhibitors. This has prompted many pre-clinical studies that have described mechanisms of PI3K monotherapy resistance in a variety of cancer cell lines including breast, ovarian, pancreatic, uveal melanoma and head and neck cancer (31–39). Gastric cancer, however has been relatively understudied despite there being several clinical trials with PI3K inhibitors ongoing. In this study, we aimed to characterize mechanisms of sensitivity and resistance to the PI3Kα inhibitor BYL719, specifically in PIK3CA mutant gastric cancers. Our data supports prior studies that have shown that PIK3CA mutant cancer cells are sensitive to single agent BYL719 and that PIK3CA mutation is the strongest predictor of response to the drug (23,30,40). Specifically, we found that BYL719 induced significant growth inhibition and cell death in PIK3CA mutant gastric cancer cell lines, whereas PIK3CA wild type gastric cancer cell lines were largely unaffected.
We sought to identify modulators of the response to BYL719 in the PIK3CA mutant gastric cancer cell lines. To accomplish this, we used a CRISPR/Cas9-based screening approach to query both wild type and PIK3CA mutant cell lines from diverse primary tissues. We identified 8 clusters of genes whose knockout conferred enriched phenotypic responses to treatment with BYL719. We found that several of the genes that scored as gastric cancer sensitizer genes, including BRD4, RAF1, AKT2 and EZH2 have been studied by previous groups in the context of PI3K inhibition. Inhibitors of the BET family of proteins, which includes BRD4, have been combined with PI3K inhibitors to overcome resistance in renal cell cancer and breast cancer cells (65,66). Similarly, many studies have shown successful combined inhibition of the PI3K/AKT/mTOR and Raf/MEK/ERK signaling pathways in a multitude of tumor types and it has been well described that Raf/MEK/ERK pathway activation is a common mechanism of resistance to PI3K pathway inhibitors (67–70). AZD5363, an inhibitor that targets all isoforms of AKT (AKT1, AKT2 and AKT3), was found to be significantly more effective in treating PIK3CA mutant tumors than wild type tumors in gastric cancer xenograft models (71). This drug also recently completed a successful phase I clinical trial in breast and gynecologic cancers harboring PIK3CA mutation (72). Lastly, a recent study found that EZH2 inhibition conferred enhanced sensitivity to PI3K inhibitors in PIK3CA mutant lung cancer xenograft models and a genome-wide gain-of-function screen completed in esophageal squamous cell carcinoma found that over-expression of EZH2 conferred resistance to PI3Kα inhibitors (73,74).
We identified a gastric cancer specific sensitizer gene, NEDD9, and found that cells with genetic knockout of NEDD9 were hyper-sensitive to BYL719 relative to the parental cell line. NEDD9 is a scaffold protein involved in RTK signaling and cell cycle regulation, and over-expression of NEDD9 has been shown to be pro-metastatic in some solid tumors (53,54). Our data show that loss of NEDD9 resulted in increased cell cycle arrest following treatment with BYL719 thus confirming the importance of NEDD9 for cell cycle progression. Loss of NEDD9 has not yet been described in the context of PI3K inhibition, however, NEDD9 depletion has been shown to confer increased sensitivity to the Src family kinase inhibitor Dasatinib and Aurora A kinase inhibitors (75,76). Given our findings that Dasatinib also re-sensitized AGS cells to BYL719, these data suggest a potential NEDD9/Src/PI3K signaling axis in PIK3CA mutant gastric cancer cells that is important in the context of PI3Kα inhibition. We additionally identified BCL-XL as a sensitizer in the AGS cell line. BCL-2 family members including BCL-2, BCL-w, BCL-XL and MCL1 are key anti-apoptotic proteins that have been shown to confer resistance to kinase inhibitors such as PI3K and MEK/ERK inhibitors in cancer cells. Thus, the combination of BCL-2 family member inhibitors with kinase inhibitors has been studied in several tumor types and shown to be synergistic in promoting apoptosis in cancer cell lines (51,52,59–62). We used pharmacological approaches to validate this finding and found that the inhibition of BCL-XL together with PI3Kα induced cell death in PIK3CA mutant gastric cancer cells.
Given the pervasiveness of resistance to monotherapy, we additionally looked for resistor genes in the PIK3CA mutant gastric cancer cells and identified CBFB. CBFB encodes the beta subunit of the core-binding factor transcriptional complex. CBFB does not directly bind DNA but heterodimerizes with the alpha core-binding factors, the RUNX family proteins RUNX1, RUNX2 and RUNX3, to enhance chromatin binding and promote transcription of RUNX target genes (77,78). Additionally, CBFB has been shown to bind hnRNPK and eIF4B on mRNAs to facilitate translation initiation (79). We found that genetic knockout of CBFB conferred resistance to BYL719 in PIK3CA mutant gastric cancer cells. Through orthogonal approaches, we found that in BYL719 resistant clones, CBFB, and its binding co-factor RUNX1, were down-regulated at both the protein and RNA levels suggesting that loss of CBFB function is a key resistance mechanism. Furthermore, we found that increased expression of the serine/threonine protein kinase PIM1 was responsible for conferring the resistant phenotype. PIM1 expression was significantly higher basally in the CBFB KO and BYL719 resistant clones, and acute treatment with 1 μM BYL719 further amplified this phenotype. PIM1 is one of three PIM kinase isoforms, including PIM2 and PIM3, which share over 60% sequence homology at the amino acid level and have comparable kinase function (80). The PIM kinase isoforms show some tissue specificity, namely PIM1 is often highly expressed in hematopoietic cells, and solid tumors including gastric, head and neck, and prostate tumors (80). Importantly, PIM kinase phosphorylates downstream targets involved in transcriptional regulation, cell cycle, and apoptosis, and shares several phosphorylation targets with AKT including PRAS40, BAD, p21 and p27 (81). There is growing evidence that PIM and AKT cooperate to activate intracellular signaling pathways and promote oncogenesis (80). We found that the CBFB KO cells were re-sensitized to BYL719 with the addition of the pan-PIM inhibitor, PIM447 or through genetic knockout of PIM1. Our data support PIM1 up-regulation as a compensatory mechanism for cell growth in the context of PI3Kα inhibition.
Interestingly, we also found that CBFB RNA was significantly over-expressed in gastric cancers compared to normal tissue and was the most highly expressed in the EBV-associated subtype, whereas PIM1 showed the inverse phenotype. Additionally, CBFB expression was significantly higher in PIK3CA mutant gastric cancers compared to wild type, with PIM1 again displaying the inverse phenotype. These data support the use of PI3Kα-specific inhibitors and PI3Kα mutant selective inhibitors in PIK3CA gastric cancer.
While our dataset is limited to gastric, colorectal, and head and neck cancers, our findings could have important implications for tumors from diverse tissue types. PI3K inhibitors are in clinical trials for many solid tumors and there are PI3Kα mutant selective inhibitors on the horizon (6,29,36,48,82). In breast cancer, 30–40% of tumors display activating mutations in PIK3CA however few inhibitors have been clinically successful (5,83). CBFB and PIK3CA mutations often occur together where CBFB mutations result in loss of protein function. Genetic knockdown of CBFB in breast cancer cell lines was shown to accelerate tumor initiation and confer resistance to a pan-PI3K inhibitor in mouse models (84). In acute myeloid leukemia (AML), CBFB forms a fusion gene with MYH11 (CBFB-MYH11) that is present in 12% of pediatric and 7% of adult AML patients. This fusion gene has been shown to act as a dominant repressor of RUNX1 and sequesters RUNX1 in the cytoplasm thereby preventing transcriptional activity of CBFB/RUNX1 heterodimers (85–87). Several PI3K pathway targeting inhibitors have been tested in clinical trials for AML and our data suggests that CBFB-MYH11 fusion gene positive AML patients may display intrinsic resistance to these inhibitors. Lastly, loss of CBFB in KRAS mutant colorectal cancer cells was shown to confer resistance to MEK/ERK inhibitors (88). The data generated in our study provide clear mechanistic insights into vulnerabilities to both increase drug sensitivity and combat drug resistance in PIK3CA mutant tumors that may be more broadly applicable to a variety of cancers.
Supplementary Material
Implications:
Loss of either NEDD9 or BCL-XL confers hyper-sensitivity to PI3K-alpha inhibition while loss of CBFB confers resistance through a CBFB/PIM1 signaling axis.
Acknowledgments:
We would like to thank members of the Luftig lab and the Wood lab for providing thoughtful feedback on the data presented in this manuscript. We would like to thank Dr. Michael Brown at Duke University for providing antibodies and immunoblotting expertise. We would like to thank Camille Krejdovsky for her assistance with wet lab experiments.
Funding:
National Institutes of Health grant R01 CA140337 (MAL)
National Institutes of Health grants R01CA266389 and R01CA207083 (both to KCW)
Duke/Duke-NUS Pilot Grant RL2016-080 and 2017/0038 (MAL, PT)
Duke Cancer Institute/Nicholas School for the Environment Pilot Grant T32-CA009111 (MAL, PT)
Footnotes
Competing interests: M.A.L. consults for Moderna, Evrys Bio, GLG, and Guidepoint Global. K.C.W. is a founder, consultant, and equity holder at Tavros Therapeutics and Celldom, an equity holder and scientific advisor at Decrypt Biomedicine and Simple Therapeutics, and a consultant for Guidepoint Global, Bantam Pharmaceuticals, and Apple Tree Partners. However, no conflicts are declared for this work.
REFERENCES:
- 1.Noorolyai S, Shajari N, Baghbani E, Sadreddini S, Baradaran B. The relation between PI3K/AKT signalling pathway and cancer. Gene 2019;698:120–8 [DOI] [PubMed] [Google Scholar]
- 2.Bilanges B, Posor Y, Vanhaesebroeck B. PI3K isoforms in cell signalling and vesicle trafficking. Nat Rev Mol Cell Biol 2019;20:515–34 [DOI] [PubMed] [Google Scholar]
- 3.Ligresti G, Militello L, Steelman LS, Cavallaro A, Basile F, Nicoletti F, et al. PIK3CA mutations in human solid tumors: role in sensitivity to various therapeutic approaches. Cell Cycle 2009;8:1352–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature 2013;502:333–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004;304:554. [DOI] [PubMed] [Google Scholar]
- 6.Yang J, Nie J, Ma X, Wei Y, Peng Y, Wei X. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer 2019;18:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yap TA, Bjerke L, Clarke PA, Workman P. Drugging PI3K in cancer: refining targets and therapeutic strategies. Curr Opin Pharmacol 2015;23:98–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wright SCE, Vasilevski N, Serra V, Rodon J, Eichhorn PJA. Mechanisms of Resistance to PI3K Inhibitors in Cancer: Adaptive Responses, Drug Tolerance and Cellular Plasticity. Cancers (Basel) 2021;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021;71:209–49 [DOI] [PubMed] [Google Scholar]
- 10.Harada K, Baba Y, Shigaki H, Ishimoto T, Miyake K, Kosumi K, et al. Prognostic and clinical impact of PIK3CA mutation in gastric cancer: pyrosequencing technology and literature review. BMC Cancer 2016;16:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim JW, Lee HS, Nam KH, Ahn S, Kim JW, Ahn SH, et al. PIK3CA mutations are associated with increased tumor aggressiveness and Akt activation in gastric cancer. Oncotarget 2017;8:90948–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014;513:202–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen D, Lin X, Zhang C, Liu Z, Chen Z, Li Z, et al. Dual PI3K/mTOR inhibitor BEZ235 as a promising therapeutic strategy against paclitaxel-resistant gastric cancer via targeting PI3K/Akt/mTOR pathway. Cell Death Dis 2018;9:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li L, Zhang S, Xie D, Chen H, Zheng X, Pan D. Dual inhibitor of PI3K and mTOR (NVP-BEZ235) augments the efficacy of fluorouracil on gastric cancer chemotherapy. Onco Targets Ther 2018;11:6111–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mueller A, Bachmann E, Linnig M, Khillimberger K, Schimanski CC, Galle PR, et al. Selective PI3K inhibition by BKM120 and BEZ235 alone or in combination with chemotherapy in wild-type and mutated human gastrointestinal cancer cell lines. Cancer Chemother Pharmacol 2012;69:1601–15 [DOI] [PubMed] [Google Scholar]
- 16.Zhang C, Awasthi N, Schwarz MA, Schwarz RE. The dual PI3K/mTOR inhibitor NVP-BEZ235 enhances nab-paclitaxel antitumor response in experimental gastric cancer. Int J Oncol 2013;43:1627–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu Y, Tian T, Zou J, Wang Q, Li Z, Li Y, et al. Dual PI3K/mTOR inhibitor BEZ235 exerts extensive antitumor activity in HER2-positive gastric cancer. BMC Cancer 2015;15:894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim MY, Kruger AJ, Jeong JY, Kim J, Shin PK, Kim SY, et al. Combination Therapy with a PI3K/mTOR Dual Inhibitor and Chloroquine Enhances Synergistic Apoptotic Cell Death in Epstein-Barr Virus-Infected Gastric Cancer Cells. Mol Cells 2019;42:448–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhattacharya B, Akram M, Balasubramanian I, Tam KK, Koh KX, Yee MQ, et al. Pharmacologic synergy between dual phosphoinositide-3-kinase and mammalian target of rapamycin inhibition and 5-fluorouracil in PIK3CA mutant gastric cancer cells. Cancer Biol Ther 2012;13:34–42 [DOI] [PubMed] [Google Scholar]
- 20.Lei Z, Tan IB, Das K, Deng N, Zouridis H, Pattison S, et al. Identification of molecular subtypes of gastric cancer with different responses to PI3-kinase inhibitors and 5-fluorouracil. Gastroenterology 2013;145:554–65 [DOI] [PubMed] [Google Scholar]
- 21.Shin JY, Kim JO, Lee SK, Chae HS, Kang JH. LY294002 may overcome 5-FU resistance via down-regulation of activated p-AKT in Epstein-Barr virus-positive gastric cancer cells. BMC Cancer 2010;10:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rasti AR, Guimaraes-Young A, Datko F, Borges VF, Aisner DL, Shagisultanova E. PIK3CA Mutations Drive Therapeutic Resistance in Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer. JCO Precis Oncol 2022;6:e2100370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fritsch C, Huang A, Chatenay-Rivauday C, Schnell C, Reddy A, Liu M, et al. Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther 2014;13:1117–29 [DOI] [PubMed] [Google Scholar]
- 24.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol 2010;11:329–41 [DOI] [PubMed] [Google Scholar]
- 25.Jessen KA, Kessler L, Kucharski J, Guo X, Staunton J, Elia M, et al. INK1117: A potent and orally efficacious PI3K alpha-selective inhibitor for the treatment of cancer. Cancer Research 2011;71 [Google Scholar]
- 26.Barlaam B, Cosulich S, Fitzek M, Germain H, Green S, Hanson LL, et al. Discovery of a novel aminopyrazine series as selective PI3Kalpha inhibitors. Bioorg Med Chem Lett 2017;27:3030–5 [DOI] [PubMed] [Google Scholar]
- 27.Zhang M, Jang H, Nussinov R. PI3K inhibitors: review and new strategies. Chem Sci 2020;11:5855–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanan EJ, Braun MG, Heald RA, MacLeod C, Chan C, Clausen S, et al. Discovery of GDC-0077 (Inavolisib), a Highly Selective Inhibitor and Degrader of Mutant PI3Kalpha. J Med Chem 2022 [DOI] [PubMed] [Google Scholar]
- 29.Ando Y, Iwasa S, Takahashi S, Saka H, Kakizume T, Natsume K, et al. Phase I study of alpelisib (BYL719), an alpha-specific PI3K inhibitor, in Japanese patients with advanced solid tumors. Cancer Sci 2019;110:1021–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Juric D, Rodon J, Tabernero J, Janku F, Burris HA, Schellens JHM, et al. Phosphatidylinositol 3-Kinase alpha-Selective Inhibition With Alpelisib (BYL719) in PIK3CA-Altered Solid Tumors: Results From the First-in-Human Study. J Clin Oncol 2018;36:1291–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leroy C, Ramos P, Cornille K, Bonenfant D, Fritsch C, Voshol H, et al. Activation of IGF1R/p110beta/AKT/mTOR confers resistance to alpha-specific PI3K inhibition. Breast Cancer Res 2016;18:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zorea J, Prasad M, Cohen L, Li N, Schefzik R, Ghosh S, et al. IGF1R upregulation confers resistance to isoform-specific inhibitors of PI3K in PIK3CA-driven ovarian cancer. Cell Death Dis 2018;9:944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Milton CK, Self AJ, Clarke PA, Banerji U, Piccioni F, Root DE, et al. A Genome-scale CRISPR Screen Identifies the ERBB and mTOR Signaling Networks as Key Determinants of Response to PI3K Inhibition in Pancreatic Cancer. Mol Cancer Ther 2020;19:1423–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Musi E, Ambrosini G, de Stanchina E, Schwartz GK. The phosphoinositide 3-kinase alpha selective inhibitor BYL719 enhances the effect of the protein kinase C inhibitor AEB071 in GNAQ/GNA11-mutant uveal melanoma cells. Mol Cancer Ther 2014;13:1044–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell 2014;26:136–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Juric D, Janku F, Rodon J, Burris HA, Mayer IA, Schuler M, et al. Alpelisib Plus Fulvestrant in PIK3CA-Altered and PIK3CA-Wild-Type Estrogen Receptor-Positive Advanced Breast Cancer: A Phase 1b Clinical Trial. JAMA Oncol 2019;5:e184475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le X, Antony R, Razavi P, Treacy DJ, Luo F, Ghandi M, et al. Systematic Functional Characterization of Resistance to PI3K Inhibition in Breast Cancer. Cancer Discov 2016;6:1134–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meister KS, Godse NR, Khan NI, Hedberg ML, Kemp C, Kulkarni S, et al. HER3 targeting potentiates growth suppressive effects of the PI3K inhibitor BYL719 in pre-clinical models of head and neck squamous cell carcinoma. Sci Rep 2019;9:9130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruicci KM, Plantinga P, Pinto N, Khan MI, Stecho W, Dhaliwal SS, et al. Disruption of the RICTOR/mTORC2 complex enhances the response of head and neck squamous cell carcinoma cells to PI3K inhibition. Mol Oncol 2019;13:2160–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim KJ, Kim JW, Sung JH, Suh KJ, Lee JY, Kim SH, et al. PI3K-targeting strategy using alpelisib to enhance the antitumor effect of paclitaxel in human gastric cancer. Sci Rep 2020;10:12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc 2017;12:828–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson GR, Winter PS, Lin KH, Nussbaum DP, Cakir M, Stein EM, et al. A Landscape of Therapeutic Cooperativity in KRAS Mutant Cancers Reveals Principles for Controlling Tumor Evolution. Cell Rep 2017;20:999–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014;343:84–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin J, Gresham J, Wang T, Kim SY, Alvarez J, Damrauer JS, et al. bcSeq: an R package for fast sequence mapping in high-throughput shRNA and CRISPR screens. Bioinformatics 2018;34:3581–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li W, Koster J, Xu H, Chen CH, Xiao T, Liu JS, et al. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol 2015;16:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong CH, Ma BB, Cheong HT, Hui CW, Hui EP, Chan AT. Preclinical evaluation of PI3K inhibitor BYL719 as a single agent and its synergism in combination with cisplatin or MEK inhibitor in nasopharyngeal carcinoma (NPC). Am J Cancer Res 2015;5:1496–506 [PMC free article] [PubMed] [Google Scholar]
- 47.Elkabets M, Vora S, Juric D, Morse N, Mino-Kenudson M, Muranen T, et al. mTORC1 inhibition is required for sensitivity to PI3K p110alpha inhibitors in PIK3CA-mutant breast cancer. Sci Transl Med 2013;5:196ra99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mayer IA, Abramson VG, Formisano L, Balko JM, Estrada MV, Sanders ME, et al. A Phase Ib Study of Alpelisib (BYL719), a PI3Kalpha-Specific Inhibitor, with Letrozole in ER+/HER2- Metastatic Breast Cancer. Clin Cancer Res 2017;23:26–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang M, Li J, Huang J, Luo M. The Predictive Role of PIK3CA Mutation Status on PI3K Inhibitors in HR+ Breast Cancer Therapy: A Systematic Review and Meta-Analysis. Biomed Res Int 2020;2020:1598037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Topatana W, Juengpanich S, Li S, Cao J, Hu J, Lee J, et al. Advances in synthetic lethality for cancer therapy: cellular mechanism and clinical translation. J Hematol Oncol 2020;13:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qian J, Zou Y, Rahman JS, Lu B, Massion PP. Synergy between phosphatidylinositol 3-kinase/Akt pathway and Bcl-xL in the control of apoptosis in adenocarcinoma cells of the lung. Mol Cancer Ther 2009;8:101–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rahmani M, Aust MM, Attkisson E, Williams DC, Jr., Ferreira-Gonzalez A, Grant S. Dual inhibition of Bcl-2 and Bcl-xL strikingly enhances PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and Bim-dependent mechanism. Cancer Res 2013;73:1340–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shagisultanova E, Gaponova AV, Gabbasov R, Nicolas E, Golemis EA. Preclinical and clinical studies of the NEDD9 scaffold protein in cancer and other diseases. Gene 2015;567:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singh M, Cowell L, Seo S, O’Neill G, Golemis E. Molecular basis for HEF1/NEDD9/Cas-L action as a multifunctional co-ordinator of invasion, apoptosis and cell cycle. Cell Biochem Biophys 2007;48:54–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bradbury P, Bach CT, Paul A, O’Neill GM. Src kinase determines the dynamic exchange of the docking protein NEDD9 (neural precursor cell expressed developmentally down-regulated gene 9) at focal adhesions. J Biol Chem 2014;289:24792–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O’Neill GM, Seo S, Serebriiskii IG, Lessin SR, Golemis EA. A new central scaffold for metastasis: parsing HEF1/Cas-L/NEDD9. Cancer Res 2007;67:8975–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang L, Doherty GA, Judd AS, Tao ZF, Hansen TM, Frey RR, et al. Discovery of A-1331852, a First-in-Class, Potent, and Orally-Bioavailable BCL-X(L) Inhibitor. ACS Med Chem Lett 2020;11:1829–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010;70:440–6 [DOI] [PubMed] [Google Scholar]
- 59.Anderson GR, Wardell SE, Cakir M, Crawford L, Leeds JC, Nussbaum DP, et al. PIK3CA mutations enable targeting of a breast tumor dependency through mTOR-mediated MCL-1 translation. Sci Transl Med 2016;8:369ra175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bojarczuk K, Wienand K, Ryan JA, Chen L, Villalobos-Ortiz M, Mandato E, et al. Targeted inhibition of PI3Kalpha/delta is synergistic with BCL-2 blockade in genetically defined subtypes of DLBCL. Blood 2019;133:70–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choudhary GS, Al-Harbi S, Mazumder S, Hill BT, Smith MR, Bodo J, et al. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis 2015;6:e1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jokinen E, Koivunen JP. Bcl-xl and Mcl-1 are the major determinants of the apoptotic response to dual PI3K and MEK blockage. Int J Oncol 2015;47:1103–10 [DOI] [PubMed] [Google Scholar]
- 63.Arrouchi H, Lakhlili W, Ibrahimi A. A review on PIM kinases in tumors. Bioinformation 2019;15:40–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Braso-Maristany F, Filosto S, Catchpole S, Marlow R, Quist J, Francesch-Domenech E, et al. PIM1 kinase regulates cell death, tumor growth and chemotherapy response in triple-negative breast cancer. Nat Med 2016;22:1303–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stratikopoulos EE, Dendy M, Szabolcs M, Khaykin AJ, Lefebvre C, Zhou MM, et al. Kinase and BET Inhibitors Together Clamp Inhibition of PI3K Signaling and Overcome Resistance to Therapy. Cancer Cell 2015;27:837–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu M, Xu L, Wang Y, Dai G, Xue B, Liu YY, et al. BRD4 inhibition sensitizes renal cell carcinoma cells to the PI3K/mTOR dual inhibitor VS-5584. Aging (Albany NY) 2020;12:19147–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Serra V, Eichhorn PJ, Garcia-Garcia C, Ibrahim YH, Prudkin L, Sanchez G, et al. RSK3/4 mediate resistance to PI3K pathway inhibitors in breast cancer. J Clin Invest 2013;123:2551–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dunn E, Chitcholtan K, Sykes P, Garrill A. The Anti-Proliferative Effect of PI3K/mTOR and ERK Inhibition in Monolayer and Three-Dimensional Ovarian Cancer Cell Models. Cancers (Basel) 2022;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ewald F, Norz D, Grottke A, Hofmann BT, Nashan B, Jucker M. Dual Inhibition of PI3K-AKT-mTOR- and RAF-MEK-ERK-signaling is synergistic in cholangiocarcinoma and reverses acquired resistance to MEK-inhibitors. Invest New Drugs 2014;32:1144–54 [DOI] [PubMed] [Google Scholar]
- 70.Haagensen EJ, Kyle S, Beale GS, Maxwell RJ, Newell DR. The synergistic interaction of MEK and PI3K inhibitors is modulated by mTOR inhibition. Br J Cancer 2012;106:1386–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li J, Davies BR, Han S, Zhou M, Bai Y, Zhang J, et al. The AKT inhibitor AZD5363 is selectively active in PI3KCA mutant gastric cancer, and sensitizes a patient-derived gastric cancer xenograft model with PTEN loss to Taxotere. J Transl Med 2013;11:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Banerji U, Dean EJ, Perez-Fidalgo JA, Batist G, Bedard PL, You B, et al. A Phase I Open-Label Study to Identify a Dosing Regimen of the Pan-AKT Inhibitor AZD5363 for Evaluation in Solid Tumors and in PIK3CA-Mutated Breast and Gynecologic Cancers. Clin Cancer Res 2018;24:2050–9 [DOI] [PubMed] [Google Scholar]
- 73.Chen F, Liu J, Song X, DuCote TJ, Byrd AL, Wang C, et al. EZH2 inhibition confers PIK3CA-driven lung tumors enhanced sensitivity to PI3K inhibition. Cancer Lett 2022;524:151–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xing H, Gao M, Wang Y, Zhang X, Shi J, Wang X, et al. Genome-wide gain-of-function screening identifies EZH2 mediating resistance to PI3Kalpha inhibitors in oesophageal squamous cell carcinoma. Clin Transl Med 2022;12:e835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Singh MK, Izumchenko E, Klein-Szanto AJ, Egleston BL, Wolfson M, Golemis EA. Enhanced genetic instability and dasatinib sensitivity in mammary tumor cells lacking NEDD9. Cancer Res 2010;70:8907–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ice RJ, McLaughlin SL, Livengood RH, Culp MV, Eddy ER, Ivanov AV, et al. NEDD9 depletion destabilizes Aurora A kinase and heightens the efficacy of Aurora A inhibitors: implications for treatment of metastatic solid tumors. Cancer Res 2013;73:3168–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Speck NA, Gilliland DG. Core-binding factors in haematopoiesis and leukaemia. Nat Rev Cancer 2002;2:502–13 [DOI] [PubMed] [Google Scholar]
- 78.Ito Y, Bae SC, Chuang LS. The RUNX family: developmental regulators in cancer. Nat Rev Cancer 2015;15:81–95 [DOI] [PubMed] [Google Scholar]
- 79.Malik N, Yan H, Moshkovich N, Palangat M, Yang H, Sanchez V, et al. The transcription factor CBFB suppresses breast cancer through orchestrating translation and transcription. Nat Commun 2019;10:2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Warfel NA, Kraft AS. PIM kinase (and Akt) biology and signaling in tumors. Pharmacol Ther 2015;151:41–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang X, Song M, Kundu JK, Lee MH, Liu ZZ. PIM Kinase as an Executional Target in Cancer. J Cancer Prev 2018;23:109–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dent S, Cortes J, Im YH, Dieras V, Harbeck N, Krop IE, et al. Phase III randomized study of taselisib or placebo with fulvestrant in estrogen receptor-positive, PIK3CA-mutant, HER2-negative, advanced breast cancer: the SANDPIPER trial. Ann Oncol 2021;32:197–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ellis H, Ma CX. PI3K Inhibitors in Breast Cancer Therapy. Curr Oncol Rep 2019;21:110. [DOI] [PubMed] [Google Scholar]
- 84.Zhang Z, Christin JR, Wang C, Ge K, Oktay MH, Guo W. Mammary-Stem-Cell-Based Somatic Mouse Models Reveal Breast Cancer Drivers Causing Cell Fate Dysregulation. Cell Rep 2016;16:3146–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu P, Liu JP, Sun SJ, Gao Y, Ai Y, Chen X, et al. CBFB-MYH11 Fusion Sequesters RUNX1 in Cytoplasm to Prevent DNMT3A Recruitment to Target Genes in AML. Front Cell Dev Biol 2021;9:675424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kundu M, Liu PP. Function of the inv(16) fusion gene CBFB-MYH11. Curr Opin Hematol 2001;8:201–5 [DOI] [PubMed] [Google Scholar]
- 87.Haferlach C, Dicker F, Kohlmann A, Schindela S, Weiss T, Kern W, et al. AML with CBFB-MYH11 rearrangement demonstrate RAS pathway alterations in 92% of all cases including a high frequency of NF1 deletions. Leukemia 2010;24:1065–9 [DOI] [PubMed] [Google Scholar]
- 88.Sustic T, Bosdriesz E, van Wageningen S, Wessels LFA, Bernards R. RUNX2/CBFB modulates the response to MEK inhibitors through activation of receptor tyrosine kinases in KRAS-mutant colorectal cancer. Transl Oncol 2020;13:201–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Experimental data and results as well as CRISPR screen data and results are available in the manuscript and supplemental material. RNA-sequencing data was uploaded to the NCBI Gene Expression Omnibus (GEO) database and is available at accession GSE235631.