

Summary
Mantle cell lymphoma (MCL) after relapse is associated with poor prognosis. No standard of care exists and available evidence for treatments is limited, particularly in patients who fail Bruton tyrosine kinase inhibitor (BTKi) therapy. This multicentre retrospective chart review study, SCHOLAR-2, addresses this knowledge gap and reports on data collected from 240 patients with relapsed/refractory MCL in Europe who were treated with BTKi-based therapy between July 2012 and July 2018 and had experienced disease progression while on BTKi therapy or discontinued BTKi therapy due to intolerance. Median overall survival (OS) from initiation of first BTKi therapy was 14.6 months (95% confidence interval [CI] 11.6–20.0) in the overall cohort, 5.5 months (95% CI 3.9–8.2) in 91 patients without post-BTKi therapy, and 23.8 months (95% CI 18.9–30.1) in 149 patients who received post-BTKi therapy (excluding chimeric antigen receptor T-cell treatment). In the latter group, patients received a median of 1 (range 1–7) line of post-BTKi therapy, with lenalidomide-containing regimens and bendamustine plus rituximab being the most frequently administered; the median OS from initiation of first post-BTKi therapy was 9.7 months (95% CI 6.3–12.7). These results provide a benchmark for survival in patients with R/R MCL receiving salvage therapy after BTKi failure.
Keywords: real-world evidence, mantle cell lymphoma, survival, Bruton tyrosine kinase inhibitor, post-BTKi
Introduction
Mantle cell lymphoma (MCL) is a rare and aggressive subtype of B-cell non-Hodgkin lymphoma, accounting for less than 10% of all non-Hodgkin lymphoma subtypes. It is more commonly diagnosed in men and typically presents as advanced stage disease in patients over 60 years of age.1–4 Although some patients have indolent MCL at diagnosis, most patients usually experience a relapsing course and require multiple lines of therapy conferring a poor prognosis, especially after initial relapse.3,5,6
No widely applicable standard therapeutic approach exists for relapsed/refractory (R/R) MCL, therefore treatment choice for these patients is influenced by age, performance status, co-morbidities and prior therapy.1 Treatment options include covalent Bruton tyrosine kinase inhibitors (BTKi), cytotoxic chemotherapy, immunomodulatory drugs, proteasome inhibitors, mammalian target of rapamycin inhibitors, and allogenic stem cell transplantation.1,7,8 In the United States (US), newer approved agents include bortezomib (proteasome inhibitor), lenalidomide (immunomodulator), ibrutinib (BTKi), acalabrutinib (BTKi), and zanubrutinib (BTKi). Currently, BTKis represent the most frequently used drug class in R/R MCL; however, early observations of patients with disease progression during or after BTKi exposure have demonstrated extremely poor survival and very limited subsequent options.9–12
Recently, brexucabtagene autoleucel (formerly KTE-X19), a CD19-directed chimeric antigen receptor (CAR) T-cell therapy, was approved by the US Food and Drug Administration (FDA) and European Medicines Agency for the treatment of R/R MCL.13,14 Approvals were based on a phase 2 multi-center, open-label trial (ZUMA-2; NCT02601313) that evaluated brexucabtagene autoleucel in patients with R/R MCL who had prior BTKi therapy.15 Currently, no direct comparison between CAR T-cell and other treatment options is available. Despite limitations, historical survival estimates for standard of care therapies may provide a benchmark for such treatment comparisons. The existing evidence base for patients with R/R MCL post-BTKi therapy comprises only a limited number of small (sample size ranging from 20 to 73), retrospective, non-comparative, observational studies.9–12 Nevertheless, these studies reported median overall survival (OS) ranging from 5.8 to 12.5 months with subsequent treatment(s) in the post-BTKi setting.9–12 To provide updated survival estimates in a larger population, we conducted a large, retrospective chart review of European patients with R/R MCL with prior BTKi therapy (SCHOLAR-2).
Methods
Study Design and Patients
SCHOLAR-2 was a retrospective, observational, multicenter, international chart review of patients aged at least 18 years with R/R MCL, previously treated with a BTKi, and with disease progression while on BTKi therapy or who discontinued due to intolerance. BTKi therapy was initiated between July 2012 and July 2018. The first date reflects the availability of ibrutinib for compassionate use in 2013, and some patients may have received ibrutinib in a clinical trial prior to this date. The second date allowed for at least 12 months of potential follow-up. Patients with central nervous system manifestation (history/current) were excluded as were those who had received CAR T-cell therapy or other genetically modified T-cell therapy.
Patients were enrolled from centers in the United Kingdom, France, Germany, Spain, Italy, Sweden, and Denmark. Eligible centers met all of the following criteria: inpatient diagnostic and treatment facilities for patients with B-cell lymphomas; belonged to a network of oncologists or hematologists who treated patients with R/R B-cell lymphomas; had been operational and treating patients with B-cell lymphoma for at least 24 months; and had clinical records available for review. Participating investigators reviewed and screened medical records of all identified patients with R/R MCL for study eligibility.
Study approval was obtained from the institutional review boards and ethics committees from all relevant institutions by the participating site investigators, and written informed consent was obtained from all patients still alive. This study complied with all applicable local laws, regulations, and guidance regarding patient protection including patient privacy.
Procedures
Data were collected between February 2020 and December 2020 using standardized case report forms to abstract demographics, disease characteristics, diagnosis, co-morbid conditions, treatment history, survival status, and hospitalizations for each eligible patient. Demographics and clinical characteristics were collected at diagnosis for all patients. For patients who received post-BTKi therapy, additional baseline data were collected at the start of their first post-BTKi therapy.
Outcomes
The primary outcome was OS among patients with R/R MCL who had progressive disease while on BTKi therapy or discontinued BTKi therapy due to intolerance. Secondary outcomes included patient demographics, disease characteristics, and treatment patterns in this population.
Statistical approach
All analyses were descriptive, and no hypothesis or significance testing was conducted to compare subgroups. Baseline characteristics and outcome measures were summarized for patients with non-missing data. Continuous variables were summarized using means (with standard deviation) and/or medians (with range or interquartile range). Categorical variables were summarized by frequencies and percentages.
Assessments of patient characteristics at initial MCL diagnosis or at initiation of first post-BTKi therapy used measurements obtained closest to the diagnosis date (within 3 months) or post-BTKi start date (maximum of 6 to 12 months prior and up to 1 month after), respectively. This maximized data availability as some patient characteristics were not routinely collected at initiation of later-line MCL therapies. Dates with missing months or years were considered missing and data were not used.
Treatment sequences by treatment class for up to five lines of therapy were illustrated using a Sankey diagram. BTKi treatments, pre-BTKi treatments, and first post-BTKi treatments received by the patients were also assessed.
Four different index dates were used to estimate OS: 1) initial MCL diagnosis, 2) initiation of first BTKi therapy, 3) discontinuation of first BTKi therapy, and 4) initiation of first post-BTKi therapy. Survival times for each patient were defined as the time from each of these dates until death from any cause. Patients who were known to be alive or lost to follow-up were censored at the last date of contact. Survival curves were estimated using the Kaplan-Meier approach and summarized using medians and two-sided 95% confidence intervals (CIs).16 Median follow-up times were calculated using the reverse Kaplan-Meier approach.17
Results
Of the 282 patients screened, 240 patients with R/R MCL met the study eligibility criteria (Fig 1). These patients were from France (n=79), Italy (n=42), the United Kingdom (n=42), Germany (n=35), Spain (n=28), Sweden (n=9), and Denmark (n=5).
Fig 1. Patient flow diagram and treatment overview.
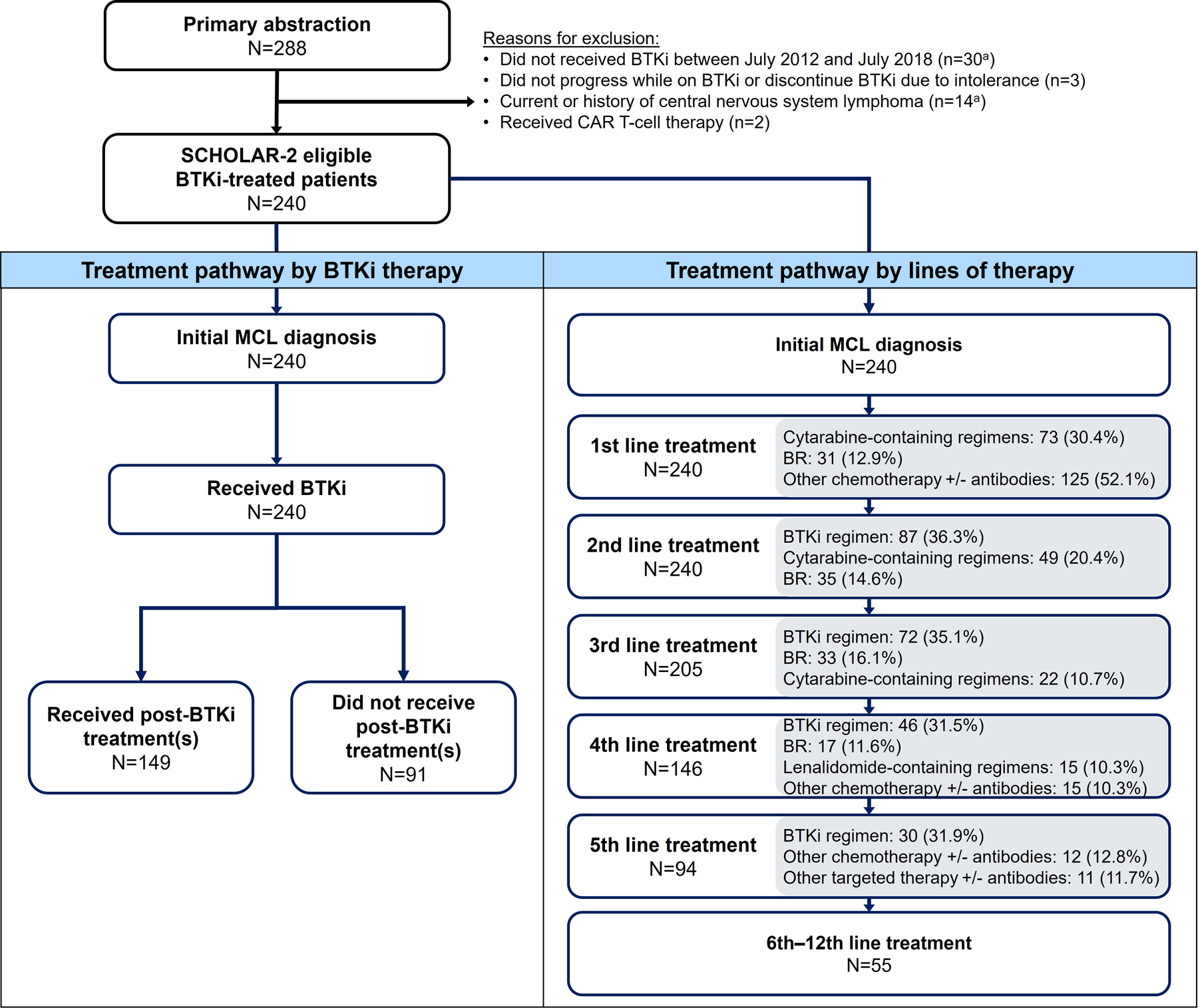
The top three most common treatments received are listed in the grey-shaded boxes; treatments were classified into 11 mutually exclusive sub-categories according to the following hierarchy: BTKi regimens, chemotherapies (BR, R-BAC, cytarabine-containing regimen, gemcitabine-containing regimens or other chemotherapy +/− antibodies), targeted therapies (lenalidomide-containing regimens, bortezomib-containing regimens, temsirolimus-containing regimens, and other targeted therapies +/− antibodies), and other treatments such as anti-metabolite, radioimmunotherapy, radiotherapy, or unknown. BR, bendamustine + rituximab; BTKi, Bruton tyrosine kinase inhibitor; CAR, chimeric antigen receptor; MCL, mantle cell lymphoma; R-BAC, rituximab + bendamustine + cytarabine aOne patient had multiple reasons for exclusion
Demographic and clinical characteristics
Patient characteristics are presented in Table I.
Table I:
Baseline characteristics
| Baseline characteristics | At initial MCL diagnosisa (N=226) | At start of first post-BTKi therapyb (N=149) |
|---|---|---|
| Age ― years | ||
| Median (range) | 68.0 (39.0–92.0) | 71.0 (43.0–91.0) |
| Mean (SD) | 67.1 (10.8) | 70.9 (9.45) |
| Male ― n (%) | 169 (74.8) | 108 (72.5) |
| Disease stage ― n (%) | ||
| I | 2 (1.0) | 5 (4.5) |
| II | 10 (5.0) | 8 (7.2) |
| III | 25 (12.5) | 20 (18.0) |
| IV | 163 (81.5) | 78 (70.3) |
| Missing | 26 | 38 |
| ECOG performance statusc ― n (%) | ||
| 0 | 67 (48.9) | 29 (27.6) |
| 1 | 54 (39.4) | 39 (37.1) |
| ≥2 | 16 (11.7) | 37 (35.2) |
| Missing | 89 | 44 |
| s-MIPI ― n (%) | ||
| Low risk (score 0–3) | 14 (13.6) | 3 (4.8) |
| Intermediate risk (score 4–5) | 18 (17.5) | 14 (22.6) |
| High risk (score ≥6) | 71 (68.9) | 45 (72.6) |
| Missing | 123 | 87 |
| Ki-67 proliferation index ― n (%) | ||
| ≥30% | 55 (58.5) | 17 (77.3) |
| ≥50% | 33 (35.1) | 13 (59.1) |
| Missing | 132 | 127 |
| Morphology ― n (%) | ||
| Blastoid | 36 (25.2) | 17 (37.0) |
| Pleomorphic | 7 (4.9) | 2 (4.3) |
| Non-blastoid or non-pleomorphic | 100 (69.9) | 27 (58.7) |
| Missing | 83 | 103 |
| LDH ― units/L | ||
| Assessed | 156 | 103 |
| Median (range) | 334.5 (10.0–2551.0) | 363.00 (3.0–2990.0) |
| Presence of B symptoms ― n/N (%) | 51/179 (28.5) | 27/100 (27.0) |
| Bulky disease, ≥10 cm ― n/N (%) | 15/179 (8.4) | 19/100 (19.0) |
| Splenic involvement ― n/N (%) | 79/179 (44.1) | 41/100 (41.0) |
| Extranodal disease ― n/N (%) | 49/179 (27.4) | 29/100 (29.0) |
| Bone marrow involvement ― n/N (%) | 130/179 (72.6) | 45/100 (45.0) |
| t(11;14) or cyclin D1 overexpression ― n/N (%) | 130/141 (92.2) | --d |
| t(11;14) ― n/N (%) | 30/35 (85.7) | 10/12 (83.3) |
| Cyclin D1 overexpression ― n/N (%) | 117/134 (87.3) | --d |
| WBC ― x109/L | ||
| Assessed | 164 | --d |
| Median (range) | 8.96 (1.00–47570.0) | --d |
| TP53 mutation ― n/N (%) | 20/37 (54.1) | --d |
| Median no. of prior lines of therapy (range) | -- | 3 (1–11) |
| No. of prior lines of therapy ― n (%) | ||
| 1 | -- | 4 (2.7) |
| 2 | -- | 52 (34.9) |
| 3 | -- | 44 (29.5) |
| ≥4 | -- | 49 (32.9) |
| Previous SCT ― n (%) | ||
| Anye | -- | 50 (33.6) |
| Autologous | -- | 48 (32.2) |
| Allogeneic | -- | 3 (2.0) |
| None | -- | 99 (66.4) |
| Prior BTKi therapy ― n (%) | ||
| Ibrutinib | -- | 145 (97.3) |
| Acalabrutinib | -- | 4 (2.7) |
| Duration on prior BTKi therapy ― months | -- | n=148 |
| Median (range) | -- | 7.1 (0.4–53.7) |
| Mean (SD) | -- | 11.7 (12.0) |
| CR or PR as best response to prior BTKi therapy ― n/N (%) | -- | 46/133 (34.6) |
| Reasons for BTKi discontinuation ― n (%) | ||
| Intolerance to BTKi therapy | -- | 22 (14.8) |
| Disease progression while on BTKi therapy | -- | 127 (85.2) |
| Time interval between BTKi discontinuation and start of post-BTKi therapy ― months | n=148 | |
| Median (range) | -- | 0.4 (0, 48.9) |
| Mean (SD) | -- | 2.5 (7.2) |
Abbreviations: BTKi, Bruton tyrosine kinase inhibitor; CR, complete response; ECOG, Eastern Cooperative Oncology Group; LDH, lactate dehydrogenase; MCL, mantle cell lymphoma; PR, partial response; SCT, stem cell transplantation; SD, standard deviation; s-MIPI, simplified Mantle Cell Lymphoma International Prognostic Index; WBC, white blood cells
Time window: reported within 3 months (before or after) of diagnosis date.
Time window: reported 6 to 12 months prior to start of first post-BTKi therapy and up to 1 month after.
Performance score was based on Karnofsky performance status in two patients at initial MCL diagnosis and in one patient at start of first post-BTKi therapy and converted to ECOG PS (Karnofsky performance status categories 90–100/70–80/50–60/30–40/10–20 corresponded to ECOG PS 0/1/2/3/4).
WBC, cyclin D1 overexpression, and TP53 mutation not available at start of first post-BTKi therapy.
One patient received both autologous SCT and allogeneic SCT.
At initial MCL diagnosis
Patient characteristics at initial MCL diagnosis were summarized for all patients with an available diagnosis date (n=226/240). Patients had a median age of 68 years (range 39–92). Most patients were male (74.8%), had stage IV disease (81.5%), Eastern Cooperative Oncology Group (ECOG) performance score (PS) of 0 or 1 (88.3%), bone marrow involvement (72.6%), and t(11;14) or cyclin D1 overexpression (92.2%). Other disease characteristics included blastoid morphology (25.2%), presence of B symptoms (28.5%), bulky disease ≥10 cm (8.4%), splenic involvement (44.1%), and extranodal disease (27.4%).
At the time of first post-BTKi treatment
At the initiation of post-BTKi therapy (n=149), there was a higher proportion of patients with ECOG PS ≥2, blastoid morphology, Ki67 >30% or >50% (albeit with high missing rates), and bulky disease, but a slightly lower proportion of patients with stage IV disease and bone marrow involvement than at initial MCL diagnosis. Median age increased from 68 to 71 years. Other disease characteristics were generally similar between the two time-points.
Fifty (33.6%) patients had prior autologous/allogeneic stem cell transplantation. The median number of prior lines of therapy including a BTKi was 3 (range 1–11). The median duration of prior BTKi therapy was 7.1 months (range 0.4–53.7). Reasons for BTKi discontinuation included intolerance (n=22, 14.8%) and disease progression while on therapy (n=127, 85.2%). The best objective response rate to BTKi was 34.6% (n=46/133, 16 complete and 30 partial responses) in evaluable patients. Of note, in the subgroup of patients without post-BTKi therapy, 73.6% of patients discontinued BTKi due to disease progression (and 26.4% due to intolerance) after a median BTKi duration of 3.8 months (range, 0.1–55.2).
Treatments
At the time of data collection, all 240 patients had received at least two lines of therapy for MCL (median 4, range 2–12), with a high proportion of patients observed receiving further treatments in third-line (3L; 85.4%), fourth-line (4L; 60.8%), and fifth or later lines (≥5L; 39.2%; Fig 1). Almost all patients (97.1%) received first-line (1L) chemotherapy +/− antibodies. Approximately one-third of patients received a BTKi in each subsequent line of treatment. Fig 2 illustrates the transition from 1L up to 5L therapy, including the proportion of patients without further treatments (due to death or censoring at last follow-up). Most patients (96.6%) had received BR (38.1%), cytarabine-containing regimens (51.7%) and other chemotherapy +/− antibodies (58.9%) prior to a BTKi (Supplementary Table SI).
Fig 2. Sankey diagram of treatment patterns (from 1L up to 5L).
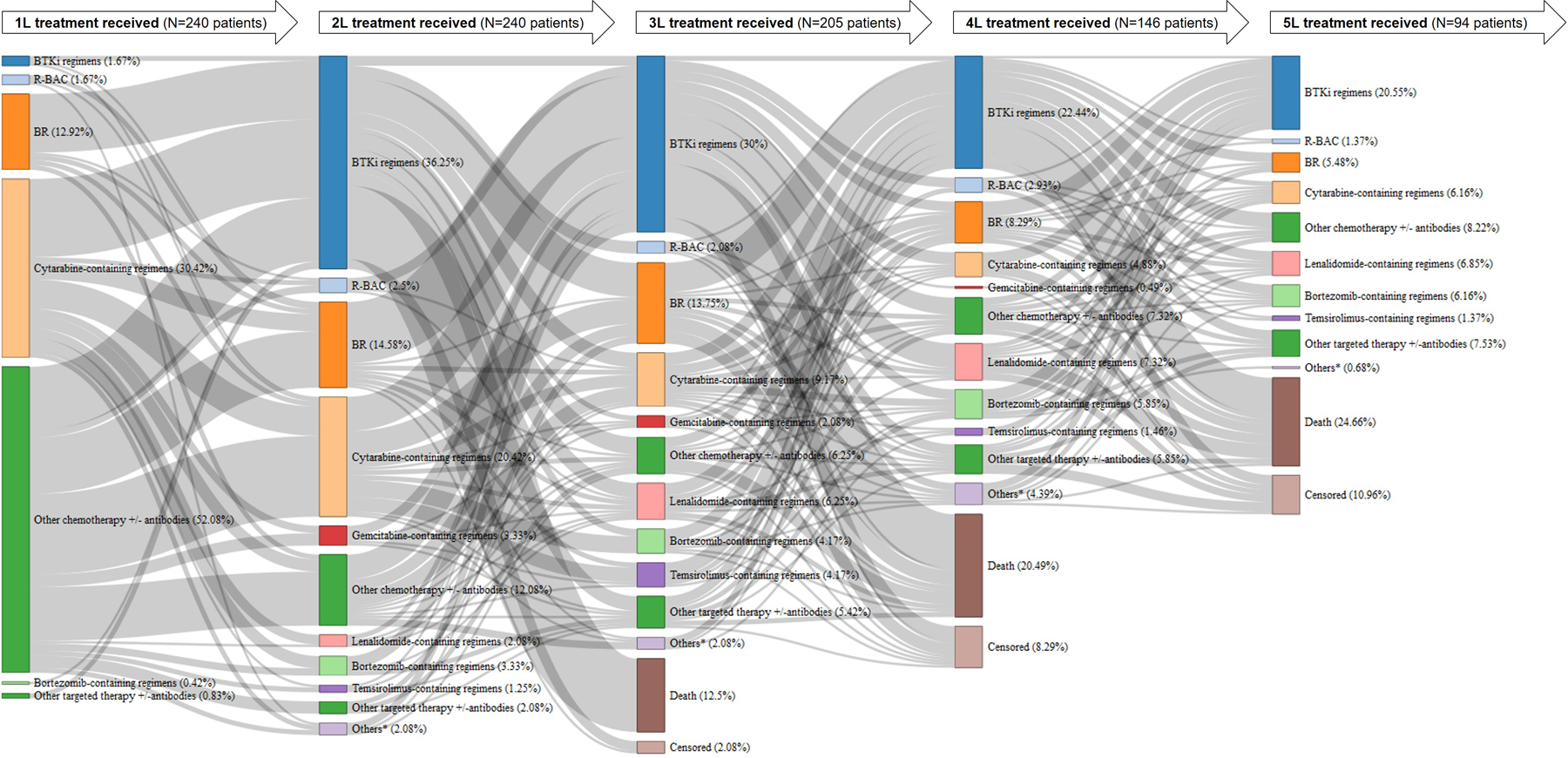
Treatment patterns of patients from 1L up to 5L (left to right) among 240 patients with relapsed/refractory mantle cell lymphoma who received BTKi between July 2012 to July 2018. The proportions (%) shown are based on total number of patients (N) receiving the preceding line of treatment (except for 1L which is based on 240 patients). 1L, first-line; 2L, second-line; 3L, third-line; 4L, fourth-line; 5L, fifth-line; BR, bendamustine + rituximab; BTKi, Bruton tyrosine kinase inhibitor; MCL, mantle cell lymphoma; R-BAC, rituximab + bendamustine + cytarabine *Others included anti-metabolite, radioimmunotherapy, radiotherapy, or unknown
Most patients (93.8%) received only one BTKi regimen, but 14 (5.8%) received two lines of BTKi regimens and one (0.4%) received three lines of BTKi regimens. Four patients (1.7%) received 1L BTKi and more than half of patients received their first BTKi therapy in second-line (2L; 35.4%) or 3L (28.3%). The remaining patients received their first BTKi in 4L (16.7%), 5L (12.1%), or later lines (5.8%). The first BTKi regimens often included ibrutinib, either as monotherapy (87.1%) or in combination with other agents (10.4%), with a small proportion of patients (2.5%) receiving acalabrutinib monotherapy (Supplementary Table SII).
After discontinuing BTKi therapy, 91 (37.9%) patients received no further systemic anti-lymphoma therapy. Causes of death and reasons for not receiving post-BTKi therapies were not collected. These patients had received their first BTKi in 2L (36.3%), 3L (26.4%), and 4L and beyond (37.4%).
Of the other 149 patients who received further systemic anti-lymphoma therapy, four patients (2.7%) had received 1L BTKi whereas the rest received their first BTKi in 2L (34.9%), 3L (29.5%), or beyond (32.9%). Most patients (61.7%) received only one post-BTKi treatment, 16.8% received a total of two post-BTKi treatments, and 14.8% received three. Lenalidomide-containing regimens (17.4%) and BR (16.8%) were the two most common types of subsequent treatment following BTKi discontinuation (Table II and Supplementary Table SIII).
Table II:
First post-BTKi therapy (N=149)
| Treatment | n (%) |
|---|---|
| Chemotherapies (+/− antibodies) | 78 (52.3) |
| R-BAC | 11 (7.4) |
| BR | 25 (15.8) |
| Cytarabine-containing regimens | 16 (10.7) |
| Gemcitabine-containing regimens | 4 (2.7) |
| Other chemotherapy (+/− antibodies) | 22 (14.8) |
| Targeted therapies (+/−antibodies) | 67 (45.0) |
| BTKi regimens | 8 (5.4) |
| Lenalidomide-containing regimens | 26 (17.4) |
| Bortezomib-containing regimens | 13 (8.7) |
| Temsirolimus-containing regimens | 1 (0.7) |
| Other targeted therapies (+/−antibodies) | 19 (12.8) |
| Others | 4 (2.7) |
| Radiotherapy | 4 (2.7) |
Abbreviations: BR, bendamustine + rituximab; BTKi, Bruton tyrosine kinase inhibitor; R-BAC, rituximab + bendamustine + cytarabine
Survival
At the time of data collection, 192/240 (80.0%) patients had died; of the 149 who received post-BTKi treatments, 106 (71.1%) had died. The median OS from initial MCL diagnosis for the 226 patients with an available diagnosis date was 64.5 months (95% CI 51.7–78.5; Fig 3). The estimated OS was 93.3% at 1 year, 78.3% at 2 years, and 53.8% at 5 years after MCL diagnosis.
Fig 3. Kaplan-Meier plots of overall survival from initial diagnosis of mantle cell lymphoma.
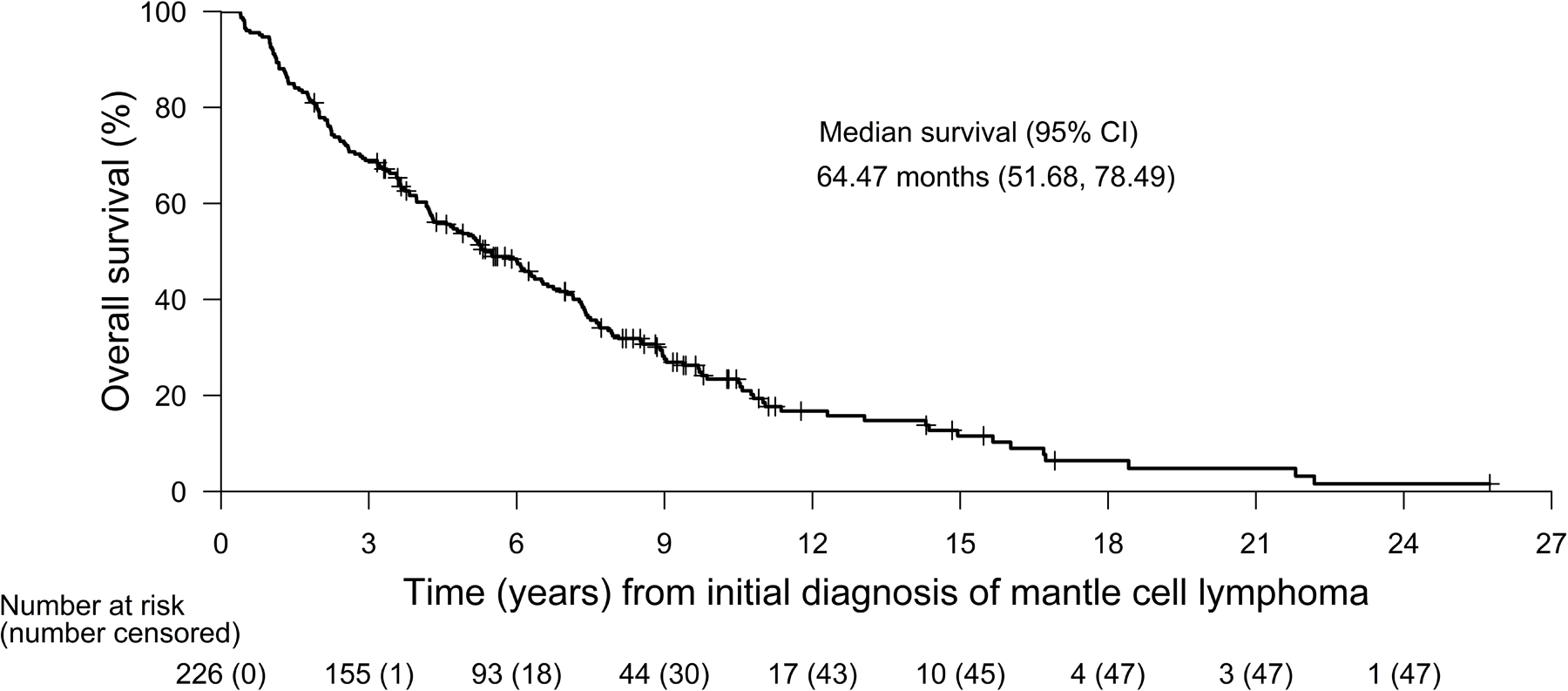
Tick marks indicate censored data. BTKi, Bruton tyrosine kinase inhibitor; CI, confidence interval
The median OS from initiation of first BTKi therapy for all 240 patients was 14.6 months (95% CI 11.6–20.0; Fig 4A). Patients who received post-BTKi therapy had longer survival from initiation of first BTKi therapy (median OS: 23.8 months [95% CI 18.9–30.1], n=149) compared with those who did not receive subsequent therapy (median OS: 5.5 months [95% CI 3.9–8.2], n=91). The estimated OS at 1 year from the initiation of BTKi therapy was 69.1% in patients receiving post-BTKi therapy and 34.4% in patients without post-BTKi therapy; at 2 years, the estimated OS was 50.0% and 17.2%, respectively.
Fig 4. Kaplan-Meier plots of overall survival from initiation or discontinuation of treatment for mantle cell lymphoma.
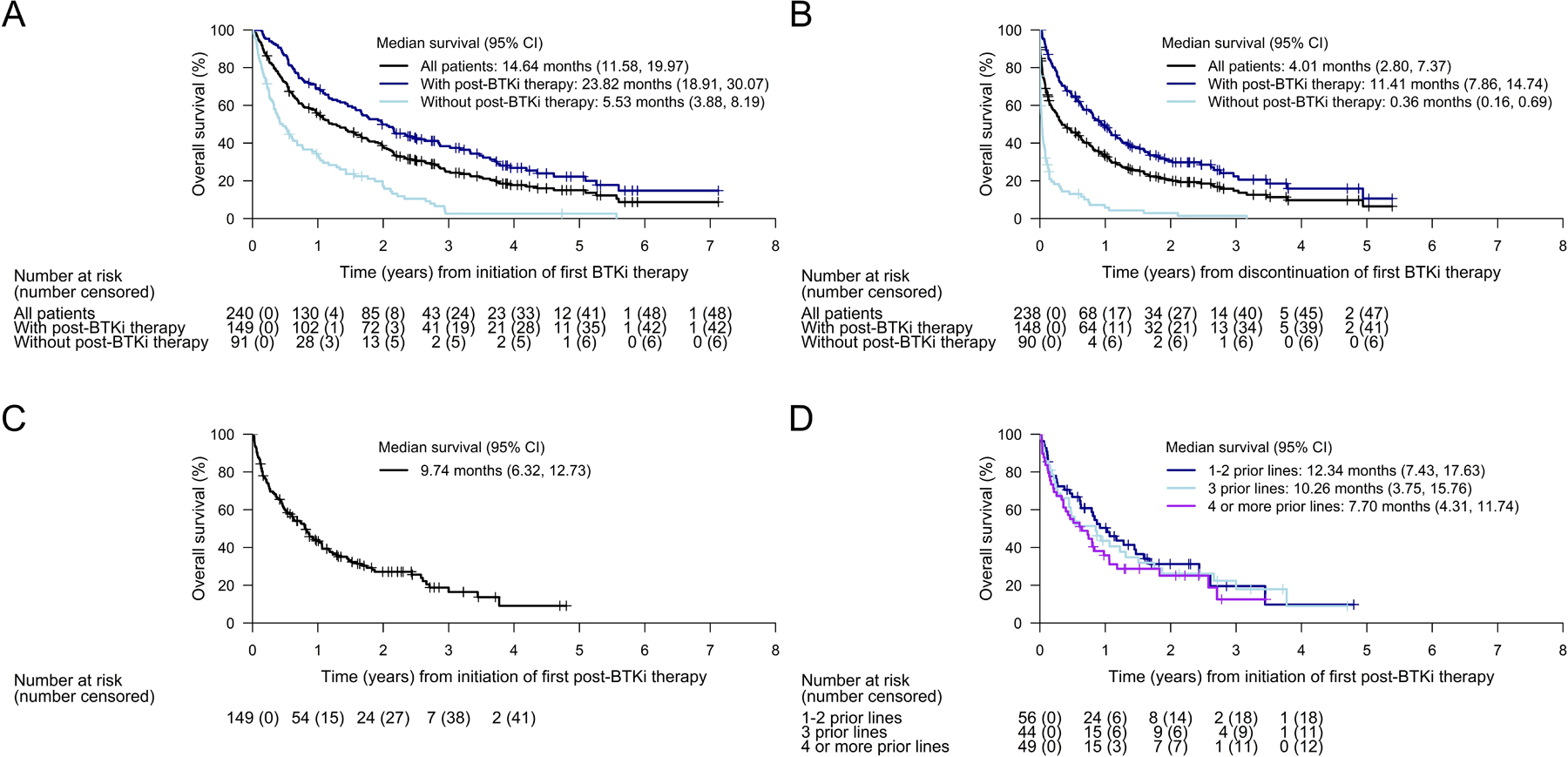
Kaplan-Meier plots of overall survival A) from date of initiation of first BTKi therapy, B) from date of discontinuation of first BTKi therapy, C) from date of initiation of first post-BTKi therapy (among all 149 patients who received post-BTKi therapy), and D) from date of initiation of first post-BTKi therapy, by subgroup based on lines of therapy. Tick marks indicate censored data. BTKi, Bruton tyrosine kinase inhibitor; CI, confidence interval; LOT, lines of prior therapy
The median OS from discontinuation of first BTKi therapy was 4.0 months (95% CI 2.8–7.4, n=238) for the entire cohort, 0.4 months (95% CI 0.2–0.7, n=90) for the subgroup of patients without post-BTKi therapy, and 11.4 months (95% CI 7.9–14.7, n=148) for the subgroup of patients with post-BTKi therapy (Fig 4B). In this latter subgroup of patients with post-BTKi therapy, OS was higher among those with longer median BTKi duration (versus shorter) and among those who discontinued BTKi due to intolerance (versus disease progression; Supplementary Fig SI).
A landmark analysis for OS from discontinuation of first BTKi therapy was performed to account for potential immortal time bias where some patients may not have survived long enough after BTKi discontinuation to receive subsequent therapy. A landmark time of 11 days, which represented the median time from BTKi discontinuation to the start of post-BTKi therapy, was selected and any patients who were lost to follow-up or died prior to this time were excluded from the analysis. Compared to the original analysis, median OS among patients who did not receive post-BTKi therapy increased from 0.4 months to 1.3 months whereas the median survival among patients with post-BTKi therapy slightly shifted from 11.4 to 11.1 months in the landmark analysis (Supplementary Table SIV).
Among the 149 patients receiving post-BTKi therapy, the median OS from initiation of first post-BTKi therapy was 9.7 months (95% CI 6.3–12.7) after a median follow-up of 27.3 months (Fig 4C), and the estimated OS was 43.4% at 1 year and 27.2% at 2 years. In this group, patients with one to two lines prior to initiation of their first post-BTKi therapy tended to have a slightly more favorable survival (median OS: 12.3 months [95% CI 7.4–17.6]) compared with patients who received three prior lines (median OS: 10.3 months [95% CI 3.8–15.8]) or four or more prior lines (median OS: 7.7 months [95% CI 4.3–11.7]; Fig 4D).
Discussion
As MCL generally follows an aggressive clinical course with patients experiencing frequent relapses, management of patients with R/R disease remains challenging as treatment response declines at each successive line of treatment.4,18 In particular, clinical outcomes are usually poor in patients with R/R MCL treated with currently available systemic therapies in the post-BTKi setting.9–12,19 Published data on patients with R/R MCL who have previously failed a BTKi are limited, and, to our knowledge, SCHOLAR-2 provides the largest real-world experience to date of subsequent salvage therapies following BTKi discontinuation. This cohort of BTKi-treated patients with R/R MCL across multiple European centers confirms the urgent unmet need for new treatment options that can improve survival in patients with R/R MCL who relapse after receiving BTKi or who are intolerant to BTKi therapies. Results from our study showed a median OS of 9.7 months (95% CI 6.3–12.7) from start of subsequent treatment post-BTKi, which is consistent with the literature of smaller retrospective studies, i.e., median OS of 5.8–12.5 months with subsequent treatments such as venetoclax11, R-BAC (rituximab, bendamustine, and cytarabine)10, and various salvage treatments.9,12,19
Comparisons across retrospective studies can be challenging due to known and unknown differences in the distributions of prognostic factors and treatment effect modifiers, with prior lines of therapy being a potential key factor.18 Nevertheless, other published studies provide context for our findings. A retrospective observational study by McCulloch et al. previously evaluated the efficacy of R-BAC (post-BTKi therapy) in 36 patients with R/R MCL who were treated within routine clinical practice in the United Kingdom and Italy.10 Most patients (n=32/36, 88.9%) had less than three prior lines of therapy at start of R-BAC, which differed from our study population (n=56/149, 37.6%). Median OS was 12.5 months (95% CI 11.0–14.0) from start of R-BAC and this was comparable to that observed in the subgroup of patients from SCHOLAR-2 with less than three prior lines of therapy (12.3 months). This is despite SCHOLAR-2 featuring a broader mix of post-BTKi interventions than just R-BAC. In another retrospective study by Martin et al., median OS from the start of first subsequent salvage treatment post-BTKi was 5.8 months (95% CI 3.7–10.4) in patients with R/R MCL who had received a median of four prior therapies,9 which again was more closely in line with that observed in the subgroup of patients with four or more prior lines in SCHOLAR-2 (7.7 months). Beyond differences in number of prior lines of therapy and the types of subsequent treatments received by the patients, variations in OS reported across existing studies and SCHOLAR-2 may have also been influenced by differences in other baseline characteristics (prognostic factors and/or treatment effect modifiers). Not taking into account potential differences in patient or treatment characteristics, SCHOLAR-2 did show more favorable post-BTKi survival outcomes in patients who had fewer prior lines of therapy, which could also reflect improved clinical management in later stages of the disease.
Our study population included heavily pre-treated, high-risk patients with R/R MCL who had failed BTKi. We recognise that the observed response rates are not representative of all patients with R/R MCL receiving BTKi in clinical practice. Almost all study patients (97.0%) received chemotherapies +/− antibodies as front-line treatment. In subsequent lines, patients mostly received BTKi regimens, and chemotherapies such as cytarabine-containing regimens or BR. About two-thirds of patients received their first BTKi regimens in 2L or 3L, and upon BTKi discontinuation, a third of patients switched to lenalidomide-containing regimens or BR. It is important to highlight that data from SCHOLAR-2 reflects recent clinical practice but did not include CAR T-cell therapies. Anti-CD19 CAR T-cell therapies belong to a new generation of cancer immunotherapies and have recently shown considerable efficacy in patients with R/R malignant B-cell lymphoma.20–22 To date, brexucabtagene autoleucel is the only CAR T-cell therapy approved in Europe for post-BTKi R/R MCL based on the positive results from the single-arm ZUMA-2 trial.15 At time of the primary efficacy analysis of ZUMA-2, the median OS had not been reached, and the 12-month OS was estimated to be 83.3% (compared to 43.4% in SCHOLAR-2). There were, however, some observed differences in baseline characteristics between ZUMA-2 and SCHOLAR-2 populations. Mostly notably, the SCHOLAR-2 population at start of post-BTKi treatments were slightly older, had more patients with Stage IV disease and included 34% patients with ECOG PS 2. When restricting to the 59 patients with ECOG PS of 0/1 (to match ZUMA-2) and who had started a post-BTKi therapy prior to July 2019 (to allow for a minimum of 12-months follow-up), the median OS increased to 15.7 (95% CI 10.0–30.9) months with an estimated 12-month OS of 57.5%.23 We recently performed an indirect comparison to ZUMA-2 using this subset of 59 patients, and results suggested that brexucabtagene autoleucel was more effective in terms of OS than other salvage treatments in this population both before and after adjusting for differences in key prognostic factors and treatment effect modifiers (unadjusted hazard ratio 0.37 [95% CI 0.20–0.66], adjusted hazard ratio 0.33 [95% CI 0.18–0.59] based on an inverse probability weighting approach).23
Of note, although the indirect treatment comparison was based on 59/240 (25%) SCHOLAR-2 patients who best-matched the patients enrolled in ZUMA-2, this does not represent the true proportion of patients who would be eligible for CAR T-cell therapy from SCHOLAR-2. For the indirect comparison, we were only interested in the subset of patients who had received post-BTKi treatment at data cut-off. It is unclear what proportion of patients who did not initiate post-BTKi therapies in SCHOLAR-2 would have been able to go on to CAR T-cell therapy if it had been available at that time. Similarly, a proportion of the patients that were removed from our analysis due to poor or missing ECOG PS may also have been eligible for CAR T-cell therapy. Nonetheless, there will still be a subset of patients who will not be able or eligible to receive CAR T-cell therapy post-BTKi failure due to various reasons. This highlights an important area for further research to improve access to CAR T-cell therapy and/or develop alternative effective treatments for those not eligible.
Several limitations should be taken into consideration given the nature of the study design of SCHOLAR-2. Retrospective studies are reliant on the quality and type of data available for collection. The median follow-up in SCHOLAR-2 was long (11.1 years from initial MCL diagnosis); however, not all baseline characteristics were available for capture at diagnosis, resulting in high missing rates for several key variables such as s-MIPI (54%), Ki67 (58%), and TP53 mutation (84%). Although disease stage was well-populated, we still observed a 12% missing rate. Moreover, patient characteristics were not routinely collected at the start date of the first post-BTKi therapy, and as such we extended the time window for collection to 6 or 12 months prior to treatment start date in order to maximise data availability, which may mean the reported values were not entirely reflective of the true characteristics at the start of treatment. It is also important to consider immortal time bias when estimating OS from the discontinuation of BTKi therapy for subgroups of patients with and without post-BTKi therapy, as their assignment to either of these groups is contingent on surviving long enough to initiate further treatment. To address this, we performed a landmark analysis including only patients who survived until the 11-day landmark time-point (based on median time to next treatment after discontinuation of BTKi therapy among the SCHOLAR-2 patients). These results suggested that the OS estimates should be interpreted with caution due to the potential immortal time bias, specifically for the subgroup with no post-BTKi therapy. However, it is important to note that the objective of this analysis was not to compare survival between groups but to summarize treatment patterns and survival at different time-points.
SCHOLAR-2 provides a detailed description of patient characteristics, treatment patterns, and survival outcomes for real-world patients with MCL who initiated BTKi treatments between July 2012 and July 2018 and had experienced disease progression while on BTKi therapy or discontinued BTKi therapy due to intolerance, and benchmarks survival in patients receiving salvage therapy (excluding CAR T-cell therapy) after BTKi failure. Among these BTKi-treated patients, 64% received BTKi therapy in 2L or 3L of therapy, and 62% received subsequent treatments after BTKi discontinuation. Based on the available data, there was a higher proportion of patients with high-risk features at start of post-BTKi therapy compared to initial MCL diagnosis, which aligned with the poorer median OS in the post-BTKi therapy setting. The OS observed in SCHOLAR-2 is consistent with that reported in the literature, which again confirms the poor clinical outcomes of patients with R/R MCL, in particular those who failed on prior BTKi therapy.
Supplementary Material
Acknowledgments
The authors would like to thank Greg Maglinte sincerely for his contributions towards the conception and design of this work. This study was supported by Kite, a Gilead Company.
Footnotes
Data sharing statement
The datasets generated and analyzed during this study can be made available upon reasonable request to the corresponding author, and decisions regarding data sharing will be made on a case-by-case basis considering data protection and other applicable regulations.
Conflicts of interest
GH has received research support for the submitted work from Kite, a Gilead Company; has received grants or contracts from Celgene, Kite, a Gilead Company, Incyte, Janssen, Morphosys, Pfizer, and Roche; has received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events from Abbvie, ADC, AstraZeneca, Genmab, Kite, a Gilead Company, Incyte, Janssen, Morphosys, Novartis, Roche, and Takeda; has received support for attending meetings and/or travel from Kite, a Gilead Company, and Janssen; and has participated in on a Data Safety Monitoring Board or Advisory Board for Miltenyie. LO has received support for attending meetings and/or travel from Roche and AstraZeneca; and has participated on a Data Safety Monitoring Board or Advisory Board for Roche and Gilead Sciences. PLZ has received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events from Roche, Takeda, Gilead Sciences, Sanofi, Incyte, Novartis, Merck, Bristol-Myers Squibb (BMS), AstraZeneca, and Kyowa Kirin. KL has received consulting fees from Genmab, Roche, Kite, a Gilead Company, and Beigene; has received payment or honoraria for educational events from Abbvie and Celgene/BMS; has received support for attending meetings from BMS and Takeda; and has leadership or fiduciary role in NCRI, EHA LyG, WiL, and Epcoritamab Global Council. MJ has received grants or contracts from Roche, Abbvie, and BMS; has received consulting fees from Gilead Sciences, Janssen, and Abbvie; has received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events from Janssen; has participated on Data Safety Monitoring board or Advisory Board for GenMab. SS has received grants or contracts; consulting fees; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events; support for attending meetings and/or travel; equipment, materials, drugs, medical, writing, gifts or other services; and payments for participation on a Data Safety Monitoring Board or Advisory Board from AbbVie, Acerta, Amgen, AstraZeneca, BeiGene, BMS, Celgene, Genentech, Gilead Sciences, GSK, Hoffmann-La Roche, Incyte, Infinity, Janssen, Novartis, Pharmacyclics, Sunesis, and Verastem. JL has received payment or honoraria for educational events and support for attending meetings from Kite, a Gilead Company. VRZ has received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or education events from Janssen; has received support for attending meetings and/or travel from Janssen; and has participated on Data Safety Monitoring board or Advisory Board for Gilead Sciences and Novartis. JMS has received honoraria for educational events from Roche, Kite, a Gilead Company, Celgene/BMS, Novartis, Janssen, Takeda, and Incyte; has received support for attending meetings and/or travel from Roche; and has received honoraria for participation in Advisory Board from Roche, Kite, a Gilead Company, Celgene/BMS, Novartis, Janssen, Incyte, Beigene, Lilly. TAE has received payments or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events from Loxo Oncology, Eli Lilly, Beigene, AstraZeneca, Janssen, Incyte, Abbvie, Roche, and Kite, a Gilead Company; has received grants or contracts from AstraZeneca and Beigene; has received consulting fees from Loxo Oncology, Eli Lilly, Beigene, AstraZeneca, Janssen, Incyte, Secura Bio, Abbvie, Roche, and Kite a Gilead Company; and has received payments for travel to scientific congress from Abbvie. JJW is an employee of Kite, a Gilead Company; has received honoraria from the Patient-Centered Outcomes Research Institute (PCORI), both as a member of the Rare Disease Advisory Panel, and as a grants reviewer for the Improving Methods Program; has received travel/meeting support from Kite, a Gilead Company, and Amgen; owns stock in Gilead Sciences, Amgen, Abbott, AbbVie, Pfizer, Roche, Curis, Avid Biosciences, Evofem, Lensar, VBI Vaccines, and Viracta Therapeutics. RS is an employee of Kite, a Gilead Company, and was previously employed by Amgen; has led advisory boarding meeting for Kite, a Gilead Company; and owns stocks in Gilead Sciences and Amgen. SW has received writing support for the submitted work from Kite, a Gilead Company; has received consulting fees from Kite, a Gilead Company, Allergan, Amgen, and Johnson & Johnson; and has received support for attending meetings from Kite, a Gilead Company. GS has received consulting fees from Abbvie, Celgene/BMS, Epizyme, Genmab, Incyte, Janssen, Kite, a Gilead Company, Loxo, Milteniy, Molecular Partners, Morphosys, Nordic Nanovector, Novartis, Rapt, and Takeda; has received payment or honoraria for speaking in symposium from Bayer, Epizyme, and Regeneron; has participated on a Data Safety Monitoring board or Advisory Board for Beigene; and has stock or stock options from Owkin. JMHC, SK, and JEP are employees of PRECISIONheor which received funding for this study from Kite, a Gilead Company. MD, EG, AV, AO, CT, AJU, LF, and JR declare no competing interests.
References
- 1.McKay P, Leach M, Jackson B, Robinson S, Rule S. Guideline for the management of mantle cell lymphoma. Br J Haematol 2018;182(1):46–62. [DOI] [PubMed] [Google Scholar]
- 2.Dreyling M, Campo E, Hermine O, Jerkeman M, Le Gouill S, Rule S, et al. Newly diagnosed and relapsed mantle cell lymphoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2017;28(suppl_4):iv62–iv71. [DOI] [PubMed] [Google Scholar]
- 3.Owen C, Berinstein NL, Christofides A, Sehn LH. Review of Bruton tyrosine kinase inhibitors for the treatment of relapsed or refractory mantle cell lymphoma. Curr Oncol 2019;26(2):e233–e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Avivi I, Goy A. Refining the mantle cell lymphoma paradigm: Impact of novel therapies on current practice. Clinical cancer research : an official journal of the American Association for Cancer Research 2015;21(17):3853–61. [DOI] [PubMed] [Google Scholar]
- 5.Wang M, Schuster SJ, Phillips T, Lossos IS, Goy A, Rule S, et al. Observational study of lenalidomide in patients with mantle cell lymphoma who relapsed/progressed after or were refractory/intolerant to ibrutinib (MCL-004). J Hematol Oncol 2017;10(1):171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eyre TA, Cheah CY, Wang ML. Therapeutic options for relapsed/refractory mantle cell lymphoma. Blood 2022;139(5):666–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dreyling M, Campo E, Hermine O, Jerkeman M, Le Gouill S, Rule S, et al. Newly diagnosed and relapsed mantle cell lymphoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2017;28(suppl_4):iv62–iv71. [DOI] [PubMed] [Google Scholar]
- 8.National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): B-Cell Lymphomas, Version 5.2021 — September 22, 2021 [ [Google Scholar]
- 9.Martin P, Maddocks K, Leonard JP, Ruan J, Goy A, Wagner-Johnston N, et al. Postibrutinib outcomes in patients with mantle cell lymphoma. Blood 2016;127(12):1559–63. [DOI] [PubMed] [Google Scholar]
- 10.McCulloch R, Visco C, Eyre TA, Frewin R, Phillips N, Tucker DL, et al. Efficacy of R-BAC in relapsed, refractory mantle cell lymphoma post BTK inhibitor therapy. Br J Haematol 2020;189(4):684–8. [DOI] [PubMed] [Google Scholar]
- 11.Eyre TA, Walter HS, Iyengar S, Follows G, Cross M, Fox CP, et al. Efficacy of venetoclax monotherapy in patients with relapsed, refractory mantle cell lymphoma after Bruton tyrosine kinase inhibitor therapy. Haematologica 2019;104(2):e68–e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jain P, Kanagal-Shamanna R, Zhang S, Ahmed M, Ghorab A, Zhang L, et al. Long-term outcomes and mutation profiling of patients with mantle cell lymphoma (MCL) who discontinued ibrutinib. Br J Haematol 2018;183(4):578–87. [DOI] [PubMed] [Google Scholar]
- 13.European Medicines Agency. Tecartus : EPAR - Public assessment report [Available from: https://www.ema.europa.eu/en/documents/assessment-report/tecartus-epar-public-assessment-report_en.pdf.
- 14.U.S Food & Drug Administration. FDA approves brexucabtagene autoleucel for relapsed or refractory mantle cell lymphoma 2020. [Available from: https://www.fda.gov/drugs/fda-approves-brexucabtagene-autoleucel-relapsed-or-refractory-mantle-cell-lymphoma.
- 15.Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2020;382(14):1331–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. Journal of the American Statistical Association 1958;53(282):457–81. [Google Scholar]
- 17.Schemper M, Smith TL. A note on quantifying follow-up in studies of failure time. Controlled clinical trials 1996;17(4):343–6. [DOI] [PubMed] [Google Scholar]
- 18.Kumar A, Sha F, Toure A, Dogan A, Ni A, Batlevi CL, et al. Patterns of survival in patients with recurrent mantle cell lymphoma in the modern era: progressive shortening in response duration and survival after each relapse. Blood Cancer Journal 2019;9(6):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheah CY, Chihara D, Romaguera JE, Fowler NH, Seymour JF, Hagemeister FB, et al. Patients with mantle cell lymphoma failing ibrutinib are unlikely to respond to salvage chemotherapy and have poor outcomes. Ann Oncol 2015;26(6):1175–9. [DOI] [PubMed] [Google Scholar]
- 20.Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol 2019;20(1):31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med 2017;377(26):2531–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. New England Journal of Medicine 2018;380(1):45–56. [DOI] [PubMed] [Google Scholar]
- 23.Hess G, Dreyling M, Oberic L, Gine E, Zinzani PL, Linton K, et al. , editors. KTE-X19 versus standard of care for relapsed/refractory mantle cell lymphoma previously treated with bruton tyrosine kinase inhibitors: real-world evidence from Europe. European Hematology Association; 2021; Presented at EHA 2021 Virtual Congress, June 9–17, 2021. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.