

Keywords: bacteremia, inflammation, liver injury, P2Y2 purinergic receptor, sepsis
Abstract
The liver plays a significant role in regulating a wide range of metabolic, homeostatic, and host-defense functions. However, the impact of liver injury on the host’s ability to control bacteremia and morbidity in sepsis is not well understood. Leukocyte recruitment and activation lead to cytokine and chemokine release, which, in turn, trigger hepatocellular injury and elevate nucleotide levels in the extracellular milieu. P2Y2 purinergic receptors, G protein-coupled and activated by extracellular ATP/UTP, are expressed at the cell surface of hepatocytes and nonparenchymal cells. We sought to determine whether P2Y2 purinergic receptor function is necessary for the maladaptive host response to bacterial infection and endotoxin-mediated inflammatory liver injury and mortality in mice. We report that P2Y2 purinergic receptor knockout mice (P2Y2−/−) had attenuated inflammation and liver injury, with improved survival in response to LPS/galactosamine (LPS/GalN; inflammatory liver injury) and cecal ligation and puncture (CLP; polymicrobial sepsis). P2Y2−/− livers had attenuated c-Jun NH2-terminal kinase activation, matrix metallopeptidase-9 expression, and hepatocyte apoptosis in response to LPS/GalN and attenuated inducible nitric oxide synthase and nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain containing 3 protein expression in response to CLP. Implicating liver injury in the disruption of amino acid homeostasis, CLP led to lower serum arginine and higher bacterial load and morbidity in the WT mice, whereas serum arginine levels were comparable to sham-operated controls in P2Y2−/− mice, which had attenuated bacteremia and improved survival. Collectively, our studies highlight the pathophysiological relevance of P2Y2 purinergic receptor function in inflammatory liver injury and dysregulation of systemic amino acid homeostasis with implications for sepsis-associated immune dysfunction and morbidity in mice.
NEW & NOTEWORTHY Our studies provide experimental evidence for P2Y2 purinergic receptor-mediated potentiation of inflammatory liver injury, morbidity, and mortality, in two well-established animal models of inflammatory liver injury. Our findings highlight the potential to target P2Y2 purinergic signaling to attenuate the induction of “cytokine storm” and prevent its deleterious consequences on liver function, systemic amino acid homeostasis, host response to bacterial infection, and sepsis-associated morbidity and mortality.
INTRODUCTION
Despite intense research over the past 30 years and numerous therapeutic trials targeting inflammatory responses to sepsis, morbidity, and mortality remain unacceptably high (1). During sepsis, liver-resident and recruited leukocytes release cytokines and chemokines, triggering hepatocellular injury and elevating nucleotide levels in the extracellular milieu (2). These nucleotides released from hepatocytes, macrophages, neutrophils, and platelets at the sites of injury, function as “danger signals” modulating inflammatory cascades and the extent of tissue injury (2–11). Extracellular ATP/UTP activates P2Y2 purinergic receptors, which are G protein-coupled receptors expressed in hepatocytes and nonparenchymal cells (12–20). Therefore, we wanted to determine whether P2Y2 purinergic signaling has the potential to modify the magnitude of inflammation (cytokine storm), and liver injury, hallmarks of early events associated with sepsis, and subsequent hepatic responses to bacteremia, which determine the severity and outcomes of sepsis.
We evaluated the role of P2Y2 purinergic receptor function in the host response to bacterial infection and endotoxin-mediated inflammatory liver injury, morbidity, and mortality, in two independent animal models of liver injury induced in response to lipopolysaccharide/galactosamine (LPS/GalN) treatment (inflammation) and cecal ligation and puncture (CLP, polymicrobial sepsis). LPS/D-GalN-induced hepatotoxicity results from increased expression of proinflammatory cytokines, for example, tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6), along with the generation of reactive oxygen species (ROS) and activation of the downstream signaling cascade, whereas cecal ligation and puncture (CLP), a well-established murine model of sepsis, leads to bacterial peritonitis, systemic cytokine storm, multiorgan injury, bacteremia, and mortality (21, 22). To gain mechanistic insights into P2Y2 purinergic receptor-mediated effects on the induction of inflammatory liver injury, experiments were designed to test the impact of P2Y2 purinergic receptor antagonist pre treatment on 1) cytokine and chemokine release in response to lipopolysaccharide-mediated toll-like receptor 4 (LPS/TLR4) activation of Raw264.7 macrophages and 2) macrophage-hepatocyte crosstalk, assessed by Raw264.7 macrophage-conditioned media treatment of AML-12 hepatocytes and evaluation of AML-12 hepatocyte apoptosis in vitro.
Arginine, a semiessential amino acid, is necessary for effective innate immune response to bacterial infection. Previous studies have identified that inducible nitric oxide synthase (iNOS) induction characterizes proinflammatory polarization and activation of macrophages, and arginine is necessary for the iNOS-mediated nitric oxide generation. Macrophages play a significant role in nitric oxide-mediated bacterial killing and phagocytosis of bacteria. Patients with lower circulating arginine had a higher risk for sepsis-associated morbidity and mortality (23–25). Liver function is critical for the maintenance of metabolic homeostasis including amino acid metabolism and plays a key role in arginine biosynthesis and metabolism. Liver dysfunction contributes to the severity and outcomes of sepsis, and it is an independent risk factor for increased morbidity and mortality in patients (26–29). Therefore, we hypothesized that P2Y2 purinergic receptor-mediated inflammatory liver injury, associated with dysregulation of systemic amino acid homeostasis and impaired host response to bacterial infection, contributes to poor survival in response to polymicrobial sepsis.
Here, we report that P2Y2 purinergic receptor gene-deleted mice (P2Y2−/−) exhibit significant attenuation of liver injury and inflammation and improved survival in response to LPS/GalN treatment as well as CLP, compared with wild-type (WT) mice. P2Y2 purinergic antagonist pretreatment of macrophages attenuated 1) LPS/TLR4-mediated cytokine and chemokine (MCP-1, IL-6, IL-10) release in Raw264.7 macrophages and 2) hepatocyte apoptosis in response to Raw264.7 macrophage-conditioned media treatment of AML-12 hepatocytes in vitro. Furthermore, P2Y2 purinergic receptor-mediated inflammatory liver injury is associated with alteration in systemic amino acid homeostasis, most notably the depletion of circulating arginine, and increased bacterial load in the peritoneal cavity and the blood after CLP of the WT mice. Our studies highlight the potential to target P2Y2 purinergic signaling to prevent liver injury and maintenance of systemic amino acid homeostasis to prevent morbidity and mortality in polymicrobial sepsis.
METHODS
Animals
Adult male C57BL6J wild-type mice (WT) were purchased from Jackson Laboratory (Bar Harbor, ME). P2Y2−/− mice in C57BL6/J background were a kind gift from Dr. Beverly Koller of the University of North Carolina Chapel Hill (30). Both WT and P2Y2−/− mice were bred and housed in a specific-pathogen-free animal facility. All experiments were conducted following the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (IACUC) of Baylor College of Medicine (Houston, TX).
LPS/Galactosamine-Mediated Model of Acute Liver Injury
Adult (10–12 wk old) male WT and P2Y2−/− mice were given a single intraperitoneal injection of galactosamine (GalN, 700 mg/kg; Sigma Chemical Co., St. Louis, MO) followed by injection with saline or LPS (E. coli 0111: B4, 100 µg/kg; Sigma Chemical Co., St. Louis, MO) with 25-gauge needle (BD Biosciences, Franklin Lakes, NJ). Liver tissues were harvested at 1 and 5 h after LPS/GalN treatment. Serum, RNA (qRT-PCR), and total protein extracts (Western blotting) isolated from liver tissues were analyzed for markers of liver injury.
Cecal Ligation and Puncture-Mediated Model of Polymicrobial Sepsis
Adult male (10–18 wk old) WT and P2Y2−/− mice were subjected to cecal ligation and puncture (CLP) to induce midgrade polymicrobial sepsis (50% cecal ligation) (22). Polymicrobial sepsis was induced by subjecting mice to CLP, as described by Rittirsch et al. (22). Briefly, WT and P2Y2−/− mice were anesthetized with isoflurane. Under aseptic conditions, a 1- to 2-cm midline laparotomy was performed, and the cecum was exteriorized. The identified cecum was tightly ligated midway between the distal pole and the base of the cecum with a 3.0-silk suture. The cecum was perforated twice (through and through) with a 21-gauge needle (BD Biosciences, Franklin Lakes, NJ) distal to the ligation. The cecum was then gently squeezed to extrude a small amount of feces from the perforation sites to ensure patency. The cecum was then relocated to the peritoneal cavity and the peritoneum, fasciae, and abdominal musculature were closed with sutures. The skin was closed using metallic clips. The mice were then returned to their cages with free access to food and water. As a control for CLP, sham surgeries were performed following the same procedures except for cecal ligation and puncture. Mice were divided into four experimental groups: 1) WT-Sham; 2) WT-CLP; 3) P2Y2−/−-Sham; and 4) P2Y2−/−-CLP. Mice were monitored daily and euthanized if moribund or at postoperative day 7, whichever was earlier. All animal experimentation was performed according to approved protocol by the IACUC of Baylor College of Medicine, Houston, TX.
Serum Analyses for Liver Injury and Inflammation
Serum alanine aminotransferase (ALT) and total bilirubin were analyzed by the Center for Comparative Medicine (BCM) Pathology Labs, Baylor College of Medicine, Houston, TX. Cytokines and chemokines were analyzed by the MILLIPLEX MAP Mouse cytokine/chemokine panel kit (Millipore) of serum samples harvested at 21 h after surgery, according to the manufacturer's instructions. The cytokines and chemokines analyzed included TNF-α, IL-1β, IL-6, IL-10, MCP-1, and MKC. Beads were assayed on a Luminex 100 xMAP technology machine (Luminex, Austin, TX) using Master PlexCT v.1.2.0.7 software (Hitachi, South San Francisco, CA). Standard curves were generated for each cytokine/chemokine using five-parameter logistic model equations. The mean fluorescence intensity for each analyte was calculated using Master PlexCT v.2.0.0.73 (Hitachi, South San Francisco, CA).
Amino Acids Analysis
The metabolites were extracted from mouse serum using the liquid-liquid extraction method and analyzed by high-throughput liquid chromatography-mass spectrometry (LC-MS) described previously (31–34). The extracted samples were resuspended into methanol-water (50:50 vol/vol). The chromatographic separation of extracted amino acids was performed through Hydrophilic Interaction Chromatography (HILIC) separation mode with XBridge Amide column (3.5 µm, 4.6 × 100 mm, Waters, Milford, MA) in ESI-positive ionization. In water and acetonitrile, formic acid (0.1%) was used as mobile phases A and B, respectively. The data were acquired via multiple reaction monitoring (MRM) using a 6495 Triple Quadrupole mass spectrometry coupled to an HPLC system (Agilent Technologies, Santa Clara, CA) through Agilent Mass Hunter Data Acquisition Software (version 10.1) (34, 35). The acquired mass spectra were analyzed and integrated into each targeted compound peak using Agilent Mass Hunter Quantitative Analysis Software. The reviewed peak area was log2 transformed and normalized by an isotopically spiked internal standard. The amino acids with a significant change in their relative abundance were determined using P values (P < 0.05) of the t test following the Benjamini–Hochberg method with less than 0.25 false-discovery rate (FDR).
Assessment of Bacterial Load in Blood and Peritoneal Fluid
Analyses of aerobic bacterial counts were performed on aseptically obtained blood and peritoneal fluid at 6, 12, and 21 h after surgery. Samples were serially diluted in sterile normal saline and plated on 5% tryptic soy agar plates. Plates were incubated at 37°C for 24 h and colony-forming units (CFUs) were manually counted. Accordingly, the results were expressed as the number of CFU per milliliter of blood or peritoneal fluid.
RNA Analysis by Real-Time qRT-PCR
RNA was isolated from the livers harvested at 21 h post-CLP, and 1 and 5 h post-LPS/GalN treatment, using Qiagen RNeasy mini kit, according to the manufacturer’s instructions (Qiagen Sciences, MD). Complementary DNA (cDNA) synthesis was performed by reverse transcription of total RNA (2 μg) with high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). The cDNA product was amplified by qRT-PCR in ABI prism 7700 sequence-detection using SYBR Green (Applied Biosystems, Foster City, CA). Quantitative expression values were determined using the ΔΔCt method as specified by the manufacturers and using cyclophilin as a control. Data are expressed as relative expressions compared with the control group (WT-Sham). DNA sequences of gene-specific primers are listed in Table 1.
Table 1.
Primers used for the qRT-PCR analysis
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| TNF-α | CATCTTCTCAAAATTCGAGTGACAA | TGGGAGTAGACAAGGTACAACCC |
| IL-6 | CCGGAGAGGAGACTTCACAGA | AGAATTGCCATTGCACAACTCTT |
| IL-1β | CAACCAACAAGTGATATTCTCCATG | GATCCACACTCTCCAGCTGCA |
| MIP-2 | GCGCCCAGACAGAAGTCATAG | AGCCTTGCCTTTGTTCAGTATC |
| MCP-1 | TTAAAAACCTGGATCGGAACCAA | GCATTAGCTTCAGATTTACGGGT |
| iNOS | TCTTGGTGAAAGTGGTGTTCTTTT | AGTAGTTGCTCCTCTTCCAAGGT |
| ICAM | GTGATGCTCAGGTATCCATCC | CACAGTTCTCAAAGCACAGCG |
| MMP-9 | CCTGGAACTCACACGACATCTTC | TGGAAACTCACACGCCAGAA |
| Cyclophilin | GGCCGATGACGAGCCC | TGTCTTGGAACTTTGTCTGCA |
TNFα, tumor necrosis factor-α; IL-6, interleukin-6; IL-1β, interleukin 1β; MCP-1, monocyte chemoattractant protein-1; iNOS, inducible nitric oxide synthase; MMP-9, matrix metallopeptidase-9.
Histology and Immunohistochemistry
Liver samples harvested at 12 and 21 h post-CLP or 5 h post-LPS/GalN treatment were fixed in 10% formaldehyde at room temperature for 24 h. After hematoxylin-eosin staining, paraffin-embedded tissue sections (5 μm) were analyzed by light microscopy with Nikon Eclipse Ni microscope equipped with a camera (DS Fi1) and NIS-Elements AR 5.20.00 (Build 1423) imaging software (Nikon, Melville, NY). To quantify the extent of leukocyte infiltration, liver sections were immunostained for CD45+ cells with anti-mouse CD45 antibody (Pan-leukocyte marker, Cat. No. 550539, BD Biosciences, Franklin Lakes, NJ; 1:25 dilution), and secondary antibody [goat anti-rat IgG antibody (H + L), Biotinylated, Cat. No. BA-9400, Vector Labs, Newark, CA; 1:200 dilution] and randomly selected fields were assessed by an unbiased stereological technique described by Howard and Reed (36, 37). Liver sections were immunostained for F4/80+ cells with anti-mouse F4/80 antibody (macrophage marker, Cat. No. MCA497, AbD Serotec-Bio Rad, Hercules, CA; 1:250 dilution) and secondary antibody (ImmPRESS HRP goat anti-rat IgG, Cat. No. MP-7444, Vector Labs, Newark, CA) to assess macrophage infiltration. Apoptosis was detected by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) using Roche in situ apoptosis detection kit (Mannheim, Germany). Percent TUNEL-positive nuclei were calculated based on the analysis of nuclei in 10 random fields of view for each liver section.
Cell Culture and Experimental Reagents
RAW 264.7 murine macrophages were purchased from the American Type and Culture Collection (Cat. No. TIB-71, ATCC, Manassas, VA). Cells were cultured in Dulbecco’s modified Eagle medium (DMEM, Cat. No. 30–2002, ATCC, Manassas, VA) supplemented with 2 mM glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin (Thermo Fisher, Waltham, MA) and 10% fetal bovine serum (FBS, Cat. No. 30–2020, ATCC, Manassas, VA) at 37°C in a humidified 5% CO2 atmosphere. LPS (LPS-SM Ultrapure, TLR4 agonist, Cat. No. Tlrl smlps) was purchased from Invivogen (San Diego, CA). To determine if P2Y2 purinergic receptor function is necessary for LPS-mediated induction of cytokine and chemokine synthesis and secretion, Raw264.7 macrophages (1 × 106 cells/well, 6-well tissue culture plates) were treated with LPS (100 ng/mL, TLR4 agonist) for 7.5 h or pretreated with AR-C118925 (AR-C, 50 µM, P2Y2 antagonist, Tocris, MN) 30 min before LPS treatment. DMSO (0.2%) served as vehicle control (Veh) for AR-C treatment. At the end of the treatment, conditioned cell culture media was centrifuged at 10,000 rpm for 10 min (to remove cell debris), and the supernatants were stored at −80°C.
AML-12 hepatocytes were purchased from the American Type and Culture Collection (Cat. No. CRL-2254, ATCC, Manassas, VA). Cells were cultured in Dulbecco’s modified Eagle medium-F12 (DMEM-F12, 1:1 mixture, Cat. No. 30–2006, ATCC, Manassas, VA) supplemented with 100 U/mL penicillin and 100 µg/mL streptomycin (Thermo Fisher, Waltham, MA) and 10% fetal bovine serum (FBS, Cat. No. -2020, ATCC, Manassas, VA) at 37°C in a humidified 5% CO2 atmosphere. To evaluate the role of P2Y2 purinergic receptor function in macrophage-mediated hepatocellular injury, AML-12 cells grown in 12-well plates (500,000 cells/well) were washed once with warm phosphate-buffered saline (PBS) and exposed to RAW264.7 culture supernatants [conditioned media obtained after treatment with 1) Veh, 0.2% DMSO, 2) 50 µM AR-C, 3) Veh + 100 ng/mL LPS, 4) 50 µM AR-C + 100 ng/mL LPS for 7.5 h] for 30 h, and total protein extracts of AML-12 cells were analyzed by Western blotting with antibodies specific for iNOS (Abcam, Cat. No. 15323), cleaved caspase-3 (CST, Cat. No. 9661), and cleaved PARP-1 (CST, Cat. No. 9541).
Mouse Cytokine Proinflammatory Focused 10-Plex Discovery Assay Array (MDF10)
Cell culture supernatants were analyzed using Luminex xMAP technology for multiplexed quantification of 10 cytokines and chemokines. The multiplexing analysis was performed using the Luminex 200 system (Luminex, Austin, TX) by Eve Technological Corp. (Calgary, Alberta, Canada). Ten markers were simultaneously measured in the samples using Eve Technologies’ Mouse Proinflammatory Focused 10-Plex Discovery Assay (MilliporeSigma, Burlington, MA) according to the manufacturer’s protocol. The 10-plex consisted of MCP-1, IL-6, IL-10, TNFα, GM-CSF, IL-2, IFNγ, IL-1β, IL-12p70, and IL-4. Assay sensitivities of these markers range from 0.4 to 10.9 pg/mL for the 10-plex.
Western Blotting
Total protein extracts obtained by homogenizing liver segments in total lysis buffer (50 mM Tris·HCl, pH 7.5, 0.5 M NaCl, 2 mM EGTA, 2 mM EDTA, 1.0% Triton X-100, 0.25% deoxycholate, 1.0 µg/mL pepstatin, 1.0 µg/mL leupeptin, 1.0 µg/mL aprotinin, 1.0 mM phenylmethylsulfonyl fluoride, 1.0 mM dithiothreitol, 2.0 mM activated Na3VO4, and 2.0 mM NaF) and centrifuging at 14,000 rpm for 10 min (4°C). As described previously, equal amounts of total proteins as determined by BCA protein assay (Pierce, Rockford, IL) were analyzed by Western blotting (38, 39). Briefly, total protein extracts were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using the Bio-Rad mini-PROTEAN Tetra Cell gel electrophoresis system and transferred to nitrocellulose membranes using the Bio-Rad Tetra blotting module (Bio-Rad, Hercules, CA). Blots were probed with antibodies specific for matrix metallopeptidase-9 (MMP-9) (Abcam, Cat. No. 38898, Waltham, MA), phospho-c-Jun NH2-terminal kinase (JNK) (CST, Cat. No. 9251, Danvers, MA), total JNK (CST, Cat. No. 9252), cleaved caspase-3 (CST, Cat. No. 9661), cleaved PARP-1 (CST, Cat. No. 9541), iNOS (Abcam, Cat. No. 15323), and nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain containing 3 (NLRP3) (CST, Cat. No. 15101). α-Tubulin (Sigma, Cat. No. T9026, St. Louis, MO), β-actin (Sigma, Cat. No. A5316), or GAPDH (CST, Cat. No. 2118) served as loading control. Relative expressions were calculated based on densitometric analysis of band intensities measured using the ImageQuant TL1 8.1 image analysis software (Cytiva, Marlborough, MA). Antibodies and dilutions are listed in Table 2.
Table 2.
Antibodies used for Western blotting
| Antibody | Source | Cat. No. | Primary Antibody Dilution | Secondary Antibody and Dilution |
|---|---|---|---|---|
| MMP-9 | Abcam | 38898 | 1:1,000 | GAR-HRP, 1:2,000 |
| TBST + 5% BSA | TBST + 5% NFDM | |||
| Phospho-JNK (Thr183/Tyr185) | Cell Signaling Technology | 9251 | 1:1,000 | GAR-HRP1, 2,000 |
| TBST + 5% BSA | TBST + 5% NFDM | |||
| JNK | Cell Signaling Technology | 9252 | 1:1,000 | GAR-HRP, 1:2,000 |
| TBST + 5% BSA | TBST + 5% NFDM | |||
| Cleaved PARP-1 (Asp214) | Cell Signaling Technology | 9541 | 1:500 | GAR-HRP, 1:2,000 |
| TBST + 5% NFDM | TBST + 5% NFDM | |||
| Cleaved Caspase-3 (Asp175) | Cell Signaling Technology | 9661 | 1:500 | GAR-HRP, 1:2,000 |
| TBST + 5% NFDM | TBST + 5% NFDM | |||
| iNOS | Abcam | 15323 | 1:1,000 | GAR-HRP, 1:2,000 |
| TBST + 5% BSA | TBST + 5% NFDM | |||
| NLRP3 (D4D8T) | Cell Signaling Technology | 15101 | 1:1,000 | GAR-HRP, 1:2,000 |
| TBST + 5% BSA | TBST + 5% NFDM | |||
| α-Tubulin | Sigma | T9026 | 1:4,000 | HAM-HRP, 1:2,000 |
| TBST + 5% BSA | TBST + 5% NFDM | |||
| β-Actin | Sigma | A5316 | 1:10,000 | HAM-HRP, 1:2,000 |
| TBST + 5% BSA | TBST + 5% NFDM | |||
| GAPDH | Cell Signaling Technology | 2118 | 1:4,000 | GAR-HRP, 1:2,000 |
| TBST + 5% BSA | TBST + 5% NFDM |
BSA, bovine serum albumin; GAR-HRP, goat anti-rabbit IgG, horseradish peroxidase-linked antibody; HAM-HRP, horse anti-mouse IgG, horseradish peroxidase-linked antibody; iNOS, inducible nitric oxide synthase; JNK, c-Jun NH2-terminal kinase; MMP-9, matrix metallopeptidase-9; NLRP3, nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain containing 3; NFDM, nonfat dry milk; TBST, tris-buffered saline.
Statistical Analysis
The comparisons between multiple groups were made by analysis of variance (ANOVA), followed by a post hoc Bonferroni test. The unpaired Student's t test (two-tailed) was applied in analyzing data comparisons between the two groups. The survival rate was analyzed using the Kaplan–Meier curve with Log-rank (Mantel-Cox) test for significance. The values are expressed as means ± SD. A P value of <0.05 was considered significant. Amino acid analysis data and bacterial colony counts are represented by Box and Whisker plots with whiskers representing the minimum and the maximum values with all data points indicated in the plots. All data were analyzed with the statistical software GraphPad Prism version 7.00 for Windows (GraphPad Software, La Jolla, CA; www.graphpad.com).
RESULTS
Acute Liver Injury, Inflammation, and Hepatocyte Apoptosis Are Attenuated in P2Y2−/− Mice in Response to LPS/GalN Administration
Extracellular nucleotides (ATP, UTP) via the activation of P2Y2 purinergic receptors influence multiple hepatic functions (19, 20, 39–45). Despite its ubiquitous expression, the pathophysiological relevance of P2Y2 purinergic receptor-mediated activation of liver-resident and infiltrating macrophages and their role in endotoxin-induced acute liver injury is currently unknown. In this study, we sought to determine if P2Y2 receptor function was necessary for the induction of acute liver injury in response to LPS/GalN treatment.
LPS-GalN treatment led to acute inflammatory liver injury as evidenced by the H&E analysis of liver sections of the WT mice. Hemorrhagic necrotic lesions and loss of hepatic parenchyma, evident in the WT-LPS/GalN liver sections, were absent in P2Y2−/− livers (Fig. 1A). Serum ALT (a key biochemical marker of liver injury) was significantly lower in P2Y2−/− mice after LPS-GalN treatment (Veh vs. LPS/GalN: WT, 12-fold, P < 0.001; P2Y2−/−, 2-fold, P < 0.05; Fig. 1B). To evaluate the influence of P2Y2 purinergic receptors on leukocyte infiltration during inflammatory liver injury, liver sections (5 h post-LPS-GalN) were analyzed by immunohistochemistry for CD45+ cells. Unbiased quantification demonstrated a significant (54%, P < 0.05) reduction in infiltrating CD45+ leukocytes in P2Y2−/− livers when compared with their WT counterparts (Fig. 1, C and D). LPS/GalN treatment led to increased macrophage infiltration, as compared with vehicle (saline)-treated control livers in the WT mice with attenuation of F4/80+ macrophage infiltration observed in P2Y2−/− livers (Fig. 1E). Immune cell infiltration and activation of endothelial cells aggravate tissue injury via the release of matrix metalloprotease-9 (MMP-9) (46). MMP-9-mediated proteolytic degradation of the extracellular matrix contributes to liver injury via leukocyte infiltration and extravasation into hepatic parenchyma (47, 48). Suggesting a role for P2Y2 purinergic receptor function in the upregulation of MMP-9 expression during inflammatory liver injury, LPS/GalN treatment led to a robust induction of MMP-9 protein expression in the WT livers at 1 h (5.2-fold, P < 0.05) and 5 h (6.3-fold, P < 0.05) with attenuated induction of MMP-9 in P2Y2−/− livers at both time points (2.8-fold, P < 0.05, 1 h; 2.4-fold, P = 0.30, 5 h; Fig. 1, F–I).
Figure 1.
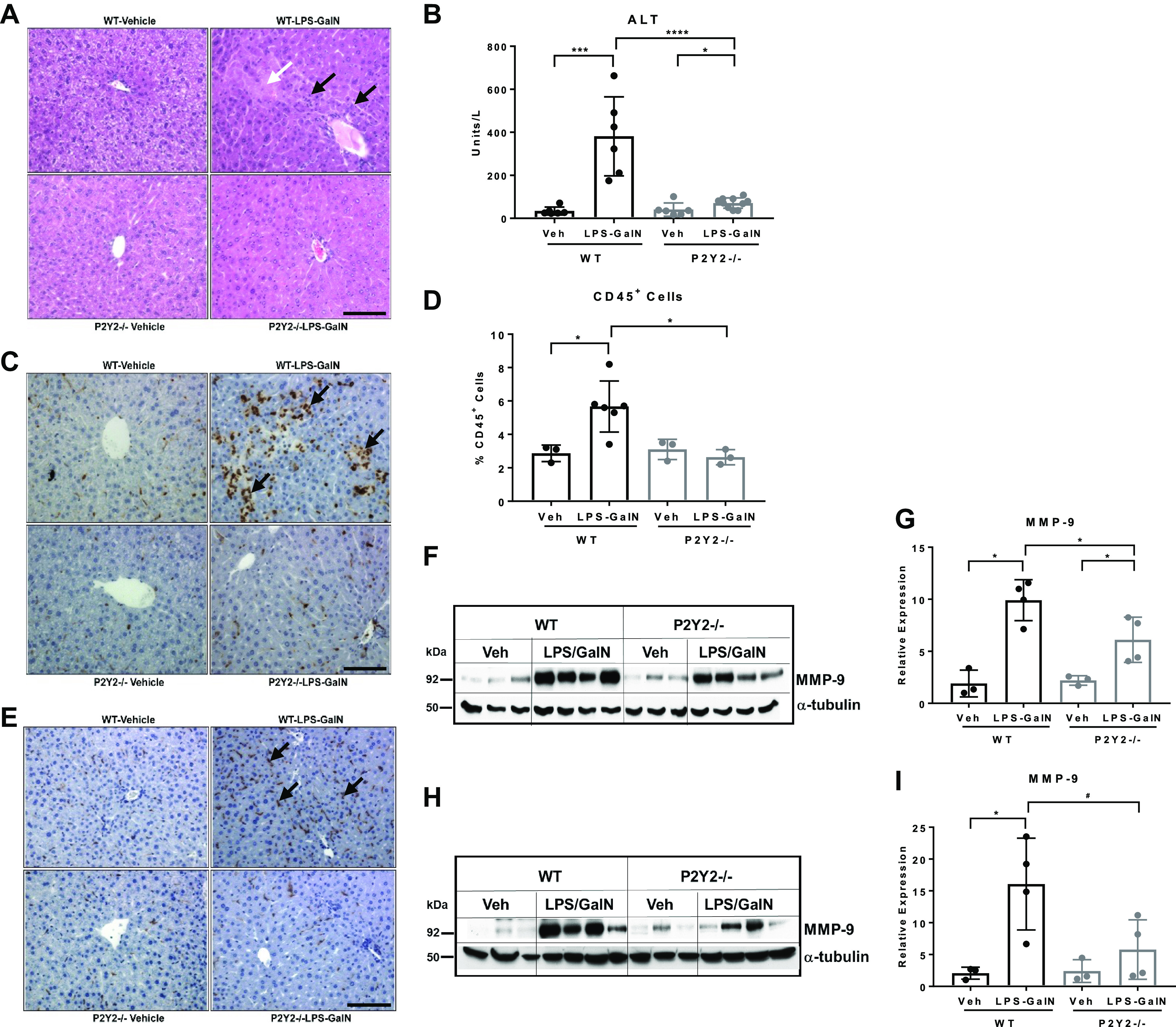
LPS/GalN-mediated acute liver injury is attenuated in P2Y2−/− mice. WT and P2Y2−/− mice treated with LPS (100 µg/kg) and galactosamine (GalN, 700 mg/kg) or saline (Veh) for 1 and 5 h. A: representative H&E-stained liver sections (×20 field of view). White arrow points to the necrotic lesion and black arrows point to leukocyte infiltration within hepatic parenchyma. Black scale bar, 100 µm. B: serum ALT (n, WT-Veh, 5, WT-LPS/GalN, 5; P2Y2-Veh, 7; P2Y2-LPS/GalN, 11). C: representative liver sections stained for CD45+ leukocytes (×20 field of view). Black arrows point to leukocyte infiltration within hepatic parenchyma. Black scale bar, 100 µm. D: quantified by a stereological method at ×60 magnification by observers blinded to treatment groups. Bar diagram represents the analysis of 15 fields of view in each liver section (n, WT-Veh, 3, WT-LPS/GalN, 6; P2Y2-Veh, 3; P2Y2-LPS/GalN, 3). E: representative F4/80-stained liver sections (×20 field of view). Black arrows point to macrophage infiltration. Black scale bar, 100 µm. Total protein extracts of livers harvested at 1 h (F–G) and 5 h (H–I) post-LPS-GalN were analyzed by Western blotting with antibodies specific for MMP-9 and α-tubulin (protein loading control; n, WT-Veh, 3, WT-LPS/GalN, 4; P2Y2-Veh, 3; P2Y2-LPS/GalN, 4). Bar diagrams represent relative expression. Means ± SD, *P < 0.05, ***P < 0.001, ****P < 0.0001, #P = 0.05. n, number of biological replicates. Veh, vehicle.
Inflammatory liver injury is associated with the activation of JNK signaling, a key mediator of proinflammatory cytokine-mediated mitochondrial dysfunction and hepatocyte apoptosis (49, 50). The temporal profile of JNK activation in hepatocytes has divergent outcomes. Whereas transient JNK activation is associated with hepatocyte proliferation, sustained JNK activation is a hallmark of drug-induced liver injury orchestrated by oxidative stress and mitochondrial dysfunction (40, 51). To gain mechanistic insights and identify potential mediators of liver injury dependent on P2Y2 purinergic receptor function, we evaluated the phosphorylation and activation status of JNK signaling by Western blotting of total liver protein extracts isolated at 1 h and 5 h after LPS/GalN treatment. LPS/GalN treatment led to sustained JNK activation in the WT (8.1-fold, 1 h, P < 0.01; 4.3-fold, P < 0.001, 5 h) with attenuated induction in P2Y2−/− livers (5.7-fold, 1 h, P < 0.01; 1.3-fold, P = 0.39, 5 h; Fig. 2, A–D). Featuring proinflammatory activation status of liver-resident and infiltrating leukocytes, mRNA expression of key inflammatory mediators (TNFα, IL-1β, MIP-2 at 1 h; and MCP-1, ICAM-1, iNOS at 5 h post-LPS-GalN) was elevated in the WT liver with significant attenuation (∼50%–80%, P < 0.05) in the P2Y2−/− livers (Fig. 2, E–J).
Figure 2.

LPS/GalN-mediated induction of JNK signaling and hepatic inflammation is attenuated in P2Y2−/− mice. Total protein extracts of livers harvested at 1 h (A and B) and 5 h (C and D) post-LPS-GalN were analyzed by Western blotting with antibodies specific for phospho-JNK, total JNK, and β-actin (protein loading control; n, WT-Veh, 3, WT-LPS/GalN, 4; P2Y2-Veh, 3; P2Y2-LPS/GalN, 4). E–J: qRT-PCR analysis of total RNA isolated from the livers post-LPS/GalN at 1 h: TNFα, IL1β, MIP2; post-LPS/GalN at 5 h: MCP1, ICAM, iNOS (n, WT-Veh, 5, WT-LPS/GalN, 5; P2Y2-Veh, 7; P2Y2-LPS/GalN, 7–9). Bar diagrams represent relative expression. Means ± SD, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. n, number of biological replicates. Veh, vehicle; WT, wild type.
TNFα-induced hepatocyte apoptosis is an early event and precedes liver failure in experimental models of inflammatory liver injury (52). Since LPS/GalN-mediated induction of TNFα mRNA expression and JNK activation were attenuated in P2Y2−/− livers, when compared with WT, we evaluated hepatocyte apoptosis by TUNEL staining. LPS/GalN treatment led to increased TUNEL-positive apoptotic nuclei in the WT with a significant reduction (76%, P < 0.001) in the number of apoptotic nuclei detected in P2Y2−/− livers (Fig. 3, A and B). Caspase-mediated PARP-1 cleavage is a surrogate marker for apoptosis. We evaluated cleaved PARP-1 (89 kDa) protein expression by Western blotting of total liver protein extracts. LPS/GalN treatment induced cleaved PARP-1 protein expression in the WT livers (7-fold, P < 0.001) with significant attenuation of induction observed in P2Y2−/− livers (0.7-fold, P = 0.88; Fig. 3, C and D). These findings highlight a role for P2Y2 purinergic signaling in the induction of hepatocyte apoptosis during inflammatory liver injury.
Figure 3.

LPS/GalN-mediated induction of hepatocyte apoptosis is attenuated in P2Y2−/− mice. A: representative images of liver sections after TUNEL staining (×40 field of view; 5 h post-LPS/GalN). White arrows, apoptotic nuclei. White scale bar, 50 µm. B: bar diagram represents the percentage of TUNEL-positive nuclei, based on the analysis of 10 fields of view (×40) in each liver section (n, WT-Veh, 5, WT-LPS/GalN, 8; P2Y2-Veh, 5; P2Y2-LPS/GalN, 8). C and D: total protein extracts of livers harvested at 5 h post-GalN/LPS were analyzed by Western blotting for Cleaved PARP-1 and α-tubulin (protein loading control). Bar diagram represents relative expression (n, WT-Veh, 3; WT-LPS/GalN, 4; P2Y2-Veh, 3; P2Y2-LPS/GalN, 4). E: Kaplan–Meier curve representing percentage survival of WT vs. P2Y2−/− after LPS/GalN treatment (n, 16–17). Means ± SD, *P < 0.05, ***P < 0.001. n, number of biological replicates.
P2Y2−/− Exhibits Improved Survival in Response to Endotoxin-Mediated Inflammatory Liver Injury
LPS-GalN-mediated lethality in mice has been attributed to liver-specific apoptotic DNA fragmentation and liver failure and mice become moribund within 12 h (21). Since P2Y2−/− mice had attenuated liver inflammation, apoptosis, and liver injury in response to LPS/GalN treatment when compared with WT mice, we evaluated the pathophysiological and clinical relevance of these observations by comparing their outcomes on morbidity and mortality. At 12 h after treatment, 63% of WT mice were moribund and required emergency euthanasia, whereas 24% of P2Y2−/− were moribund during this time (P < 0.05). Based on the Kaplan–Meyer analysis, the percentage survival, comparing the WT and P2Y2−/− mice in response to LPS/GalN treatment, is presented in Fig. 3E.
Collectively, these findings (Figs. 1–3) on the attenuation of liver injury, inflammation, and apoptosis in P2Y2−/− livers based on the biochemical, histologic, and RNA analysis, and our observation of improved survival of P2Y2−/− mice in response to LPS/GalN treatment has uncovered an essential role of P2Y2 purinergic receptor function in endotoxin-induced acute liver failure in mice. Next, we sought to determine whether P2Y2 receptor function is necessary for liver inflammation, bacteremia, and host survival in a well-established and clinically relevant mouse model of CLP-induced polymicrobial sepsis.
Liver Injury and Leukocyte Infiltration Is Attenuated in P2Y2−/− Mice during Polymicrobial Sepsis
Light microscopic analysis of the hematoxylin and eosin (H&E) liver sections of P2Y2−/− after CLP showed marked attenuation of histological markers of liver injury (leukocyte infiltration and hemorrhagic necrosis) when compared with WT (Fig. 4A). On biochemical analysis, serum ALT and bilirubin concentrations were significantly lower in P2Y2−/− mice at 21 h post-CLP (CLP vs. sham, ALT: 6.3-fold, P < 0.0001, WT; 1.4-fold, P = 0.43, P2Y2−/−; bilirubin: 3.3-fold, P < 0.05, WT; 1.0-fold, P2Y2−/−; Fig. 4, B and C). To evaluate the influence of P2Y2 purinergic receptor-mediated effects on leukocyte infiltration during sepsis-induced liver injury, liver sections (21 h post-CLP) were analyzed by immunohistochemistry for CD45+ cells. Unbiased quantification demonstrated a significant (59%, P < 0.01) reduction in infiltrating CD45+ leukocytes in P2Y2−/− livers when compared with their WT counterparts (Fig. 4, D and E). Sepsis-induced liver injury (21 h post-CLP) is associated with increased macrophage infiltration in the WT livers with attenuation of F4/80+ macrophage infiltration observed in P2Y2−/− livers (Fig. 4F).
Figure 4.
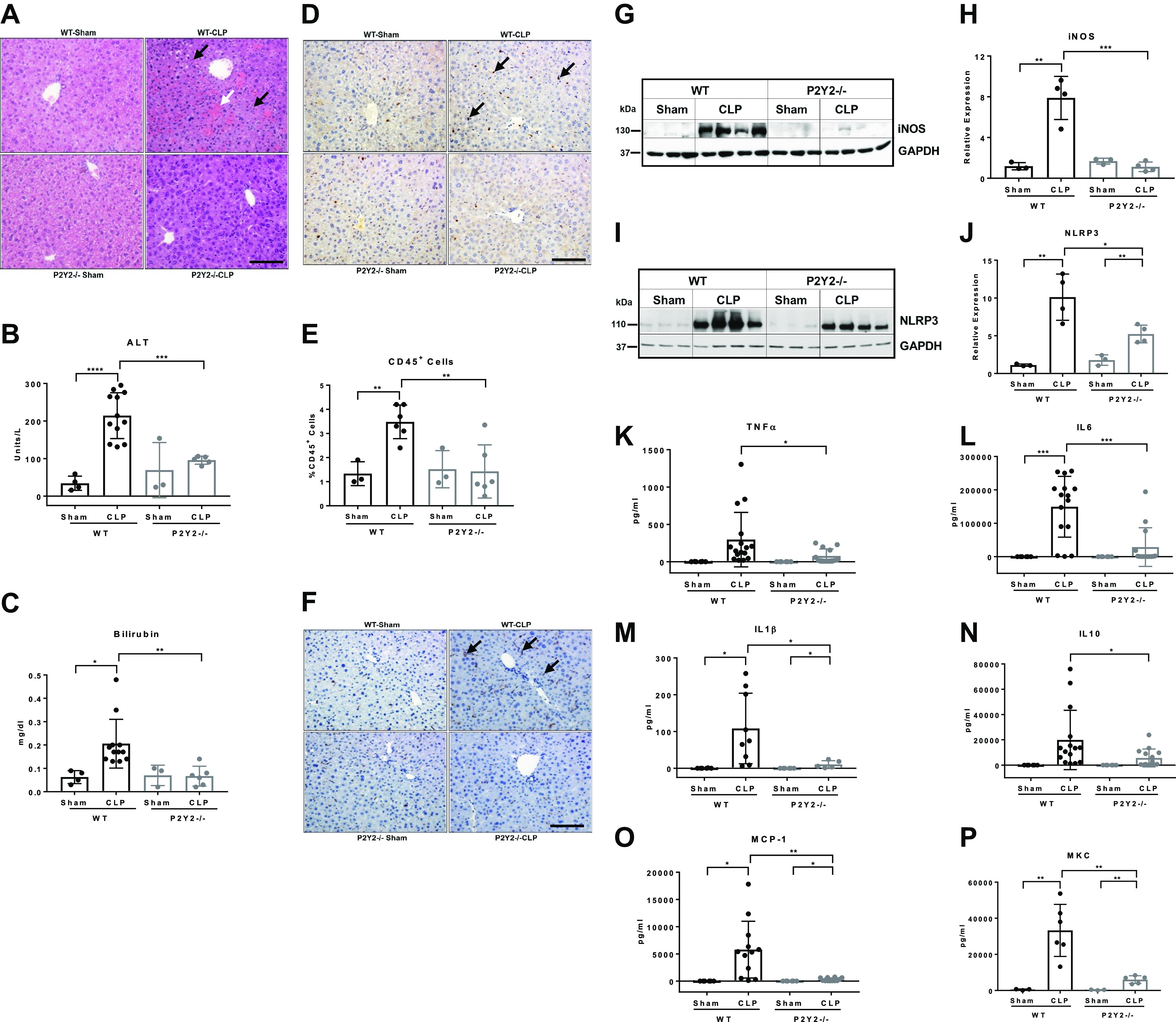
P2Y2−/− mice are protected from sepsis-induced systemic inflammation and liver injury. WT and P2Y2−/− mice were subjected to CLP or sham surgery, and the liver and blood were harvested at 21 h post-CLP. A: representative images of H&E-stained liver sections, ×20 field of view. White arrow points to hemorrhagic necrosis and black arrows point to immune cell infiltrates. Black scale bar, 100 µm. B: serum ALT (n, WT-Sham, 4; WT-CLP, 12; P2Y2-Sham, 3; P2Y2-CLP, 5). C: serum bilirubin (n, WT-Sham, 4; WT-CLP, 12: P2Y2-Sham, 3; P2Y2-CLP, 6). D: representative liver sections stained for CD45+ leukocytes (×20 field of view). Black arrows point to leukocyte infiltration within hepatic parenchyma. Black scale bar, 100 µm. E: quantified by a stereological method at ×20 magnification by observers blinded to treatment groups. Bar diagram represents the analysis of five fields of view in each liver section (n, WT-Sham, 3, WT-CLP, 6; P2Y2-Sham, 3; P2Y2-CLP, 6). F: representative F4/80-stained liver sections (×20 field of view). Black arrows point to macrophage infiltration. Black scale bar, 100 µm. G–J: total protein extracts isolated from the livers harvested after CLP were analyzed by Western blotting with antibodies specific for iNOS (12 h post-CLP), NLRP3 (21 h post-CLP), GAPDH (protein loading control; n, WT-Sham, 3; WT-CLP, 4; P2Y2-Sham, 3; P2Y2-CLP, 4). Bar diagrams represent relative expression. K–P: multiplex assay of serum cytokines, TNFα, IL6, IL1β, IL10, MCP1, MKC (n, WT-Sham, 6; WT-CLP, 9–16; P2Y2-Sham, 6; P2Y2-CLP, 5–14). Means ±SD, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. n, number of biological replicates. CLP, cecal ligation and puncture; WT, wild type.
Bioenergetic failure due to the overproduction of nitric oxide has been identified as an important pathophysiological mechanism responsible for multiorgan dysfunction in sepsis (53). During inflammatory liver injury, inducible nitric oxide synthase (iNOS), which generates nitric oxide from arginine, is upregulated in hepatocytes and nonparenchymal cells. CLP led to robust iNOS protein induction in the WT livers (6.7-fold, P < 0.01) with no appreciable induction in P2Y2−/− livers (0.7-fold, P = 0.13; Fig. 4, G and H). Inflammatory liver injury is enhanced by the activation of the NLR family pyrin domain containing 3 (NLRP3) inflammasome, responsible for the maturation of proinflammatory cytokines IL-1β and IL-18 and hepatocyte death. Corresponding to the extent of liver injury observed in the WT and P2Y2−/− livers, CLP induced NLRP3 protein expression in the WT (9.1-fold, P < 0.01) with attenuated induction in P2Y2−/− livers (2.9-fold, P < 0.01; Fig. 4, I and J).
Systemic Proinflammatory Cytokine/Chemokine Induction in Response to CLP Is Attenuated in P2Y2−/− Mice
We evaluated systemic inflammatory responses to sepsis by measuring serum cytokine/chemokine levels in mice 21 h after CLP by MILLIPLEX MAP assay. CLP led to robust induction of all the cytokines and chemokines tested and serum concentrations of proinflammatory (TNFα, IL-6, IL-1β), anti-inflammatory (IL-10) cytokines, and chemokines (MCP-1, MKC) were significantly attenuated in P2Y2−/− mice in response to CLP compared with WT-CLP mice (Fig. 4, K–P). These cytokines and chemokines were either undetectable or comparable between the Sham-operated P2Y2−/− and WT mice.
P2Y2 Purinergic Receptor Antagonist Pretreatment Attenuates LPS-Mediated Induction of Cytokine (IL-6, IL-10) and Chemokine (MCP1/CCL2) Release from Raw264.7 Macrophages In Vitro
To gain mechanistic insights into the role of P2Y2 receptor function in macrophage activation and cytokine and chemokine release, Raw264.7 macrophages were treated with LPS (TLR4 agonist, 100 ng/mL, 7.5 h) or pretreated with Vehicle (0.2% DMSO) or AR-C118925 (P2Y2 antagonist, 50 µM, 30 min) before LPS treatment. Cell culture supernatants were analyzed by multiplex cytokine and chemokine assay. LPS treatment led to a robust induction chemokine (MCP-1) and cytokine (IL-6, IL-10, TNFα, GM-CSF) release. Whereas P2Y2 purinergic receptor antagonist pretreatment led to a significant attenuation of MCP-1, IL-6 and IL-10 release, LPS-mediated induction of TNFα, GM-CSF, and IL-2 was comparable between LPS versus AR-C + LPS treatment groups (Fig. 5, A–F). Multiplex cytokine assay did not detect appreciable INFγ, IL-1β, IL-4, and IL-12p70 release under the above experimental conditions. Our results suggest that intact P2Y2 purinergic receptor function is necessary for LPS-mediated induction of select cytokines (IL-6, IL-10) and chemokine (MCP-1) release in Raw 264.7 macrophages in vitro.
Figure 5.

P2Y2 purinergic receptor antagonist pretreatment attenuates LPS-mediated induction of cytokine (IL-6, IL-10) and chemokine (MCP1/CCL2) release from Raw264.7 macrophages in vitro. Raw264.7 macrophages were treated with LPS (100 ng/mL) for 7.5 h or pretreated with Vehicle (0.2% DMSO) or AR-C (50 µM, P2Y2 purinergic receptor antagonist) for 30 min before LPS treatment. A–F: multiplex assay for the cytokine and chemokine levels in the cell culture supernatants, MCP-1, IL-6, IL-10, TNFα, GM-CSF, IL-2 (n, Veh, 3; AR-C, 3; Veh + LPS, 3; AR-C+LPS, 3). Means ±SD, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. n, number of biological replicates.
P2Y2 Purinergic Receptor Antagonist Pretreatment of Raw264.7 Macrophages Inhibits iNOS Induction and Apoptosis in AML12 Hepatocytes Exposed to Macrophage-Conditioned Media In Vitro
Raw264.7 macrophages were treated with LPS (100 ng/mL) for 7.5 h or pretreated with Vehicle (Veh, 0.2% DMSO) or AR-C118925 (50 µM, P2Y2 purinergic receptor antagonist) for 30 min before LPS treatment. AML-12 hepatocytes were treated with macrophage culture supernatant (conditioned media) for 30 h, and total protein extracts of AML-12 hepatocytes were analyzed by Western blotting. AML-12 hepatocytes exposed to LPS-treated Raw264.7 macrophage culture supernatant had a robust induction of iNOS protein expression and cleaved caspase-3 and cleaved PARP-1, hallmarks of apoptosis in hepatocytes. Suggesting a role for P2Y2 purinergic receptor effects on macrophage secretion of humoral factors responsible for the induction of hepatocyte apoptosis, P2Y2 purinergic antagonist pretreatment before LPS treatment of Raw264.7 macrophages inhibited iNOS protein expression (Fig. 6, A and B) and induction cleaved caspase-3 and cleaved PARP-1 in AML-12 cells exposed to macrophage-conditioned media in vitro (Fig. 6, C–E).
Figure 6.
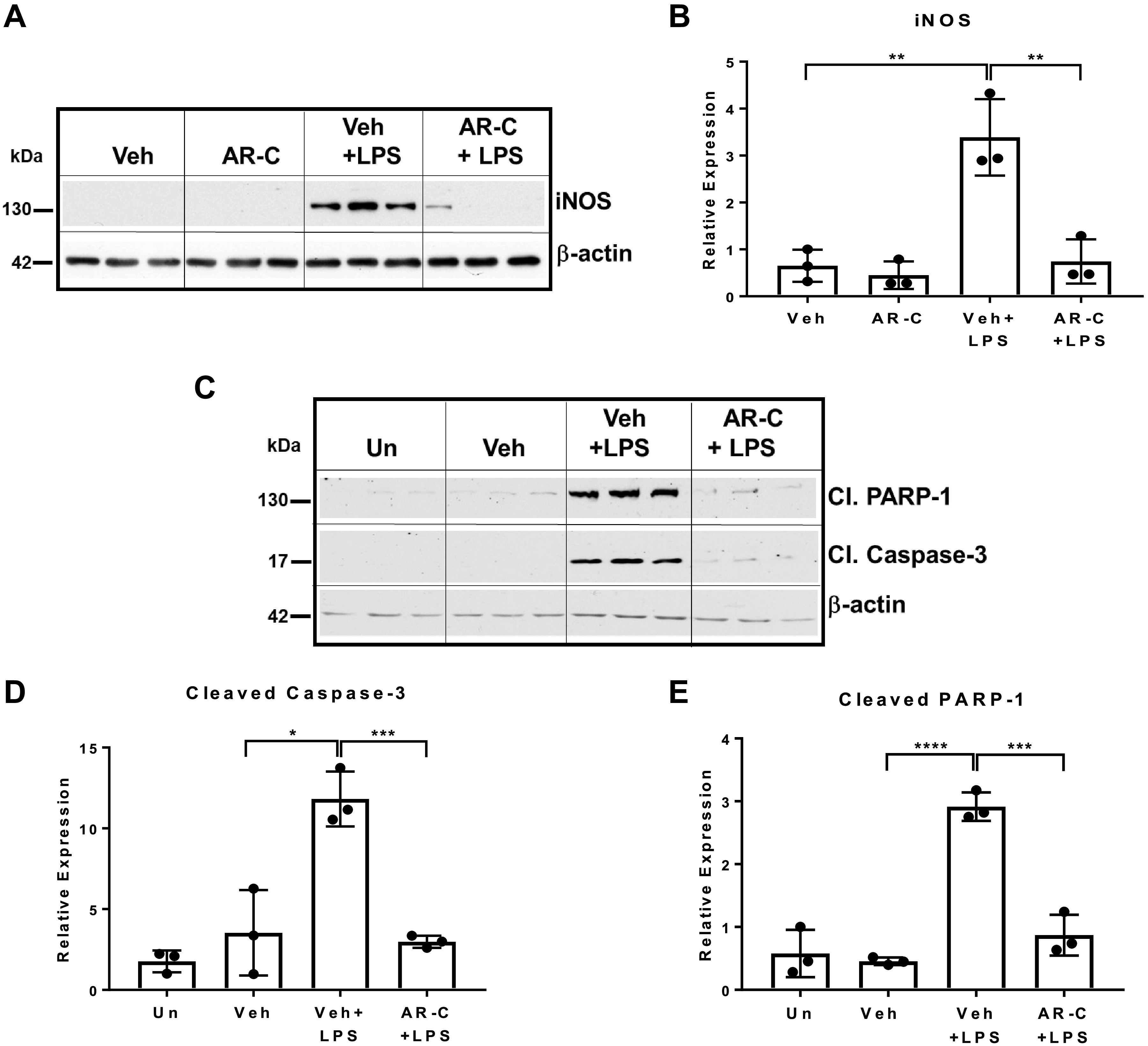
P2Y2 purinergic receptor antagonist pretreatment of Raw264.7 macrophages inhibits macrophage-mediated induction of iNOS and apoptosis in AML12 hepatocytes in vitro. Raw264.7 macrophages were treated with LPS (100 ng/mL) for 7.5 h or pretreated with Vehicle (Veh, 0.2% DMSO) or AR-C (50 µM, P2Y2 purinergic receptor antagonist) for 30 min before LPS treatment. AML-12 hepatocytes were treated with macrophage culture supernatant (conditioned media) for 30 h, and total protein extracts of AML-12 hepatocytes were analyzed by Western blotting with antibodies (A and B) specific for iNOS and β-actin (protein loading control; n, Veh, 3; AR-C, 3; Veh+LPS, 3; AR-C+LPS, 3). C–E: antibodies specific for cleaved PARP-1, cleaved Caspase-3, and β-actin [n, Untreated (Un), 3; Veh, 3; Veh +LPS, 3; AR-C+LPS, 3]. Bar diagrams represent relative expression. Means ± SD, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. n, number of biological replicates.
P2Y2 Purinergic Receptor-Mediated Liver Injury Is Associated with the Dysregulation of Systemic Amino Acid Homeostasis
To evaluate whether P2Y2 purinergic receptor-mediated potentiation of inflammation and liver injury leads to dysregulation of systemic amino acid homeostasis, specifically arginine depletion in response to CLP in mice, serum samples harvested after 21 h. CLP or sham surgery of WT and P2Y2−/− mice was analyzed by liquid chromatography-mass spectrometry (LC-MS). Our LC-MS analysis of serum samples revealed profound dysregulation of systemic amino acid homeostasis, as evident from the divergent serum amino acid profiles observed between the WT and P2Y2−/− mice. The heatmaps depicting the relative levels of serum amino acids of all four groups (WT-Sham, P2Y2−/− Sham, WT-CLP, P2Y2−/−CLP) and the relative levels of serum amino acids altered in response to CLP between the WT and P2Y2−/− mice are presented in Supplemental Fig. S1, A and B. Representative bar diagrams of amino acid levels that varied between the WT-CLP versus P2Y2−/−CLP are shown in Fig. 7, A–E. Despite arginine levels being comparable between the WT and P2Y2−/− mice subjected to Sham surgery, CLP led to reduced serum arginine levels in the WT. Serum arginine levels were comparable between the CLP and sham-operated P2Y2−/− mice (Fig. 7A).
Figure 7.

P2Y2−/− mice are protected from sepsis-induced depletion of serum arginine and dysregulation of amino acid homeostasis. WT and P2Y2−/− mice were subjected to CLP or sham surgery and blood harvested at 21 h post-CLP was subjected to serum amino acid profiling by liquid chromatography-mass spectrometry (LC-MS). Data are represented by Box and Whisker plots representing the amino acids with a significant change in their relative abundance (P < 0.05) arginine (A), ornithine (B), proline (C), isoleucine/leucine (D), and asparagine (E). Data were log2 transformed and normalized by an isotopically spiked internal standard (n, WT-Sham, 3; WT-CLP, 9; P2Y2-Sham, 3; P2Y2-CLP, 9), *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. n, number of biological replicates. CLP, cecal ligation and puncture; WT, wild type.
During Polymicrobial Sepsis, Bacterial Translocation and Bacteremia Are Attenuated in P2Y2−/− Mice
Septic patients have low serum arginine (25). Clinical studies show that arginine depletion in sepsis leads to decreased ability of the host to clear bacterial pathogens (23, 54). This impaired ability to mount an effective immune response to infection eventually leads to morbidity and mortality in sepsis. Based on our observation that CLP reduced serum arginine levels in the WT and the arginine levels were not altered in the P2Y2−/− mice (Fig. 7A), we reasoned that the potential to mount an effective antibacterial defense and control bacterial infection will be impaired in the WT mice. To test this, peritoneal fluid and blood harvested at 6, 12, and 21 h time point post-CLP or Sham surgeries of WT and P2Y2−/− mice were plated on 5% tryptic soy agar plates and incubated at 37°C for 24 h. Bacterial load in the peritoneal cavity and blood increased over time in the WT mice after CLP at 6, 12, and 21 h. Bacterial growth from the peritoneal fluid and blood harvested after 6, 12, and 21 h of CLP of P2Y2−/− mice decreased when compared with WT mice (peritoneal fluid: 84%–93%; blood: 93%–96%, Fig. 8, A and B). Bacterial growth was not observed from the samples collected after Sham surgeries (data not shown).
Figure 8.

Bacterial translocation, bacteremia, and mortality are attenuated in P2Y2−/−. Comparison of bacterial load expressed in colony-forming units per milliliter (CFU/mL) in peritoneal lavage fluid (A) and blood (B) at 6, 12, and 21 h post-CLP. (Peritoneal lavage fluid: n, WT, 4–9; P2Y2−/−, 6–10; Blood: n, WT, 6–8; P2Y2−/−, 5–8.) Data are represented by Box and Whisker plots, *P < 0.05, #P = 0.07, ##P = 0.06, ###P = 0.10. C: Kaplan–Meier 7-day survival curves. Log-rank (Mantel-Cox) analysis ****P < 0.0001 (n, WT-sham, 6, WT-CLP, 10; P2Y2-Sham, 6, P2Y2-CLP, 10). n, number of biological replicates. CLP, cecal ligation and puncture; WT, wild type.
P2Y2−/− Mice Are Protected from Sepsis-Induced Mortality
The above data reflecting significantly attenuated liver injury, and inflammation, along with maintenance of serum arginine levels, and efficient bacterial clearance in P2Y2−/− mice after CLP prompted us to evaluate the overall effect of the P2Y2 gene deletion on morbidity and mortality. WT and P2Y2−/− mice after sham surgery did not exhibit any signs of morbidity and all mice were viable when followed for 7 days. WT mice subjected to CLP developed severe symptoms of systemic infection and were moribund (100%) between 21 and 50 h. After 48 h of CLP, 70% of the WT mice were moribund and showed signs of systemic infection, as compared with 20% of P2Y2−/− mice. At 7 days post-CLP, 100% of WT mice were moribund and euthanized, whereas only 30% of P2Y2−/− mice were moribund and euthanized (P < 0.0001). The Kaplan–Meier analysis of survival data is presented in Fig. 8C.
Collectively, our findings presented in Figs. 4–8 based on the interrogation of a murine model of polymicrobial sepsis highlight the pathophysiological relevance of P2Y2 purinergic receptor function in inflammatory liver injury, dysregulation of systemic amino acid homeostasis, bacteremia, and morbidity leading to moribund mice requiring euthanasia.
DISCUSSION
Pathologic inflammatory response to bacterial infection leads to septic shock, multiorgan failure, and death. The onset of liver injury further worsens patient survival, perhaps due to the critical role played by the liver in immune regulation and metabolic homeostasis (26, 27). Using two distinct clinically relevant mouse models of liver injury (endotoxemia and sepsis), we evaluated the pathophysiological relevance of P2Y2 purinergic receptor function in the induction of inflammatory liver injury. We report that P2Y2 purinergic receptors play a key role in the induction of liver injury, inflammation, and arginine depletion in response to bacterial infection and regulation of bacterial load with a substantial impact on host morbidity and mortality.
Our studies suggest that the P2Y2 purinergic receptor, activated by extracellular ATP and UTP (damage-associated molecular patterns, DAMP), and TLR4 receptor, activated by bacterial endotoxin (pathogen-associated molecular patterns, PAMP), under their shared intracellular Ca2+ signaling, may regulate, potentiate, and “fine-tune” the functions of the respective DAMP and PAMP signaling during liver injury. TLR4 signaling, via activation of PLCγ, leads to IP3-mediated ER Ca++ release and elevation of intracellular Ca2+ (55). Extracellular nucleotide-mediated activation of P2Y2 purinergic receptors, Gq-coupled G proteins via activation of PLCβ and inositol trisphosphate (IP3) and activation of IP3R-mediated endoplasmic (ER) calcium (Ca2+) release, leads to elevation of cytoplasmic free Ca2+ and impact the activation of cell signaling (56). Moreover, TLR4 activation is known to induce connexin 43-mediated ATP release and has the potential to activate multiple purinergic receptor subtypes expressed in macrophages, including P2Y2. Recent studies have identified a role for connexin 43-mediated ATP release and P2Y1-mediated macrophage activation during sepsis (4, 57, 58). Lipopolysaccharide (LPS), a component of Gram-negative bacterial cell wall, via activation of toll-like receptor 4 (TLR4) signaling reprograms macrophage metabolism and proinflammatory polarization of macrophages (59). TLR4 activation alone is sufficient to induce nucleotide release and upregulation of P2Y2 purinergic receptor expression in macrophages (60, 61). Administration of suramin, a P2Y2 receptor antagonist with a broad specificity toward multiple P2 purinergic receptors, including P2Y2, prevented fulminant hepatic failure and mortality in WT mice (62). Suramin effects were attributed to the attenuation of TNF-α and IL-6 production through the suppression of NF-kB activity in macrophages with therapeutic benefits preventing acute liver damage.
We reasoned that LPS/TLR4-mediated ATP release in macrophages, as well as elevated nucleotide levels in the extracellular milieu, via activation of P2Y2 purinergic signaling in liver-resident and infiltrated macrophages, has the potential to modify the bacterial endotoxin-mediated inflammation and hepatocellular injury in mice. Since macrophage activation is central to inflammatory liver injury, first, we evaluated the importance of P2Y2 receptor function in the induction of endotoxin-mediated inflammatory liver injury. LPS/GalN-mediated acute liver injury selectively targets the liver (21). In this model, liver injury is initiated via the activation of liver-resident macrophages with low-dose LPS (100 µg/kg), and GalN-mediated sensitization of TNFα-mediated apoptosis for low-dose LPS, minimizing LPS effects on other vital organs (52). LPS-GalN-mediated inflammatory liver injury is abolished after clodronate-mediated macrophage depletion in the liver further validating the suitability of this experimental model for the evaluation of macrophage-mediated inflammatory liver injury in mice (63). TNFα is a well-known mediator of hepatocyte apoptosis and hepatocellular injury (52).
We report that endotoxin-mediated sustained activation of JNK signaling, hepatocyte apoptosis, and liver injury observed in the WT livers were significantly attenuated in the P2Y2−/− livers (Figs. 1, A and B, 2, A–D, and 3, A–D). Activated liver-resident macrophages release chemokines such as MCP1 and MIP2, known chemotactic agents for leukocyte infiltration into hepatic parenchyma. Elevated ICAM expression at the endothelial cell surface facilitates neutrophil adhesion and extravasation and MMP9 released from activated endothelial cells and leukocytes further aggravates liver injury via the dissolution of extracellular matrix. Our studies have uncovered the importance of P2Y2 purinergic receptor function in chemokine induction (MIP2 and MCP1; Fig. 2, G and H), leukocyte infiltration (Fig. 1, C and D), ICAM (Fig. 2I), and MMP9 induction (Fig. 1, F–I) and their pathophysiological role in the induction of inflammatory liver injury.
Although the pathophysiological role of nitric oxide in mitochondrial dysfunction, oxidative stress, and multiorgan injury as well as its vasodilatory function culminating in hypoperfusion and septic shock are widely known, the potential contribution of purinergic receptor function in the modulation of iNOS induction is currently unknown. In this study, we report that induction of iNOS mRNA (LPS/GalN treatment; Fig. 2J) and protein (CLP; Fig. 4, G and H) was attenuated in P2Y2−/− livers when compared with WT. TLR4-mediated proinflammatory polarization of macrophages is characterized by induction of iNOS and cytokine mRNA and NLRP3 inflammasome-mediated posttranslational processing and maturation of proinflammatory cytokines such as IL1β and IL-18. Our observation of attenuated iNOS (Figs. 2J and 4, G and H), TNFα (Figs. 2E and 4K), IL1β (Fig. 4M), and NLRP3 (Fig. 4, I and J) expression in P2Y2−/− livers may, in part, result from P2Y2 purinergic receptor-mediated potentiation macrophage activation in the WT livers. In this study, we provide evidence for P2Y2 purinergic receptor-mediated potentiation of endotoxin-induced hepatic inflammation, liver injury, and lethality in WT mice. P2Y2−/− mice had attenuated liver inflammation, hepatocellular apoptosis, liver injury, and improved survival, in response to LPS/GalN administration, a well-established animal model of fulminant hepatitis (Figs. 1–3).
P2Y2 purinergic receptor-mediated upregulation of neutrophil infiltration and hepatocyte injury has been reported. P2Y2−/− mice sustained an attenuated liver injury in response to concanavalin A treatment due to attenuated CD4+ and CD8+ lymphocyte activation and neutrophil infiltration into hepatic parenchyma (19). Although these findings highlight the importance of P2Y2 purinergic signaling in inflammatory liver injury, they may not replicate the dynamic clinical course of bacterial infection and sepsis. Neutrophil recruitment and lung injury were attenuated in P2Y2−/− with improved survival observed in a mouse model of sepsis (64). However, the pathophysiological role of P2Y2 receptor function in sepsis-induced liver injury and long-term morbidity and mortality had not been previously evaluated. Hence, we utilized cecal ligation and puncture, a clinically relevant model of polymicrobial sepsis, to investigate P2Y2 purinergic receptor function in the pathogenesis of sepsis. Cecal ligation and puncture of mice mimic human sepsis progression (65). A key feature of this experimental model is that the degree of sepsis development can be controlled based on the size of the needle puncture and the length of the cecum ligated (66). To delineate the mechanisms underlying sepsis-associated liver injury and mortality, we chose to induce midgrade sepsis (50% cecal ligation) as opposed to severe-grade sepsis, as defined by Rittirsch et al. (22). Our studies demonstrate that P2Y2−/− mice had significant attenuation of liver injury and morbidity and showed improved survival when compared with WT mice, which sustained elevated liver injury and poor survival (Figs. 4 and 8C).
Our survival data at 24 h post-CLP are consistent with Inoue et al. (64) who showed that P2Y2−/− mice subjected to CLP had significantly improved short-term survival (24 h) when compared with WT mice. This improved outcome in P2Y2−/− mice has been attributed to P2Y2 receptor function in neutrophil activation and lung injury, both attenuated in P2Y2−/−. We have extended these studies, evaluating the long-term survival (7 days) in response to CLP, and identified that the P2Y2 receptor is a key determinant of the magnitude of systemic “cytokine storm,” hepatic inflammation, and hepatocellular injury. However, our observations on the peritoneal and blood bacterial load and the extent of early mortality in P2Y2−/− differ from the observations of Inoue et al. (64) where, in response to CLP, bacterial load was either higher (blood, 8 h) or comparable (peritoneal lavage fluid, 8 h) in P2Y2−/− with improved survival (37.5%), whereas 98% of WT mice were moribund by 48 h after CLP. These differences can be explained based on the differences in the ligation of the cecum (length) to moderate the outcomes of polymicrobial sepsis—a key inherent strength of the CLP procedure. We choose to study midgrade sepsis (50% ligation) when compared with the study published by Inoue et al. (100% ligation; severe sepsis), which likely facilitated our observation of significant attenuation of bacterial load at 6, 12, and 21 h post-CLP in the peritoneum and blood, as well as improved survival of P2Y2−/− mice at 7 days post-CLP. Nevertheless, both studies highlight the significance of P2Y2 purinergic receptor function in the pathogenesis of polymicrobial sepsis and that the overall long-term survival is determined by multiple factors including initial bacterial load in mice. We suggest that attenuation of systemic cytokine storm and inflammatory liver injury observed in P2Y2−/− mice have implications for the maintenance of systemic amino acid homeostasis necessary to prevent immune dysfunction and bacteremia in response to CLP.
Excessive induction of proinflammatory cytokines (TNFα, IL-1β, IFN, IL-6) is associated with multiorgan dysfunction and death (67). Previous studies have implicated the P2Y2 receptor in the synthesis of prostaglandins, cell adhesion molecules, and cytokines (60, 68–70). P2Y2 activation induces these proinflammatory responses through several pathways including activation of protein kinase C (PKC); phosphorylation of c-Jun NH2-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), and p38; and transactivation of integrins (71). P2Y2 receptor activation induced the release of IL-8 and IL-6 in human renal epithelial cell line A498 (72). P2Y2 receptors have been shown to facilitate the migration of neutrophils and monocytes to inflammatory tissues via the mediation of IL-8 and MCP-1 secretion, respectively (73, 74). The data reported here clearly show that serum levels of chemotactic signals such as MCP-1 and MKC (mouse analog of IL-8) are markedly suppressed in P2Y2−/− mice (Fig. 4, O–P). The loss of P2Y2 purinergic receptor function resulted in the attenuation of systemic inflammation in response to CLP in P2Y2−/− mice.
P2Y2 purinergic receptor expression and function in hepatocytes has been validated by immunohistochemical analysis of mice livers and Western blotting of total protein extracts isolated from hepatocytes (43). Ayata et al. (19) reported that P2Y2 purinergic receptor expression in bone marrow-derived cells was required for liver infiltration by neutrophils and subsequent liver damage. Proinflammatory cytokines and chemokines such as TNFα, IL-1β, and MCP-1 released by activated macrophages have been implicated in the induction of hepatocyte apoptosis in vitro and animal models of inflammatory liver injury in vivo (52, 75, 76). Previous studies have highlighted the importance of LPS/TLR4-mediated macrophage activation for inflammatory liver injury in response to LPS/GalN treatment as wells as CLP (77–79). In this study, we report that attenuation of hepatic and systemic inflammation is associated with attenuation of liver injury in response to LPS/GalN treatment and CLP of P2Y2−/− mice as compared with WT mice. Based on the above observations, we reasoned that P2Y2 purinergic receptor function is necessary for LPS/TLR4-mediated macrophage activation and exacerbation of inflammatory cytokines and chemokine and release. Our in vitro findings with LPS treatment (or P2Y2 antagonist pretreatment) of Raw264.7 cells support the notion that intact P2Y2 receptor function is necessary for LPS/TLR4-mediated secretion of key cytokines (IL-6, IL-10) and chemokines (MCP-1) in vitro (Fig. 5, A–C). These observations extend and validate previous findings on P2Y2 purinergic receptor-mediated induction of MCP-1/CCL2 synthesis and release from rat alveolar and peritoneal macrophages (74).
Monocyte chemoattractant protein-1 (MCP-1) also known as CC motif chemokine ligand 2 (CCL2) is a key mediator of monocyte chemotaxis and regulation of leukocyte function. Elevated MCP-1 level is associated with organ dysfunction and mortality in sepsis patients and animal models of sepsis (80–82). In this study, we have identified that elevated MCP-1 mRNA (liver) in response LPS/GalN treatment and elevated systemic MCP-1 levels (serum) in response to CLP in the WT mice were attenuated in P2Y2−/− mice. It is likely that the P2Y2 purinergic receptor-mediated potentiation of LPS/TLR4 activation of macrophages led to elevated MCP-1 release, leukocyte recruitment, and inflammation in response to LPS/GalN treatment and CLP in the WT mice, which was attenuated in P2Y2−/− mice.
Previous studies suggest that MCP-1/CCL-2 sensitized hepatocytes to LPS-induced liver injury (75). An increasing array of macrophage-derived humoral factors such as proinflammatory cytokines and HMGB1 are known to induce iNOS induction, excessive nitric oxide generation, nitrosative stress, and mitochondrial dysfunction in hepatocytes culminating in caspase-3-mediated apoptosis and hepatocellular injury (83–86). Immunohistochemical analysis of liver sections of mice subjected to carbon-tetrachloride (CCL4)-induced acute liver injury revealed elevated iNOS expression and peroxynitrite formation in centrilobular hepatocytes (87). Elevation of intracellular Ca2+ and iNOS-mediated nitric oxide-induced cell death in primary rat hepatocytes in vitro (83).
Our results suggest that the conditioned medium (cell culture supernatant) obtained after LPS/TLR4 activation of Raw264.7 macrophages alone is sufficient to induce iNOS protein expression and apoptosis of AML-12 hepatocytes in vitro, as assessed by the induction of cleaved caspase-3 and cleaved PARP-1 (Fig. 6). Most notably, P2Y2 purinergic receptor antagonist pretreatment of Raw264.7 macrophages, while attenuating LPS/TLR4-mediated MCP-1, IL-6, and IL-10 release, potently inhibited iNOS protein induction and apoptosis in AML-12 cells observed in response to Raw264.7-conditioned media treatment in vitro (Fig. 6).
Although our in vitro findings with 1) P2Y2 purinergic receptor antagonist treatment of Raw264.7 macrophages and 2) macrophage-conditioned media treatment of AML-12 hepatocytes (macrophage-hepatocyte cross talk) highlight the importance of P2Y2 purinergic receptor function in macrophage-mediated cytokine and chemokine release and hepatocyte apoptosis in vitro, further studies are warranted to fully comprehend the impact of P2Y2 purinergic signaling in the liver-resident and recruited macrophages, neutrophils, lymphocytes and their interaction with hepatocytes and endothelial cells in the exacerbation of inflammatory liver injury.
In response to CLP, circulating IL-6 and IL-10 accurately predicted mortality resulting from experimental sepsis (88, 89). We observed that the anti-inflammatory cytokine IL-10 was significantly elevated in the serum of WT-CLP mice when compared with P2Y2−/− at 21 h (Fig. 4N). Multiple reports evaluating the role of inflammation in sepsis have consistently shown that elevated IL-10 is a poor prognostic factor in sepsis and likely indicates a state of immune suppression (90–92). Inflammation is necessary to contain the bacterial infection, however, hyperinflammation, referred to as “cytokine storm” results from exacerbation of proinflammatory cytokine synthesis and release from innate immune cells including macrophages (53). Sepsis-associated cytokine storm has far-reaching consequences on systemic homeostasis and vital organ functions and, when uncontrolled, contributes to multiorgan injury, shock, and morbidity in patients and animal models of sepsis (93). Paradoxically, sepsis-associated inflammation manifests as a biphasic response (94, 95). Cytokine storm, an early event, is followed by immune dysfunction in macrophages and lymphocytes which contribute to morbidity and mortality in late sepsis (96–99).
Sepsis-associated immune dysfunction and inability to fight bacterial infection and control bacteremia may result from multiple pathophysiological perturbations. Amino acid metabolism plays an essential role in innate and adaptive immunity and regulates the activation of immune cells (100). Plasma arginine levels are reduced in sepsis patients and preclinical models of sepsis and acute liver injury (24, 101). l-Arginine is a semiessential amino acid and the substrate for iNOS-mediated nitric oxide generation in activated macrophages (23). Macrophage activation is associated with enhanced arginine utilization to support elevated iNOS activity and nitric oxide generation. It is likely that the early induction of cytokine storm, in response to CLP, may contribute to subsequent systemic arginine depletion observed in WT mice. Supporting this notion, we noticed that WT mice, with pronounced “cytokine storm” (Fig. 4, K–P) in response to CLP, had lower serum arginine levels (Fig. 7A) when compared with P2Y2−/− mice after CLP. P2Y2−/− mice had attenuated “cytokine storm,” and the serum arginine levels of P2Y2−/− mice were comparable to control mice subjected to Sham surgery (Fig. 7A).
Moreover, liver injury has been identified as an independent risk factor for morbidity and mortality in a subset of patients (102, 103). The liver plays a vital role in the maintenance of intermediary metabolism and synthesis and secretion of factors; both are necessary for the elicitation of effective antibacterial defenses (27). Arginine biosynthesis is impaired in acute liver injury (101). The impaired function of arginosuccinate synthase 1 (ASS1), a key rate-limiting enzyme in the biosynthesis of arginine, has been reported in thioacetamide-induced acute hepatitis (101). During the urea cycle, the liver catalyzes the conversion of ornithine to citrulline, which is the substrate for de novo synthesis of arginine via ASS1 and arginosuccinate lyase (ASL). Interestingly, P2Y2−/− mice subjected to CLP had lower serum ornithine, when compared with WT after CLP. This may be secondary to enhanced urea cycle function in P2Y2−/− livers to meet the metabolic demands associated with elevated amino acid catabolism during sepsis (Fig. 7B). Furthermore, liver injury and elevation of liver-derived arginase II in the systemic circulation can contribute to the depletion of arginine observed in sepsis patients and animal models of liver injury (104, 105).
Our studies with murine sepsis provide important insights into previous observations of arginine depletion reported in sepsis patients. Our data support the notion that excessive arginine utilization during cytokine storm and compromised liver function due to inflammatory liver injury, independently and collectively, has the potential to aggravate systemic arginine depletion and immune dysfunction with devastating consequences in mice, i.e., bacteremia, morbidity, and mortality. Nevertheless, the impact of CLP on intestine and kidney function, and their role in the de novo synthesis of arginine, needs further evaluation.
Previous studies suggest that septic patients had characteristic alterations in the levels of aromatic, branch chain, and sulfur-containing amino acids (106, 107). Branched-chain amino acids (leucine, isoleucine, and valine) have been reported to be elevated in the nonsurvivors, differentiating septic survivors from nonsurvivors (108). Most notably, serum proline levels were elevated in sepsis patients and reported as an excellent indicator of mortality (107). In agreement with these previous observations in patients, we report that the serum proline and Isoleucine/leucine levels were elevated in the WT mice, which exhibit poor survival in response to CLP, as compared with P2Y2−/− mice. However, the serum isoleucine/leucine and asparagine levels were notably higher in the sham-operated P2Y2−/− mice, as compared with sham-operated WT mice, despite a significant reduction of these amino acids being observed in the P2Y2−/− mice, after CLP. Interestingly, WT and P2Y2−/− mice exhibit distinctive differences in serum amino acids (Fig. 7, A–E), implicating P2Y2 purinergic receptor function in the regulation of amino acid metabolism during sepsis. Since amino acid metabolism plays a vital role in innate and adaptive immunity and immune cell activation, our observations underscore the importance of P2Y2 purinergic signaling in the pathogenesis of liver injury and impaired host response to bacterial infection during sepsis.
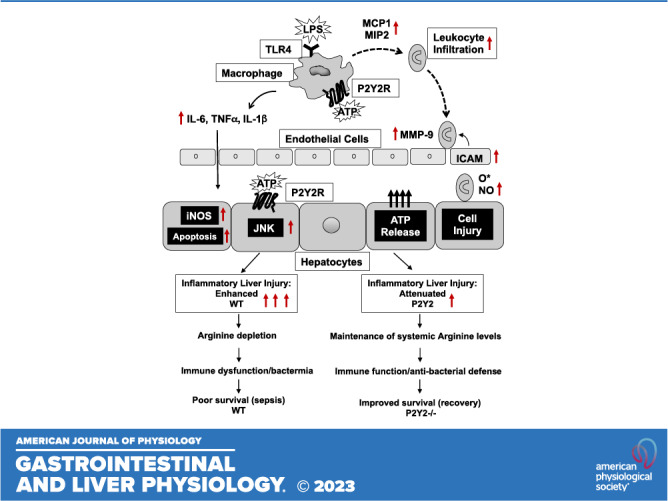
The schematic representation (Fig. 9) summarizes our findings and highlights the potential to target P2Y2 purinergic signaling to “fine-tune” the inflammatory response, necessary for the activation of innate immune cells including macrophages while preventing the early induction of “cytokine storm” and its deleterious consequence on liver injury, systemic arginine depletion, bacteremia, morbidity, and mortality.
Figure 9.

Schematic representation of P2Y2 purinergic receptor-mediated potentiation of inflammatory liver injury, serum arginine depletion, immune dysfunction, bacteremia, and poor survival in polymicrobial sepsis.
In conclusion, our studies provide experimental evidence for P2Y2 purinergic receptor-mediated potentiation of inflammatory liver injury, morbidity, and mortality, in two well-established animal models of inflammatory liver injury, i.e., LPS/GalN-mediated sterile inflammation and liver injury and polymicrobial sepsis in response cecal ligation and puncture. Our studies highlight the importance of P2Y2 receptor function in the sepsis-associated dysregulation of amino acid homeostasis, which limits systemic arginine availability necessary for the elicitation of efficient immune response and antibacterial defense during sepsis (Fig. 9). A comprehensive understanding of the molecular mechanisms underlying P2Y2-mediated inflammatory and immune response may lead to the development of potential new therapeutics in the management of sepsis.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.24030426.v1.
GRANTS
This work was supported, in part, by the Whitlock Grant from Baylor College of Medicine (to A.R.A.), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grant NIH/NIDDK RO1 DK069558 (to S.T.) and P30DK56338 which funds the Texas Medical Center Digestive Diseases Center. The metabolomics core was supported by the CPRIT Core Facility Support Award RP210227 “Proteomic and Metabolomic Core Facility,” NCI Cancer Center Support Grant P30CA125123, NIH/NCI R01CA220297, and NIH/NCI R01CA216426 intramural funds (to A.H.M.K., C.S.R.A., N.P.) from the Dan L. Duncan Cancer Center (DLDCC). The study sponsors were not involved in the study design, data collection, analysis, interpretation, or writing of the manuscript.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.T. conceived and designed research; A.R.A., S.S.S., A.M., J.P.M., K.M.S., E.D., S.N., A.B., S.P., K.S., A.H.M.K., C.S.R.A., N.P., M.S.D., and S.T. performed experiments; A.R.A., S.S.S., A.M., J.P.M., K.M.S., E.D., S.N., A.B., S.P., K.S., A.H.M.K., C.S.R.A., N.P., M.S.D., and S.T. analyzed data; A.R.A., S.S.S., A.H.M.K., C.S.R.A., N.P., M.S.D., and S.T. interpreted results of experiments; A.R.A., S.S.S., A.H.M.K., C.S.R.A., N.P., M.S.D., and S.T. prepared figures; A.R.A., S.S.S., and S.T. drafted manuscript; M.S.D. and S.T. edited and revised manuscript; A.R.A., S.S.S., A.M., J.P.M., K.M.S., E.D., S.N., A.B., S.P., K.S., A.H.M.K., C.S.R.A., N.P., M.S.D., and S.T. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Pamela Parsons, Angela Major (Tissue analysis and Molecular Imaging Core, TMC-DDC), and Sue Venable (Functional Genomics and Microbiome Core, TMC-DDC) for their help with immunohistochemistry and Multiplex cytokine assay.
REFERENCES
- 1. Reinhart K, Daniels R, Kissoon N, Machado FR, Schachter RD, Finfer S. Recognizing sepsis as a global health priority - a WHO resolution. N Engl J Med 377: 414–417, 2017. doi: 10.1056/NEJMp1707170. [DOI] [PubMed] [Google Scholar]
- 2. Ledderose C, Bao Y, Kondo Y, Fakhari M, Slubowski C, Zhang J, Junger WG. Purinergic signaling and the immune response in sepsis: a review. Clin Ther 38: 1054–1065, 2016. doi: 10.1016/j.clinthera.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Corriden R, Insel PA. Basal release of ATP: an autocrine-paracrine mechanism for cell regulation. Sci Signal 3: re1, 2010. doi: 10.1126/scisignal.3104re1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dosch M, Gerber J, Jebbawi F, Beldi G. Mechanisms of ATP release by inflammatory cells. Int J Mol Sci 19: 1222, 2018. doi: 10.3390/ijms19041222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol 11: 201–212, 2011. doi: 10.1038/nri2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schwiebert EM, Fitz JG. Purinergic signaling microenvironments: an introduction. Purinergic Signal 4: 89–92, 2008. doi: 10.1007/s11302-007-9091-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Paalme V, Rump A, Mädo K, Teras M, Truumees B, Aitai H, Ratas K, Bourge M, Chiang CS, Ghalali A, Tordjmann T, Teras J, Boudinot P, Kanellopoulos JM, Rüütel Boudinot S. Human peripheral blood eosinophils express high levels of the purinergic receptor P2X4. Front Immunol 10: 2074, 2019. doi: 10.3389/fimmu.2019.02074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Le Guilcher C, Garcin I, Dellis O, Cauchois F, Tebbi A, Doignon I, Guettier C, Julien B, Tordjmann T. The P2X4 purinergic receptor regulates hepatic myofibroblast activation during liver fibrogenesis. J Hepatol 69: 644–653, 2018. doi: 10.1016/j.jhep.2018.05.020. [DOI] [PubMed] [Google Scholar]
- 9. Besnard A, Gautherot J, Julien B, Tebbi A, Garcin I, Doignon I, Péan N, Gonzales E, Cassio D, Grosse B, Liu B, Safya H, Cauchois F, Humbert L, Rainteau D, Tordjmann T. The P2X4 purinergic receptor impacts liver regeneration after partial hepatectomy in mice through the regulation of biliary homeostasis. Hepatology 64: 941–953, 2016. doi: 10.1002/hep.28675. [DOI] [PubMed] [Google Scholar]
- 10. Csóka B, Németh ZH, Törő G, Koscsó B, Kókai E, Robson SC, Enjyoji K, Rolandelli RH, Erdélyi K, Pacher P, Haskó G. CD39 improves survival in microbial sepsis by attenuating systemic inflammation. FASEB J 29: 25–36, 2015. doi: 10.1096/fj.14-253567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Csóka B, Németh ZH, Törő G, Idzko M, Zech A, Koscsó B, Spolarics Z, Antonioli L, Cseri K, Erdélyi K, Pacher P, Haskó G. Extracellular ATP protects against sepsis through macrophage P2X7 purinergic receptors by enhancing intracellular bacterial killing. FASEB J 29: 3626–3637, 2015. doi: 10.1096/fj.15-272450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beldi G, Enjyoji K, Wu Y, Miller L, Banz Y, Sun X, Robson SC. The role of purinergic signaling in the liver and in transplantation: effects of extracellular nucleotides on hepatic graft vascular injury, rejection and metabolism. Front Biosci 13: 2588–2603, 2008. doi: 10.2741/2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fausther M, Gonzales E, Dranoff JA. Role of purinergic P2X receptors in the control of liver homeostasis. Wiley Interdiscip Rev Membr Transp Signal 1: 341–348, 2012. doi: 10.1002/wmts.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feranchak AP, Lewis MA, Kresge C, Sathe M, Bugde A, Luby-Phelps K, Antich PP, Fitz JG. Initiation of purinergic signaling by exocytosis of ATP-containing vesicles in liver epithelium. J Biol Chem 285: 8138–8147, 2010. doi: 10.1074/jbc.M109.065482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dutta AK, Woo K, Doctor RB, Fitz JG, Feranchak AP. Extracellular nucleotides stimulate Cl- currents in biliary epithelia through receptor-mediated IP3 and Ca2+ release. Am J Physiol Gastrointest Liver Physiol 295: G1004–G1015, 2008. doi: 10.1152/ajpgi.90382.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Graubardt N, Fahrner R, Trochsler M, Keogh A, Breu K, Furer C, Stroka D, Robson SC, Slack E, Candinas D, Beldi G. Promotion of liver regeneration by natural killer cells in a murine model is dependent on extracellular adenosine triphosphate phosphohydrolysis. Hepatology 57: 1969–1979, 2013. doi: 10.1002/hep.26008. [DOI] [PubMed] [Google Scholar]
- 17. Beldi G, Wu Y, Sun X, Imai M, Enjyoji K, Csizmadia E, Candinas D, Erb L, Robson SC. Regulated catalysis of extracellular nucleotides by vascular CD39/ENTPD1 is required for liver regeneration. Gastroenterology 135: 1751–1760, 2008. doi: 10.1053/j.gastro.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gonzales E, Julien B, Serrière-Lanneau V, Nicou A, Doignon I, Lagoudakis L, Garcin I, Azoulay D, Duclos-Vallée JC, Castaing D, Samuel D, Hernandez-Garcia A, Awad SS, Combettes L, Thevananther S, Tordjmann T. ATP release after partial hepatectomy regulates liver regeneration in the rat. J Hepatol 52: 54–62, 2010. doi: 10.1016/j.jhep.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ayata CK, Ganal SC, Hockenjos B, Willim K, Vieira RP, Grimm M, Robaye B, Boeynaems JM, Di Virgilio F, Pellegatti P, Diefenbach A, Idzko M, Hasselblatt P. Purinergic P2Y2 receptors promote neutrophil infiltration and hepatocyte death in mice with acute liver injury. Gastroenterology 143: 1620–1629.e4, 2012. doi: 10.1053/j.gastro.2012.08.049. [DOI] [PubMed] [Google Scholar]
- 20. Schulien I, Hockenjos B, van Marck V, Ayata CK, Follo M, Thimme R, Hasselblatt P. Extracellular ATP and purinergic P2Y2 receptor signaling promote liver tumorigenesis in mice by exacerbating DNA damage. Cancer Res 80: 699–708, 2020. doi: 10.1158/0008-5472.CAN-19-1909. [DOI] [PubMed] [Google Scholar]
- 21. Dong W, Song E, Song Y. Co-administration of lipopolysaccharide and D-galactosamine induces genotoxicity in mouse liver. Sci Rep 11: 1733, 2021. doi: 10.1038/s41598-021-81383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc 4: 31–36, 2009. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wijnands KA, Castermans TM, Hommen MP, Meesters DM, Poeze M. Arginine and citrulline and the immune response in sepsis. Nutrients 7: 1426–1463, 2015. doi: 10.3390/nu7031426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davis JS, Anstey NM. Is plasma arginine concentration decreased in patients with sepsis? A systematic review and meta-analysis. Crit Care Med 39: 380–385, 2011. doi: 10.1097/CCM.0b013e3181ffd9f7. [DOI] [PubMed] [Google Scholar]
- 25. Freund H, Atamian S, Holroyde J, Fischer JE. Plasma amino acids as predictors of the severity and outcome of sepsis. Ann Surg 190: 571–576, 1979. doi: 10.1097/00000658-197911000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Woźnica EA, Inglot M, Woźnica RK, Łysenko L. Liver dysfunction in sepsis. Adv Clin Exp Med 27: 547–551, 2018. doi: 10.17219/acem/68363. [DOI] [PubMed] [Google Scholar]
- 27. Strnad P, Tacke F, Koch A, Trautwein C. Liver - guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol 14: 55–66, 2017. doi: 10.1038/nrgastro.2016.168. [DOI] [PubMed] [Google Scholar]
- 28. Dhainaut JF, Marin N, Mignon A, Vinsonneau C. Hepatic response to sepsis: interaction between coagulation and inflammatory processes. Crit Care Med 29, Suppl 7: S42–S47, 2001. doi: 10.1097/00003246-200107001-00016. [DOI] [PubMed] [Google Scholar]
- 29. Geier A, Fickert P, Trauner M. Mechanisms of disease: mechanisms and clinical implications of cholestasis in sepsis. Nat Clin Pract Gastroenterol Hepatol 3: 574–585, 2006. doi: 10.1038/ncpgasthep0602. [DOI] [PubMed] [Google Scholar]
- 30. Homolya L, Watt WC, Lazarowski ER, Koller BH, Boucher RC. Nucleotide-regulated calcium signaling in lung fibroblasts and epithelial cells from normal and P2Y(2) receptor (−/−) mice. J Biol Chem 274: 26454–26460, 1999. doi: 10.1074/jbc.274.37.26454. [DOI] [PubMed] [Google Scholar]
- 31. Vantaku V, Donepudi SR, Piyarathna DWB, Amara CS, Ambati CR, Tang W, Putluri V, Chandrashekar DS, Varambally S, Terris MK, Davies K, Ambs S, Bollag R, Apolo AB, Sreekumar A, Putluri N. Large-scale profiling of serum metabolites in African American and European American patients with bladder cancer reveals metabolic pathways associated with patient survival. Cancer 125: 921–932, 2019. doi: 10.1002/cncr.31890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Putluri N, Shojaie A, Vasu VT, Vareed SK, Nalluri S, Putluri V, Thangjam GS, Panzitt K, Tallman CT, Butler C, Sana TR, Fischer SM, Sica G, Brat DJ, Shi H, Palapattu GS, Lotan Y, Weizer AZ, Terris MK, Shariat SF, Michailidis G, Sreekumar A. Metabolomic profiling reveals potential markers and bioprocesses altered in bladder cancer progression. Cancer Res 71: 7376–7386, 2011. doi: 10.1158/0008-5472.CAN-11-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vantaku V, Donepudi SR, Ambati CR, Jin F, Putluri V, Nguyen K, Rajapakshe K, Coarfa C, Battula VL, Lotan Y, Putluri N. Expression of ganglioside GD2, reprogram the lipid metabolism and EMT phenotype in bladder cancer. Oncotarget 8: 95620–95631, 2017. [Erratum in Oncotarget 10: 6843–6844, 2019]. doi: 10.18632/oncotarget.21038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Amara CS, Ambati CR, Vantaku V, Badrajee Piyarathna DW, Donepudi SR, Ravi SS, Arnold JM, Putluri V, Chatta G, Guru KA, Badr H, Terris MK, Bollag RJ, Sreekumar A, Apolo AB, Putluri N. Serum metabolic profiling identified a distinct metabolic signature in bladder cancer smokers: a key metabolic enzyme associated with patient survival. Cancer Epidemiol Biomarkers Prev 28: 770–781, 2019. doi: 10.1158/1055-9965.EPI-18-0936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gohlke JH, Lloyd SM, Basu S, Putluri V, Vareed SK, Rasaily U, Piyarathna DWB, Fuentes H, Rajendiran TM, Dorsey TH, Ambati CR, Sonavane R, Karanam B, Bhowmik SK, Kittles R, Ambs S, Mims MP, Ittmann M, Jones JA, Palapattu G, Putluri N, Michailidis G, Sreekumar A. Methionine-homocysteine pathway in African-American prostate cancer. JNCI Cancer Spectr 3: pkz019, 2019. doi: 10.1093/jncics/pkz019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Howard CV, Reed MG. Estimation of Component Volume and Volume Fraction. Oxford, UK: Bios Scientific, 1998. [Google Scholar]
- 37. Desai MS, Mariscalco MM, Tawil A, Vallejo JG, Smith CW. Atherogenic diet-induced hepatitis is partially dependent on murine TLR4. J Leukoc Biol 83: 1336–1344, 2008. doi: 10.1189/jlb.0607390. [DOI] [PubMed] [Google Scholar]
- 38. Maynard JP, Lee JS, Sohn BH, Yu X, Lopez-Terrada D, Finegold MJ, Goss JA, Thevananther S. P2X3 purinergic receptor overexpression is associated with poor recurrence-free survival in hepatocellular carcinoma patients. Oncotarget 6: 41162–41179, 2015. doi: 10.18632/oncotarget.6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tackett BC, Sun H, Mei Y, Maynard JP, Cheruvu S, Mani A, Hernandez-Garcia A, Vigneswaran N, Karpen SJ, Thevananther S. P2Y2 purinergic receptor activation is essential for efficient hepatocyte proliferation in response to partial hepatectomy. Am J Physiol Gastrointest Liver Physiol 307: G1073–G1087, 2014. doi: 10.1152/ajpgi.00092.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thevananther S, Sun H, Li D, Arjunan V, Awad SS, Wyllie S, Zimmerman TL, Goss JA, Karpen SJ. Extracellular ATP activates c-jun N-terminal kinase signaling and cell cycle progression in hepatocytes. Hepatology 39: 393–402, 2004. doi: 10.1002/hep.20075. [DOI] [PubMed] [Google Scholar]
- 41. Liu ZN, Su QQ, Wang YH, Wu X, Lv XW. Blockade of the P2Y2 receptor attenuates alcoholic liver inflammation by targeting the EGFR-ERK1/2 signaling pathway. Drug Des Devel Ther 16: 1107–1120, 2022. doi: 10.2147/DDDT.S346376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang T, Takikawa Y, Watanabe A, Kakisaka K, Oikawa K, Miyamoto Y, Suzuki K. Proliferation of mouse liver stem/progenitor cells induced by plasma from patients with acute liver failure is modulated by P2Y2 receptor-mediated JNK activation. J Gastroenterol 49: 1557–1566, 2014. [Erratum in J Gastroenterol 49: 1588, 2014]. doi: 10.1007/s00535-013-0927-6. [DOI] [PubMed] [Google Scholar]
- 43. Velázquez-Miranda E, Molina-Aguilar C, González-Gallardo A, Vázquez-Martínez O, Díaz-Muñoz M, Vázquez-Cuevas FG. Increased purinergic responses dependent on P2Y2 receptors in hepatocytes from CCl4-treated fibrotic mice. Int J Mol Sci 21: 2305, 2020. doi: 10.3390/ijms21072305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Merz J, Albrecht P, von Garlen S, Ahmed I, Dimanski D, Wolf D, Hilgendorf I, Härdtner C, Grotius K, Willecke F, Heidt T, Bugger H, Hoppe N, Kintscher U, von Zur Mühlen C, Idzko M, Bode C, Zirlik A, Stachon P. Purinergic receptor Y2 (P2Y2)- dependent VCAM-1 expression promotes immune cell infiltration in metabolic syndrome. Basic Res Cardiol 113: 45, 2018. doi: 10.1007/s00395-018-0702-1. [DOI] [PubMed] [Google Scholar]
- 45. Woo K, Sathe M, Kresge C, Esser V, Ueno Y, Venter J, Glaser SS, Alpini G, Feranchak AP. Adenosine triphosphate release and purinergic (P2) receptor-mediated secretion in small and large mouse cholangiocytes. Hepatology 52: 1819–1828, 2010. doi: 10.1002/hep.23883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zou D, Hu W, Qin J, Wei Z, Wang D, Li L. Rapid orderly migration of neutrophils after traumatic brain injury depends on MMP9/13. Biochem Biophys Res Commun 579: 161–167, 2021. doi: 10.1016/j.bbrc.2021.09.044. [DOI] [PubMed] [Google Scholar]
- 47. Hori T, Uemoto S, Walden LB, Chen F, Baine AM, Hata T, Kogure T, Nguyen JH. Matrix metalloproteinase-9 as a therapeutic target for the progression of fulminant liver failure with hepatic encephalopathy: A pilot study in mice. Hepatol Res 44: 651–662, 2014. doi: 10.1111/hepr.12161. [DOI] [PubMed] [Google Scholar]
- 48. Wang X, Walkey CJ, Maretti-Mira AC, Wang L, Johnson DL, DeLeve LD. Susceptibility of Rat steatotic liver to ischemia-reperfusion is treatable with liver-selective matrix metalloproteinase inhibition. Hepatology 72: 1771–1785, 2020. doi: 10.1002/hep.31179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev 90: 1165–1194, 2010. doi: 10.1152/physrev.00061.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem 281: 12093–12101, 2006. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- 51. Win S, Than TA, Kaplowitz N. The regulation of JNK signaling pathways in cell death through the interplay with mitochondrial SAB and upstream post-translational effects. Int J Mol Sci 19: 3657, 2018. doi: 10.3390/ijms19113657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Leist M, Gantner F, Bohlinger I, Tiegs G, Germann PG, Wendel A. Tumor necrosis factor-induced hepatocyte apoptosis precedes liver failure in experimental murine shock models. Am J Pathol 146: 1220–1234, 1995. [PMC free article] [PubMed] [Google Scholar]
- 53. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, Angus DC. The Third International Consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315: 801–810, 2016. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Reizine F, Grégoire M, Lesouhaitier M, Coirier V, Gauthier J, Delaloy C, Dessauge E, Creusat F, Uhel F, Gacouin A, Dessauge F, Le Naoures C, Moreau C, Bendavid C, Daniel Y, Petitjean K, Bordeau V, Lamaison C, Piau C, Cattoir V, Roussel M, Fromenty B, Michelet C, Le Tulzo Y, Zmijewski J, Thibault R, Cogné M, Tarte K, Tadié JM. Beneficial effects of citrulline enteral administration on sepsis-induced T cell mitochondrial dysfunction. Proc Natl Acad Sci USA 119: e2115139119, 2022. doi: 10.1073/pnas.2115139119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chiang CY, Veckman V, Limmer K, David M. Phospholipase Cγ-2 and intracellular calcium are required for lipopolysaccharide-induced Toll-like receptor 4 (TLR4) endocytosis and interferon regulatory factor 3 (IRF3) activation. J Biol Chem 287: 3704–3709, 2012. doi: 10.1074/jbc.C111.328559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. del Rey A, Renigunta V, Dalpke AH, Leipziger J, Matos JE, Robaye B, Zuzarte M, Kavelaars A, Hanley PJ. Knock-out mice reveal the contributions of P2Y and P2X receptors to nucleotide-induced Ca2+ signaling in macrophages. J Biol Chem 281: 35147–35155, 2006. doi: 10.1074/jbc.M607713200. [DOI] [PubMed] [Google Scholar]
- 57. Rodjakovic D, Salm L, Beldi G. Function of connexin-43 in macrophages. Int J Mol Sci 22: 1412, 2021. doi: 10.3390/ijms22031412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dosch M, Zindel J, Jebbawi F, Melin N, Sanchez-Taltavull D, Stroka D, Candinas D, Beldi G. Connexin-43-dependent ATP release mediates macrophage activation during sepsis. eLife 8: e42670, 2019. doi: 10.7554/eLife.42670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity 23: 344–346, 2005. doi: 10.1016/j.immuni.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 60. Eun SY, Seo J, Park SW, Lee JH, Chang KC, Kim HJ. LPS potentiates nucleotide-induced inflammatory gene expression in macrophages via the upregulation of P2Y2 receptor. Int Immunopharmacol 18: 270–276, 2014. doi: 10.1016/j.intimp.2013.11.026. [DOI] [PubMed] [Google Scholar]
- 61. Kawamura H, Kawamura T, Kanda Y, Kobayashi T, Abo T. Extracellular ATP-stimulated macrophages produce macrophage inflammatory protein-2 which is important for neutrophil migration. Immunology 136: 448–458, 2012. doi: 10.1111/j.1365-2567.2012.03601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Goto T, Takeuchi S, Miura K, Ohshima S, Mikami K, Yoneyama K, Sato M, Shibuya T, Watanabe D, Kataoka E, Segawa D, Endo A, Sato W, Yoshino R, Watanabe S. Suramin prevents fulminant hepatic failure resulting in reduction of lethality through the suppression of NF-kappaB activity. Cytokine 33: 28–35, 2006. doi: 10.1016/j.cyto.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 63. Xia Y, Wang P, Yan N, Gonzalez FJ, Yan T. Withaferin A alleviates fulminant hepatitis by targeting macrophage and NLRP3. Cell death Dis 12: 174, 2021. doi: 10.1038/s41419-020-03243-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Inoue Y, Chen Y, Hirsh MI, Yip L, Junger WG. A3 and P2Y2 receptors control the recruitment of neutrophils to the lungs in a mouse model of sepsis. Shock 30: 173–177, 2008. doi: 10.1097/shk.0b013e318160dad4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dejager L, Pinheiro I, Dejonckheere E, Libert C. Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol 19: 198–208, 2011. doi: 10.1016/j.tim.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 66. Ruiz S, Vardon-Bounes F, Merlet-Dupuy V, Conil JM, Buléon M, Fourcade O, Tack I, Minville V. Sepsis modeling in mice: ligation length is a major severity factor in cecal ligation and puncture. Intensive care Med Exp 4: 22, 2016. doi: 10.1186/s40635-016-0096-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pinsky MR, Vincent JL, Deviere J, Alegre M, Kahn RJ, Dupont E. Serum cytokine levels in human septic shock. Relation to multiple-system organ failure and mortality. Chest 103: 565–575, 1993. doi: 10.1378/chest.103.2.565. [DOI] [PubMed] [Google Scholar]
- 68. Xu J, Chalimoniuk M, Shu Y, Simonyi A, Sun AY, Gonzalez FA, Weisman GA, Wood WG, Sun GY. Prostaglandin E2 production in astrocytes: regulation by cytokines, extracellular ATP, and oxidative agents. Prostaglandins Leukot Essent Fatty Acids 69: 437–448, 2003. doi: 10.1016/j.plefa.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 69. Seye CI, Yu N, Jain R, Kong Q, Minor T, Newton J, Erb L, González FA, Weisman GA. The P2Y2 nucleotide receptor mediates UTP-induced vascular cell adhesion molecule-1 expression in coronary artery endothelial cells. J Biol Chem 278: 24960–24965, 2003. doi: 10.1074/jbc.M301439200. [DOI] [PubMed] [Google Scholar]
- 70. Hochhauser E, Cohen R, Waldman M, Maksin A, Isak A, Aravot D, Jayasekara PS, Müller CE, Jacobson KA, Shainberg A. P2Y2 receptor agonist with enhanced stability protects the heart from ischemic damage in vitro and in vivo. Purinergic Signal 9: 633–642, 2013. doi: 10.1007/s11302-013-9374-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Weisman GA, Wang M, Kong Q, Chorna NE, Neary JT, Sun GY, González FA, Seye CI, Erb L. Molecular determinants of P2Y2 nucleotide receptor function: implications for proliferative and inflammatory pathways in astrocytes. Mol Neurobiol 31: 169–183, 2005. doi: 10.1385/MN:31:1-3:169. [DOI] [PubMed] [Google Scholar]
- 72. Kruse R, Säve S, Persson K. Adenosine triphosphate induced P2Y2 receptor activation induces proinflammatory cytokine release in uroepithelial cells. J Urol 188: 2419–2425, 2012. doi: 10.1016/j.juro.2012.07.095. [DOI] [PubMed] [Google Scholar]
- 73. Ben Yebdri F, Kukulski F, Tremblay A, Sévigny J. Concomitant activation of P2Y(2) and P2Y(6) receptors on monocytes is required for TLR1/2-induced neutrophil migration by regulating IL-8 secretion. Eur J Immunol 39: 2885–2894, 2009. doi: 10.1002/eji.200939347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Stokes L, Surprenant A. Purinergic P2Y2 receptors induce increased MCP-1/CCL2 synthesis and release from rat alveolar and peritoneal macrophages. J Immunol 179: 6016–6023, 2007. doi: 10.4049/jimmunol.179.9.6016. [DOI] [PubMed] [Google Scholar]
- 75. Ambade A, Lowe P, Kodys K, Catalano D, Gyongyosi B, Cho Y, Iracheta-Vellve A, Adejumo A, Saha B, Calenda C, Mehta J, Lefebvre E, Vig P, Szabo G. Pharmacological inhibition of cCR2/5 signaling prevents and reverses alcohol-induced liver damage, steatosis, and inflammation in mice. Hepatology 69: 1105–1121, 2019. doi: 10.1002/hep.30249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Trautwein C, Rakemann T, Brenner DA, Streetz K, Licato L, Manns MP, Tiegs G. Concanavalin A-induced liver cell damage: activation of intracellular pathways triggered by tumor necrosis factor in mice. Gastroenterology 114: 1035–1045, 1998. doi: 10.1016/s0016-5085(98)70324-5. [DOI] [PubMed] [Google Scholar]
- 77. Deng M, Scott MJ, Loughran P, Gibson G, Sodhi C, Watkins S, Hackam D, Billiar TR. Lipopolysaccharide clearance, bacterial clearance, and systemic inflammatory responses are regulated by cell type-specific functions of TLR4 during sepsis. J Immunol 190: 5152–5160, 2013. doi: 10.4049/jimmunol.1300496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hasuzawa N, Tatsushima K, Wang L, Kabashima M, Tokubuchi R, Nagayama A, Ashida K, Ogawa Y, Moriyama Y, Nomura M. Clodronate, an inhibitor of the vesicular nucleotide transporter, ameliorates steatohepatitis and acute liver injury. Sci Rep 11: 5192, 2021. doi: 10.1038/s41598-021-83144-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Engelmann C, Sheikh M, Sharma S, Kondo T, Loeffler-Wirth H, Zheng YB, Novelli S, Hall A, Kerbert AJC, Macnaughtan J, Mookerjee R, Habtesion A, Davies N, Ali T, Gupta S, Andreola F, Jalan R. Toll-like receptor 4 is a therapeutic target for prevention and treatment of liver failure. J Hepatol 73: 102–112, 2020. doi: 10.1016/j.jhep.2020.01.011. [DOI] [PubMed] [Google Scholar]
- 80. Bossink AW, Paemen L, Jansen PM, Hack CE, Thijs LG, Van Damme J. Plasma levels of the chemokines monocyte chemotactic proteins-1 and -2 are elevated in human sepsis. Blood 86: 3841–3847, 1995. [PubMed] [Google Scholar]
- 81. Bozza FA, Salluh JI, Japiassu AM, Soares M, Assis EF, Gomes RN, Bozza MT, Castro-Faria-Neto HC, Bozza PT. Cytokine profiles as markers of disease severity in sepsis: a multiplex analysis. Crit Care 11: R49, 2007. doi: 10.1186/cc5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Matsukawa A, Hogaboam CM, Lukacs NW, Lincoln PM, Strieter RM, Kunkel SL. Endogenous MCP-1 influences systemic cytokine balance in a murine model of acute septic peritonitis. Exp Mol Pathol 68: 77–84, 2000. doi: 10.1006/exmp.1999.2296. [DOI] [PubMed] [Google Scholar]
- 83. Canová NK, Kmonícková E, Martínek J, Zídek Z, Farghali H. Thapsigargin, a selective inhibitor of sarco-endoplasmic reticulum Ca2+ -ATPases, modulates nitric oxide production and cell death of primary rat hepatocytes in culture. Cell Biol Toxicol 23: 337–354, 2007. doi: 10.1007/s10565-007-0185-6. [DOI] [PubMed] [Google Scholar]
- 84. Kurose I, Miura S, Higuchi H, Watanabe N, Kamegaya Y, Takaishi M, Tomita K, Fukumura D, Kato S, Ishii H. Increased nitric oxide synthase activity as a cause of mitochondrial dysfunction in rat hepatocytes: roles for tumor necrosis factor alpha. Hepatology 24: 1185–1192, 1996. [Erratum in Hepatology 25: 1549, 1997]. doi: 10.1002/hep.510240534. [DOI] [PubMed] [Google Scholar]
- 85. Murakami T, Suzuki K, Tamura H, Nagaoka I. Suppressive action of resolvin D1 on the production and release of septic mediators in D-galactosamine-sensitized endotoxin shock mice. Exp Ther Med 2: 57–61, 2011. doi: 10.3892/etm.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Liu Q, Rehman H, Krishnasamy Y, Schnellmann RG, Lemasters JJ, Zhong Z. Improvement of liver injury and survival by JNK2 and iNOS deficiency in liver transplants from cardiac death mice. J Hepatol 63: 68–74, 2015. doi: 10.1016/j.jhep.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Tipoe GL, Leung TM, Liong E, So H, Leung KM, Lau TY, Tom WM, Fung ML, Fan ST, Nanji AA. Inhibitors of inducible nitric oxide (NO) synthase are more effective than an NO donor in reducing carbon-tetrachloride induced acute liver injury. Histol Histopathol 21: 1157–1165, 2006. doi: 10.14670/HH-21.1157. [DOI] [PubMed] [Google Scholar]
- 88. Remick DG, Bolgos GR, Siddiqui J, Shin J, Nemzek JA. Six at six: interleukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock 17: 463–467, 2002. doi: 10.1097/00024382-200206000-00004. [DOI] [PubMed] [Google Scholar]
- 89. Song M, Kellum JA. Interleukin-6. Crit Care Med 33, Suppl 12: S463–S465, 2005. doi: 10.1097/01.ccm.0000186784.62662.a1. [DOI] [PubMed] [Google Scholar]
- 90. Gogos CA, Drosou E, Bassaris HP, Skoutelis A. Pro- versus anti-inflammatory cytokine profile in patients with severe sepsis: a marker for prognosis and future therapeutic options. J Infect Dis 181: 176–180, 2000. doi: 10.1086/315214. [DOI] [PubMed] [Google Scholar]
- 91. Yende S, D'Angelo G, Kellum JA, Weissfeld L, Fine J, Welch RD, Kong L, Carter M, Angus DC; GenIMS Investigators. Inflammatory markers at hospital discharge predict subsequent mortality after pneumonia and sepsis. Am J Respir Crit Care Med 177: 1242–1247, 2008. doi: 10.1164/rccm.200712-1777OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Latifi SQ, O'Riordan MA, Levine AD. Interleukin-10 controls the onset of irreversible septic shock. Infect Immun 70: 4441–4446, 2002. doi: 10.1128/IAI.70.8.4441-4446.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kumar V. Toll-like receptors in sepsis-associated cytokine storm and their endogenous negative regulators as future immunomodulatory targets. Int Immunopharmacol 89: 107087, 2020. doi: 10.1016/j.intimp.2020.107087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD 2nd, Kreisel D, Krupnick AS, Srivastava A, Swanson PE, Green JM, Hotchkiss RS. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 306: 2594–2605, 2011. doi: 10.1001/jama.2011.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis 13: 260–268, 2013. doi: 10.1016/S1473-3099(13)70001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Liu W, Liu T, Zheng Y, Xia Z. Metabolic reprogramming and its regulatory mechanism in sepsis-mediated inflammation. J Inflamm Res 16: 1195–1207, 2023. doi: 10.2147/JIR.S403778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Xu J, Gao C, He Y, Fang X, Sun D, Peng Z, Xiao H, Sun M, Zhang P, Zhou T, Yang X, Yu Y, Li R, Zou X, Shu H, Qiu Y, Zhou X, Yuan S, Yao S, Shang Y. NLRC3 expression in macrophage impairs glycolysis and host immune defense by modulating the NF-κB-NFAT5 complex during septic immunosuppression. Mol Ther 31: 154–173, 2023. doi: 10.1016/j.ymthe.2022.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Nascimento DC, Viacava PR, Ferreira RG, Damaceno MA, Piñeros AR, Melo PH, Donate PB, Toller-Kawahisa JE, Zoppi D, Veras FP, Peres RS, Menezes-Silva L, Caetité D, Oliveira AER, Castro IMS, Kauffenstein G, Nakaya HI, Borges MC, Zamboni DS, Fonseca DM, Paschoal JAR, Cunha TM, Quesniaux V, Linden J, Cunha FQ, Ryffel B, Alves-Filho JC. Sepsis expands a CD39+ plasmablast population that promotes immunosuppression via adenosine-mediated inhibition of macrophage antimicrobial activity. Immunity 54: 2024–2041.e8, 2021. doi: 10.1016/j.immuni.2021.08.005. [DOI] [PubMed] [Google Scholar]
- 99. Meisel C, Schefold JC, Pschowski R, Baumann T, Hetzger K, Gregor J, Weber-Carstens S, Hasper D, Keh D, Zuckermann H, Reinke P, Volk HD. Granulocyte-macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression: a double-blind, randomized, placebo-controlled multicenter trial. Am J Respir Crit Care Med 180: 640–648, 2009. doi: 10.1164/rccm.200903-0363OC. [DOI] [PubMed] [Google Scholar]
- 100. Halaby MJ, McGaha TL. Amino acid transport and metabolism in myeloid function. Front Immunol 12: 695238, 2021. doi: 10.3389/fimmu.2021.695238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Hong W, Peng X, Zhou X, Li P, Ye Z, Liang W. FXR/ASS1 axis attenuates the TAA-induced liver injury through arginine metabolism. Biochem Biophys Res Commun 611: 31–37, 2022. doi: 10.1016/j.bbrc.2022.04.073. [DOI] [PubMed] [Google Scholar]
- 102. Cui Y, Shan Y, Chen R, Wang C, Zhang Y. Elevated serum total bilirubin level is associated with poor outcomes in pediatric patients with sepsis-associated liver injury. Can J Infect Dis Med Microbiol 2018: 4591729, 2018. doi: 10.1155/2018/4591729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Liu Y, Sun R, Jiang H, Liang G, Huang Z, Qi L, Lu J. Development and validation of a predictive model for in-hospital mortality in patients with sepsis-associated liver injury. Ann Transl Med 10: 997, 2022. doi: 10.21037/atm-22-4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ikemoto M, Tsunekawa S, Awane M, Fukuda Y, Murayama H, Igarashi M, Nagata A, Kasai Y, Totani M. A useful ELISA system for human liver-type arginase, and its utility in diagnosis of liver diseases. Clin Biochem 34: 455–461, 2001. doi: 10.1016/s0009-9120(01)00254-5. [DOI] [PubMed] [Google Scholar]
- 105. Ikemoto M, Tsunekawa S, Toda Y, Totani M. Liver-type arginase is a highly sensitive marker for hepatocellular damage in rats. Clin Chem 47: 946–948, 2001. [PubMed] [Google Scholar]
- 106. Hassett J, Border JR. The metabolic response to trauma and sepsis. World J Surg 7: 125–131, 1983. doi: 10.1007/BF01655920. [DOI] [PubMed] [Google Scholar]
- 107. Cerra FB, Caprioli J, Siegel JH, McMenamy RR, Border JR. Proline metabolism in sepsis, cirrhosis and general surgery. The peripheral energy deficit. Ann Surg 190: 577–586, 1979. doi: 10.1097/00000658-197911000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Liu Z, Yin P, Amathieu R, Savarin P, Xu G. Application of LC-MS-based metabolomics method in differentiating septic survivors from non-survivors. Anal Bioanal Chem 408: 7641–7649, 2016. doi: 10.1007/s00216-016-9845-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.24030426.v1.
Data Availability Statement
Data will be made available upon reasonable request.