

Abstract
Microfluidic devices have been used for decades to isolate cells, viruses, and proteins using on-chip immunoaffinity capture using biotinylated antibodies, proteins, or aptamers. To accomplish this, the inner surface is modified to present binding moieties for the desired analyte. While this approach has been successful in research settings, it is challenging to scale many surface modification strategies. Traditional polydimethylsiloxane (PDMS) devices can be effectively functionalized using silane-based methods; however, it requires high labor hours, cleanroom equipment, and hazardous chemicals. Manufacture of microfluidic devices using plastics, including cyclic olefin copolymer (COC), allows chips to be mass produced, but most functionalization methods used with PDMS are not compatible with plastic. Here we demonstrate how to deposit biotin onto the surface of a plastic microfluidic chips using aryl-diazonium. This method chemically bonds biotin to the surface, allowing for the addition of streptavidin nanoparticles to the surface. Nanoparticles increase the surface area of the chip and allow for proper capture moiety orientation. Our process is faster, can be performed outside of a fume hood, is very cost-effective using readily available laboratory equipment, and demonstrates higher rates of capture. Additionally, our method allows for more rapid and scalable production of devices, including for diagnostic testing.
Keywords: microfluidics, surface-chemistry, extracellular vesicles, cyclic olefin copolymer (COC), immunoaffinity capture
Graphical Abstract
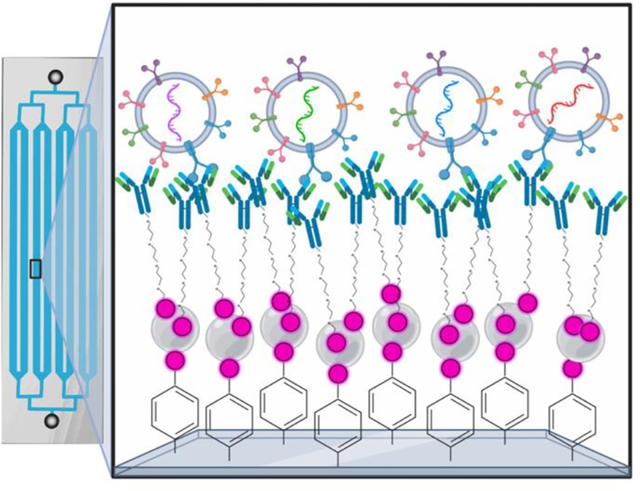
In this study, the authors demonstrate that aryl-diazonium salts can be used to coat the interior surface of injection molded plastic microfluidic devices with biotin for immunocapture. This methodology can be done outside of cleanroom facilities, is very stable at ambient temperatures, and shows improved immunocapture to previous methods of surface functionalization of plastic and PDMS microfluidic devices.
1. Introduction
Numerous microfluidic strategies have been developed to quickly and efficiently isolate or enrich numerous types of biomarkers, including cells, extracellular vesicles, and viruses using affinity-based capture approaches [1–6]. Most lab-made microfluidic devices are comprised of the pliable elastomer, polydimethylsiloxane (PDMS)[7] bonded to a glass slide. While they are relatively easy to create, they can be limiting when considering scaling of devices for clinical or commercial use. Generating PDMS-glass devices is very labor-intensive and often requires production in a cleanroom environment. Because of the hydrophobic properties of PDMS, they must be chemically modified or functionalized, to avoid adsorption of materials into the PDMS device, potentially resulting in biofouling of materials inside the device[7]. By contrast, devices made using thermoplastics, including polymethylmethacrylate (PMMA), polycarbonate (PC), cyclic olefin polymers (COP), or cyclic olefin copolymers (COC) can be manufactured at a rate of thousands per day. Thermoplastic devices can be created using a variety of methods including embossing, injection molding, or thermoforming. This brings the per chip cost down significantly for production in point of care or clinical diagnostics. COC plastics are often chosen because of their high optical transparency and ability to withstand many solvents[8,9].
Once created, a variety of techniques are used to isolate or enrich rare nanoparticles from biofluids. They include acoustofluidics, electrophoresis, filtration, deterministic lateral displacement (DLD), inertial microfluidics, optofluidics, and affinity selection[1]. Affinity based selection methods rely on the ability to bind capture molecules to the surface of portions or the entire internal surface of the microfluidic device. To chemically bond molecules onto the inner surface of devices, several strategies are undertaken. To create stable, covalent bonding to the inside surface of the device, most studies create a free carboxyl group on the surface of the device. This is achieved either through oxygen plasma, ultraviolet/ozone, or piranha solution (H2SO4 and H2O2 at a ratio of 3:1 to 7:1) treatment of the surface prior to bonding of the device[10]. The free carboxyl group is then reacted with silane immediately following bonding of the device to allow further functionalization of proteins to the surface of the device[10–12]. Depending on the form of silane used, the surface can then be functionalized with NeutrAvidin[11], proteins[13], or nanoparticles[12]. However, silane is a highly toxic compound which must be handled in a nitrogen filled glove box with a fume hood to avoid reaction with water prior to use [14–16]. Recent methods have been developed to utilize ultraviolet/ozone treatment of plastics to create a free carboxyl group which can be reacted to the primary amine of a linker, not requiring the use of silane[17]. However, these strategies all must be performed on devices after molding, prior to bonding. Microfluidic devices that are currently commercially available and already bonded or devices 3D printed with internal features could not utilize these methods that require surface treatment of interior features.
Many gels and polymers have also been used in microfluidic devices to increase the ability to add functional groups to the inner surface[7,10,19,20]. Thermoresponsive and layer-by-layer deposition approaches are a potential solution for PDMS-based devices[21–25], but have yet to be applied to plastic-based chips. However, the inherent thickness of gel and polymer coating cannot conform to devices that have precise three-dimensional features, masking device features and reducing performance. Physical adsorption (or physisorption) of proteins onto the surface of PDMS and plastic devices has been utilized[18,26,27]. To account for the non-specific nature of this process, often reagents are used at significantly higher concentrations (often 10X) to account for the less efficient deposition process[19,28,29]. This results in higher costs, higher variation in coating density, and less stable surfaces[18]. However, translating these methods to injection molded plastics can be challenging for several reasons. Ozone treatment can create higher rates of bonding; however, special ventilation must be considered due to the carcinogenic nature of ozone[8]. If plastic devices are already bonded upon receipt, the use of oxygen plasma or ultraviolet/ozone to hydroxylate the devices prior to silane treatment isn’t possible. Additionally, silane and piranha solution methods are time consuming, toxic, and require use of specialized equipment, some of which requires a class 1000 cleanroom[18].
Aryl diazonium salts are an attractive method to modify surfaces because of their ease of use and preparation, strong covalent bonding, and wide variety of chemical groups available in the initial phenolic starting compound[30]. They are created chemically by reacting a phenolic compound (NH2-C6H4-R) with sodium nitrite to form an aryl diazonium salt (+N2-C6H4-R). Then with an electron donor source supplied through either an electric current, UV-light, or ultrasonic stimulation, N2 gas is released, and a C6H4-R radical is formed. This radical can then readily react with a variety of inert substances including metal, carbon, or silicon to form a covalent bond[31–34]. They have been used to covalently coat carbon surfaces[35], fibers[36], nanotubes[37], and biosensors[34]. Both carbon and metal electrodes have been coating using aryl-diazonium salts for the use in sensors[38]. They have also been utilized to coat the interior of surfaces of devices to prevent biofouling[39]. Because of their ability to form a covalent bond, compared to transient physisorption, they are now a strategy for functionalization of nanoparticles that can be used for a variety of applications including targeted drug delivery[40]. They have also been used to nanopattern silicon microfluidic devices with biotin for immobilization of proteins as an application for biosensors[32]. However, to date they have not been utilized to functionalize the surface of thermoplastics or in immuncapture.
Here we demonstrate that aryl-diazonium salts can function as a relatively inexpensive, stable, and consistent source of surface functionalization of already bonded COC plastic microfluidic devices using a UV-light bed. Building upon previous literature[32], we identified a new reaction strategy to efficiently react biotin-NHS-esters to aryl-diazonium. We utilized a p-phenylenediamine, that when reacted with sodium nitrite, left a free NH2 group to react with a biotin-NHS-ester (Figure 1A). Prior methods have added further modifications, such as biotin only after aryl-diazonium has been added the surface. We found pre-reacting biotin-NHS-ester with aryl-diazonium prior to addition to our device greatly increased surface functionalization with biotin. This strategy effectively and evenly coats the entire surface of our microfluidic device within one hour. The process is highly reproducible and does not require access to a cleanroom. The absence of highly volatile chemicals, such as silane or piranha solution, allows for the protocol to be completed on a laboratory bench, if desired. When compared to physisorption on plastic devices or silane based functionalization in PDMS-Silane devices, we see a dramatic increase the surface binding capacity of our device functionalized with aryl diazonium, leading to higher levels of analyte capture. In addition to being an effective strategy for chemically modifying plastic microfluidic devices, we demonstrate that it is also compatible with PDMS-glass devices. Overall, we have shown an easy, effective, and inexpensive method for functionalizing plastic COC microfluidic devices, leading to higher capture rates and longer stability at room temperature, thereby allowing for easier distribution and scaling.
Figure 1: Aryl-diazonium reaction strategy.
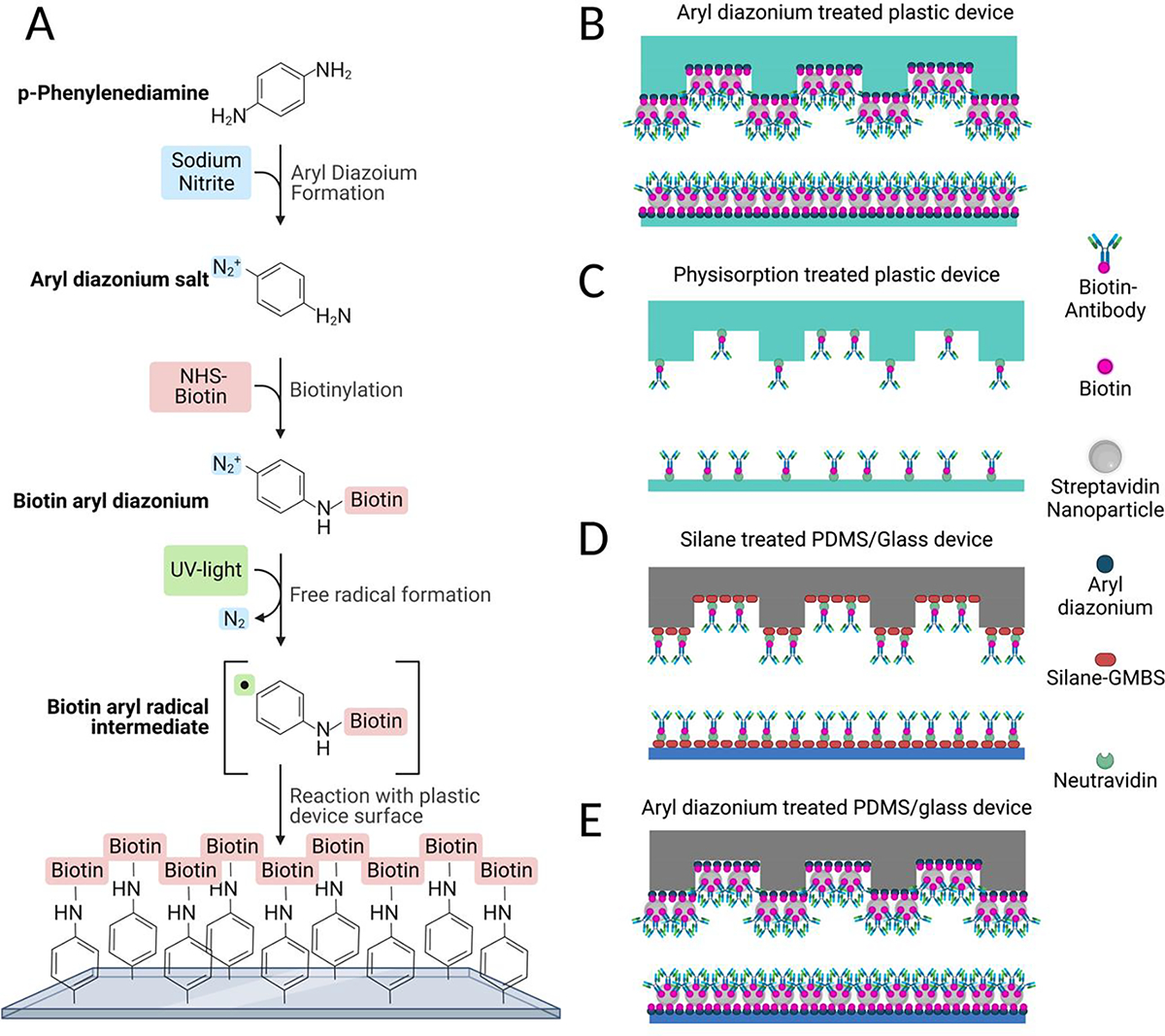
(A) Reaction strategy for aryl-diazonium functionalization. (B-E) Schematic comparing different methods of functionalizing the surface of herringbone devices to allow binding of biotinylated antibodies. (B) Biotin-aryl-diazonium coated plastic devices with streptavidin nanoparticles. (C) Physisorption of NeutrAvidin to plastic devices. (D) Silane-GMBS treatment of PDMS devices with NeutrAvidin E) Biotin-aryl-diazonium coated PDMS devices with streptavidin nanoparticles. Created using BioRender.com
2. Results
2.1. Aryl-diazonium reaction strategy
Further, the lack of covalent binding results in a less stable surface coating. To overcome this, we developed a new reaction strategy for depositing a biotinylated aryl-diazonium directly onto the surface of the cyclic olefin copolymer (COC) plastic devices. Briefly, p-phenylenediamine and sodium nitrite are reacted with biotin-NHS-ester to create a biotin-aryl-diazonium salt. This solution is then flowed into the devices, where UV light is used to produce a biotin-aryl radical through introduction of electrons and loss of N2 gas. This radical is then able to efficiently react with all plastic surfaces of the herringbone device, coating it in biotin (Figure 1).
2.2. Optimization of Aryl-Diazonium Reaction
To optimize our functionalization of biotin to surfaces within our device, we used a visual assay to assess the binding capacity of our surface coatings (Figure 2A). After biotin functionalization of the surface, streptavidin nanoparticles are added to the device. Following this, a fluorescently tagged biotin molecule (biotin R-phycoerythrin, R-PE) was flown through our devices, such that this red fluorescent reporter would bind to available streptavidin coated nanoparticles on the surface. Once tagged, nine random points were then imaged to determine average fluorescent intensity of the devices. During early optimization of our functionalization strategy, we found that allowing the biotin-NHS-ester to react with the aryl diazonium in solution led to a dramatic increase in the coating of biotin across the surface of the device. Reaction of biotin-NHS-ester inside the chip after addition of aryl-diazonium resulted in poor coverage of the chip surface. Additionally, we found that increasing the concentration of aryl diazonium above 20 mM led to a decrease in biotin functionalization to the surface (Figure S1). To further test our reaction efficiency, we tested the UV source, UV exposure time, molar ratio of biotin to aryl-diazonium, solution volume, as well as nanoparticles vs NeutrAvidin. When we compared functionalization using a UV light box vs a UV light bed, we found that the UV bed produced the most robust and consistent reaction efficiency (Figure 2B, S2). Exposure of the devices to UV light for ten minutes significantly increased binding capacity of the devices compared to a shorter five-minute exposure, with further exposure time showing no increase in binding capacity (Figure 2C, S3). This result suggests that while shorter times of five minutes are not sufficient to provide enough electrons for the reaction and result in incomplete coating of the surface, a ten-minute UV treatment provides enough electron donors for an even coating of the entire inner surface of the chip. Further treatment time showed no benefit in surface coating, suggesting the reaction had reached saturation within ten minutes. We then tested the effect of the molar ratio of biotin to aryl-diazonium in solution prior to reaction of the chip. We found that a 1:1 molar ratio showed the best reaction efficiency with increased biotin showing no increase in binding, suggesting the reaction was saturated at this molar ratio (Figure 2D, S4). Previous work from our group had shown that utilizing streptavidin coated nanoparticles increased the binding capacity of our device to both cells[12] as well as extracellular vesicles[25]. When we compared binding capacity with NeutrAvidin to streptavidin nanoparticles, we found that like previous studies, streptavidin coated nanoparticles produced increased levels of binding capacity (Figure 2E, S6). Finally, we wanted to test the amount of solution required to flow through the device for optimum binding without chemical waste. Flowing just one round of 200 μL showed the least surface functionalization. Because of the nitrogen gas released during the reaction, some areas of the chip will form bubbles, resulting in unreacted patches if only one round is used. Flowing 100 μL through twice showed increased levels of binding capacity, but increased inconsistency between and within devices, likely due to insufficient amounts of reactants. Flowing 200 μL of biotin-aryl-diazonium through the device twice produced the highest and most consistent levels of binding (Figure 2F, S5). [12][25]
Figure 2: Optimization of aryl-diazonium reaction.
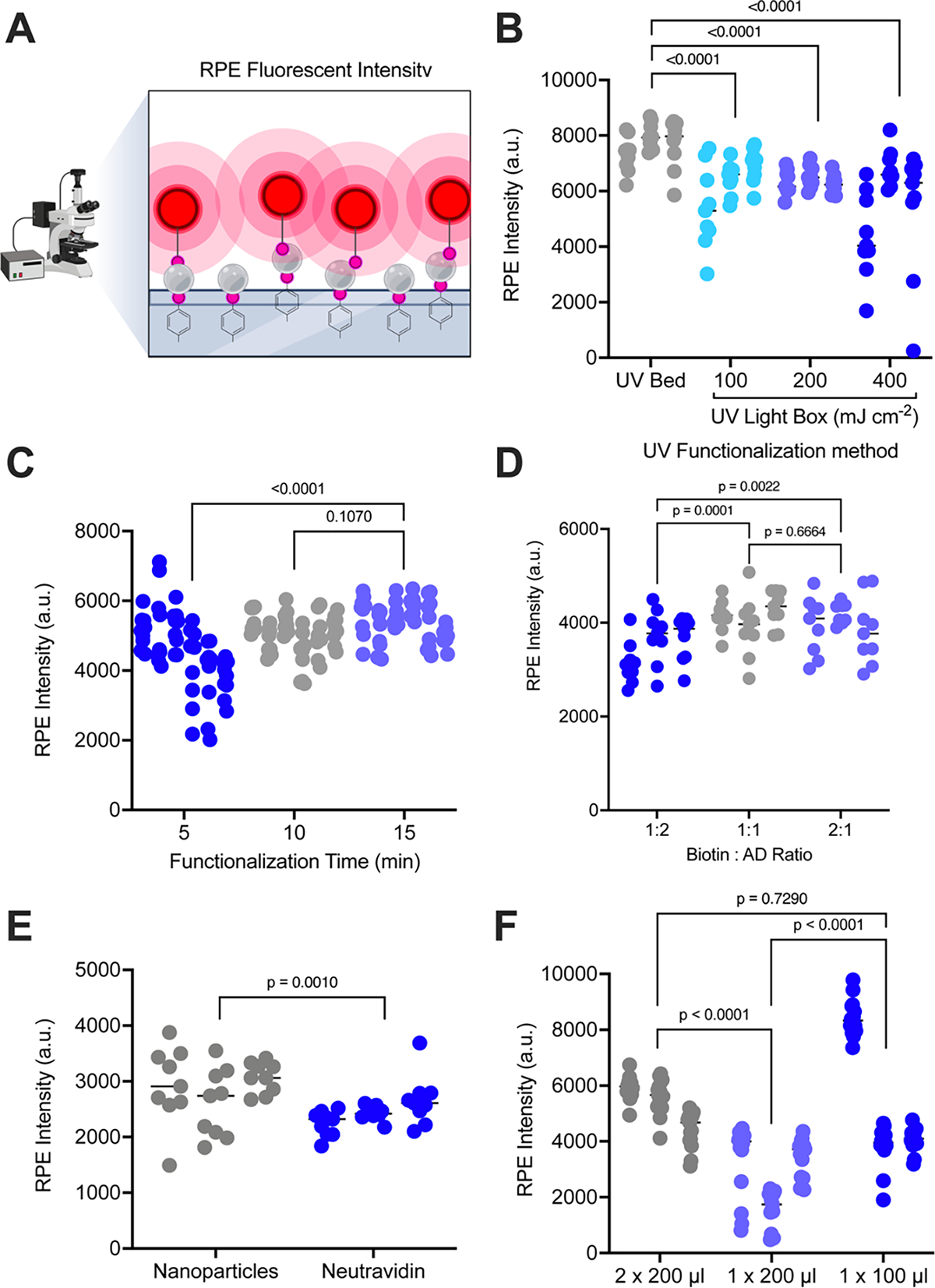
(A) Schematic depicting our Biotin-(R-PE) assay for determining binding capacity of functionalized devices. Created using BioRender.com (B-F) Average fluorescent R-PE intensity is shown for nine areas per device functionated. (B) Devices were functionalized for 15 minutes using a UV light bed at high (grey) or a UV light box with differing energies: 100 mJ cm−2 (aqua), 200 mJ cm−2 (lilac), or 400 mJ cm−2 (blue); (C) UV light box set at high for 5 minutes (blue), 10 minutes (grey), or 15 minutes (lilac); (D) a biotin to aryl-diazonium ratio of 1:2 (blue), 1:1 (grey), or 2:1 (lilac); (E) with streptavidin nanoparticles (grey) or neutravidin (blue); or (F) reaction volume of 200 μL flown through the device twice (grey), 200 μL flown through the device once (lilac), or 100 μL flown through the device once (blue). For all experiments (N=3) devices were used per condition and average fluorescent intensity values are shown for (N=9) images across each device surface. P-values were determined using a two-way ANOVAs, with correction for multiple comparisons (B-D,F) or a one-way ANOVA (E).
2.3. Device stability
Previous methods of functionalizing plastic devices, namely physisorption, relied on cold storage in PBS at 4 °C[41]. However, aryl diazonium functionalization does not require cold storage, and storage with water has the potential to cause hydrolysis of biotin-aryl-diazonium functionalized to the surface. With this in mind, we moved forward testing device stability over time stored in a desiccator. When we examined devices at week one after functionalization compared to week 0, we saw a 29% decay in binding capacity. However, subsequent weekly measurements saw no significant decay in binding capacity of devices stored dry in desiccant at 25 °C (Figure 3A, S7). It is likely that chips initially stored in a desiccator allowed for some early hydrolysis of biotin-aryl-diazonium from the surface, as they had not fully dried. Chips stored for longer showed no subsequent loss, likely because they had completely dried and could not undergo further hydrolysis. With these concerns in mind, for long-term storage experiments, dried devices were stored in a vacuum desiccator for the number of months shown before nanoparticles were added and surface binding was assessed by our R-PE assay. When we examined devices stored in a vacuum desiccator up to six months, we saw no loss in binding capacity of devices (Figure 3B, S8). This further supports that when stored free of moisture, our devices are stable at room temperature for extended periods of time. Using this strategy of dry packing, our method for functionalizing devices could be mass produced and shipped to labs for use either as research products or diagnostic tools in clinical labs. It is a widely used method to ship moisture sensitive products in vacuum sealed packaging with desiccant[42,43], which could replicate our lab storage in a vacuum desiccator box. We have previously used PDMS-glass devices functionalized with silane, dried, and stored in vacuum sealed packaging with desiccant ten years later (Figure S9). Because of their stability at room temperature, production and shipping of these devices will be much easier, allowing for more rapid and cost-effective dissemination.
Figure 3: Device stability.
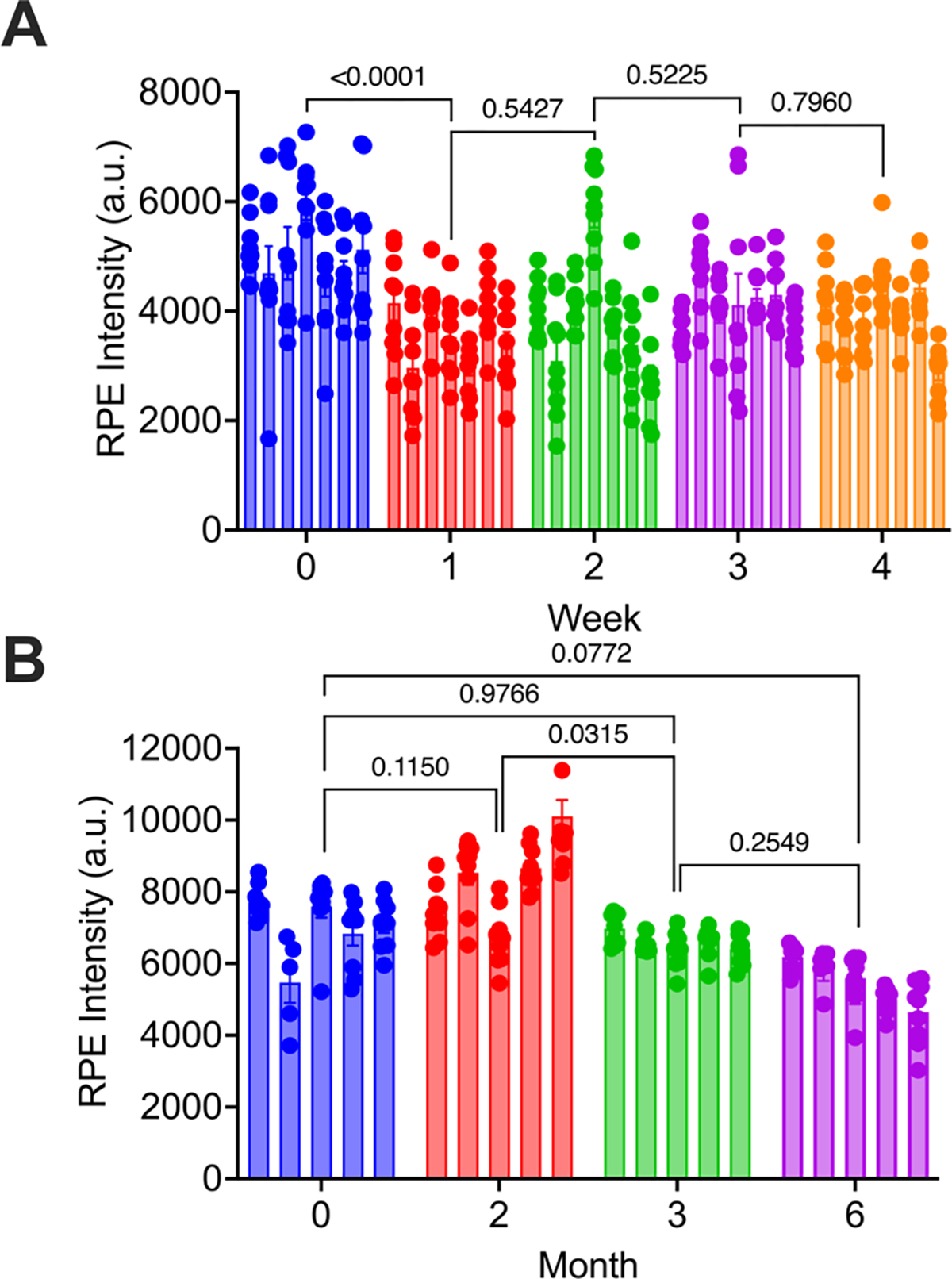
For each device, average fluorescent intensity values are shown for (N=9) images across the device surface. R-PE intensity is shown for devices functionalized and stored (A) dry in desiccant at 25 °C for up to 4 weeks (N=6 per time point) or (B) dry in a vacuum desiccator at 25 °C for up to six months (N=5 per time point). P-values were determined using a two-way ANOVA with correction for multiple comparisons.
2.4. Device cost and chemical safety
While the per device cost is not that high for making PDMS based microfluidic devices, the specialized equipment and time required to create them and functionalize the surface for capture makes them prohibitively expensive at scale. In comparison, microfluidic devices made through injection molding of plastic become cheaper at scale, lowering the per-device cost. Additionally, our procedure for functionalizing devices through aryl diazonium salts requires no expensive equipment purchase by research labs and uses inexpensive chemical reagents (Table 1). This would allow research labs to functionalize the inner surface of plastic microfluidic chips that are commercially available. This method only requires a low-cost UV bed (frequently available in research labs). Both silane and aryl diazonium wastes require special disposal (Table 2).
Table 1.
Time, cost, and stability of different methods (list prices in US dollars). Cost considerations are only factoring in the cost of devices, device materials, and chemicals. Because of differences in equipment, maintenance, and facility costs between different institutions, these were not factored in. #Clean room equipment we typically need for herringbone chips include plasma asher, oven, hot plate, and chemical hood with glove box.
| Plastic Aryl Diazonium |
Plastic Physisorption |
PDMS-Glass Aryl Diazonium |
PDMS-Glass Silane |
|
|---|---|---|---|---|
| Device Cost (chip−1) | $11 | $11 | $30 | $30 |
| Reagent Cost (chip−1) | $5.89 | $11.45 | $5.89 | $1.06 |
| Equipment needed | UV light bed | − | UV light bed #Clean Room Equip. |
#Clean Room Equip. |
| Units day−1 (production) | >10,000 | >10,000 | <100 | <100 |
| Clean Room Time (oxygen plasma bonding) | − | − | 1 hour | 1 hour |
| Functionalizing Time | 2 hours | 2 hours | 2 hours | 3.5 hours |
| Batch size (functionalize, 2 users) | 100 | 100 | 100 | 40 |
| Stability (months) | 6 months, 25 °C | 1 month, 4 °C | Not tested | 6 months, 25 °C |
Table 2.
Toxicity of reagents in aryl-diazonium vs silane
| Functionalization Strategy | Chemical | NFPA/HMIS Ratings | Shipping Restrictions |
||
|---|---|---|---|---|---|
| Health | Flammability | Physical | |||
| Silane | 3-mercaptopropyl trimethoxysilane[15,44] | 4 | 2 | 1 | Class 9 |
| dimethyl sulfoxide[45] | 2 | 2 | 1 | Group III | |
| Aryl-diazonium | p-phenylenediamine[46] | 3 | 1 | 0 | Group III |
| sodium nitrite[47] | 3 | 0 | 2 | Group III | |
| hydrochloric acid[48] | 3 | 0 | 1 | Class 8 | |
When considering the chemical safety of functionalization, silane treatment requires the use of a nitrogen filled glove box to prepare the silane solution, and subsequent steps to functionalize the surface of the devices must be prepared in a chemical fume hood. Additionally, silane functionalization must be performed immediately after oxygen-plasma bonding of PDMS devices. In contrast, use of aryl diazonium only requires a fume hood to weigh powders. The process takes approximately an hour and can be done on the bench. It can be done at a separate time from the bonding procedure, allowing for more flexibility at production. This method works on a variety of surfaces including glass, PDMS, carbon, and plastic. Because it is amenable to a variety of surfaces, it is a convenient method for functionalizing plastic devices. Traditionally, physisorption has been used to add molecules to the surface of devices. However, this process results in a relatively low binding capacity of the device and poor nanoparticle recovery.
2.5. Aryl-diazonium devices have increased binding compared to previous methods
To determine how our new reaction strategy performed, we compared it to previously used methods (Figure 1B–E). Our lab had historically used PDMS devices functionalized with silane to coat the surface of the device with NeutrAvidin. These devices showed high rates of capture, uniform coating, and high stability stored dry in desiccant at 25 °C. They additionally require the use of glove boxes and highly toxic chemicals for functionalization. To move to a scalable version of the herringbone device, COC plastic devices were produced using injection molding techniques. One strategy developed for the use of plastic devices involved direct deposition of NeutrAvidin to the surface (physisorption) (Figure 1D). While these devices work for some applications[18,29], the method typically requires significantly higher concentrations of reagents (i.e. NeutrAvidin) to compensate for the less efficient process. This method showed uneven binding at significantly lower levels than our new aryl-diazonium method (Figure 4A, S10). When we compared the binding capacity of PDMS devices functionalized with GMBS and NeutrAvidin to both PDMS and plastic devices functionalized with Aryl Diazonium and streptavidin nanoparticles, we saw no difference in binding capacity in our R-PE assay (Figure 4B–C, S10). Overall, our new method shows more consistency of binding across the device surface, between devices, and between different batches of devices, compared to currently available methods of functionalizing plastic devices. Additionally, we see similar levels of binding to widely used PDMS silane and GMBS functionalization methods, suggesting equivalency for use with immunoaffinity capture applications with patient biofluids. Because of the ability to mass produce plastic devices, the ease of functionalization, and the long-term stability at ambient temperatures, these devices are likely to serve as an improved method for analyzing small biomolecules and nanoparticles from patient samples.
Figure 4: Aryl-diazonium plastic devices demonstrate increased binding to physisorption devices and similar binding to Silane PDMS devices.

For each device, average fluorescent intensity values are shown for (N=9) images across the device surface. R-PE intensity is shown for (A) plastic devices functionalized by physisorption of neutravidin (blue circles) or by aryl-diazonium and streptavidin nanoparticles (red circles); (B) PDMS devices functionalized with silane, GMBS, and neutravidin (lavender boxes) or with aryl-diazonium and streptavidin nanoparticles (light blue triangles); or (C) PDMS devices functionalized with silane, GMBS, and neutravidin (lavender boxes) or plastic devices functionalized aryl-diazonium and streptavidin nanoparticles (red circles); and For all experiments (N=4) devices were used per condition and P-values were determined using an unpaired t-test.
2.6. Aryl-diazonium EVHB binds tumor EVs at a higher rate
Because our new functionalization method showed higher rates of surface capture in our R-PE assay, we next sought to determine the capability of aryl diazonium functionalized devices to bind tumor EVs compared to silane-GMBS or physisorption functionalized devices. To do this we flowed serum-free concentrated conditioned media from MDA-MB-231-BM1 tumor cells, containing palmitoylated tdTomato tagged EVs, through our functionalized devices using a syringe pump. RNA was extracted directly from the devices and analyzed for known EV RNA markers by ddPCR (Figure 5A, S11). When we examined non-specific binding of EVs by an IgG antibody, we found that only the aryl diazonium PDMS devices exhibited non-specific capture of EVs (Figure 5B, S11). Using specific capture of EGFR+ EVs with the EGFR/EGFRvIII targeting antibody Cetuximab, we found that aryl diazonium functionalized plastic and PDMS devices had higher rates of EV capture compared to GMBS-silane functionalized PDMS and physisorption functionalized plastic devices (Figure 5C, S11). Specifically, the aryl diazonium functionalized plastic device had strong captured fluorescent EV signal compared to nearly none in the physisorption devices. Compared to silane functionalized PDMS, plastic aryl diazonium devices had a 13 per-cent increase in captured fluorescent EV signal (Figure S11). By contrast, ddPCR showed physisorption based EV capture using plastic devices was equivalent to EV capture with silane PDMS devices. Our new aryl diazonium coated plastic devices showed a two to five-fold increase in RNA copies depending on the gene examined compared to either strategy (Figure 5).
Figure 5: Aryl-diazonium devices bind tumor EVs at a higher rate.
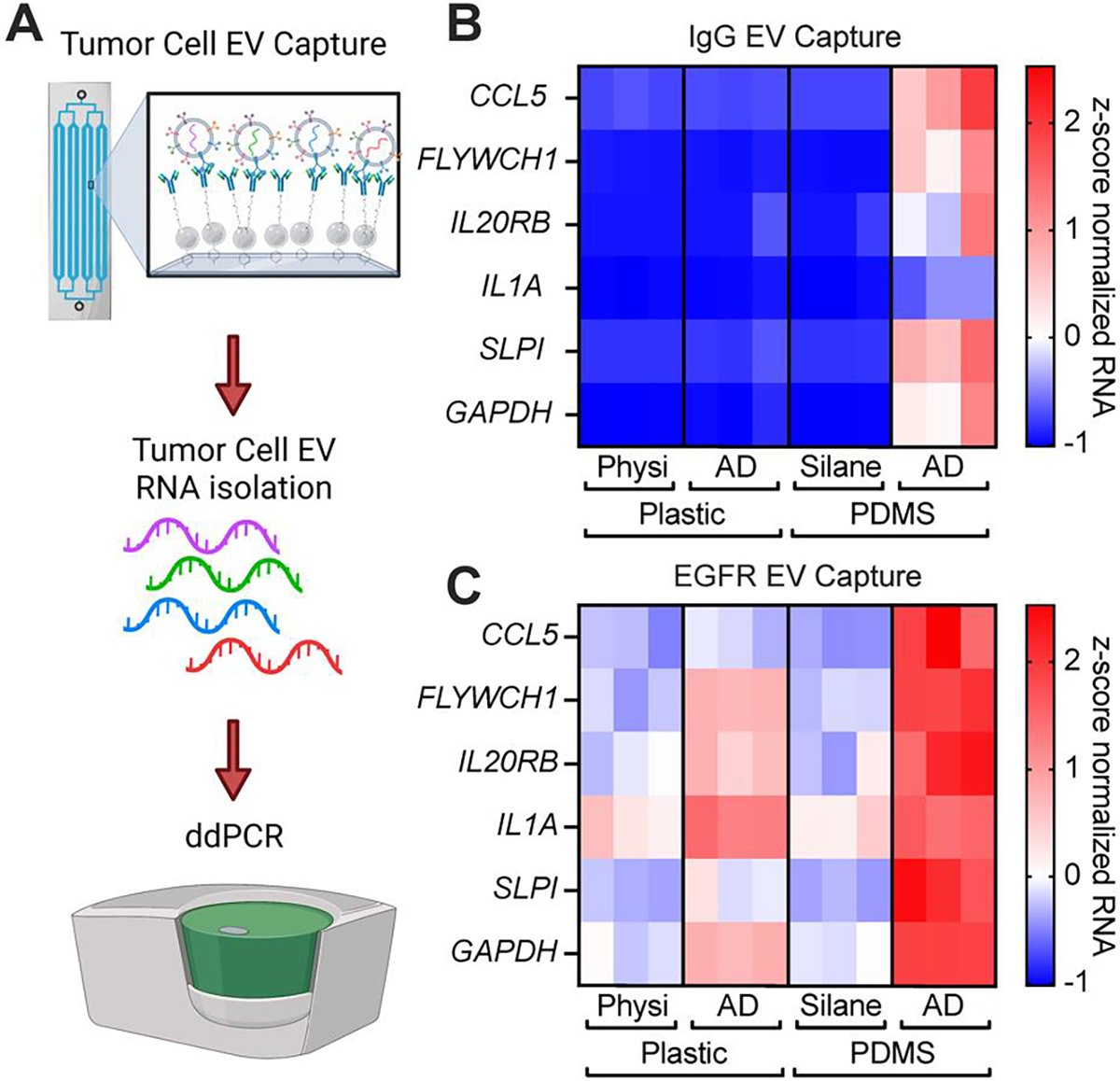
(A) Schematic describing our experimental setup. Concentrated tumor EVs are flown through the device with a syringe pump. EVs captured on our microfluidic devices are then detected by ddPCR following RNA extraction. Created using BioRender.com (B-C) Concentrated serum-free conditioned media containing tdTomato labelled EVs were flown through and captured on devices containing an IgG antibody (B) or an anti-EGFR antibody (Cetuximab) (C). RNA was extracted and measured by ddPCR with (N=3) devices per condition.
Because of issues with scalability as well as the higher rates of non-specific capture of EVs on the surface of PDMS aryl diazonium devices, we moved forward with characterizing just plastic aryl diazonium functionalized devices further. We spiked concentrated media containing palmitoylated tdTomato tagged EVs into normal patient plasma (Figure 6A). We compared RNA signal from captured tumor cell EVs between plasma alone or plasma spiked with tumor EVs captured on devices with either IgG or anti-EGFR antibody. Only anti-EGFR containing devices captured MDA-MB-231-BM1 or MDA-MB-468 tumor EVs spiked into normal plasma (Figure 6B–C, S12–13). This demonstrates that our functionalization strategy can capture tumor specific EVs from complex biofluids, such as plasma, further demonstrating its potential use in clinical diagnostic assays.
Figure 6: Aryl-diazonium devices specifically capture tumor EVs from plasma.
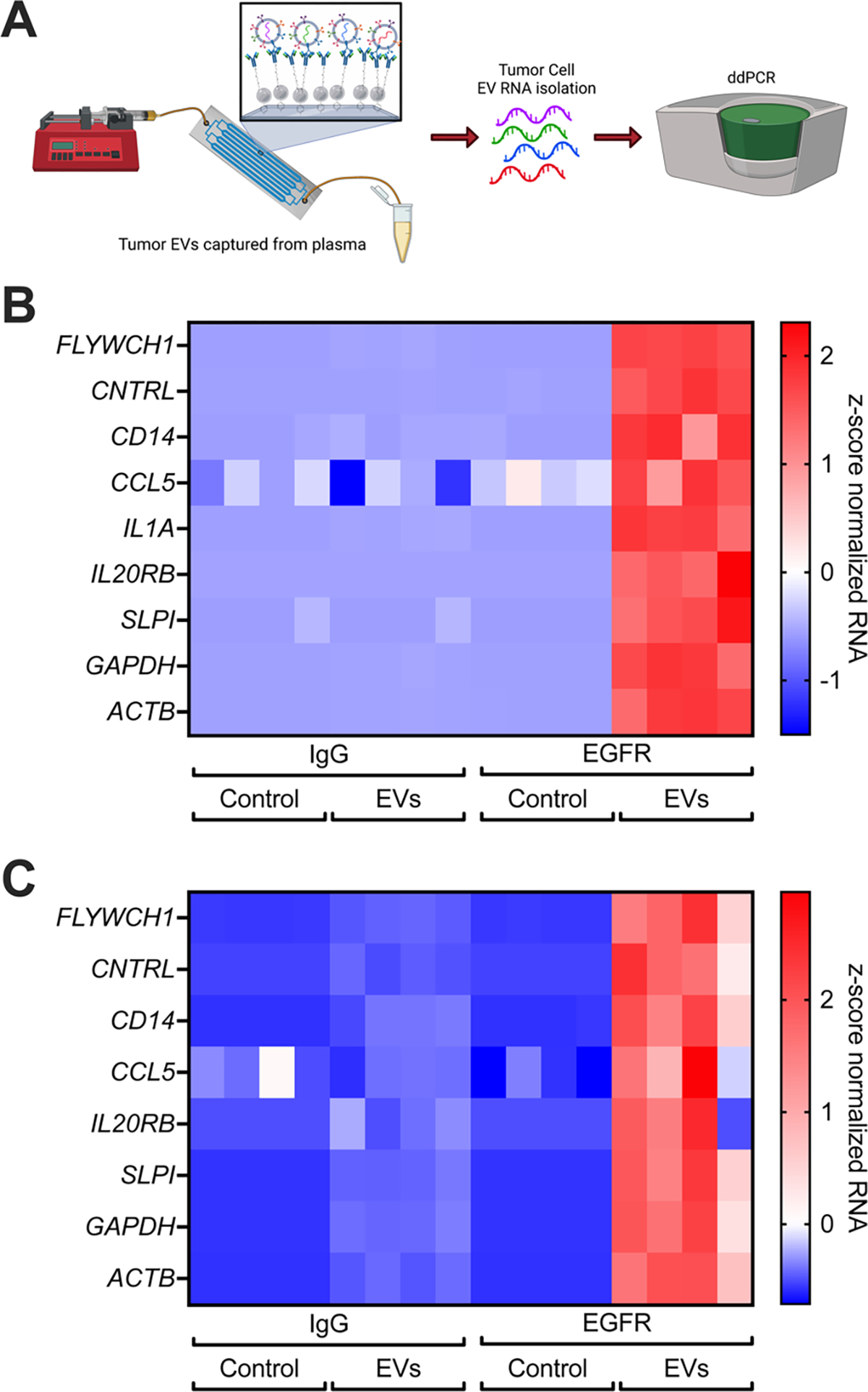
(A) Schematic describing our experimental setup. Concentrated tumor EVs or PBS are spiked into normal plasma and then flown through the device with a syringe pump. EVs captured on our microfluidic devices are then detected by ddPCR following RNA extraction. Created using BioRender.com (B-C) Concentrated serum-free conditioned media from either MDA-MB-231-BM1 (B) or MDA-MB-468 (C) tumor cells containing tdTomato labelled EVs was spiked into normal plasma, then flown through and captured on devices containing an IgG antibody or an anti-EGFR antibody (Cetuximab). RNA was extracted and measured by ddPCR.
3. Conclusion
Use of PDMS silane-GMBS functionalized microfluidic devices requires high up-front equipment costs as well as access to a clean room for production of silicon wafers and pouring and bonding PDMS devices to glass slides. Because of the specialized equipment and time required for functionalization per device, PDMS devices have many limitations when considering scaling of microfluidics for clinical assays. Rapid, mass production of microfluidic devices for clinical assays will require the use of molded plastic devices. When microfluidic devices are manufactured in plastic, they are often produced as complete devices, sealed and bonded. For this reason, surface modification is often done as part of the process, but can also be highly labile over time. Post-production modification prior to bonding is difficult, unless it is done using complex equipment, often in a cleanroom environment. When modification is done downstream, post-manufacturing, it is very difficult to uniformly apply oxygen plasma to inner surfaces without the use of complex equipment. If the devices are produced without bonding, additional challenges result when trying to bond these devices outside the cleanroom. The watertight seal required to withstand the pressures needed for most microfluidic assays is difficult to obtain. For these reasons, most manufactured and bonded devices utilize physisorption onto the surface, or forms of gel or polymer coating. However, physisorption requires 10-fold increased capture analyte and is highly unstable due to the lack of covalent bonding. Forms of gel coating often mask detailed device features and require cold storage. To produce an easily translatable method of functionalizing the surface of plastic COC devices, we utilized an aryl diazonium salt reacted with Biotin-NHS ester. This produced a robust, even, stable, and inexpensive method of deposition of biotin across the entire surface of our devices. Because these devices are stable for at least six months after functionalization at room temperature, they will be much easier to produce and then distribute (in vacuum sealed packages with desiccant) to clinical labs for analysis. Additionally, because the instrumentation and chemicals needed for this method are relatively inexpensive and readily available, it will allow research labs to functionalize plastic devices purchased through various vendors. Further, this method has a much higher rate of EV capture from plasma with our new device coating compared to plastic physisorption devices and PDMS-Glass devices functionalized with Silane. This new method for functionalizing plastic microfluidic devices for immuno-affinity capture will allow for future analysis of rare cells, vesicles, viruses, or other particles from blood, plasma, or other complex patient biofluids using mass-produced injection molded plastic devices. Our increased capture efficiency rates with this method will allow for the detection of rarer RNAs, DNAs, or proteins from EVs than compared with previously published methods using silane or physisorption.
4. Experimental Section/Methods
Microfluidic device:
For this study, we utilized a multichannel, single inlet and outlet, microfluidic device that we refer to as the ‘herringbone chip’ (EVHB-Chip)[11,25,29,41,49]. We selected this device due to the higher aspect ratio of its inner features (>1), complex three-dimensional geometry, and knowledge that it quickly highlights the limitations of any surface modification strategy. Further, our laboratory can produce this device with identical features using PDMS-glass methods[11,12,25,49] and injection molding[29,41]. Injection molded EVHB-Chips were commercially produced by thinXXS Microtechnology (Germany).
Plastic aryl-diazonium devices:
Plastic herringbone chips (Figure 1B) were inspected for debris and imperfections. 20mM p-phenylenediamine (Sigma, P6001) in 1 M hydrochloric acid (HCl, Sigma, 258148) and 20 mM sodium nitrite (Sigma, 237213) solution was reacted with EZ-link biotin-NHS-ester (final concentration of 10 mM, Pierce, 20217) for 30 minutes at room temperature to form a biotin aryl-diazonium salt (Figure 1A). Devices were then flushed with two device volumes of the biotin aryl-diazonium solution through the inlet and exposed to UV light using a UV light bed (UVP 95042001) set to high for 10 minutes. UV light allows for creation of biotin aryl radical intermediates that then react with the plastic surface of the device. Devices were then flushed with five device volumes of ethanol (EtOH, Sigma, 493546) to removed bubbles, followed by five device volumes of PBS (Corning, MT21040CV) through the inlet. Another two device volumes of the biotin aryl diazonium intermediate solution were flushed through the outlet of each device followed by another 10-minute UV exposure. Devices were flushed with five device volumes of ethanol to remove bubbles, followed by 10 device volumes of air to dry them. Herringbone chips were then stored at 25 °C in a vacuum desiccator until used. Prior to use, devices were flushed with two device volumes of a 0.01667% solution of streptavidin nanoparticles in PBS through the inlet of the devices. After a 15-minute incubation, two device volumes of streptavidin nanoparticles were flown through the outlet of the device. Chips were then used immediately or capped and stored up to one week at 4 °C.
Plastic physisorption devices:
Plastic herringbone chips (Figure 1C) were inspected for debris and imperfections. Devices were then flushed with five device volumes of EtOH through the inlet, followed by ten device volumes of PBS through the inlet. Four device volumes of 1 mg/ml NeutrAvidin diluted in PBS were flushed through the inlet of the devices, followed by a 30-minute incubation at room temperature. The process was repeated by flushing another four device volumes of NeutrAvidin through the outlet of each device and incubating at room temperature for 30 minutes.
PDMS-glass device silane functionalization:
Glass-polydimethylsiloxane (PDMS) microfluidic chips were produced using our previously described protocol[11,50,51]. Briefly, in a class 1000 cleanroom, the PDMS and glass surfaces were exposed to oxygen plasma for seven minutes (March Instruments, PX-250) then placed together and put on a hot plate for ten minutes. Within 30 minutes of bonding, the devices were brought into a chemical hood where a 4% (w∕v) solution of 3-mercaptopropyl trimethoxysilane (Silane, Gelest, SIM6476.0) in EtOH (Figure 1C) was manually pushed through the chip using a syringe. Four device volumes of 100 μg/ml N-γ-maleimidobutyryl-oxysuccinimide ester (GMBS, Pierce, 22309) diluted in EtOH were flushed through the inlet and outlet of each device. After a 15-minute incubation period, the GMBS addition was repeated. Following, devices were washed with five device volumes of EtOH, alternating between the device inlet and outlet. Devices were then flushed with five device volumes of 20 μg/mL NeutrAvidin (Thermo Scientific, 31050) diluted in phosphate buffered saline (PBS), again alternating between the inlet and outlet. After a 30-minute incubation at room temperature, the NeutrAvidin addition was repeated.
PDMS-glass device: aryl-diazonium functionalization:
Following device bonding (see above), [11,12,25,49] 20mM p-phenylenediamine in 1 M HCl and 20mM sodium nitrite solution were reacted with biotin-NHS-ester (final concentration of 10 mM) for 30 minutes at room temperature to form a biotin aryl diazonium salt (Figure 1D). Devices were then flushed with two device volumes of the biotin aryl diazonium solution through the inlet and exposed to UV light using a UV light bed (UVP 95042001) set to high for 10 minutes. UV light allows for creation of biotin aryl radical intermediates that then react with the plastic surface of the device. Devices were then flushed with five device volumes of ethanol to removed bubbles, followed by five device volumes of PBS through the inlet. Another two device volumes of the biotin aryl diazonium intermediate solution were flushed through the outlet of each device followed by another 10-minute UV exposure. Devices were flushed with five device volumes of ethanol to remove bubbles. Devices were flushed with five device volumes of PBS. Then two device volumes of a 0.01667% solution of streptavidin nanoparticles were flown through the inlet of the devices. After a 15-minute incubation, two device volumes of streptavidin nanoparticles were flown through the outlet of the device.
R-phycoerythrin (R-PE) assay:
Devices were flushed with five device volumes of PBS per side and blocked with five device volumes of Intercept (TBS) Blocking Buffer (LICOR, 927-60001). For each device, 10 μL of R-Phycoerythrin (R-PE), Biotin-XX Conjugate (ThermoFisher, P811) in 990 μL 1% BSA (Sigma, A3059) in PBS was flown through each device at 2 mL/hour using a PhD ULTA syringe pump (Harvard Apparatus) protected from light. Devices were incubated at room temperature, protected from light for 30 minutes. Devices were then flushed with 2.5 mL PBS at 2.5 mL/hour using a syringe pump. Nine representative images were taken per device using a Nikon Eclipse 90i microscope with a 10X lens and Andor camera [Model #DR-328G0C01-SIL] with a neutral density 4 (ND4) filter. A TexasRed filter was used, and 100 millisecond (ms) exposure images were taken. Using NIS-Elements, the average fluorescent intensity of each image was measured. Background fluorescence (with no device present) was recorded and subtracted from all values.
Extracellular Vesicle (EV) Capture on Devices
Cell Culture:
MDA-MB-231-BM1 (BM1) cells were generously provided by Dr Marsha Rosner and MDA-MB-468 cells were generously provided by Dr Lief Ellison. BM1 and HEK-293T cells (ATCC, CRL-3216) were propagated in Dulbecco’s Modified Eagle Media with glutamine and 4.5 g/L glucose (Corning, 10-013-CV) supplemented with fetal bovine serum (FBS), qualified (Gibco, 26140-079) at a final concentration of 10% and penicillin, streptomycin (P/S, Gibco, 15140163) at a final concentration of 1% at 37 °C with 5% CO2. MDA-MB-468 Cells were propagated in RMPI-1640 media with glutamine (Corning, 10-040-CV) supplemented with FBS at a final concentration of 10% and P/S at a final concentration of 1% at 37 °C with 5% CO2. Cells authentication was performed by short tandem repeat analysis compared to the primary MDA-MB-231, HEK-293, and MDA-MB-468 genotypes respectively and cells were checked for mycoplasma prior to use and every 6 weeks following using the MycoAlert test (Lonza, LT07-318).
Lentiviral Transductions:
To fluorescently label EVs, MDA-MB-231-BM1 and MDA-MB-468 cells were transduced with a palmitoylated-tdTomato fluorescent reporter using lentivirus. Third generation lentiviruses were propagated under BL2+ conditions as approved by the Mass General Brigham Institutional Biosafety Committee. pMDLg/pRRE, pRSV-Rev, pVSV-G lentiviral packaging plasmids were combined with pCSCGW2-PalmtdTomato[52] lentiviral vector and TransIT-Lenti (Mirrus, MIR6600) to transfect HEK-293T cells per the manufacturer’s protocol. Lentiviral media was collected after 48 hours and filtered through a 0.45 μm filter. 1 μL of TransduceIT Transduction Reagent (Mirrus, MIR6620) was added per 1 mL of lentiviral media. 1.5 mL of viral containing media was then added to transduce cells over 24 hours. Transduced cells were then selected for viral expression of palmitoylated-tdTomato by flow cytometry following transduction.
Antibody biotinylation for EV Capture:
Antibodies were incubated at room temperature while rotating with Biotin PEG SCM 2kDa (Creative PEGworks, PJK-1900) for two hours at a molar ratio of biotin linker:antibody of 20:1. Excess biotin linker was removed using Zeba Desalting Columns (Thermo Scientific, 89882). Antibodies were then aliquoted for single use and stored at −80 °C.
Extracellular Vesicle (EV) Capture:
MDA-MB-231-BM1, and MDA-MB-468 cells were grown to 90% confluence in 15-cm dishes. They were then washed three times with PBS, to remove any media containing FBS. Cells were incubated in serum free media containing 1% P/S for 48 hours in a 37 °C incubator to collect secreted EVs. Conditioned media was removed from the cells and spun at 2,000 × g for 10 minutes to remove any cells, debris, or apoptotic bodies. Media was then concentrated 10-fold using 10-kDa Amicon Ultra-15 filters (Millipore, UFC901024).
For all devices, 2 device volumes of a 20 μg ml−1 solution of either an anti-EGFR antibody (Eli Lilly, Cetuximab) or non-specific IgG (BioLegend, 401402) were added to inlet of each device, and incubated at room temperature for 30 minutes. Then two device volumes of the same antibody were then flown through the outlet of the same device. After a 30 min incubation, devices are blocked with two device volumes of Intercept (TBS) Blocking Buffer (LICOR, 927-60001).
For EV capture alone, 500 μL of 10x concentrated conditioned media was flown through each device. For experiments with normal plasma, 50 μL of 30x concentrated EVs were added per 500 μL of single donor human plasma (Blood derived, Innovative Research, IPLASK2EDTAUNIT). Plasma was collected by Innovative Research with patient consent under a Food and Drug Administration approval 3003372368 in an ISO 9001:2015 certified environment. 500 μL of EV-spiked plasma was flown through a herringbone capture device at 1 mL hour−1. Devices were then washed with 1.5 mL of PBS flown through at 1.5 mL hour−1.
EV Imaging:
After washing with PBS, devices were capped and then imaged on a Nikon Eclipse 90i microscope with a 10X lens and Andor camera [Model #DR-328G0C01-SIL], with ND4 and TexasRed filters. Nine representative images were taken per device with a one-second (s) exposure time. Using NIS-Elements, the total fluorescent intensity of each image was measured. Fluorescent intensity values were then normalized to the IgG, no EV devices to show ΔtdTomato signal from background chip fluorescence.
RNA Extraction:
RNA was extracted from devices using the MagMAX mirVana Total RNA Isolation Kit (Applied Biosystems, A27828). For each device 99 μL Lysis Buffer (from A27828) + 100 μL Isopropanol (Fisher Chemical, A451SK-1) + 1 μL β-mercaptoethanol (Sigma Aldrich, M3148) was flown through 12 times by manually pushing between syringes attached to the inlet and exit port of devices. RNA was then isolated with DNase treatment per the manufacturer’s manual extraction protocol (Applied Biosystems, A27828).
One-Step Reverse Transcription and ddPCR:
RNA levels were measured using the 1-Step RT-ddPCR Advanced Kit for Probes (Bio-Rad, 1864021) and pre-designed primer/probe mixes for each gene (Integrated DNA Technologies, IDT). Reactions were performed using 5.5 μL RNA per reaction with 500 nM of primers (final concentration) and a primer:probe ratio of 4:1. Droplet generation was performed on the QX200 AutoDG, PCR amplification on the C1000 Touch Thermal Cycler, droplet reading on the QX200 Droplet Reader, and analysis using QX Manager (Bio-Rad).
Primer and Probe Sequences (Integrated DNA Technologies, IDT)
ACTB Assay: Hs.PT.39a.22214847
ATCB-F: CCTTGCACATGCCGGAG
ACTB-R: ACAGAGCCTCGCCTTTG
ACTB-Probe: TCATCCATGGTGAGCTGGCGG
CCL5 Assay: Hs.PT.58.40305992
CCL5-F: GACTCTCCATCCTAGCTCATCT
CCL5-R: GAGTATTTCTACACCAGTGGCA
CCL5-Probe: ATGTACTCCCGAACCCATTTCTTCTCTG
CD14 Assay: Hs.PT.56a.3118607.g
CD14-F: AATCTTCATCGTCCAGCTCAC
CD14-R: CAGAGGTTCGGAAGACTTATCG
CD14-Probe: CGCAGAGACGTGCACCAGC
CNTRL Assay: Hs.PT.58.1241761
CNTRL-F: CATTTTCCACCTCCGTTCATTG
CNTRL-R: GTCTCTTTCCAGTCTTTCTACCTC
CNTRL-Probe: TTGGAAGGTCAGCCAGTAACCACTC
IL1A Assay: Hs.PT.58.2851435
IL1A-F: TCTTCATCTTGGGCAGTCAC
IL1A-R: GCTGCTGCATTACATAATCTGG
IL1A-Probe: TGAAGCAGTGAAATTTGACATGGGTGC
IL20RB Assay: Hs.PT.58.39994983
IL20RB-F: GACCTTCAGTGAGTGAGCAC
IL20RB-R: ACCAACATGAAGCATCTCTTGA
IL20RB-Probe: AGCCTGTACACGAGCCACATCTG
GAPDH Assay: Hs.PT.39a.22214836
GAPDH-F: TGTAGTTGAGGTCAATGAAGGG
GAPDH-R: ACATCGCTCAGACACCATG
GAPDH-Probe: AAGGTCGGAGTCAACGGATTTGGTC
SLPI Assay: Hs.PT.58.3977822
SLPI-F: TGTGGAAGGCTCTGGAAAG
SLPI-R: TGGCACTCAGGTTTCTTGTATC
SLPI-Probe: TGGGCAGATTTCTTAGGAGGACAGACT
FLYWCH1 Assay: Hs.PT.58.40054436.g
FLYWCH1-F: CCAGCCAGCCCTAGAGAT
FLYWCH1-R: CACTGCCTTCTCCTGCTTG
FLYWCH1-Probe: AGGAAGGACTCCAGCACCAGGA
Supplementary Material
Acknowledgements
This study was funded by National Cancer Institute grants R01-CA226871 (to SLS) and F32-CA236417 (to DCR), National Center for Advancing Translational Sciences grant U18-TR003793 (to SLS), and American Cancer Society Grant 132030-RSG-18-108-01-TBG (to SLS). We would like to thank Dr. Marsh Rosner for providing MDA-MB-231-BM1 cells, Dr. Leif Ellison for providing MDA-MB-468 cells, Dr. John Walsh for providing his protocol for physisorption, and Drs. Xandra Breakefield and Charles Lai for providing the pCSCGW2-PalmtdTomato plasmid.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the corresponding author upon reasonable request.
References
- [1].Xie Y, Rufo J, Zhong R, Rich J, Li P, Leong KW, Huang TJ, ACS Nano 2020, 14, 16220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Squires TM, Quake SR, Rev Mod Phys 2005, 77, 977. [Google Scholar]
- [3].Whitesides GM, Nature 2006, 442, 368. [DOI] [PubMed] [Google Scholar]
- [4].Basiri A, Heidari A, Nadi MF, Fallahy MTP, Nezamabadi SS, Sedighi M, Saghazadeh A, Rezaei N, Rev Med Virol 2021, 31, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Berkenbrock JA, Grecco-Machado R, Achenbach S, Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences 2020, 476, DOI 10.1098/rspa.2020.0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Du K, Park M, Griffiths A, Carrion R, Patterson J, Schmidt H, Mathies R, Anal Chem 2017, 89, 12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shakeri A, Khan S, Didar TF, Lab Chip 2021, 21, 3053. [DOI] [PubMed] [Google Scholar]
- [8].Tsao C-W, DeVoe DL, Microfluid Nanofluidics 2009, 6, 1. [Google Scholar]
- [9].Agha A, Waheed W, Alamoodi N, Mathew B, Alnaimat F, Abu-Nada E, Abderrahmane A, Alazzam A, Macromol Mater Eng 2022, 307, 2200053. [Google Scholar]
- [10].Wong I, Ho C-M, Microfluid Nanofluidics 2009, 7, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Stott SL, Hsu C-H, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, Rothenberg SM, Shah AM, Smas ME, Korir GK, Floyd FP, Gilman AJ, Lord JB, Winokur D, Springer S, Irimia D, Nagrath S, v Sequist L, Lee RJ, Isselbacher KJ, Maheswaran S, Haber DA, Toner M, Proc Natl Acad Sci U S A 2010, 107, 18392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Park M-H, Reátegui E, Li W, Tessier SN, Wong KHK, Jensen AE, Thapar V, Ting D, Toner M, Stott SL, Hammond PT, J Am Chem Soc 2017, 139, 2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Casadei L, Choudhury A, Sarchet P, Mohana Sundaram P, Lopez G, Braggio D, Balakirsky G, Pollock R, Prakash S, J Extracell Vesicles 2021, 10, e12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mojsiewicz-Pieńkowska K, Jamrógiewicz M, Szymkowska K, Krenczkowska D, Front Pharmacol 2016, 7, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].“Silane | SiH4 - PubChem,” can be found under https://pubchem.ncbi.nlm.nih.gov/compound/Silane, n.d.
- [16].O’Grady BJ, Geuy MD, Kim H, Balotin KM, Allchin ER, Florian DC, Bute NN, Scott TE, Lowen GB, Fricker CM, Fitzgerald ML, Guelcher SA, Wikswo JP, Bellan LM, Lippmann ES, Lab Chip 2021, 21, 4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pahattuge TN, Jackson JM, Digamber R, Wijerathne H, Brown V, Witek MA, Perera C, Givens RS, Peterson BR, Soper SA, Chem Commun (Camb) 2020, 56, 4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shakeri A, Jarad NA, Khan S, F Didar T, Anal Chim Acta 2022, 1209, 339283. [DOI] [PubMed] [Google Scholar]
- [19].Rusmini Federica, *,† and Zhong Zhiyuan, Feijen J, 2007, DOI 10.1021/BM061197B. [DOI] [PubMed] [Google Scholar]
- [20].Shah AM, Yu M, Nakamura Z, Ciciliano J, Ulman M, Kotz K, Stott SL, Maheswaran S, Haber DA, Toner M, Anal Chem 2012, 84, 3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Richardson JJ, Björnmalm M, Caruso F, Science (1979) 2015, 348, DOI 10.1126/science.aaa2491. [DOI] [PubMed] [Google Scholar]
- [22].Tan W, Desai TA, Biomaterials 2004, 25, 1355. [DOI] [PubMed] [Google Scholar]
- [23].Chen C, Mehl BT, Munshi AS, Townsend AD, Spence DM, Martin RS, Analytical Methods 2016, 8, 6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ao Z, Parasido E, Rawal S, Williams A, Schlegel R, Liu S, Albanese C, Cote RJ, Agarwal A, Datar RH, Lab Chip 2015, 15, 4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Reátegui E, Aceto N, Lim EJ, Sullivan JP, Jensen AE, Zeinali M, Martel JM, Aranyosi AJ, Li W, Castleberry S, Bardia A, Sequist L. v., Haber D. a., Maheswaran S, Hammond PT, Toner M, Stott SL, Adv Mater 2015, 27, n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Giridharan GA, Nguyen M-D, Estrada R, Parichehreh V, Hamid T, Ismahil MA, Prabhu SD, Sethu P, Anal Chem 2010, 82, 7581. [DOI] [PubMed] [Google Scholar]
- [27].van Engeland NCA, Pollet AMAO, den Toonder JMJ, Bouten CVC, Stassen OMJA, Sahlgren CM, Lab Chip 2018, 18, 1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kim D, Herr AE, Biomicrofluidics 2013, 7, 41501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sundaresan TK, v Sequist L, v Heymach J, Riely GJ, Jänne PA, Koch WH, Sullivan JP, Fox DB, Maher R, Muzikansky A, Webb A, Tran HT, Giri U, Fleisher M, Yu HA, Wei W, Johnson BE, Barber TA, Walsh JR, Engelman JA, Stott SL, Kapur R, Maheswaran S, Toner M, Haber DA, Clin Cancer Res 2016, 22, 1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chehimi M. Mehdi, Aryl Diazonium Salts : New Coupling Agents in Polymer and Surface Science, John Wiley & Sons, 2012. [Google Scholar]
- [31].Mahouche-Chergui S, Gam-Derouich S, Mangeney C, Chehimi MM, Chem Soc Rev 2011, 40, 4143. [DOI] [PubMed] [Google Scholar]
- [32].Flavel BS, Gross AJ, Garrett DJ, Nock V, Downard AJ, ACS Appl Mater Interfaces 2010, 2, 1184. [DOI] [PubMed] [Google Scholar]
- [33].and JL, Bélanger D*, 2006, DOI 10.1021/CM060752D. [DOI] [Google Scholar]
- [34].Hetemi D, Noël V, Pinson J, Biosensors (Basel) 2020, 10, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Delamar M, Hitmi R, Pinson J, Saveant JM, J Am Chem Soc 1992, 114, 5883. [Google Scholar]
- [36].Delamar M, Désarmot G, Fagebaume O, Hitmi R, Pinsonc J, Savéant J-M, Carbon NY 1997, 35, 801. [Google Scholar]
- [37].Bahr Jeffrey L., Yang Jiping, Kosynkin Dmitry V., Bronikowski Michael J., and Smalley Richard E., Tour JM*, 2001, DOI 10.1021/JA010462S. [DOI] [PubMed] [Google Scholar]
- [38].Gooding JJ, Electroanalysis 2008, 20, 573. [Google Scholar]
- [39].Mrabet B, Mejbri A, Mahouche S, Gam-Derouich S, Turmine M, Mechouet M, Lang P, Bakala H, Ladjimi M, Bakhrouf A, Tougaard S, Chehimi MM, Surface and Interface Analysis 2011, 43, 1436. [Google Scholar]
- [40].Li D, Luo Y, Onidas D, He L, Jin M, Gazeau F, Pinson J, Mangeney C, Adv Colloid Interface Sci 2021, 294, 102479. [DOI] [PubMed] [Google Scholar]
- [41].Reátegui E, van der Vos KE, Lai CP, Zeinali M, Atai NA, Aldikacti B, Floyd FP, H Khankhel A, Thapar V, Hochberg FH, v Sequist L, v Nahed B, S Carter B, Toner M, Balaj L, T Ting D, Breakefield XO, Stott SL, Nat Commun 2018, 9, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Asghar W, Yuksekkaya M, Shafiee H, Zhang M, Ozen MO, Inci F, Kocakulak M, Demirci U, Sci Rep 2016, 6, 21163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Stevens DY, Petri CR, Osborn JL, Spicar-Mihalic P, McKenzie KG, Yager P, Lab Chip 2008, 8, 2038. [DOI] [PubMed] [Google Scholar]
- [44].3-MERCAPTOPROPYLTRIMETHOXYSILANE Safety Data Sheet, n.d. [Google Scholar]
- [45].SAFETY DATA SHEET (Dimethyl Sulfoxide), n.d. [Google Scholar]
- [46].P-Phenylenediamine SAFETY DATA SHEET, n.d. [Google Scholar]
- [47].Safety Data Sheet Sodium Nitrite, Reagent, n.d. [Google Scholar]
- [48].Safety Data Sheet (Hydrochloric Acid), n.d. [Google Scholar]
- [49].Stott SL, Lee RJ, Nagrath S, Yu M, Miyamoto DT, Ulkus L, Inserra EJ, Ulman M, Springer S, Nakamura Z, Moore AL, Tsukrov DI, Kempner ME, Dahl DM, Wu C-L, Iafrate AJ, Smith MR, Tompkins RG, v Sequist L, Toner M, Haber DA, Maheswaran S, Sci Transl Med 2010, 2, 25ra23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, Concannon KF, Donaldson MC, Sequist L. v., Brachtel E, Sgroi D, Baselga J, Ramaswamy S, Toner M, Haber DA, Maheswaran S, Science (1979) 2013, 339, 580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sundaresan TK, Sequist L. v., Heymach J. v., Riely GJ, Jänne PA, Koch WH, Sullivan JP, Fox DB, Maher R, Muzikansky A, Webb A, Tran HT, Giri U, Fleisher M, Yu HA, Wei W, Johnson BE, Barber TA, Walsh JR, Engelman JA, Stott SL, Kapur R, Maheswaran S, Toner M, Haber DA, Clinical Cancer Research 2016, 22, 1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lai CP, Kim EY, Badr CE, Weissleder R, Mempel TR, Tannous BA, Breakefield XO, Nat Commun 2015, 6, 7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.