

ABSTRACT
Mitophagy, the process of removing damaged mitochondria to promote cell survival, plays a crucial role in cellular functionality. However, excessive, or uncontrolled mitophagy can lead to reduced mitochondrial content that burdens the remaining organelles, triggering mitophagy-mediated cell death. FBXL4 mutations, which affect the substrate-binding adaptor of the CUL1 (cullin 1)-RING ubiquitin ligase complex (CRL1), have been linked to mitochondrial DNA depletion syndrome type 13 (MTDPS13) characterized by reduced mtDNA content and impaired energy production in affected organs. However, the mechanism behind FBXL4 mutation-driven MTDPS13 remain poorly understood. In a recent study, we demonstrate that the CRL1-FBXL4 complex promotes the degradation of BNIP3 and BNIP3L, two key mitophagy cargo receptors. Deficiency of FBXL4 results in a strong accumulation of BNIP3 and BNIP3L proteins and triggers high levels of BNIP3- and BNIP3L-dependent mitophagy. Patient-derived FBXL4 mutations do not affect its interaction with BNIP3 and BNIP3L but impair the assembly of an active CRL1-FBXL4 complex. Furthermore, excessive mitophagy is observed in knockin mice carrying a patient-derived FBXL4 mutation, and in cortical neurons generated from human patient induced pluripotent stem cells (hiPSCs). These findings support the model that the CRL1-FBXL4 complex tightly restricts basal mitophagy, and its dysregulation leads to severe symptoms of MTDPS13.
KEYWORDS: Lysosome, mitochondria, mitophagy, multi-system disorder, ubiquitination
Main text
Mitophagy, the process by which damaged, dysfunctional, or superfluous mitochondria are selectively eliminated through autophagy, plays a crucial role in maintaining cellular homeostasis. Dysregulation of mitophagy has been implicated in the pathogenesis of numerous human diseases. The most studied and understood regulator of mitophagy is the PRKN/parkin-PINK1 pathway, which is responsible for initiating the clearance of defective mitochondria. However, mitophagy can also be initiated by various mitochondrial outer membrane receptors, such as BNIP3, BNIP3L/NIX, FUNDC1, and BCL2L13, which bind to phagophore membranes and facilitate the recruitment of mitochondria for degradation. Mitophagy can occur even in seemingly normal conditions, without the presence of any overt stress. The exact function and regulation of this so-called basal mitophagy remain poorly understood, but accumulating evidence suggests that that autophagy cargo receptors, rather than the PRKN-PINK1 pathway, play a crucial role in this process.
FBXL4 mutations are associated with MTDPS13, a rare genetic disorder characterized by significant reduction in the amount of mtDNA in affected tissues. MTDPS13 patients exhibit a range of severe symptoms, including hypotonia, developmental delay, muscle weakness, seizures, neuropathy, and optic atrophy. Studies have shown that FBXL4 deficiency leads to excessive mitophagy activity, resulting in the depletion of mtDNA in patients. FBXL4 possesses an F-Box domain and thus is thought to function as an adaptor protein, binding to substrate proteins and facilitating their recognition by the CUL1-RBX1-SKP1 E3 ubiquitin ligase complex.
In a recent study, we attempted to delineate the molecular mechanism underlying FBXL4- mutated MTDPS13 [1]. We suspected that FBXL4 May restrict mitophagy by targeting a specific mitophagy-related protein(s) for ubiquitination and degradation. To test this hypothesis, we utilized affinity purification coupled with mass spectrometry methods to identify FBXL4-interacting proteins, and unexpectedly found that BNIP3 and BNIP3L are present in the purified FBXL4 complex. Through co-immunoprecipitation and in vitro binding assays, we demonstrated that FBXL4 directly interacts with BNIP3 and BNIP3L in cells through its leucine-rich repeat domain. Depletion of FBXL4 through siRNA-mediated knockdown (KD) or CRISPR-Cas9-mediated knockout (KO) both lead to a strong accumulation of BNIP3 and BNIP3L proteins in multiple cancer cell lines. Moreover, the CRL1-FBXL4 complex mainly catalyzes K48-linked ubiquitination and subsequent proteasomal degradation of BNIP3 and BNIP3L. This degradation mechanism is not reliant on macroautophagy, because depletion of FBXL4 in ATG7 KO cells still elevates the protein levels of BNIP3 and BNIP3L.
More than 50 FBXL4 mutation types are reported in MTDPS13 patients. Approximately half of these mutations are frameshift or nonsense, which typically predict loss of function. Thus, we focused our analysis on the functional impact of missense mutations on FBXL4 function. We observed that FBXL4 missense mutants are generally less stable than their wild-type counterpart. Interestingly, although the majority of missense mutations of FBXL4 are located on the leucine-rich repeat domain, which is responsible for binding BNIP3 and BNIP3L, we were surprised to observe that these mutants exhibit a similar binding capacity to BNIP3 and BNIP3L as WT FBXL4. In contrast, these mutants display a diminished ability to bind RBX1, SKP1, and CUL1, suggesting that the formation of a functional CRL1-FBXL4 complex is hindered by the FBXL4 mutations. Hence, MTDPS13-associated FBXL4 mutants are inherently incapable of facilitating the ubiquitination and degradation of BNIP3 and BNIP3L. Furthermore, the overexpression of these mutant proteins fails to restore mitophagy induced by FBXL4 deficiency.
To ascertain the causal relationship between BNIP3 and BNIP3L accumulation and FBXL4 KO-induced mitophagy, we conducted individual or combined KD of BNIP3 and BNIP3L in FBXL4-deficient cells. Our results indicated that KD of either BNIP3 or BNIP3L partially rescues FBXL4 KO-induced mitophagy and metabolic remodeling, whereas combined KD of BNIP3 and BNIP3L completely rescues it, indicating that both proteins are the downstream effectors of FBXL4. In addition to mitochondria, BNIP3 and BNIP3L are also localized to peroxisomes to promote pexophagy, a selective autophagy that targets peroxisomes and is essential for the maintenance of homeostasis of peroxisomes. However, FBXL4 KO does not have any effect on pexophagy. The possible reason for this could be that FBXL4 is mainly found in the mitochondria and may only regulate the levels of BNIP3 and BNIP3L specifically within the mitochondria, thus playing a specific role in mitophagy.
Previously, we reported a homozygous variant, FBXL4(c.993insA), in an affected patient. To better understand the pathophysiological impact of FBXL4 mutations on BNIP3 and BNIP3L proteins and mitophagy in vivo, we generated a corresponding mouse model with FBXL4 c.994insA (p.L332Tfs *20) knockin (KI) mutation (FBXL4 KI mice). We observed that multiple organs of FBXL4 KI mice exhibit varying degrees of upregulation in BNIP3 and BNIP3L, as well as downregulation of mitochondrial content. FBXL4 KI mouse embryonic fibroblasts show a reduced oxygen consumption rate and ATP production, as well as an increase in lactate production, compared to wild-type mouse embryonic fibroblasts. Notably, these effects are largely reversed by co-depletion of BNIP3 and BNIP3L.
Neurons are particularly susceptible to mitochondrial dysfunction, primarily because of their high energy reliance and their post-mitotic nature. To examine whether FBXL4 mutations result in hyperactive mitophagy in neurons, we modeled the disease through a series of neural induction experiments utilizing an hiPSC clone (FBXL4 MUT) derived from an MTDPS13 patient. FBXL4 mutations do not appear to affect neuronal differentiation; however, we observe a significant accumulation of BNIP3 and BNIP3L proteins and high levels of basal mitophagy in FBXL4 MUT hiPSC-induced neural progenitor cells and cortical neurons.
Overall, this study highlights the important role of FBXL4 in the regulation of basal mitophagy through its interaction with BNIP3 and BNIP3L. The findings provide insights into the pathogenic mechanisms underlying FBXL4 mutation-driven MTDPS13 (Figure 1). Based on our findings, we propose that the development and exploration of small-molecule PROTAC BNIP3 and BNIP3L degraders can be pursued as a novel therapeutic strategy to overcome the accumulation of BNIP3 and BNIP3L in MTDPS13 patients.
Figure 1.
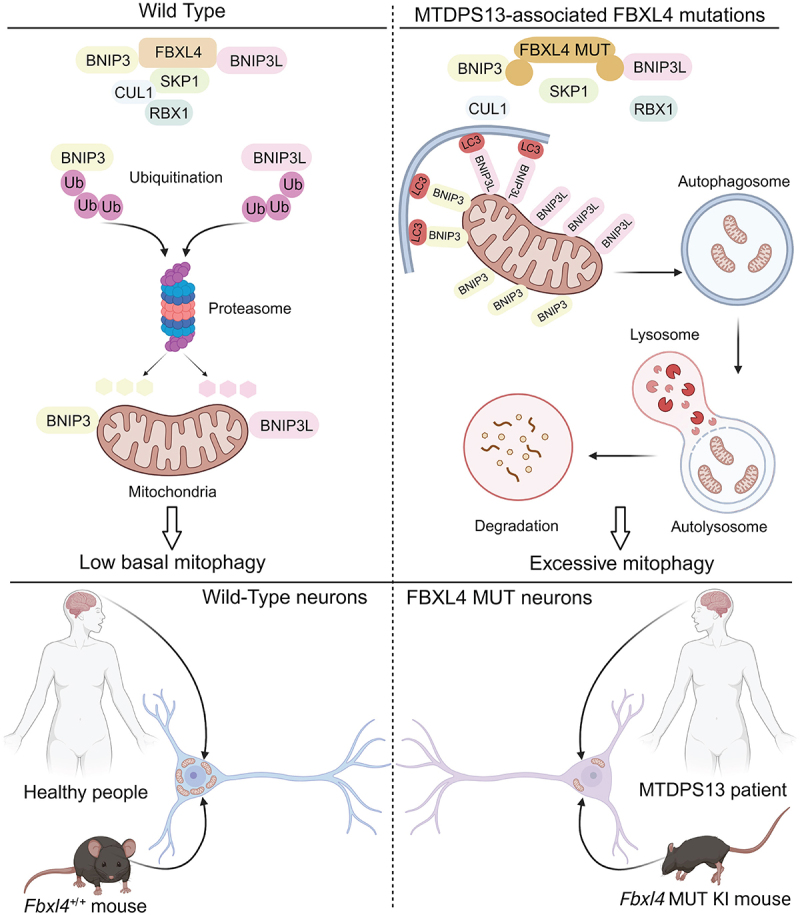
A schematic diagram depicting a model in which FBXL4 mutations lead to excessive mitophagy in MTDPS13 patients and a KI mouse model.
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Funding Statement
This work was in part supported by the National Natural Science Foundation of China (No. 82272992,91954106,and 81872109 to K.G.; No. 32370726, 91957125, 81972396 to C.W.), the State Key Development Programs of China (No. 2022YFA1104200 to C.W.), the Natural Science Foundation of Shanghai (No. 22ZR1449200 to K.G; 22ZR1406600 to C.W.), and the Open Research Fund of State Key Laboratory of Genetic Engineering, Fudan University (No. SKLGE-2111 to K.G.), the Central Guidance on Local Science and Technology Development Foundation (No. 2021ZY0037 to R.M.), Science and Technology Research Program of Shanghai (No. 9DZ2282100).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Reference
- [1].Chen Y, Jiao D, Liu Y, et al. FBXL4 mutations cause excessive mitophagy via BNIP3/BNIP3L accumulation leading to mitochondrial DNA depletion syndrome. Cell Death Differ. 2023 Aug 11;30(10):2351–2363. doi: 10.1038/s41418-023-01205-1 [DOI] [PMC free article] [PubMed] [Google Scholar]