

Abstract
Objectives
The objective of this study was to perform a proof-of-concept experiment that validates a precision medicine approach to identify variants associated with hypertrophic cardiomyopathy (HCM). We hypothesized that whole-genome sequencing would identify variant(s) associated with HCM in two affected Maine Coon/Maine Coon cross cats when compared with 79 controls of various breeds.
Methods
Two affected and two control Maine Coon/Maine Coon cross cats had whole-genome sequencing performed at approximately × 30 coverage. Variants were called in these four cats and 77 cats of various breeds as part of the 99 Lives Cat Genome Sequencing Initiative (http://felinegenetics.missouri.edu/99lives) using Platypus v0.7.9.1, annotated with dbSNP ID, and variants’ effect predicted by SnpEff. Strict filtering criteria (alternate allele frequency >49%) were applied to identify homozygous-alternate or heterozygous variants in the two HCM-affected samples when compared with 79 controls of various breeds.
Results
A total of four variants were identified in the two Maine Coon/Maine Coon cross cats with HCM when compared with 79 controls after strict filtering. Three of the variants identified in genes MFSD12, BTN1A1 and SLITRK5 did not segregate with disease in a separate cohort of seven HCM-affected and five control Maine Coon/Maine Coon cross cats. The remaining variant MYBPC3 segregated with disease status. Furthermore, this gene was previously associated with heart disease and encodes for a protein with sarcomeric function.
Conclusions and relevance
This proof-of-concept experiment identified the previously reported MYBPC3 A31P Maine Coon variant in two HCM-affected cases. This result validates and highlights the power of whole-genome sequencing for feline precision medicine.
Keywords: HCM, WGS, individualized medicine, next-generation sequencing
Introduction
Hypertrophic cardiomyopathy (HCM) is the most commonly diagnosed cardiomyopathy in cats. 1 A previous study reported that a colony of Maine Coon (MC) cats with HCM closely resembles the inheritance pattern of human familial HCM. 2 This colony ultimately led to the identification of the MYBPC3 A31P variant in MC cats via a candidate gene approach. 3 Furthermore, the R820W MYBPC3 HCM variant in cats from the Ragdoll breed was also identified through a candidate gene approach. 4 Even though candidate gene studies have previously identified HCM variants in known genes encoding for proteins with a sarcomeric function, investigators are limited to previously described gene-encoded protein functions and known gene-to-disease relationships when utilizing this method.
As a result of this limitation, whole-genome sequencing (WGS) has emerged as a powerful tool to identify variants associated with inherited diseases. In recent years, new next-generation sequencing (NGS) technology has resulted in a reduction in WGS cost and led to higher integration of its use for precision medicine.5,6 Furthermore, the reduced cost of NGS has allowed genomes from multiple species, including cats, to be assembled and annotated. 7 Therefore, conducting an NGS study in cats is beneficial for the detection of novel disease-causing genetic variants, such as single nucleotide polymorphism (SNP) and/or insertion/deletions (indels). 8 The 99 Lives Cat Genome Sequencing Initiative (http://felinegenetics.missouri.edu/99lives) is a consortium that provides a large control cohort of cats that have had their whole genome sequenced, and has already led to huge advances in the healthcare of cats.9,10 A recent publication by Aberdein et al further illustrates how the 99 Lives Cat Genome Sequencing control database can be used to identify a variant associated with disease in two affected and 82 control cats. 11
In humans, HCM is a familial genetic heart disease affecting 1 in 500 individuals. 12 HCM is also highly prevalent in cats, with recent estimates showing a rate as high as one in seven. 13 Despite the high prevalence of this condition, genetic discoveries are limited to just two previously reported breed-specific variants in MYBPC3.3,4 In particular, the correlation of the MYBPC3 A31P variant and HCM development in MC cats has been in the center of discussion after Wess et al reported that testing positive or negative for this variant was not an indicator for HCM development in a cohort of MC cats phenotyped by a single echocardiogram. 14 Nevertheless, multiple clinical investigations have continued to demonstrate the correlation between the MYBPC3 A31P variant and development of HCM in MC cats.15,16
For this proof-of-concept study, we aimed to demonstrate the efficacy of WGS by identifying the MYBPC3 A31P variant in two cats affected by HCM, one MC and one MC/domestic shorthair cross (MCX), that were distantly related. This proof of concept is an important step toward validating this methodology for future genetic studies that aim to identify novel HCM variants in the cat population. Previous publications have identified that the MC/MCX cat MYBPC3 A31P variant may not be the sole HCM-associated variant in this breed. Therefore, further genetic studies are warranted to define the presence or absence of other biologically relevant variants associated with HCM in MC/MCX cats.17–19
Materials and methods
Sample phenotyping
Extensive cardiac phenotyping was provided to the four MC/MCX cat samples submitted for WGS and for 12 cats used in variant segregation analysis. These cats received annual echocardiograms by a board-certified veterinary cardiologist and necropsy at the time of death. HCM status was defined as a diastolic left ventricular (LV) segment exceeding 6 mm on two-dimensional (2D) or M-Mode imaging, as well as necropsy findings, which confirmed histopathological evidence of HCM and gross appearance of significant LV hypertrophy. 13
Blood collection and DNA extraction
Whole blood and/or tissue samples were collected for all MC/MCX cats following protocols approved by the University of California Davis Institutional Animal Care and Use Committee. DNA was extracted from whole blood and/or tissue using a commercially available kit (ArchivePure; 5Prime), following the manufacturer’s protocol.
Sanger sequencing
PCR was used to verify the MYBPC3 A31P genotype of the four MC/MCX cats submitted for WGS. We used previously designed primers for the MYBPC3 A31P variant PCR reaction following the manufacturer’s protocol for LA Taq with GC Buffer (Takara). 3 After, ExoSAP-IT (Affymetrix) was used to purify PCR amplicons following the manufacturer’s protocol and PCR products were submitted to the UC Davis DNA Sequencing Facility. For cycle sequencing, ABI BigDye Terminator v3.1 kit (ThermoFisher) was used prior to sequencing on an ABI 3730 Capillary Electrophoresis Genetic Analyzer (ThermoFisher). The sequences were aligned to the Felis catus 6.2 reference genome (https://www.ncbi.nlm.nih.gov/assembly/GCF_000181335.1/) and analyzed using Lasergene Evolution Suite (DNASTAR).
WGS analysis
High-quality DNA (2 µg with a 260/280 ratio of 1.80–1.90) was submitted to BGI@UC Davis (Sacramento, CA) for WGS. Quality control of DNA samples was also completed by BGI@UC Davis via a Quibit Fluorometer (ThermoFisher) and 2% agarose gel electrophoresis. If DNA samples passed quality control and were not degraded, 350 bp and 550 bp insert PCR-free libraries were prepared using an Illumina TruSeq DNA PCR-free sample preparation kit. The libraries were pooled and sequenced on 12 lanes – six lanes per library insert – using an Illumina HiSeq 2500 sequencing platform. This generated paired-end 100 bp reads to achieve a desired coverage of × 30.
As part of the 99 Lives Cat Genome Sequencing Initiative, generated fastq.gz sequence files were submitted to MaverixBiomics to be processed and analyzed as described previously. 20 In short, sequence reads were trimmed with Trimmomatic v0.32, and the quality of reads was evaluated with FastQC v0.10.1 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). 21 Reads were aligned to Felis catus 6.2 genome (https://www.ncbi.nlm.nih.gov/assembly/GCF_000181335.1/) with bwa-mem v0.7.6a. 22 Duplicate reads were detected and removed with SAMBLASTER v0.1.9. 23 Depth of coverage was analyzed with GATK Depth Of Coverage tool v2.3. 24 Variants were called simultaneously on all samples with Platypus v0.7.9.1 and annotated with dbSNP ID.25,26 SnpEff was used to predict variant effects based on the Ensembl gene model.27,28 Variant calls were uploaded to MaverixBiomics Variant Explorer (www.maverix.com).
Detection of candidate variants
Results from SnpEff classified variants as severe (ie, frameshift, splice site, stop gain, etc), moderate (ie, indels, non-synonymous variants, etc) and low (ie, intronic and/or intergenic). 24 These results were incorporated into the Maverix Bioinformatics Variant Explorer interface and only variants classified as severe and/or moderate were used in our filtering analysis. Therefore, to identify variants correlated with an autosomal dominant disease in either a gene encoding for a protein with sarcomeric function and/or a gene previously associated with heart disease the following filtering criteria was used: alternate allele frequency >49% in the two HCM-affected MC/MCX cats and wild type in 79 control cat samples. This stringent filtering protocol would thus identify all heterozygous or homozygous variants that were present in the two cases, whereas all of the control cats were wild type.
Variant segregation analysis
We performed variant segregation analysis using seven MC/MCX cats with HCM and five unaffected MC/MCX cats. We used previously designed primers for the MYBPC gene variant identified. 3 Primers were designed for the additional three variants in genes MFSD12, BTN1A1 and SLTRK5 (see Table 1 in the supplementary material). The PCR reaction was optimized using LA Taq with GC Buffer (Takara) and PCR amplicons were purified with ExoSAP-IT (Affymetrix), following the manufacturer’s protocol. All PCR products were submitted to the UC Davis DNA Sequencing Facility for Sanger sequencing, as described above. Results were analyzed with Lasergene Evolution Suite (DNASTAR).
Results
Sample phenotypes
The MC/MCX control cats were free of LV hypertrophy by echocardiography within 6 months of their deaths and had a normal cardiac necropsy with no evidence of LV hypertrophy or histopathological features of HCM. The control cats were >10 years old at time of necropsy. The MC/MCX HCM-affected cats had a diastolic LV segment exceeding 6 mm via 2D or M-Mode imaging during their annual echocardiogram examinations. Cardiac necropsy examination of the affected cats illustrated the gross appearance of LV hypertrophy. The phenotype of the parents of both HCM-affected MC/MCX submitted for WGS is unknown. The 77 control cats from the consortium were not phenotyped cardiovascularly for this study and represent samples from collaborating institutions. However, none of the additional control cats used for comparison in this study were from the MC breed.
Sanger sequencing to verify genotype
Sanger sequencing verified the MYBPC3 A31P genotype of the four experimental MC/MCX cat samples submitted for WGS (Table 1). We did not have DNA for the 77 additional control cats from the 99 Lives Consortium; thus, we were unable to verify their genotype via Sanger sequencing.
Table 1.
Sanger sequencing verification results for the MYBPC 3A31P variant for two cats with hypertrophic cardiomyopathy and two control Maine Coon (MC)/MC domestic shorthair cross (MCX) cats
| Sample | A31P genotype |
|---|---|
| Reference genome | CC |
| MC/MCX1 | CG |
| MC/MCX2 | GG |
| MC/MCX control 1 | CC |
| MC/MCX control 2 | CC |
| Control (n = 77) | n/a |
n/a = not available
WGS
Maverix Variant Explorer interface was used to identify possible HCM-associated variants in two MC/MCX cases when compared with 79 controls from the 99 Lives Cat Genome Sequencing Initiative (Figure 1). The median percentage of reads mapped to the genome was 98.4% (interquartile range 98.1–98.8%) with a mean ± SD of 56.1 ± 15.0 Gbp for all 81 cats in this study. Additionally, mean ± SD coverage for all 81 cats was 25.28 ± 6.56. In particular, the coverage for the two HCM-affected samples was 27.39 and 24.54, and coverage for the two control samples was 23.89 and 24.07. The reads for all 83 cats were aligned to Felis catus 6.2 genome (https://www.ncbi.nlm.nih.gov/assembly/GCF_000181335.1/) assembly.
Figure 1.
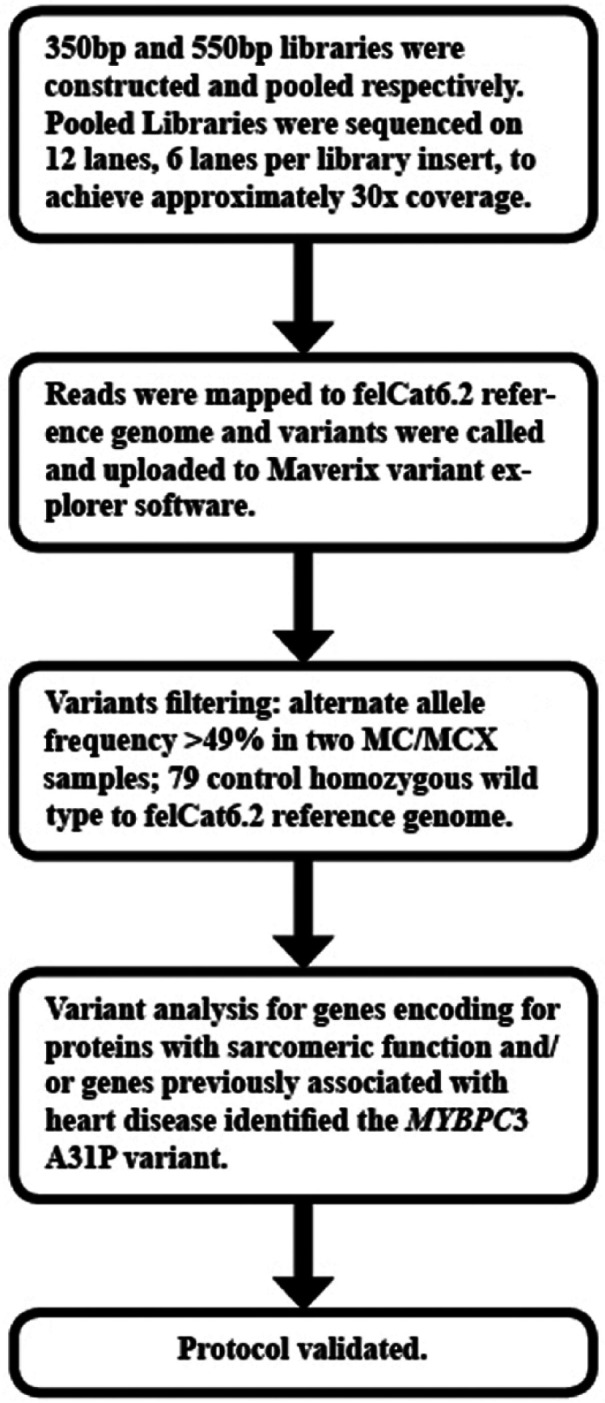
Schematic representation for the identification of the A31P variant in MYBPC3 gene.
MC = Maine Coon; MCX = MC domestic shorthair cross
Variants classified as severe and/or moderate were used in this analysis. A total of 228,598 variants were detected for all 81 WGS cat samples prior to filtering. The two affected MC/MCX cat samples shared 15,205 variants, regardless of the 79 control cats’ variant genotype (Table 2). Of those 15,205 variants, all 81 cats shared 2710 variants that were not further evaluated as these variants would be considered common in the general cat population.
Table 2.
Maverix Biomics Interface variant results for two affected Maine Coon (MC)/MC domestic shorthair cross (MCX) samples
| Effect severity | MC/MCX-1 | MC/MCX-2 | Shared variants in both cases | Shared variants in both cases + 79 controls |
|---|---|---|---|---|
| High | 3158 | 3119 | 2709 | 1632 |
| Moderate | 19,131 | 17,709 | 12,496 | 1078 |
| Low | 36,319 | 34,808 | 24,041 | None |
| Lowest | 2,214,341 | 2,099,722 | 1,461,207 | None |
Variants’ effect severity was predicted by SnpEff. 24 High effect severity refers to frameshift, stop gained and splice site donor/acceptor variants. Moderate refers to single nucleotide polymorphisms, indels and non-synonymous variants. Lowest refers to synonymous variants and lowest effect severity are intron and intergenic variants
Four variants were identified after stringent filtering criteria, described in the ‘Materials and methods’, in the two affected MC/MCX cats, and the four variants have a predicted moderate effect severity (Table 3). Variant segregation analysis was performed in a cohort of 12 MC/MCX cats (seven affected and five controls) to determine if the variants segregated with disease status (Table 4). The M3VYP3:c.112C>G(p.A38P) variant MYBPC3 was the only variant that segregated with HCM affection status. Furthermore, MYBPC3 is a sarcomeric protein-encoding gene that was previously associated with HCM.3,4 The identified variants M3VVL0:c.109C>T(p.V37M) and M3WR01:c.73C>G(p.D25H) in MFSD12 and BTNA1A, respectively, did not segregate with disease and these genes have not been previously associated with heart disease. The other variant identified was M3X7P5:c.1199C>A(p.T400K) in SLITRK5. This gene is involved in neurite modulation and its protein is expressed abundantly in neural tissue. 29 While no other variants in SLITRK5 are directly linked to HCM, this gene was found to be linked to coronary heart disease in humans, as well as an active myocardial signaling pathway known as Slit-Robo.30,31 Alterations to this signaling pathway are associated with congenital heart defects. 31 Regardless, segregation analysis illustrated that the M3X7P5:c.1199C>A(p.T400K) variant in SLITRK5 did not segregate with disease.
Table 3.
Maverix Biomics Variant results for an autosomal dominant trait
| Chromosome | Position | Reference | Alt | AA change | Gene | MC/MCX1 | MC/MCX2 | Control cats (n = 81) |
|---|---|---|---|---|---|---|---|---|
| ChrD1 | 101261107 | C | G | A38P | MYBPC3 | C/G | G/G | C/C |
| ChrA1 | 595545773 | C | A | T400K | SLITRK5 | A/C | A/C | C/C |
| ChrA2 | 2678390 | C | T | V37M | MFSD12 | C/C | T/T | C/C |
| ChrB2 | 3483782 | C | G | D25H | BTN1A1 | C/C | G/G | C/C |
Alternate allele frequency >49% in two cats with hypertrophic cardiomyopathy (Maine Coon [MC]/MC domestic shorthair cross [MCX] samples); homozygous to reference (wild type) for 79 controls
Alt = alternate allele; AA = amino acid
Table 4.
Variant segregation analysis genotype results for seven Main Coon (MC)/MC domestic shorthair cross (MCX) cats with hypertrophic cardiomyopathy (HCM) and five control MC/MCX cats
| Diagnosis | MC/MCX sample number | Variants | |||
|---|---|---|---|---|---|
| MYBPC3 A38P | SLITRK5 T400K | MFSD12 V37M | BTN1A1 D25H | ||
| HCM affected | 1 | HO | HET | HET | WT |
| 2 | HO | HET | HO | WT | |
| 3 | HO | HET | WT | WT | |
| 4 | HO | HET | HET | HO | |
| 5 | HO | WT | WT | WT | |
| 6 | HET | HET | WT | WT | |
| 7 | HET | HET | WT | HET | |
| Normal cardiac evaluation | 8 | WT | HO | WT | WT |
| 9 | WT | HET | HET | WT | |
| 10 | WT | HET | HO | WT | |
| 11 | WT | HET | HO | WT | |
| 12 | WT | HET | WT | WT | |
HO = homozygous; HET = heterozygous; WT = wild type
The previously identified MYBPC3 A31P variant by Meurs et al utilized human reference sequence to design their oligonucleotides. 3 The variant M3VYP3:c.112C>G (p.A38P) identified in this study utilized the cat reference genome Felis catus 6.2 (https://www.ncbi.nlm.nih.gov/assembly/GCF_000181335.1/), which became available after the publication of Meurs et al. 3 Therefore, to determine if these two variants are the same, we analyzed nucleotide and amino acid sequences surrounding both variants, using Ensembl (http://uswest.ensembl.org/index.html). Nucleotide sequence and amino acid alignment for both of these variants verified that M3VYP3:c.112C>G (p.A38P) variant was the previously reported MYBPC3 A31P MC cat variant. Difference in annotation is due to an additional seven amino acid in the cat MYBPC3 gene transcript ENSFCAT00000002530.3 that is not found on the human MYBPC3 gene transcript ENST00000256993.8. The additional seven amino acids are most likely caused by a transcript annotation error as the most recent MYBPC3 gene cat transcript ENSFCAT00000002530.4 also places this variant in position 31.
Discussion
This proof-of-concept study illustrates the advantages of using a WGS approach for precision medicine in cats. The identification of M3VYP3:c.112C>G(p.A38P) variant, which is the same variant previously identified by Meurs et al, further supports and validates this technique. 3 The study design relies on the assumption that the disease of interest is breed specific, as is previously reported in cats with HCM.3,4 Additionally, this study further illustrates that utilizing a small, strictly phenotype-affected sample cohort and a large control population of unknown phenotype allows for the identification of a variant associated with disease. 11
This study’s WGS methodology also revealed three additional variants that met our filtering criteria. The variants M3VVL0:c.109C>T(p.V37M) and M3WR01:c.73C>G (p.D25H) in the MFSD12 and BTN1A1 genes, respectively, were omitted as these are not genes that encode for a protein with sarcomeric function and/or previously associated with heart disease. Published alterations in MFSD12 were linked to modification in melanocytes in both humans and mice. 32 Alterations in BTN1A1 were associated with immune regulation and/or milk fat globule secretion and not cardiac disease.33,34 Furthermore, variant segregation analysis in a cohort of seven affected and five control MC/MCX cats illustrated that these variants did not segregate with HCM status.
The only additional candidate variant was M3X7P5: c.1199C>A(p.T400K) in SLITRK5. This gene was previously associated with heart disease, although not a classical HCM-associated gene.30,31,35 Nevertheless, segregation analysis of the SLITRK5 variant demonstrated that it did not segregate with HCM status. Even though the three additional variants identified did not segregate with disease, the role of compound variants and modifier genes warrants consideration in feline HCM. 36 This can be accomplished by sequencing additional HCM-affected MC/MCX cats to identify novel HCM variants that segregate with disease.
Two different HCM variants in MYBPC3 have been identified in two feline breeds, MC and Ragdoll.3,4 MYBPC3 encodes cardiac myosin binding protein-C (cMyBP-C), a critical regulator of cardiac contraction that is necessary for normal heart function, as well as for increased contractility in response to inotropic stimuli. 37 In people, variants in MYBPC3 are one of the top two most common causes of HCM.38,39
In cats, variants in MYBPC3 were initially reported to cause disease in an autosomal dominant fashion, although evidence has since been presented that the MYBPC3 A31P variant more commonly behaves as autosomal recessive.2,3,16,17,40 It was recently shown that cats heterozygous for the A31P variant express very little of the A31P mutant protein and instead express mainly wild-type protein. 41 One possibility is that structural instability of the mutant protein leads to faster turnover of the A31P cMyBP-C relative to the wild-type protein. However, in homozygous cats that express only the A31P mutant protein, the mutant protein could directly cause disease through a poison polypeptide mechanism or, indirectly, by affecting protein degradation pathways. Regardless, disease penetrance in homozygous A31P cats is higher than in cats heterozygous for the variant.16,17,42
In this proof-of-concept study, we rapidly identified the previously reported MYBPC3 A31P HCM MC cat variant. 3 The difference in amino acid numbering is due to different reference genomes and transcripts being utilized, and most likely due to an transcription error in the cat Ensembl transcript (ENSFCAT00000002530.3). To our knowledge, this is the first WGS study that utilized two distantly related HCM cases for the identification of a disease-causing variant. A limitation to this study was that apart from the two control MC/MCX cats, the remaining 77 control cats were not rigorously phenotyped via echocardiogram. Therefore, we do not know the age of these 77 cats or if they were diagnosed with HCM. Variants in MYBPC3 have low penetrance and are likely breed specific. Utilizing the additional 77 control unphenotyped samples could allow for the identification of novel variants in future studies when only a small HCM-affected sample cohort is available. This is owing to the control samples from the 99 Lives Consortium consisting of various cat breeds, including nine wild felids. Therefore, using this control population allows for rapid identification of common variants in the general cat population that would not be associated with disease.
Coverage for the known HCM genes that encode for proteins with sarcomeric function was not specifically reported in this study. Given that the mean ± SD coverage obtained for all samples in this study was 25.28 ± 6.56, we are confident with the reliability of variants identified in this group of cats. While targeted resequencing study designs specifically targeting sarcomeric genes may be able to provide a greater coverage depth, our study design has a distinct advantage of allowing the identification of non-sarcomeric protein coding variants associated with HCM across the entire genome. For this reason, WGS is commonly utilized for variant discovery in human HCM. 42
A limitation of this study is that a pedigree analysis to definitively determine pattern of inheritance was not performed and, as such, a definitive call on variant causation cannot be conclusively determined. However, the MYBPC3 A31P variant identified in the two cases of this study represented one heterozygous cat and one homozygous mutant cat. Additionally, the segregation analysis revealed two additional cats, which had HCM and an MYBPC3 A31P heterozygous genotype. These findings support the previously reported autosomal-dominant pattern of inheritance for the MYBPC3 A31P variant and warrant future evaluation with large pedigree analysis studies. 2
In the absence of a well-annotated genome, WGS data can be difficult to interpret. Specifically, disease association with variants of unknown significance (VUS) is sometimes observed. This issue has been reported in human HCM sequencing studies and underscores the possibility that many cases of HCM may not be attributed to a single variant.43–45 This problem also applies to cat sequencing studies where the annotation of the current feline reference genome is incomplete. In this study, the authors evaluated all VUS to determine if they segregate with disease status and for biological relevance based on what is currently known for gene function and disease association across species. Additionally, the authors intend to revisit sequencing data in the future as genome annotations improve.
The efficient variant filtering and successful identification of the known MYBPC3 A31P variant, as well as a second candidate variant, highlights the power of this methodology and value of the 99 Lives Cat Genome Sequencing Initiative. Our results clearly support continued precision medicine efforts to study the genetic basis of HCM in cats.
Conclusions
In this proof-of-concept study we confirmed that our methodology would rapidly identify the MYBPC3 A31P variant in two HCM-affected MC/MCX cats when compared with 79 controls of various breeds. Furthermore, variant segregation analysis demonstrated that the additional variants identified in this study did not segregate with disease in a cohort of seven HCM-affected and five control MC/MCX cats. HCM is one of the most common diagnoses in cats, and thus far only two variants have been reported to be associated with this disease in cats. We demonstrate how this methodology can be utilized to discover variants associated with HCM in a small sample cohort when compared with a large control population. Future studies can utilize this approach to identify novel variants associated with HCM, which can lead to a genetic test to reduce the prevalence of this disease in the cat population.
Supplemental Material
Primers used in segregation analysis for the four identified variants in two affected and two control MC/MCX cats
Acknowledgments
Besides two of the authors (JAS and YU), we would like to acknowledge the 99 Lives Sequencing Consortium for providing a large control sample cohort for this manuscript. Each member of the consortium has reviewed and approved this manuscript for publication. The 99 Lives Consortium consists of the following members: Leslie A Lyons and Barbara Gandolfi (Department of Veterinary Medicine and Surgery, College of Veterinary Medicine, University of Missouri, USA); Danielle Aberdein, Dorian J Garrick and John S Munday (Institute of Veterinary, Animal and Biomedical Sciences, Massey University, New Zealand); Paulo C Alves (CIBIO/InBIO, University of Portugal, Portugal; Wildlife Biology Program, University of Montana, USA); Gregory S Barsh and Christopher B Kaelin (HudsonAlpha Institute for Biotechnology, USA; Department of Genetics, Stanford University, USA); Holly C Beale and Patricia P Chan (Maverix Biomics, USA); Adam R Boyko (Department of Biomedical Sciences, College of Veterinary Medicine, Cornell University, USA); Jeffrey A Brockman (Hill’s Pet Nutrition, USA); Marta G Castelhano, Rory J Todhunter and Elizabeth A Wilcox (Department of Clinical Sciences, College of Veterinary Medicine, Cornell University, USA); N Matthew Ellinwood, Max F Rothschild and Dorian J Garrick (Department of Animal Science, College of Agriculture and Life Sciences, Iowa State University, USA); Jonathan E Fogle (College of Veterinary Medicine, North Carolina State University, USA); Christopher R Helps (Langford Veterinary Services, University of Bristol, UK); Marjo K Hytönen and Maria Kaukonen (Department of Veterinary Biosciences and Research Programs Unit, Molecular Neurology, University of Helsinki and Folkhälsan Research Center, Finland); Emilie Leclerc (Diana Pet Food, France); Maria Longeri (Department of Veterinary Medicine, University of Milan, Italy); Richard Malik (Centre for Veterinary Education, University of Sydney, Australia); Michael J Montague and Wesley C Warren (The McDonnell Genome Institute, Washington University School of Medicine, USA); William J Murphy (Department of Veterinary Integrative Biosciences, College of Veterinary Medicine, Texas A&M University, USA); Niels C Pedersen (Department of Medicine and Epidemiology, School of Veterinary Medicine, University of California at Davis, USA); William F Swanson (Center for Conservation and Research of Endangered Wildlife [CREW], Cincinnati Zoo & Botanical Garden, USA); Karen A Terio (Zoological Pathology Program, University of Illinois, USA); and Julia H Wildschutte (Bowling Green State University, Department of Biological Sciences, USA).
Footnotes
Accepted: 6 November 2018
Supplementary material: The following file is available online: Table 1: Primers used in segregation analysis for the four identified variants in two affected and two control MC/MCX cats.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: Funding support for this study was provided by BGI@UCDavis.
ORCID iD: Joshua A Stern  https://orcid.org/0000-0001-5611-5745
https://orcid.org/0000-0001-5611-5745
References
- 1. Ferasin L, Sturgess C, Cannon M, et al. Feline idiopathic cardiomyopathy: a retrospective study of 106 cats (1994–2001). J Feline Med Surg 2003; 5: 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kittleson M, Meurs K, Munro M, et al. Familial hypertrophic cardiomyopathy in maine coon cats: an animal model of human disease. Vet Surg 2001; 21: 56–62. [DOI] [PubMed] [Google Scholar]
- 3. Meurs KM, Sanchez X, David RM, et al. A cardiac myosin binding protein C mutation in the Maine Coon cat with familial hypertrophic cardiomyopathy. Hum Mol Genet 2005; 14: 3587–3593. [DOI] [PubMed] [Google Scholar]
- 4. Meurs KM, Norgard MM, Ederer MM, et al. A substitution mutation in the myosin binding protein C gene in ragdoll hypertrophic cardiomyopathy. Genomics 2007; 90: 261–264. [DOI] [PubMed] [Google Scholar]
- 5. Christensen KD, Dukhovny D, Siebert U, et al. Assessing the costs and cost-effectiveness of genomic sequencing. J Pers Med 2015; 5: 470–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gonzaga-Jauregui C, Lupski JR, Gibbs RA. Human genome sequencing in health and disease. Annu Rev Med 2012; 63: 35–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Montague MJ, Li G, Gandolfi B, et al. Comparative analysis of the domestic cat genome reveals genetic signatures underlying feline biology and domestication. Proc Natl Acad Sci U S A 2014; 111: 17230–17235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Muzzey D, Evans EA, Lieber C. Understanding the basics of NGS: from mechanism to variant calling. Curr Genet Med Rep 2015; 3: 158–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mauler D, Gandolfi B, Reinero C, et al. Precision medicine in cats: novel Niemann-Pick type C1 diagnosed by whole-genome sequencing. J Vet Intern Med 2017; 31: 539–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oh A, Pearce JW, Gandolfi B, et al. Early-onset progressive retinal atrophy associated with an IQCB1 variant in African black-footed cats (Felis nigripes). Sci Rep 2017; 7: 43918. DOI: 10.1038/srep43918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aberdein D, Munday JS, Gandolfi B, et al. A FAS-ligand variant associated with autoimmune lymphoproliferative syndrome in cats. Mamm Genome 2017; 28: 47–55. [DOI] [PubMed] [Google Scholar]
- 12. Chung M-w, Tsoutsman T, Semsarian C. Hypertrophic cardiomyopathy: from gene defect to clinical disease. Cell Res 2003; 13: 9–20. [DOI] [PubMed] [Google Scholar]
- 13. Payne JR, Brodbelt DC, Fuentes VL. Cardiomyopathy prevalence in 780 apparently healthy cats in rehoming centres (the CatScan study). J Vet Cardiol 2015; 17: S244–S257. [DOI] [PubMed] [Google Scholar]
- 14. Wess G, Schinner C, Weber K, et al. Association of A31P and A74T polymorphisms in the myosin binding protein C3 gene and hypertrophic cardiomyopathy in Maine Coon and other breed cats. J Vet Intern Med 2010; 24: 527–532. [DOI] [PubMed] [Google Scholar]
- 15. Mary J, Chetboul V, Sampedrano CC, et al. Prevalence of the MYBPC3-A31P mutation in a large European feline population and association with hypertrophic cardiomyopathy in the Maine Coon breed. J Vet Cardiol 2010; 12: 155–161. [DOI] [PubMed] [Google Scholar]
- 16. Godiksen MT, Granstrøm S, Koch J, et al. Hypertrophic cardiomyopathy in young Maine Coon cats caused by the p. A31P cMyBP-C mutation-the clinical significance of having the mutation. Acta Vet Scand 2011; 53: 7. DOI: 10.1186/1751-0147-53-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Longeri M, Ferrari P, Knafelz P, et al. Myosin-binding protein C DNA variants in domestic cats (A31P, A74T, R820W) and their association with hypertrophic cardiomyopathy. J Vet Intern Med 2013; 27: 275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Casamian-Sorrosal D, Chong S, Fonfara S, et al. Prevalence and demographics of the MYBPC3-mutations in ragdolls and Maine Coons in the British Isles. J Small Anim Pract 2014; 55: 269–273. [DOI] [PubMed] [Google Scholar]
- 19. Meurs K, Norgard M, Kuan M, et al. Analysis of 8 sarcomeric candidate genes for feline hypertrophic cardiomyopathy mutations in cats with hypertrophic cardiomyopathy. J Vet Intern Med 2009; 23: 840–843. [DOI] [PubMed] [Google Scholar]
- 20. Gandolfi B, Grahn RA, Creighton EK, et al. COLQ variant associated with Devon Rex and Sphynx feline hereditary myopathy. Anim Genet 2015; 46: 711–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014; 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009; 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Faust GG, Hall IM. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 2014; 30: 2503–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rimmer A, Phan H, Mathieson I, et al. Integrating mapping-, assembly-and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet 2014; 46: 912–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sherry ST, Ward M-H, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001; 29: 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cingolani P, Platts A, Wang LL, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012; 6: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cunningham F, Amode MR, Barrell D, et al. Ensembl 2015. Nucleic Acids Res 2014; 43: D662–D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aruga J, Yokota N, Mikoshiba K. Human SLITRK family genes: genomic organization and expression profiling in normal brain and brain tumor tissue. Gene 2003; 315: 87–94. [DOI] [PubMed] [Google Scholar]
- 30. De Las Fuentes L, Yang W, Dávila-Román VG, et al. Pathway-based genome-wide association analysis of coronary heart disease identifies biologically important gene sets. Eur J Hum Genet 2012; 20: 1168–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Payne S, Burney MJ, McCue K, et al. A critical role for the chromatin remodeller CHD7 in anterior mesoderm during cardiovascular development. Dev Biol 2015; 405: 82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Crawford NG, Kelly DE, Hansen ME, et al. Loci associated with skin pigmentation identified in African populations. Science 2017; 358. DOI: 10.1126/science.aan8433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abeler-Dörner L, Swamy M, Williams G, et al. Butyrophilins: an emerging family of immune regulators. Trends Immunol 2012; 33: 34–41. [DOI] [PubMed] [Google Scholar]
- 34. Ogg SL, Weldon AK, Dobbie L, et al. Expression of butyrophilin (Btn1a1) in lactating mammary gland is essential for the regulated secretion of milk–lipid droplets. Proc Natl Acad Sci U S A 2004; 101: 10084–10089. DOI: 10.1073/pnas.0402930101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mommersteeg MT, Yeh ML, Parnavelas JG, et al. Disrupted Slit-Robo signalling results in membranous ventricular septum defects and bicuspid aortic valves. Cardiovasc Res 2015; 106: 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maron BJ, Maron MS, Semsarian C. Double or compound sarcomere mutations in hypertrophic cardiomyopathy: a potential link to sudden death in the absence of conventional risk factors. Heart Rhythm 2012; 9: 57–63. [DOI] [PubMed] [Google Scholar]
- 37. Sadayappan S, de Tombe PP. Cardiac myosin binding protein-C: redefining its structure and function. Biophys Rev 2012; 4: 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. da Rocha AM, Guerrero-Serna G, Helms A, et al. Deficient cMyBP-C protein expression during cardiomyocyte differentiation underlies human hypertrophic cardiomyopathy cellular phenotypes in disease specific human ES cell derived cardiomyocytes. J Mol Cell Cardiol 2016; 99: 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McNally EM, Barefield DY, Puckelwartz MJ. The genetic landscape of cardiomyopathy and its role in heart failure. Cell Metab 2015; 21: 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Borgeat K, Casamian-Sorrosal D, Helps C, et al. Association of the myosin binding protein C3 mutation (MYBPC3 R820W) with cardiac death in a survey of 236 Ragdoll cats. J Vet Cardiol 2014; 16: 73–80. [DOI] [PubMed] [Google Scholar]
- 41. van Dijk SJ, Kooiker KB, Mazzalupo S, et al. The A31P missense mutation in cardiac myosin binding protein C alters protein structure but does not cause haploinsufficiency. Arch Biochem Biophys 2016; 601: 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Walsh R, Buchan R, Wilk A, et al. Defining the genetic architecture of hypertrophic cardiomyopathy: re-evaluating the role of non-sarcomeric genes. Eur Heart J 2017: 38: 3451–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Landstrom AP, Ho CY, Ackerman MJ. Mutation type is not clinically useful in predicting prognosis in hypertrophic cardiomyopathy response to Landstrom and Ackerman. Circulation 2010; 122: 2441–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Andersen PS, Havndrup O, Hougs L, et al. Diagnostic yield, interpretation, and clinical utility of mutation screening of sarcomere encoding genes in Danish hypertrophic cardiomyopathy patients and relatives. Hum Mutat 2009; 30: 363–370. [DOI] [PubMed] [Google Scholar]
- 45. Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012; 60: 705–715. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers used in segregation analysis for the four identified variants in two affected and two control MC/MCX cats