

Abstract
Diffuse large B-cell lymphoma (DLBCL) is a heterogenous group of lymphoid malignancies. Based on gene expression profiling, it has been subdivided into germinal center (GC)-derived and activated B-cell (ABC) types. Advances in molecular methodologies have further refined the subclassification of DLBCL, based on recurrent genetic abnormalities. Here, we describe a distinct case of DLBCL that presented in leukemic form. DNA sequencing targeting 275 genes revealed pathogenically relevant mutations of CD79B, MyD88, TP53, TBL1XR1, and PIM1 genes, indicating that this lymphoma would be best classified as MCD/C5 DLBCL, an ABC subtype. Despite an initial good clinical response to BTK inhibitor ibrutinib, anti-CD20 antibody rituxan, alkylating agent bendamustine, and hematopoietic stem-cell transplant, the lymphoma relapsed, accompanied by morphologic and molecular evidence of disease progression. Specifically, the recurrent tumor developed loss of TP53 heterozygosity (LOH) and additional chromosomal changes central to ABC DLBCL pathogenesis, such as PRDM1 loss. Acquired resistance to ibrutinib and rituxan was indicated by the emergence of BTK and FOXO1 mutations, respectively, as well as apparent activation of alternative cell-activation pathways, through copy-number alterations (CNAs), detected by high-resolution chromosomal microarrays. In vitro, studies of relapsed lymphoma cells confirmed resistance to standard BTK inhibitors but sensitivity to vecabrutinib, a noncovalent inhibitor active against both wild-type as well as mutated BTK. In summary, we provide in-depth molecular characterization of a de novo leukemic DLBCL and discuss mechanisms that may have contributed to the lymphoma establishment, progression, and development of drug resistance.
Keywords: B-cell lymphoma, hematological neoplasm
INTRODUCTION
Diffuse large B-cell lymphoma (DLBCL) comprises 25%–35% of adult non-Hodgkin lymphomas and is a highly heterogenous group of lymphoid malignant disorders. Although the involvement of bone marrow with DLBCL cells circulating in peripheral blood is not uncommon in progressive or stage IV disease, initial presentation in the leukemic phase is rare, mostly having been documented in case reports or small study populations (Muringampurath-John et al. 2012; Suresh et al. 2020). Previous attempts to define distinctive features of leukemic DLBCL by flow cytometry have been inconclusive (Muringampurath-John et al. 2012).
Advances in molecular methodologies have been enabling refinement in the subclassification of DLBCL. Gene expression profiling has demonstrated that most DLBCLs can be categorized as either the germinal center B cell (GCB) or activated B cell (ABC) subtype (Alizadeh et al. 2000). Recent comprehensive DNA sequencing studies have shown that DLBCL can be further subdivided into distinct genomic subtypes based on recurrent mutations and chromosomal abnormalities. This is not only of academic interest as the subtypes display significant differences in progression-free and overall survival (Schmitz et al. 2018) and therapeutic response to BTK inhibition (Wilson et al. 2021). One of the subtypes, the MCD/C5, showed an especially poor outcome, with five-year overall survival of 42% (Lacy et al. 2020) but seemingly exceptional response when the standard immunochemotherapy is combined with BTK inhibition (Wilson et al. 2021).
Here, we describe a unique case of a de novo mature B-cell neoplasm, which presented in the leukemic phase and displayed progressive genomic alterations. Based on the World Health Organization (WHO) Revised Fourth Edition criteria, the most up-to-date edition at the time of the presentation, the malignancy was initially diagnosed as B-cell prolymphocytic leukemia (B-PLL).
Initial treatment with the BTK inhibitor ibrutinib, chemotherapeutic agent bendamustine, and anti-CD20 antibody rituxan resulted in complete clinical remission. However, the tumor relapsed multiple times, with recurrent disease exhibiting changed, large-cell morphology and additional genomic alterations commonly seen in the ABC DLBCL subtype as well as evidence of the development of ibrutinib and rituxan resistance.
RESULTS
Clinical History
A 59-yr-old female without significant medical history presented in August of 2018 with hepatosplenomegaly, anemia, thrombocytopenia, and leukocytosis with the spleen being markedly enlarged, measuring 22.5 cm caudally, based on a computed tomography (CT) scan. No lymphadenopathy was detected. Peripheral blood white cell count was 55 K/μL and included lymphocyte count of 39 K/μL with the predominance of medium-sized atypical lymphocytes (Fig. 1A). Flow cytometry detected a clonal B-cell population comprising 90% of the lymphocytes (Supplemental Fig. 1) and 57% of all white cells. Based on clinical presentation and, as described below, cell morphology, and phenotype, a diagnosis of B-PLL was rendered based on the WHO Fourth Edition criteria.
Figure 1.

Morphology and immunophenotype of diffuse large B-cell lymphoma (DLBCL) at presentation and relapse. (A) Peripheral blood smear showing atypical lymphocytes with medium-sized nuclei containing moderately condensed chromatin (100× magnification). (B) Peritoneal fluid cytology showing infiltrate of highly atypical lymphocytes with large nuclei containing open chromatin and occasionally prominent nucleoli (100×).
Initial treatment with ibrutinib, bendamustine, and rituxan resulted in the absence of measurable residual disease. In December of 2018, the tumor reappeared in the tibia for which the patient was treated with fludarabine/cytoxan total body irradiation (TBI)-conditioned hematopoietic stem-cell transplant (HSCT); follow-up imaging showed complete disease remission. Six months later, while on maintenance ibrutinib, the patient developed malignant ascites with enlarged abdominal lymph nodes. The ascites and lymphadenopathy resolved after discontinuing ibrutinib and initiating treatment with bendamustine and rituxan. The patient was then placed again on ibrutinib. Two months later, in September 2019, the patient presented with thrombocytopenia, subcutaneous nodules, cerebrospinal fluid involvement, and recurrent ascites. She passed away before starting therapy with the BCL-2 inhibitor venetoclax.
Histologic, Flow Cytometry, and Immunohistochemical Studies
At presentation in August 2018, the peripheral smear examination revealed atypical medium-sized lymphoid cells with prolymphocytic features, including nuclei containing moderately condensed chromatin (Fig. 1A). Bone marrow demonstrated hypercellularity (∼70%; Supplemental Fig. 2A) and contained interstitial infiltrates of the atypical lymphoid cells comprising ∼20% of marrow cellularity, highlighted by the CD20 immunostain (Supplemental Fig. 2B).
Flow cytometry analysis of the early disease peripheral blood from August 2018 identified clonal, κ light chain-restricted B-lymphocytes, positive for CD19, CD20, and CD79B and negative for CD5, CD10, CD23, CD38, CD34, and FMC7 (Supplemental Table 1; Supplemental Fig. 1A). Immunostains performed on the bone marrow sections showed a population of lymphocytes positive for CD20, CD79A, BCL-2, and MUM1. They were negative for cyclin D1, SOX11, TDT, TP53, c-MYC, and the other markers listed in Supplemental Table 2 with BCL-6 showing staining in a few cells (Supplemental Fig. 2C).
In mid-2019, the patient developed ascites and the ascitic fluid from both July and September of 2019 contained malignant cells. However, in contrast to early disease, the malignant lymphocytes displayed marked pleomorphism and markedly increased size (Fig. 1B), resulting in the diagnosis of large B-cell lymphoma. They expressed a similar phenotype as seen initially with somewhat diminished expression of CD20 revealed by flow cytometry (Supplemental Fig. 1B). Additionally, immunohistochemistry detected c-MYC and TP53 expression, which became quite pronounced in the September 2019 sample (Supplemental Table 2).
Cytogenetic and Fluorescence In Situ Hybridization (FISH) Analysis
Cytogenetics of the August 2018 bone marrow showed a complex karyotype, with gains of Chromosomes X and 5; interstitial deletions 8p, 15q, and 17q, rearrangements of 5q, 6p, and 9q; and partial interstitial duplications 1q and 11q. Cytogenetics was not performed at relapse.
August 2018 FISH studies on the peripheral blood using probes for BCL-6, 6p, 6q21, MYC, t(11;14), t(14;18), and MALT1 showed only trisomy 6p. At recurrence in July 2019, FISH probes for 12, 13q, 17p, 11q, MYC, BCL-2, BCL-6, and deletion 17p13, performed on ascites, showed deletion of 17p13 (including the TP53 gene locus), 11q23.3 (including the ATM locus), and 13q14.2-14.3 (the D135319 locus).
High-density chromosomal microarray (CMA) and panel-based DNA mutational and genome-scale RNA expression profiling were performed at presentation and relapsed late disease. They detected genomic alterations that can be categorized as described below.
Abnormalities in B-Cell Receptor (BCR) Signaling and Constitutive NF-κB Activation
At presentation and relapse, hallmark genomic changes affecting chronic active B-cell and Toll-like receptor (TLR) signaling pathways were present. These changes promote constitutive activation of NF-κB, a transcription factor that regulates many genes that drive B-cell proliferation and survival (Baldwin 2001).
B-cell clonality studies identified a dominant B-cell clone with heavy chain IGHV4-34; IGHD3-16; IGHJ4. This variant is present in one-third of ABC DLBC cases and plays an important role in the initiation and perpetuation of BCR signaling. The signaling initiation is proposed to result from the binding of self-antigens such as glycoproteins with N-acetyl-lactosamine sugars, driving chronic active BCR signaling (Young et al. 2019).
Mutation of the BCR component CD79B Y196H was seen initially. At relapse, this CD79B mutant displayed also gene copy-number gain (Table 1) associated with increased gene expression (see Fig. 3), strongly suggestive of its oncogenic role. Accordingly, mutations affecting the ITAM motifs of either CD79A or CD79B are present in ∼30% of ABC DLBCL (Young et al. 2019), with the CD79B Y196H mutation being one of the most frequent. The Y196H mutation eliminates the phosphorylation site of the first ITAM, which is thought to impair BCR internalization, resulting in sustained BCR signaling (Baldwin 2001) and the proliferation of malignant B cells. Copy-number gain of CD79A seen at both presentation and relapse (Fig. 2A,B, respectively) and increased expression at relapse (Fig. 3) provided additional indication of the critical pathogenic role of the BCR complex in our case.
Table 1.
Gene mutational profile at presentation (8/2018) and relapse (9/2019)
| Gene | 8/2018 Peripheral blood | 2018 Mutation effect | 2018 Mutation VAF (%) | 9/2019 Ascitic fluid | 2019 Mutation VAF (%) | 2019 Mutation effect | Pathogenic |
|---|---|---|---|---|---|---|---|
| CD79B | c.586T > C p.Y196H | Missense | 35 | c.586T > C p.Y196H | 43 | Missense | Yes |
| MYD88 | c.794T > C p.L265P | Missense | 35 | c.794T > C p.L265P | 63 | Missense | Yes |
| TBL1XR1 | c.1337A > G p.Y446C | Missense | 30 | c.1337A > G p.Y446C | 34 | Missense | Yes |
| TBL1XR1 | c.1192A > T p.K398a | Nonsense | 34 | c.1192A > T p.K398a | 60 | Nonsense | Yes |
| TP53 | c.742C > T p.R248W | Missense | 39 | c.742C > T p.R248W | 90 | Missense | Yes |
| PIM1 | c.373C > T p.P125S | Missense | 51 | c.373C > T p.P125S | 64 | Missense | Uncertain |
| PIM1 | – | c.149G > A p.G50D | 28 | Missense | Uncertain | ||
| PIM1 | – | c.97C > T p.P33S | 29 | Missense | Uncertain | ||
| ETV6 | c.33 + 1G > A | Splice donor | 53 | – | 0 | – | Yes |
| BTK | – | c.1442G > C p.C481S | 45 | Missense | Yes | ||
| FOXO1 | – | c.482G > A p.G161D | 47 | Missense | Yes | ||
| NF1 | – | c.7064G > T p.S2355I | 50 | Missense | Uncertain | ||
| NOTCH1 | – | c.2856C > A p.D952E | 47 | Missense | Uncertain | ||
| ABL2 | – | c.2503G > T p.A835S | 30 | Missense | Uncertain | ||
| BRAF | c.1600G > A p.G534S | Missense | 37 | c.1600G > A p.G534S | 41 | Missense | Uncertain |
(VAF) Variant allele frequency.
Figure 3.
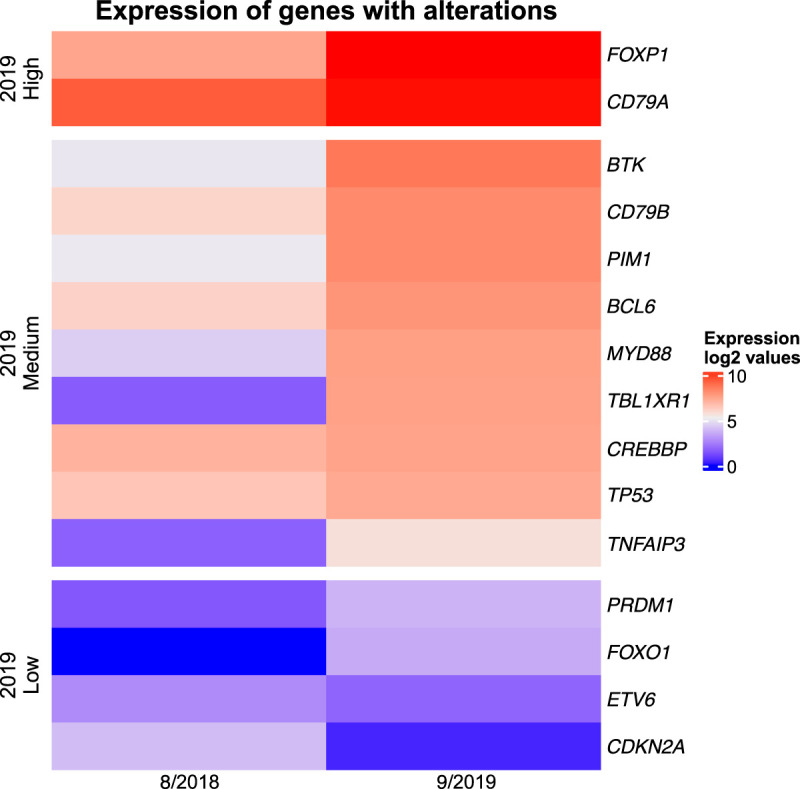
Expression of key altered genes. Relative expression of the depicted genes in the 8/2018 and 9/2019 samples.
Figure 2.

Genomic gains and losses detected by high-density chromosomal microarray (CMA). (A) Specimen from 8/2018 (Peripheral blood). (B) 9/2019 (Ascites); new alterations are marked by thick green arrows.
MYD88 is one of the most frequently altered genes in ABC DLBCL, with mutations occurring in some series of up to 39% of cases (Ngo et al. 2011; Dubois et al. 2017). MYD88 is an essential signaling adaptor protein that interacts with TLRs and kinases IRAK1 and IRAK4 to form a “myddosome complex.” The MYD88 (L265P) was detected in our tumor at presentation and with increased variant allele frequency (VAF) (Table 1), copy-number variation (CNV) (Fig. 2), and expression (Fig. 3) at recurrence. The L265P mutation is a gain-of-function oncogenic mutation resulting in spontaneous assembly of the myddosome complex (Ngo et al. 2011) and ultimately NF-κB pathway activation.
TNFAIP3 encodes A20, a ubiquitin-modifying enzyme required for the termination of NF-κB responses. Compagno et al. (2009) demonstrated monoallelic deletions were observed in 23% of ABC DLBCL. Inactivation through deletions, as seen in recurrent disease in this tumor (Fig. 2B), are more common in ABC DLBCL (30%) and often accompany MYD88 L265P mutations in ABC DLBCL, as is also the case here.
BTK is an essential component of the BCR-activated signaling pathway. At presentation, this B-cell malignancy had an increased copy number of BTK (Fig. 2), likely another indication of dependency on BCR signaling. A recurrent tumor developed a BTK C481S mutation associated with BTK inhibitor resistance (discussed separately, below).
Impairment of B-Cell Maturation and Differentiation Mechanisms
Derailment of orderly B-cell differentiation is another factor contributing to lymphomagenesis (Novak et al. 2015). Chromosomal abnormalities affecting it were detected, mainly in relapsed disease.
BCL-6 is a transcriptional repressor that functions to attenuate or prevent expression of genes that interfere with the germinal center reaction. Up-regulation of BCL-6 is common in DLBCL and occurs through mutations, translocations (Miao et al. 2019), or genes whose products interact with BCL-6. Although mutations in the BCL-6 gene were not detected in this tumor, evidence of up-regulation in relapse was detected through copy-number gain (Table 1) and enhanced expression of the gene (Fig. 3). The gain of BCL-6 expression was also confirmed on the protein level (Supplemental Fig. 2). Although bone marrow at the disease presentation showed only a few BCL-6-positive cells (Supplemental Fig. 2C) in contrast to the robust staining for B-cell marker CD20 (Supplemental Fig. 2B), the majority of the CD20+ malignant cells (Supplemental Fig. 2D,E) in the ascitic fluid upon recurrence displayed various degree of BCL-6 staining (Supplemental Fig. 2F).
The function of TBL1XR1 is not well characterized. It is a core component of the SMRT/NCOR1 transcriptional repressor complexes. TBL1XR1 Y446C missense and TBL1XR1 K398 nonsense mutations were present initially and at relapse, with doubled VAF at relapse for the latter (Table 1). These TBL1XR1 alterations result in the loss of interaction of the SMRT/NCOR1 complex with the germinal center transcriptional repressor BCL-6. Instead, BCL-6 binding to the transcription factor BACH2 complex is induced. This binding realignment blocks GCBs from developing into plasma cells by repressing PRDM1, a transcriptional repressor. Additionally, at relapse, this tumor had an arm-level loss of 6q, which contains PRDM1 (Fig. 2). Inactivation of PRDM1 is critical to the pathogenesis of ABC DLBCL (Pasqualucci et al. 2006; Calado et al. 2010; Mandelbaum et al. 2010). Collectively, TBL1XR1 mutations and loss of PRDM1 prevent B-cell differentiation toward plasma cells and drive them toward the memory B-cell program (Venturutti and Melnick 2020; Venturutti et al. 2020), most likely contributing to malignant cell transformation.
Loss of Key Epigenetic Regulator
At presentation and relapse, CMA detected loss of 16p13.3 region (Fig. 2), which harbors CREBBP, a gene encoding acetyltransferase. Loss of one CREBBP allele occurs in >80% of DLBCL, with expression of the residual wild-type allele, akin to a haploinsufficient tumor suppressor. CREBBP regulates germinal center physiology, possibly contributing to lymphomagenesis through blocking B-cell differentiation, facilitating immune escape, impairing BCL-6 inactivation, and causing defects in TP53 activation (Pasqualucci and Dalla-Favera 2018; Miao et al. 2019; Bakhshi and Georgel 2020).
Impairment of TP53 Pathway
The TP53 tumor suppressor plays a crucial role in cell proliferation and survival. Dysregulation through different mechanisms is associated with aggressive biologic and clinical behavior, including only partial or no response to CHOP-based chemotherapy (Young et al. 2007; Xu-Monette et al. 2012). TP53 mutations frequently involve the DNA binding domain, exons 5–8, impairing TP53-mediated transcriptional transactivation (Miao et al. 2019). Indeed, the TP53 (R248W) mutation detected in this tumor at presentation and recurrence causes structural defects in the region responsible for DNA binding. Furthermore, the second allelic, wild-type copy of the gene has been lost at relapse (Fig. 2), stressing further the pathogenic role of TP53 alterations in this case. TP53 mutation seems uncommon in the MYD88 cluster but, when present, it confers an extremely poor prognosis (Lacy et al. 2020).
Deletion of the CDKN2A gene, detected at presentation and recurrence (Fig. 2), occurs in 30%–40% of all DLBCLs (Miao 640). The locus encodes two proteins p16INK4A and p14ARF involved in G1/S phase transition and in stabilizing TP53, respectively. Loss of CKDN2A inactivates these pathways and may provide a selective growth advantage of the affected malignant cells (Jardin et al. 2010).
Mutations of PIM1 and ETV6
PIM1 encodes a serine/threonine protein kinase overexpressed in many human cancers, including DLBCL. Its oncogenic role remains to be fully clarified (Amin et al. 2017; Zhang et al. 2022). In a study of 188 DLBCL patients, Zhang et al. (2022) demonstrated that PIM1 mutations co-occurred significantly with MYD88 mutations. These patients displayed shorter progression-free and overall survival. Additionally, PIM1 mutations based on C to T transition were the most frequent in DLBCL, with almost half of them predicted to be pathogenic. Initially and at recurrence, our tumor had a PIM mutation c.373C > T (P125S). At recurrence, additional PIM1 mutations—G50D, and P33S, both also resulting from C to T substitution—were present. Interestingly, in a series of six patients with sequential DLBCL biopsies, it was found that relapsed samples were enriched in PIM1 mutations (Nijland et al. 2018), as was the case here.
ETV6 is a transcriptional factor with a critical role in hematopoiesis and embryonic development, frequently mutated in hematologic malignancies. Mutant ETV6 was present at initial presentation but was not detected in relapsed tumor. Although alterations in ETV6 are one of the hallmark features of the MCD/C5 group, its oncogenic role in DLBCL remains undefined (Marino et al. 2022).
Trisomy of Chromosome 3
The relapsed tumor had trisomy 3 (Fig. 2), which appears to occur almost exclusively in ABC DLBCL and has been associated with inferior outcome (Lenz et al. 2008). One of the candidate genes on Chromosome 3 is FOXP1, which is highly expressed in ABC DLBCL (Lenz et al. 2008). This has been described as occurring concurrently with BCL-2 expression in the absence of translocation t(14;18) (Barrans et al. 2004), as seen also in our case (Supplemental Table 2). It has also been demonstrated that aberrant, high expression of FOXP1 can block B-cell differentiation (van Keimpema et al. 2015). FOXP1-expressing DLBCL seem also to be prone to therapy resistance and early relapses with subsequent treatments being ineffective.
Development of Drug Resistance
ABC DLBCL may initially yield a good therapeutic response. Tumors dependent on BCR activity and the MyD88 mutation are particularly sensitive to BTK inhibition (Wilson et al. 2021). However, resistance to both the anti-CD20 antibody rituximab and BTK inhibitor ibrutinib frequently develop (Pyrzynska et al. 2018) and result in recurrent tumors.
The dominant genetic mechanism of BTK inhibitor resistance is the emergence of the BTK mutation C481S, which alters the binding site for ibrutinib, rendering the drug ineffective (George et al. 2020; Ondrisova and Mraz 2020). This has been well-described in chronic lymphocytic leukemia and other lymphoid malignancies (Nakhoda et al. 2023). The effect of BTK C481S mutation-based resistance to ibrutinib seemed in our case to be further amplified by CNV gain of the BTK gene through tetraploidy of the Chromosome X (Table 1 and Fig. 2). Interestingly, the BTK-encoding region of the Chromosome X partially lost its amplification at relapse (Fig. 2). This may represent a mechanism to decrease BTK signaling dependence as a possible additional mechanism of ibrutinib resistance. However, the strong expression of BTK at relapse (Fig. 3) indicated that the BTK mutation, not BTK independence, was overall responsible for ibrutinib becoming ineffective. Another possible mechanism of ibrutinib resistance is through activation of alternative pathways bypassing BTK. In our tumor this was indicated by CNV gain of genes, including those affecting the MYD8-TLR9-BCR (My-T-BCR) super-complex (Phelan et al. 2018). Accordingly, these genes have been implicated in the activation of an alternative B-cell signaling pathway (Shaffer et al. 2021). Finally, up-regulation of CD79B, as seen in our relapsed tumor, was associated with strong activation of the BTK-independent AKT and MAPK pathways (Kim et al. 2016).
In addition to ibrutinib resistance, there is evidence in our case suggesting the development of rituxan resistance. Mutations in FOXO1, a transcription factor, which was present at tumor relapse, have been described in 10% of DLBCL (Sablon et al. 2022), and correlate with decreased overall survival in DLBCL treated with immunochemotherapy regimen containing anti-CD20 antibody. Novak et al. (2015) found that gene losses localized to 6q21–6q24.2 and gains to 3q13.12–3q9 (both were observed in the recurrent tumor) and 11q23.1–11q23.3, when combined with FOXO1 mutations, identified 77% of cases that failed to achieve event-free survival at 24 mo. The specific FOXO1 mutation in our relapsed tumor (G161D) has not been described before in DLBCL, but it affects DNA binding domain. Pyrzynska et al. (2018) showed FOXO1 activating mutations possibly affected its binding to the promoter of the MS4A1 gene encoding the CD20 protein and resulted in down-regulation of CD20 expression; impairment of CD20 expression occurred in our case at relapse (Supplemental Table 2).
Examination of BTK Inhibition on DLBCL Cell Growth
Lymphoma cells collected from the peritoneal fluid at the time of relapse were cultured ex vivo in a tissue microenvironment model, as previously described (Lu et al. 2021; Wu et al. 2023). As shown in Figure 4, viability and proliferation of lymphoma cells remained unaffected by the covalent BTK inhibitors (BTKi) ibrutinib and acalabrutinib that rely on BTK C481 residue for binding. However, the cells were markedly affected by a third-generation, noncovalent BTK inhibitor vecalabrutinib, known to be reactive against the native BTK as well as the C481S mutant.
Figure 4.

Effects of different BTK inhibitors on viability and proliferation of diffuse large cell lymphoma (DLCL) cells. Diffuse large B-cell lymphoma (DLBCL) cells from the July 2019 ascetic fluid were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) and cultured for 7 d with the depicted BTK inhibitors at indicated doses, using the ex vivo cell culture model (Rosenwald et al. 2022; Sablon et al. 2022) and evaluated for cell viability (left) and cell proliferation by the CFSE assay (right) with the latter gated on the cells remaining live. Drug vehicle (DMSO) served as a control.
DISCUSSION
Initially, this lymphoid malignancy was thought to be a de novo B-PLL. At the time of presentation, the WHO Fourth Edition was the most updated version, which based the diagnosis of B-PLL on morphology, phenotype, clinical presentation, and exclusion of other leukemic lymphomas. Accordingly, our immunohistochemistry and FISH studies showed this tumor was not leukemic mantle cell lymphoma, chronic lymphocytic leukemia, or a “double”-hit DLBCL. Although in-depth DNA sequencing and CMA studies performed on the presentation and disease-relapse specimens had the hallmark mutations and chromosomal abnormalities of the DLBCL MCD/C5 subtype, their relationship to this entity was not recognized until later. This is in part because the landmark studies by Schmitz et al. (2018) and Chapuy et al. (2018), which demonstrated that DLBCL could potentially be categorized by recurrent mutations, had been published only shortly before this patient presented; the Lacy et al. report did not appear until 2020. Once recognized, these molecular studies provided undisputable evidence that this malignancy should be classified as DLBCL, ABC type, MCD/C5 subtype. BCR signaling pathway activation and dysregulated TP53 were central in the pathogenesis and progression of this lymphoma, as demonstrated by markedly increased VAFs and gene expression in mutated CD79B, MyD88, and TP53 in late disease (Fig. 3). Overall, the final classification of our case supports the removal of de novo B-PLL as a distinct category in the recent WHO Fifth Edition classification and supports the notion that cases like ours are a leukemic variant of DLBCL.
The morphologic changes that accompanied tumor progression were striking. The lymphoma cells seen initially in 2018 were medium-sized with prolymphocytic morphology. In 2019, the malignant cells displayed enlarged nuclei and marked pleomorphism, typical for aggressive DLBCL. Although a second de novo B-cell malignancy could have been considered (Juskevicius et al. 2016), the multiple shared genomic alterations between the initial and relapsed tumor indicated they were the same malignant clone that had acquired additional alterations with the pathologic and clinical progression.
Although still a rather novel concept, at least at the current patient-care level, progression within the DLBCL subcategories is becoming firmly conceptualized (Nijland et al. 2018; Los-deVries et al. 2023). In principle, progression/relapse is thought to occur through two patterns: “early divergent,” with clones at initial presentation and relapse clones diverging early in development, and “late divergent,” with clones retaining a high degree of similarity (Juskevicius et al. 2016; Bakhshi and Georgel 2020). As previously noted, the initial and relapsed tumors shared many genomic abnormalities and imply this is an example of a late divergent pattern.
The origin of MCD tumors is still under investigation. Distinctive canonical activation-induced deaminase (cAID)-driven genomic footprint and associated aberrant somatic hypermutations (SHM) suggest the MCD/C5 tumors originate from GC transited B cells (Chapuy et al. 2018). Because TBL1XR1 mutations are present in approximately one-third of MCD tumors, Venturutti et al. (2020) propose they are derived from memory B cells that have cyclically reentered germinal center reactions. Recently, Pindzola et al. (2022) concluded that the cells of origin of MCD DLBCL are aberrant splenic GCBs. In that study, mice with MYD88/CD79B/PRDM1 mutations and overexpression of BCL-2 showed increased IRF4 expression, had a >50-fold expansion of germinal centers in the splenic white pulp, and developed cells with features of DLBCL. Of note, our patient presented with massive splenomegaly, supporting the notion that MCD DLBCL can be derived from spleen, at least in some cases. Antibody-based immunotherapy and small molecule–based BTK-targeting therapy have revolutionized the treatment of lymphomas. However, as this case shows, drug resistance is a significant impediment, through different mechanisms no less. In vitro studies, performed on relapsed tumor, confirmed resistance to ibrutinib and second-generation BTK inhibitors but indicated tumor sensitivity to the third-generation inhibitor vecabrutinib (Fig. 4). It is foreseeable that this sort of testing may be one way to address resistance.
From a practical perspective, this case illustrates that although the related landmark studies were published three to five years ago, the classification of DLBCL by recurrent mutations and the concept of DLBCL progression remain novel concepts for most practicing pathologists. This is likely in part because the most current WHO classification and International Consensus Classification agree that clinical trials and harmonization of the different molecular-based DLBCL classifications are required, before they are formally recognized as defined entities. Furthermore, the classifications recognize the transformation of indolent B-cell lymphomas into aggressive lymphoma, but do not directly address progression within DLBCL. It is hoped that cases such as ours illustrate how molecular analysis, at the investigational and clinical level, can refine DLBCL diagnosis, and potentially guide treatments through understanding the pathways involved in tumor pathogenesis, progression, and drug resistance.
MATERIALS AND METHODS
Diagnostic Studies
Part of the data was generated as a component of the routine diagnostic workup, including histology, cytogenetics, and FISH studies performed in the Fox Chase Cancer Center laboratories using standard methods. The other studies are listed below.
Flow Cytometry
Cells were analyzed in the clinical flow cytometry laboratory using the standard antilymphoma antibody panel. The marker expression was analyzed using directly conjugated and control IgG antibodies (BD Biosciences & BioLegend). Flow analyses were done on Canto 10 Color Flow Cytometry instrument (BD Biosciences). Data were analyzed using FlowJo software v9.9.6 (BD Biosciences).
Immunohistochemical Tissue Analysis
Immunohistochemical staining of formalin-fixed paraffin-embedded biopsy tissues was performed on a Leica Bond instrument using the Bond Polymer Refine Detection System. In brief, the slides were heat-treated for antigen retrieval in 10 mM citrate buffer and sections were incubated with the diluted standard primary antibodies routinely used in the diagnostic antibodies listed in Supplemental Table 2 at the pretested concentrations. Heat-induced epitope retrieval was done for 20 min with ER1 solution (Leica). For interpretation, the immunostained slides were evaluated by light microscopy and the images captured by the attached camera (Leica).
High-Density CMA Analysis
Thermo Fisher CytoScan high-density (HD) arrays were used, which contain >2.6 million copy-number markers of which 750,000 are “genotype–able” SNPs and 1.9 million are nonpolymorphic probes. The genomic DNA was amplified, fragmented, biotin-labeled, and hybridized to a CytoScan HD array according to the manufacturer's recommendations. The hybridized array was washed with Thermo Fisher GeneChip Fluidics 450 and scanned with the GeneChip Scanner 3000 7G. The intensities of probe hybridization were analyzed by the GeneChip Command Console (GCC), and the copy-number and genotyping analyses were performed using Thermo Fisher Chromosome Analysis Suite (ChAS) software with default settings.
DNA Sequence Analysis
Targeted DNA sequencing of 235 gene panels was performed by Genoptix using a standard protocol. In brief, isolated and prepared genomic DNA was analyzed by next-generation sequencing. The genomic alterations within each of these genes were detected through proprietary bioinformatic analysis software and interpreted in conjunction with reference databases such as COSMIC and dbSNP with the exclusion of benign sequence variants. Quality control metrics included a minimum input of 20 ng, with an optimal input of 100 ng of genomic DNA, and average mean sequencing depth of 500× coverage. The limit of detection for SNV was 5%. The DNA sequencing data have been provided as the vcf files in Supplemental Table 3.
RNA Sequence (RNA-seq) Analysis
An RNA sequencing library was prepared using the DuoSeq assay (Data Driven Bioscience). The libraries were sequenced on the Illumina NovaSeq platform, and bioinformatics analysis was performed using the included DuoSeq suite software. STAR v2.7.0 (https://doi.org/10.1093/bioinformatics/bts635) was used to align RNA-seq reads to the GRCh38.p12 transcriptome. Salmon v1.2.1 (https://doi.org/10.1038/nmeth.4197) was used to quantify the expression of transcripts, and tximport (https://doi.org/10.12688/f1000research.7563.2) was used to obtain the transcripts per million (TPM) counts by normalizing transcript counts within each sample. TPM counts were base 2 log-scaled and the log2 TPM values for chosen genes of interest were plotted for both samples (8/2018 and 9/2019) from the patient.
Examination of Drug Effect on DLBCL Cell Growth
RosetteSep Human B-Cell Enrichment Cocktail (Stemcell Technologies) was used for the isolation and enrichment of malignant B cells. In the cell culture model and drug testing procedure, the isolated B cells were labeled with CFSE by CellTrace Violet Cell Proliferation Kit (Thermo Fisher Scientific) and were seeded into a 48-well plate with a human bone marrow fibroblast (BMF) monolayer in each well. CpG (2 µg/mL) and recombinant human IL-15 (10 ng/mL) were also added for B-cell activation and proliferation. Drugs were added 24 h later along with DMSO as the control. After 7 d, cells were collected and stained for anti-CD3, anti-CD19, and anti-CD20 and live B cells were gated as PI− and CD19+/CD20+ subpopulations. CpG (ODN2006) was purchased from InvivoGen, and recombinant human Interleukin-15 (IL-15) was purchased from Gemini Bio-Products. All drugs tested were purchased from Selleckchem. PerCP anti-human CD20 (302324), PE anti-human CD19 (302254), and FITC-anti CD3 (300306) antibodies were purchased from BioLegend. Propidium iodide was purchased from Sigma-Aldrich.
ADDITIONAL INFORMATION
Data Deposition and Access
The DNA sequencing data have been provided as part of this publication in the form of the vcf files presented in Supplemental Table 3. In addition, pathogenic variants were deposited to ClinVar (https://www.ncbi.nlm.nih./gov/clinvar/) under accession numbers SCV004020298– SCV004020300 and SCV004020302–SCV004020307.
Ethics Statement
Testing was done mostly as part of routine clinical laboratory workup. The additional studies were done after obtaining written patient's consent under the FCCC IRB19-9038 protocol. The portion of the testing that was not clinically validated was not used to guide clinical care and did not interfere with clinical care.
Acknowledgments
This work was supported in part by Fox Chase Cancer Center Academic Research Fund, Daniel B. Allanoff Foundation, and National Cancer Institute (NCI) grants R21CA263415 (to Y.L.W.) and R01CA268601 (to M.A.W.).
Author Contributions
P.M.K., R.N., Y.L.W., and M.A.W. designed the study; P.L., D.T., and J.P. performed experiments and analyzed the results; S.N. took care of the patient and provided clinical correlation; P.M.K., R.N., N.M., and M.A.W. performed diagnostic evaluation of the biopsies; P.M.K., R.N., Y.L.W., D.A.B., S.S.D., and M.A.W. analyzed the data; V.D. reviewed and prepared the manuscript; and P.M.K. and M.A.W. wrote the manuscript.
Competing Interest Statement
The authors have declared no competing interest.
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. 2000. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403: 503–511. 10.1038/35000501 [DOI] [PubMed] [Google Scholar]
- Amin AD, Peters TL, Li L, Rajan SS, Choudhari R, Puvvada SD, Schatz JH. 2017. Diffuse large B-cell lymphoma: can genomics improve treatment options for a curable cancer? Cold Spring Harb Mol Case Stud 3: a001719. 10.1101/mcs.a001719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhshi TJ, Georgel PT. 2020. Genetic and epigenetic determinants of diffuse large B-cell lymphoma. Blood Cancer J 10: 123. 10.1038/s41408-020-00389-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin AS. 2001. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-κB. J Clin Invest 107: 241–246. 10.1172/JCI11991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrans SL, Fenton JA, Banham A, Owen RG, Jack AS. 2004. Strong expression of FOXP1 identifies a distinct subset of diffuse large B-cell lymphoma (DLBCL) patients with poor outcome. Blood 104: 2933–2935. 10.1182/blood-2004-03-1209 [DOI] [PubMed] [Google Scholar]
- Calado DP, Zhang B, Srinivasan L, Sasaki Y, Seagal J, Unitt C, Rodig S, Kutok J, Tarakhovsky A, Schmidt-Supprian M, et al. 2010. Constitutive canonical NF-κB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell–like diffuse large cell lymphoma. Cancer Cell 18: 580–589. 10.1016/j.ccr.2010.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, Lawrence MS, Roemer MGM, Li AJ, Ziepert M, et al. 2018. Author correction: molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 24: 1290–1291. 10.1038/s41591-018-0097-4 [DOI] [PubMed] [Google Scholar]
- Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, Bertoni F, Ponzoni M, Scandurra M, Califano A, et al. 2009. Mutations of multiple genes cause deregulation of NF-κB in diffuse large B-cell lymphoma. Nature 459: 717–721. 10.1038/nature07968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois S, Viailly PJ, Bohers E, Bertrand P, Ruminy P, Marchand V, Maingonnat C, Mareschal S, Picquenot JM, Penther D, et al. 2017. Biological and clinical relevance of associated genomic alterations in MYD88 L265P and non-L265P-mutated diffuse large B-cell lymphoma: analysis of 361 cases. Clin Cancer Res 23: 2232–2244. 10.1158/1078-0432.CCR-16-1922 [DOI] [PubMed] [Google Scholar]
- George B, Chowdhury SM, Hart A, Sircar A, Singh SK, Nath UK, Mamgain M, Singhal NK, Sehgal L, Jain N. 2020. Ibrutinib resistance mechanisms and treatment strategies for B-cell lymphomas. Cancers (Basel) 12: 1328. 10.3390/cancers12051328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardin F, Jais JP, Molina TJ, Parmentier F, Picquenot JM, Ruminy P, Tilly H, Bastard C, Salles GA, Feugier P, et al. 2010. Diffuse large B-cell lymphomas with CDKN2A deletion have a distinct gene expression signature and a poor prognosis under R-CHOP treatment: a GELA study. Blood 116: 1092–1104. 10.1182/blood-2009-10-247122 [DOI] [PubMed] [Google Scholar]
- Juskevicius D, Lorber T, Gsponer J, Perrina V, Ruiz C, Stenner-Liewen F, Dirnhofer S, Tzankov A. 2016. Distinct genetic evolution patterns of relapsing diffuse large B-cell lymphoma revealed by genome-wide copy number aberration and targeted sequencing analysis. Leukemia 30: 2385–2395. 10.1038/leu.2016.135 [DOI] [PubMed] [Google Scholar]
- Kim JH, Kim WS, Ryu K, Kim SJ, Park C. 2016. CD79B limits response of diffuse large B cell lymphoma to ibrutinib. Leuk Lymphoma 57: 1413–1422. 10.3109/10428194.2015.1113276 [DOI] [PubMed] [Google Scholar]
- Lacy SE, Barrans SL, Beer PA, Painter D, Smith AG, Roman E, Cooke SL, Ruiz C, Glover P, Van Hoppe SJL, et al. 2020. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: a haematological malignancy research network report. Blood 135: 1759–1771. 10.1182/blood.2019003535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, Carty S, Lam LT, Shaffer AL, Xiao W, et al. 2008. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci 105: 13520–13525. 10.1073/pnas.0804295105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los-de Vries GT, Stathi P, Rutkens R, Hijmering NJ, Luijks JACW, Groenen PJTA, de Jong D, Ylstra B, Roemer MGM. 2023. Large B-cell lymphomas of immune-privileged sites relapse via parallel clonal evolution from a common progenitor B-cell. Cancer Res 83: 1917–1927. 10.1158/0008-5472.CAN-22-3814 [DOI] [PubMed] [Google Scholar]
- Lu P, Wang S, Franzen CA, Venkataraman G, McClure R, Li L, Wu W, Niu N, Sukhanova M, Pei J, et al. 2021. Ibrutinib and venetoclax target distinct subpopulations of CLL cells: implication for residual disease eradication. Blood Cancer J 11: 39. 10.1038/s41408-021-00429-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelbaum J, Bhagat G, Tang H, Mo T, Brahmachary M, Shen Q, Chadburn A, Rajewsky K, Tarakhovsky A, Pasqualucci L, et al. 2010. BLIMP1 is a tumor suppressor gene frequently disrupted in activated B cell–like diffuse large B cell lymphoma. Cancer Cell 18: 568–579. 10.1016/j.ccr.2010.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino D, Pizzi M, Kotova I, Schmidt R, Schröder C, Guzzardo V, Talli I, Peroni E, Finotto S, Scapinello G, et al. 2022. High ETV6 levels support aggressive B lymphoma cell survival and predict poor outcome in diffuse large B-cell lymphoma patients. Cancers (Basel) 14: 338. 10.3390/cancers14020338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y, Medeiros LJ, Li Y, Li J, Young KH. 2019. Genetic alterations and their clinical implications in DLBCL. Nat Rev Clin Oncol 16: 634–652. 10.1038/s41571-019-0225-1 [DOI] [PubMed] [Google Scholar]
- Muringampurath-John D, Jaye DL, Flowers CR, Saxe D, Chen Z, Lechowicz MJ, Weisenburger DD, Bast M, Arellano ML, Bernal-Mizrachi L, et al. 2012. Characteristics and outcomes of diffuse large B-cell lymphoma presenting in leukaemic phase. Br J Haematol 158: 608–614. 10.1111/j.1365-2141.2012.09209.x [DOI] [PubMed] [Google Scholar]
- Nakhoda S, Vistarop A, Wang YL. 2023. Resistance to Bruton tyrosine kinase inhibition in chronic lymphocytic leukaemia and non-Hodgkin lymphoma. Br J Haematol 200: 137–149. 10.1111/bjh.18418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, et al. 2011. Oncogenically active MYD88 mutations in human lymphoma. Nature 470: 115–119. 10.1038/nature09671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijland M, Seitz A, Terpstra M, van Imhoff GW, Kluin PM, van Meerten T, Atayar Ç, van Kempen LC, Diepstra A, Kok K, et al. 2018. Mutational evolution in relapsed diffuse large B-cell lymphoma. Cancers (Basel) 10: 459. 10.3390/cancers10110459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak AJ, Asmann YW, Maurer MJ, Wang C, Slager SL, Hodge LS, Manske M, Price-Troska T, Yang ZZ, Zimmermann MT, et al. 2015. Whole-exome analysis reveals novel somatic genomic alterations associated with outcome in immunochemotherapy-treated diffuse large B-cell lymphoma. Blood Cancer J 5: e346. 10.1038/bcj.2015.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondrisova L, Mraz M. 2020. Genetic and non-genetic mechanisms of resistance to BCR signaling inhibitors in B cell malignancies. Front Oncol 10: 591577. 10.3389/fonc.2020.591577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Dalla-Favera R. 2018. Genetics of diffuse large B-cell lymphoma. Blood 131: 2307–2319. 10.1182/blood-2017-11-764332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Compagno M, Houldsworth J, Monti S, Grunn A, Nandula SV, Aster JC, Murty VV, Shipp MA, Dalla-Favera R. 2006. Inactivation of the PRDM1/BLIMP1 gene in diffuse large B cell lymphoma. J Exp Med 203: 311–317. 10.1084/jem.20052204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan JD, Young RM, Webster DE, Roulland S, Wright GW, Kasbekar M, Shaffer AL, Ceribelli M, Wang JQ, Schmitz R, et al. 2018. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature 560: 387–391. 10.1038/s41586-018-0290-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pindzola GM, Razzaghi R, Tavory RN, Nguyen HT, Morris VM, Li M, Agarwal S, Huang B, Okada T, Reinhardt HC, et al. 2022. Aberrant expansion of spontaneous splenic germinal centers induced by hallmark genetic lesions of aggressive lymphoma. Blood 140: 1119–1131. 10.1182/blood.2022015926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyrzynska B, Dwojak M, Zerrouqi A, Morlino G, Zapala P, Miazek N, Zagozdzon A, Bojarczuk K, Bobrowicz M, Siernicka M, et al. 2018. FOXO1 promotes resistance of non-Hodgkin lymphomas to anti-CD20-based therapy. Oncoimmunology 7: e1423183. 10.1080/2162402X.2017.1423183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenwald A, Delabie J, Medeiros L, Klapper W, Guijral S, Naresh KN, Calaminici M, Lenz G, Gopal A, Nair R, et al. 2022. Diffuse large B cell lymphoma, NOS. In WHO Classification of Tumours Editorial Board. Haematolymphoid tumours (WHO classification of tumours series, 5th ed.; Vol. 11). International Agency for Research on Cancer, Lyon, France. [Google Scholar]
- Sablon A, Bollaert E, Pirson C, Velghe AI, Demoulin JB. 2022. FOXO1 forkhead domain mutants in B-cell lymphoma lack transcriptional activity. Sci Rep 12: 1309. 10.1038/s41598-022-05334-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, Roulland S, Kasbekar M, Young RM, Shaffer AL, et al. 2018. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med 378: 1396–1407. 10.1056/NEJMoa1801445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer AL, Phelan JD, Wang JQ, Huang D, Wright GW, Kasbekar M, Choi J, Young RM, Webster DE, Yang Y, et al. 2021. Overcoming acquired epigenetic resistance to BTK inhibitors. Blood Cancer Discov 2: 630–647. 10.1158/2643-3230.BCD-21-0063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh PK, Basavaiah SH, Goel M, Dsouza S, Rai S. 2020. Diffuse large B-cell lymphoma in blood and bone marrow: a rare presentation of leukemic phase at diagnosis. Hematol Transfus Cell Ther 42: 180–183. 10.1016/j.htct.2019.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Keimpema M, Grüneberg LJ, Mokry M, van Boxtel R, van Zelm MC, Coffer P, Pals ST, Spaargaren M. 2015. The forkhead transcription factor FOXP1 represses human plasma cell differentiation. Blood 126: 2098–2109. 10.1182/blood-2015-02-626176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturutti L, Melnick AM. 2020. The dangers of déjà vu: memory B cells as the cells of origin of ABC-DLBCLs. Blood 136: 2263–2274. 10.1182/blood.2020005857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturutti L, Teater M, Zhai A, Chadburn A, Babiker L, Kim D, Béguelin W, Lee TC, Kim Y, Chin CR, et al. 2020. TBL1XR1 mutations drive extranodal lymphoma by inducing a pro-tumorigenic memory fate. Cell 182: 297–316.e27. 10.1016/j.cell.2020.05.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WH, Wright GW, Huang DW, Hodkinson B, Balasubramanian S, Fan Y, Vermeulen J, Shreeve M, Staudt LM. 2021. Effect of ibrutinib with R-CHOP chemotherapy in genetic subtypes of DLBCL. Cancer Cell 39: 1643–1653.e3. 10.1016/j.ccell.2021.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Lu P, Patel P, Ma J, Cai KQ, Mallikarjuna VS, Poureghbali S, Nakhoda SR, Nejati R, Lynn Wang Y. 2023. SHP1 loss augments DLBCL cellular response to ibrutinib: a candidate predictive biomarker. Oncogene 42: 409–420. 10.1038/s41388-022-02565-7 [DOI] [PubMed] [Google Scholar]
- Xu-Monette ZY, Wu L, Visco C, Tai YC, Tzankov A, Liu WM, Montes-Moreno S, Dybkaer K, Chiu A, Orazi A, et al. 2012. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood 120: 3986–3996. 10.1182/blood-2012-05-433334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KH, Weisenburger DD, Dave BJ, Smith L, Sanger W, Iqbal J, Campo E, Delabie J, Gascoyne RD, Ott G, et al. 2007. Mutations in the DNA-binding codons of TP53, which are associated with decreased expression of TRAILreceptor-2, predict for poor survival in diffuse large B-cell lymphoma. Blood 110: 4396–4405. 10.1182/blood-2007-02-072082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RM, Phelan JD, Wilson WH, Staudt LM. 2019. Pathogenic B-cell receptor signaling in lymphoid malignancies: new insights to improve treatment. Immunol Rev 291: 190–213. 10.1111/imr.12792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Lu Y, Zhang T, Guan Q, Wang X, Guo Y, Li L, Qiu L, Qian Z, Zhou S, et al. 2022. PIM1 genetic alterations associated with distinct molecular profiles, phenotypes and drug responses in diffuse large B-cell lymphoma. Clin Transl Med 12: e808. 10.1002/ctm2.808 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The DNA sequencing data have been provided as part of this publication in the form of the vcf files presented in Supplemental Table 3. In addition, pathogenic variants were deposited to ClinVar (https://www.ncbi.nlm.nih./gov/clinvar/) under accession numbers SCV004020298– SCV004020300 and SCV004020302–SCV004020307.