

Abstract
The rate of RNA polymerase II (RNAPII) transcriptional elongation plays a critical role in mRNA biogenesis, from transcription initiation to alternative splicing. As RNAPII moves along the DNA, it must read the DNA sequences wrapped up as chromatin. Thus, the structure of chromatin impedes the movement and speed at which RNAPII moves, presenting a crucial regulation to gene expression. Therefore, factors that bind and regulate the structure of chromatin will impact the rate of RNAPII elongation. We previously showed that PARP1 (poly-ADP-ribose polymerase 1) is one of such factors that bind and alter chromatin dynamics. We also showed that its alteration of chromatin structure modulates RNAPII processivity during transcriptional elongation. Here, we aim to understand how PARP1 alters RNAPII elongation kinetics genome wide.
Keywords: Nascent RNA-Seq, Splicing, RNA polymerase II elongation, Chromatin structure, RNA transcription
1. Introduction
PARP1 (poly-ADP-ribose polymerase 1), a well-known DNA repair protein, plays crucial roles in transcription and RNA biogenesis. PARP1 has been implicated in transcriptional initiation, splicing, RNA degradation, and RNA export. While the mechanism by which PARP1 alters the chromatin structure to initiate transcription has been a hot topic for decades, the complete picture of how PARP1 affects the rate of RNA polymerase II during elongation is still not fully understood. We previously showed that depletion of PARP1 or its enzymatic activity mediated alternative splicing outcomes of specific PARP1-targeted genes. These results suggested that PARP1 plays a crucial role in co-transcriptional splicing decisions [1, 2], by regulating RNAPII elongation kinetics. Indeed, we showed that knockdown of PARP1 resulted in some transcripts being longer while others were shorter [2]. These results suggest that the knowledge of how PARP1-chromatin binding affects the transcriptional elongation rate is critical to fully understand its regulation of gene expression and co-transcriptional splicing. In this work, we investigate the consequences of PARP1 depletion and inhibition of its activity on the optimal rate/processivity of RNA polymerase II by measuring the length of isolated nascent RNA transcripts with thiouridine labeling using nascent RNA sequencing. Our protocol is a combination of several NET-seq protocols [2–4] modified to obtain the best protocol for PARP1-mediated RNAPII elongation kinetics in S2 Drosophila cells.
2. Materials
2.1. Buffer Stocks
1 M NaCl (58.44 g of NaCl dissolved in 1000 mL of sterile distilled water).
1 M Tris–HCl (181.7 g of Tris–HCl dissolved in 900 mL of sterile distilled water. Adjust the pH to 7.5 with HCl, then add sterile distilled water gradually to a total volume of 1000 mL). This buffer should be stored at 4 °C.
1 M NaOAc (5.772 g of sodium acetate dissolved in 900 mL of sterile distilled water, then add 1.778 g of acetic acid to the solution. Adjust the solution with 10 N HCl to a pH of 5, then add sterile distilled water until a final volume of 1000 mL).
10X TBE buffer (121.1 g of Tris base, 61.8 g of boric acid, and 7.4 g of EDTA dissolved in 1000 mL of H2O). To make gel running buffer, this should be diluted 1:10.
10X MnCl2 (BioLabs, B0786A).
50 mM MgCl2 (Bio-Rad, 1708872).
10X EDTA (Novus Biologicals, NB900–66730).
TE buffer (10 μL of 1 M Tris-HCl at pH 8 is mixed with 2 μL of 0.5 M EDTA at pH 8, then add 988 μL of sterile distilled water).
DMSO (Sigma, D8418).
2.2. Culture of S2 Drosophila Cells
Drosophila melanogaster embryonic S2 cells (ATCC, CRL-1963).
Schneider’s Drosophila Medium (Gibco, 21720024).
Penicillin-streptomycin (10,000 units/mL of penicillin and 10,000 μg/mL of streptomycin) (Thermo Fisher, 15140122).
Heat inactivated fetal bovine serum (HI FBS) (Sigma, F4135).
Cell culture flask T75 cm2 with vent cap.
Incubator at 37 °C and 0 % CO2.
Dubecco’s phosphate-buffered saline (DPBS) (Lonza, BE17–512F).
Sterile cell scrapers.
Serological pipettes and sterile filter tips.
15 mL conical bottom centrifuge tubes.
Centrifuge with temperature control function.
Vacuum aspiration system.
Cell culture hood.
Glass disposable Pasteur pipets.
2.3. PARP1 Knockdown
DNeasy Blood & Tissue Kits (Qiagen, 69504).
-
Primers to amplify PARP1 gene (Integrated DNA Technologies – IDT)
Forward Primer: 5′-GAATTAATACGACTCACTATAGG GATGATGCCTACTTCAGGTTTCGC-3′ P
Reverse Primer: 5′- GAATTAATACGACTCACTATAGG GATTGGCACTATGCATGCCGATCT-3′).
1.5 mL RNase-free microcentrifuge tubes.
TempAssure 0.2 mL PCR 8-Tube Strips and Dome Strip caps.
Taq DNA polymerase (Syd Labs, MB042-EUT-M).
10X Taq DNA buffer (Genscript, E00007).
10 mM dNTP (Bio-Rad, 1708874).
QIAquick PCR Purification Kit (Qiagen, 28104).
Thermal cycler instrument.
MEGAscript RNAi Kit instructions (Thermo Fisher, AM1626).
ShortCut RNase III Digestion (NEB, M0245).
3 M Sodium Acetate (NaOAc) (dissolve 246.1 g of sodium acetate in 800 mL of sterile distilled water and adjust the pH to 5.5 with glacial acetic acid. Let the solution cool down overnight. Once again, adjust the pH to 5.5 the next day with glacial acetic acid. Then, add distilled water until the volume is 1000 mL and filter-sterilize the solution).
RNase-free glycogen (NEB, B1564S).
Ethyl alcohol 200 proof, absolute.
Absorbance readers such as Nanodrop spectrophotometer or Qubit fluorometer instrument.
2.4. PARylation Inhibition
10 mM PJ-34 hydrochloride hydrate (Sigma, P4365–5MG) (dissolve in 1507 μL of sterile distilled water)
2.5. Transcription Inhibition
5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole (DRB) (Milipore Sigma, D1916)
2.6. Nascent RNA Labeling
4-Thiouridine (Milipore Sigma, T4509)
TRIzol Reagent (ThermoFisher, 15596026)
15 mL canonical tubes
2.7. Total RNA Extraction
Chloroform (Sigma Aldrich, 67663)
75% ethanol (mix 75 mL of absolute ethyl alcohol and 25 mL of sterile distilled water)
Nuclease-free water (Invitrogen, Lot. No: 01188519)
2.8. Biotinylation of Labeled RNA
2.0 mL RNase-free tube.
100 mM Tris–HCl (18.17 g of Tris–HCl dissolved in 900 mL of sterile distilled water. Adjust the pH to 7.4 with HCl, then add sterile distilled water gradually to a total volume of 1000 mL).
10 mM EDTA (dissolve 0.372 g of disodium edetate in 90 mL of distilled water. Add NaOH to adjust the pH to 8. Then continue diluting the solution to 100 mL with distilled water).
Biotinylation buffer (100 mM Tris-HCl pH 7.4, 10 mM EDTA in nuclease-free water).
50 mg of EZ-Link HPDP Biotin (Thermo Fisher, 21341).
Dimethylformamide (DMF).
Rotating vertical mixer.
25:24:1 Phenol:Chloroform:isoamy alcohol (pH 8.0).
1.5 mL MaXtract high-density phase-lock tubes (Qiagen, Lot No. 56901540).
5 M NaCl (Dissolve 292 g of NaCl in 900 mL of H2O. Adjust the volume to 1000 mL with sterile H2O).
Isopropanol.
2.9. Separation of Nascent RNA
MyOne Streptavidin T1 Dyna Beads
2 M NaCl (dissolve 116.88 g of NaCl in 1000 mL of autoclaved water)
1 mM EDTA
Wash buffer (10 mM Tris–HCl pH 7.5, 1 mM EDTA, 2 M NaCl in nuclease-free H2O)
DynaMag Magnetic Rack
100 mM DTT (dissolve 15.4 mg of DTT in 1 mL water)
Qiagen RNeasy Mini Kit (Qiagen, 217004)
2.10. Gel Electrophoresis
NuSieve 3:1 low melting agarose (Lonza, 50094)
Microwave
Ethidium bromide
DNA ladder
Gel electrophoresis system
3. Methods
3.1. SiRNA Preparation for PARP1 Knockdown in S2 Drosophila Cell Line
Genomic DNA was extracted from S2 cells using DNeasy Blood & Tissue Kits, and its concentration was quantified with nanodrop.
Add TE buffer to each primer to a final concentration of 100 pmol/μL.
In a 1.5 mL microcentrifuge tube, prepare a primer mix solution by mixing 10 μL of forward primer, 10 μL of reverse primer, and 20 μL of nuclease-free water H2O.
Prepare PCR reaction to amplify PARP1 gene according ot the instructions on Table 1.
Purify the amplified DNA following the protocol in QIAquick PCR Purification.
Quantify the DNA concentration using Nanodrop or Qubit.
Transcribe the dsDNA to dsRNA using the MEGAscript RNAi Kit instructions.
Ensure that the RNA is double-stranded by heating the purified samples above at 75 °C for 5 min, then cool to room temperature on the bench for ~1 h.
To remove DNA and single-stranded RNA, digest the above reaction with nuclease enzymes. Specifically, assemble the digestion reaction on ice as instructed in MEGAscript RNAi Kit protocol, add RNase, DNase I, and 10X digestion buffer (all these reagents are included in the kit). Then, incubate the mixture at 37 °C for 1 h.
- After digestion with nuclease enzymes, purify the dsRNA per MEGAscript RNAi Kit protocol (pages 11–13). At this stage, the dsRNA can be stored in −80 °C for months if desired, or directly proceed to the next step.
- Critical step: After quantification, run approximately 500 ng of the amplified DNA and 500 ng of purified and anneled dsRNA transcript on 4 % low melting point agarose gel to check for proper amplification and transcription (Fig. 1a).
To convert long dsRNA into a heterogeneous mix of short siRNAs, follow the guidelines in ShortCut RNase III Digestion Protocol. Digest 10 μg of dsRNA with ShortCut RNase II used in the ShortCut reaction assembly to make siRNA.
- Purify the siRNA by ethanol precipitation protocol (NEB, M0245) and quantify by microplate reader. This can be aliquoted and stored at −80 °C (see Note 1).
- Critical step: Take around 500 ng of purified long dsRNA and 500 ng of siRNA and run them on 4 % low melting point agarose gel to check for proper ShortCut RNase digestion (Fig. 1b).
Table 1.
PCR amplification of PARP1 amplicon form genomic DNA
| Preparation of PCR reaction | PCR program setup (runtime ~ 02:19:00) |
|---|---|
|
| |
| Per 1x reaction, mix in PCR tube in the following order: A. 5 μL of 10x Taq buffer B. 1.5 μL of MgCl2 C. 0.5 μL of DMSO D. 1 μL of 10 mM dNTP E. 0.25 μL of Taq polymerase F. 2 μL of Primer mix G. 1 μg of Genomic DNA H. Add H2O to a total volume of50 μL |
Lid: 105 °C, volume: 50 μL A. 94 °C, 3:00 B. 94 °C, 0:45 C. 62 °C, 0:45 D. 72 °C, 1:00 E. Go to step B, repeat six times F. 94 °C, 0:45 G. 57 °C, 0:45 H. 72 °C, 1:00 I. Go to step F, repeat six times J. 94 °C, 0:45 K. 45 °C, 0:45 L. 72 °C, 0:45 M. Go to step J, repeat 25 times N. Infinite hold at 4 °C |
Fig. 1.
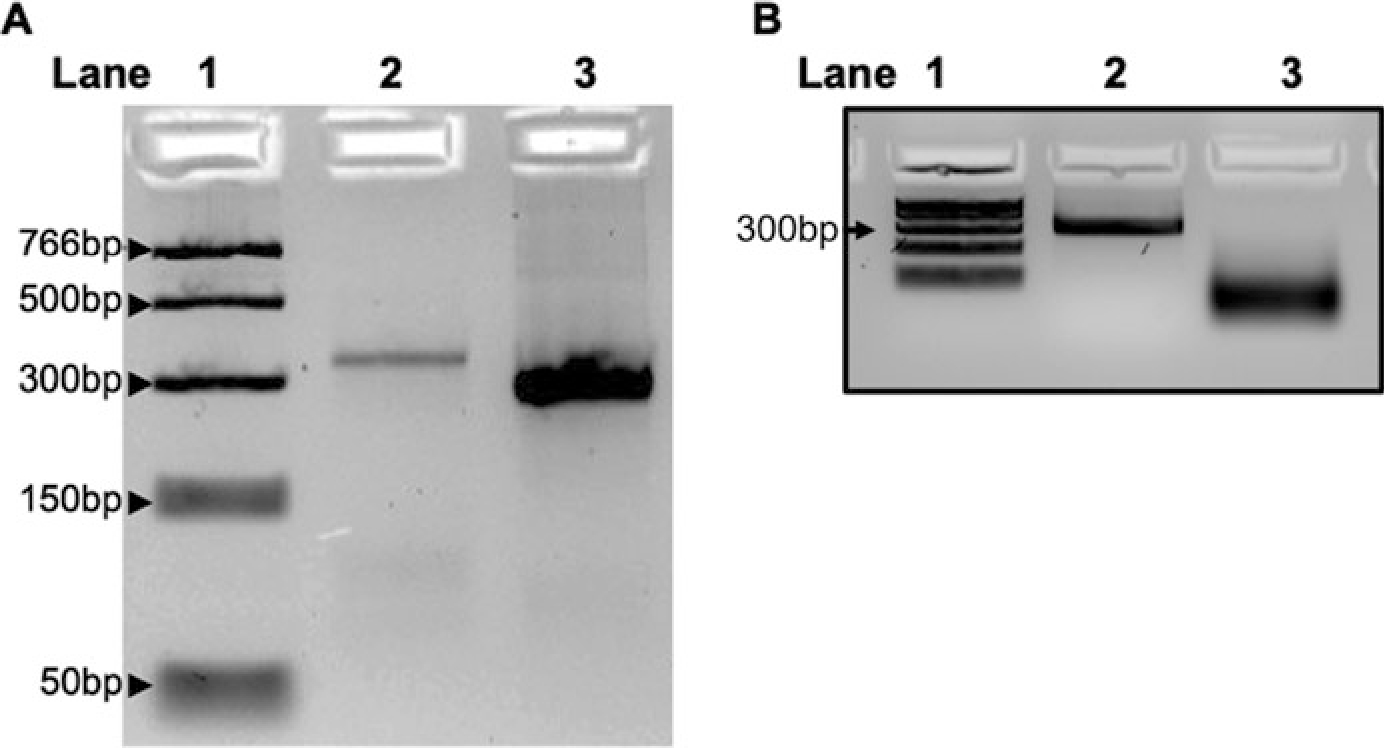
Generation of siRNA for PARP1 knockdown. (a) Lane 1, PCR ladder; Lane 2, amplified DNA of PARP1 gene from Drosophila genomic DNA; Lane 3, annealed and purified dsRNA. (b) Lane 1, PCR ladder; Lane 2, dsRNA as in Lane 3 of panel A; Lane 3, dsRNA digested to yield small siRNAs for PARP1 knockdown. Short run of samples on a 4% Nusieve agarose gel to capture the shorter siRNA samples. A longer run results in diffused bands
3.2. PARP1 Knockdown in S2 Drosophila Cells
For knockdown of PARP1, use two rounds of knockdown to ensure about 30–40 % reduction in PARP1.
First Round
-
1
Grow S2 cells in T75 flasks with complete media (Schneider’s Drosophila Medium containing 10 % heat inactivated FBS and 0.1 % penicillin-streptomycin) until ~90 % confluent.
-
2
Scrape S2 cells out of T75 flask with cell scraper, pellet cells at 4 °C for 5 min with 2000 × g, then remove the supernatant (see Note 2).
-
3
Resuspend cell pellet in Schneider’s Drosophila media without HI FBS or penicillin-streptomycin.
-
4
Split cells into four to six new flasks (split at a lighter density as they will grow in the same flask for about 5 days).
-
5
Add 8 mL of Schneider’s Drosophila media without HI FBS and penicillin-streptomycin to a new T75 flask. Resuspend the cells in 2 mL of media and add it to the flask. Swirl flask to mix.
-
6
Add 1 μg of siRNA in T75 flask. Swirl the flask to mix siRNA with the cells, then let the flask sit in the hood for 30 min.
-
7
Add 5 mL Schneider’s Drosophila media with HI FBS and penicillin-streptomycin. Then, add 1000 μL of heat inactivated FBS and 10 μL of penicillin-streptomycin. Swirl the flask to mix.
-
8
Put in incubator at 27 °C and 0 % CO2 for 2 days.
Second Round (Reinforcement of PARP1 Knockdown)
-
9
Scrape the cells from the T75 flask into a clean 15 mL canonical tube.
-
10
Pellet cells by centrifugation at a speed of 2000 × g. Remove old media by aspiration and starve the cells by resuspending in media without HI FBS and penicillin-streptomycin as in steps 5 and 6 above.
-
11
Put each tube of cells into a new flask and bring to 10 mL with media without HI FBS and penicillin-streptomycin.
-
12
Add 1 μg of siRNA and swirl the flask well to mix. Then, let the flask sit in the hood for 30 min.
-
13
Add 5 mL Schneider’s Drosophila media with heat-inactivated FBS and penicillin-streptomycin. Then, add 1000 μL heat-inactivated FBS and 10 μL of penicillin-streptomycin. Swirl the flask to mix.
- 14
Fig. 2.
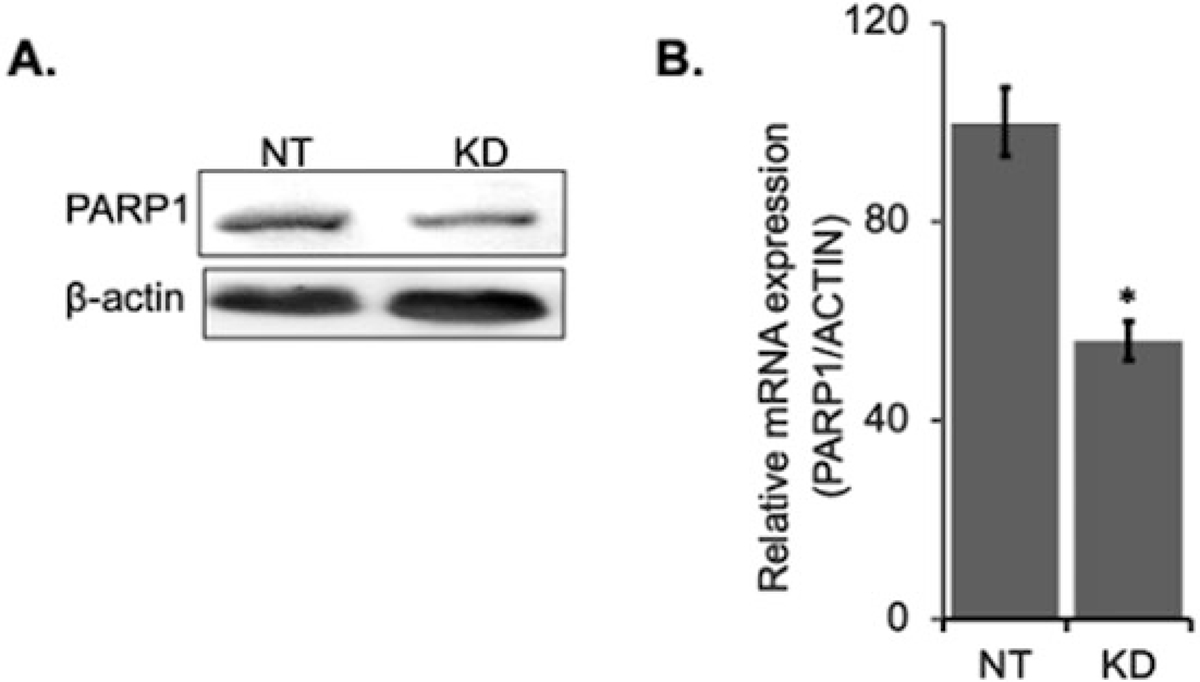
Validation of PARP1 knockdown. PARP1 was knocked down with siRNA as above. (a) Protein extracts were quantified, run on SDS PAGE and western blotted for PARP1 antibodies. (b) RNA was purified from cells and converted to cDNA, and the level of PARP1 transcript expression analyzed by qRT-PCR. *Mean of n = 3 ± SEM, p < 0.05 by T-test
3.3. PARylation Inhibition
- Add PJ-34 hydrochloride hydrate to S2 cells in complete media to a final concentration of 300 nM and incubate further for 16 h in the incubator at 27 °C and 0 % CO2.
- Critical step: Check for proper PARylation inhibition by western blot analysis using PAR antibodies (see Note 3).
3.4. Transcription Inhibition (Synchronize Cell Transcription Before Nascent RNA Labeling)
To inhibit transcription, grow cells on T75 flasks to ~90% confluency and treat them with 300 μM of 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole (DRB) in complete media for 4 h.
Stop transcription inhibition by transferring the cells in 15 mL conical tube, pellet the cells by centrifugation, and aspire the old media.
Wash the cells twice with phosphate-buffered saline (PBS) to remove the DRB.
3.5. 4-Thiouridine (4sU) Labeling of Nascent mRNA
Resuspend the cells in a minimum of 10 mL complete media containing 3 mM of 4-sU. Pipette up and down with a serological pipette to mix thoroughly and incubate for the desired amount of time. Depending on the cell type, transcription can be allowed to proceed for 0, 4, 12, and 16 min (see Note 4).
At the desired time points, collect the cells by pelleting the cells and removing the media that contains 4sU.
Immediately add 5 mL of TRIzol directly to cells, homogenize the cells by pipetting up and down vigorously. Then, incubate the sample for 3 min in the hood for complete cell lysis.
The TRIzol homogenized samples can be stored at −80 °C for months or proceed to next step.
3.6. Extraction of Total RNA
Add 1 mL of chloroform to the TRIzol lysate and mix vigorously by vortexing and incubate the samples at room temperature for 3 min.
Centrifuge the samples for 15 min with a speed of 12,000 × g at 4 °C to separate the samples into a phenol-chloroform lower red phase, an interphase (white layer), and a colorless upper aqueous phase containing the RNA.
Transfer the aqueous phase into a clean 15 mL canonical tube and mix it with 2.5 mL of isopropyl alcohol, then incubate at room temperature for 10 min. Precipitate the RNA by centrifugation at a high speed of 12,000 × g for 10 min at 4 °C.
Remove the supernatant and wash the RNA pellet with 5 mL of 75% ethanol. Mix the sample by vortexing and centrifuge samples at 6000 × g for 5 min at 4 °C.
Decant the supernatant and partially air-dry the RNA pellet for 5 min. Air-drying samples is easier for redissolving the pellet.
Redissolve the RNA in 100 μL of nuclease-free water. The total RNA can be stored at −80 °C or proceed to the RNA biotinylation (see Note 5).
3.7. Nascent RNA Biotinylation
Assemble the following in a 2.0 mL RNase-free tube: 100 μL of RNA in 700 μL of nuclease-free water, 100 μL of biotinylation buffer (100 mM Tris–HCl pH 7.4, 10 mM EDTA in nuclease-free water), and 200 μL of EZ-Link HPDP Biotin dissolved to 1 mg/mL in DMF. Pipette the solution up and down to mix.
Incubate the mixture in a rotating vertical mixer for 2 h at room temperature.
After incubation, add one volume of phenol:chloroform:isoamyl alcohol to the mixture and vortex the sample.
Spin 2.0 mL lock tubes for 30 s in a microcentrifuge at highest speed. Then, add the reaction mixture to the prespun phase lock tubes and let sit at room temperature for 5 min to allow phase separation.
Centrifuge the lock tube for 5 min at 4 °C with a speed of 12,000 × g.
Repeat steps 4 and 5 of the Nascent RNA Biotinylation to ensure complete removal of unreacted biotin.
Transfer the upper phase containing RNA into clean RNase-free 2.0 mL tube and mix with one-tenth volume of 5 M NaCl and 850 μL of Isopropanol.
Centrifuge for 45 min at 4 °C at 16000 × g. White RNA pellet will form following this centrifugation step.
Remove supernatant and resuspend pellet in 850 μL of cold 75 % ethanol.
Centrifuge for 10 min at 4 °C at 16,000 × g and carefully remove supernatant.
Decant the tube to remove supernatant and air-dry the RNA sample at room temperature.
Dissolve RNA pellet in 100 μL of nuclease-free H2O, and then transfer to a clean 1.5 mL RNase-free tube.
Quantify RNA concentration with microplate reader. The RNA sample can be stored at −80 °C or proceed to the next step.
3.8. Bead Preparation and 4sU-Tagged RNA Enrichment
Vortex MyOne Streptavidin T1 Dyna Beads until the bead is completely resuspended (approximately time: 30 s to 1 min).
Dilute 150 μL of MyOne Streptavidin T1 Dyna Beads in 1.5 mL RNase-free tube.
Prepare 13 mL of wash buffer (10 mM Tris–HCl pH 7.5, 1 mM EDTA, 2 M NaCl in nuclease-free H2O) in a 15 mL canonical tube.
Add 1.0 mL of wash buffer to beads.
Incubate on rotating vertical mixer for 2 min at room temperature.
Place tube containing resuspended beads on Dynamag Magnetic Rack for at least 1 min to separate beads from supernatant. Then, carefully remove the supernatant.
Repeat steps 4–6 three more times.
Resuspend streptavidin beads in 150 μL of wash buffer.
Add 150 μL of prepared streptavidin beads to 100 μL of RNA-biotin solution.
- Incubate in a cold room or at 4 °C for 15 min in rotation rack (see Note 6).
- Critical step: In 5 mL tube, aliquot 4 mL of wash buffer and warm buffer to 60 °C.
Wash four times with 1 mL of warm wash buffer.
Wash four times with 1 mL of room temperature wash buffer.
Add 100 μL of nuclease-free water to beads and mix the sample vigorously. Let it sit for 10 min at room temperature.
To elute the 4-thiouridine-labeled nascent RNA from beads, resuspend beads in 100 μL of 100 mM DTT and incubate on rotating vertical mixer for 10 min at room temperature.
Use Qiagen Rneasy Mini Kit (Qiagen, 217004) to purify the eluate. Start by adding 300 μL of buffer RLT to the beads resuspended in DTT.
Incubate the suspension for 10 min at room temperature.
With Dynamag Magnetic Rack, separate the beads from the supernatant for 2 min. Then, transfer the supernatant to a 1.5 mL clean RNase-free tube.
Proceed with nascent RNA purification by eliminating DNA with gDNA eliminator column and continue with purification according to the manufacturer’s instructions.
- Elute the purified RNA with 20 μL of nuclease-free water.
- Critical step: Due to low concentration of nascent RNA, we recommend using Qubit for more accurate quantification (see Note 7).
3.9. Reverse Transcription to Validate Enrichment of Nascent mRNA Prep by qRT-PCR
Take 1 μL of 4sU-tagged eluted RNA (step 19 of Subheading 3.8 above) and dilute it in 12.5 μL of nuclease-free water.
Generate cDNA by assembling the cDNA reaction as described in Table 2 (see Note 8).
Due to low amount of starting RNA material, do not dilute the cDNA product. Use as it is, for qRT-PCR experiments. Before sequencing, validate successful enrichment of nascent mRNA with primers for rRNA, pre-mRNA (should have introns) and mRNA (mature mRNA) and a DRB-sensitive gene (see Note 9).
Table 2.
cDNA generation from Nascent mRNA
| Reaction assembly | Thermocycler program |
|---|---|
|
| |
| Per 1x reaction, mix in PCR tube in the following order: A. 4 μL of 5x iScript Advanced Reaction Mix B. 1.0 μL of iScript Advanced Reverse Transcriptase C. 12.5 uL of the 4sU-tagged RNA D. Add H2O to a total volume of 20 μL |
Lid: 105 °C, Volume: 20 μL A. 46 °C, 20:00 (Reverse transcription) B. 95 °C, 01:00 (RT inactivation) |
3.10. Library Preparation and Parallel Sequencing
Reduce the volume of the nascent mRNA eluted above to about 5 μL using a vacuum concentrator.
Proceed to RNA fragmentation, cDNA synthesis, second-strand synthesis, DNA end repair, 3′-end adenylation, adaptor ligation, and PCR amplification using sample preparation kit according to the manufacturer’s instructions. Due to the low RNA yield from nascent mRNA, we recommend using the NEB Ultra II Directional RNA Kit (NEB, E7765).
Libraries can now be stored at −20 °C.
Subject libraries to next-generation sequencing on the Illumina HiSeq platform according to the manufacturer’s instructions.
The overall flow chart for the protocol is illustrated in Fig. 3.
Fig. 3.
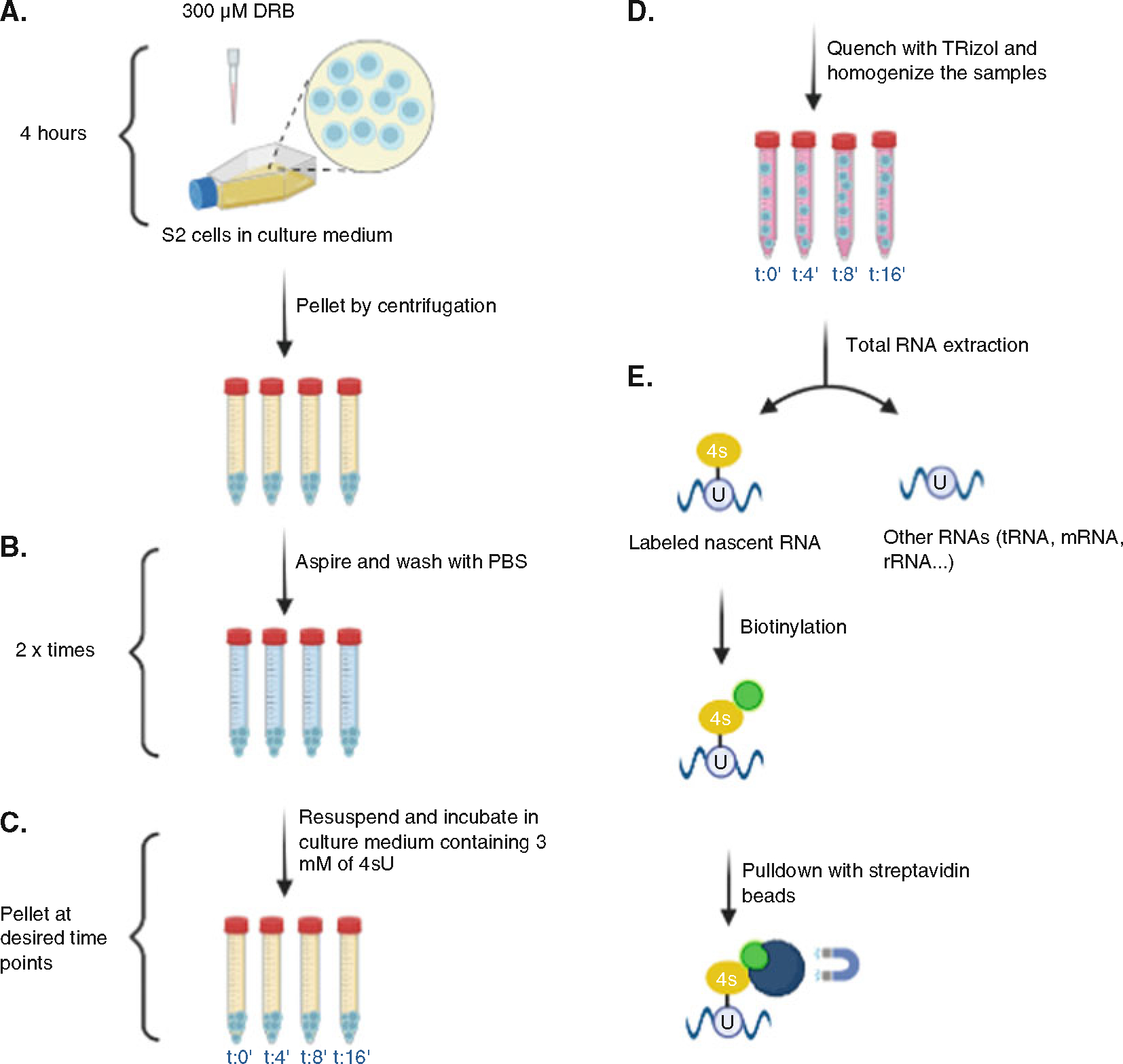
Overall workflow of protocol. (a) S2 Drosophila cells were grown in T75 flasks and treated with DRB for 4 h to stop transcription. (b) Cells were washed with 1X PBS to remove the DRB. (c) To label newly synthesized (nascent) RNA, cells now in growth media were treated with 4sU before being collected. Time point 0 min = 4 h of DRB and collected immediately after DRB removal. Time point 4 min = 4 h of DRB and collection 4 min after DRB removal. Time point 8 min = 4 h of DRB and collection 8 min after DRB removal. Time point 16 min = 4 h of DRB and collection after 16 min. (d) Samples were quenched with Trizol and total RNA purified. (e) 4sU-labeled RNA was biotinylated, purified with streptavidin beads, and subjected to deep sequencing
4. Notes
Aliquot 20 μL per tube and store at −80 °C; avoid multiple freeze-thaw cycles.
To minimize contamination, the Pasteur pipet tubing should be bleached before and after aspiring the media.
Check for PARylation inhibition by running a Western blot analysis. In wild-type conditions, using PARP1 antibody, a smear (between 160 KDa - 240 KDa) is observed above the PARP1 band and absent from the PARylation inhibition sample. Then confirm by using a PAR antibody, this smear in wild-type conditions will be clearly visible and intense and lack thereof in PARylation inhibition.
4-thiouridine (4sU) is light sensitive. Thus, cells incubated with culture medium containing 4sU and nucleic acid extracted from these cells should be kept in the dark and protected from light throughout the following procedures.
Alternatively, if high concentration is needed, the total RNA can be dissolved in 50 μL of nuclease-free water instead of a 100 μL. Also aliquotes can be made to reduce freeze-thaw cycles.
Longer incubation time in step 10 can be optimal for binding of biotinylated nascent RNA binding to streptavidin beads.
Nascent mRNA yields can be extremely low (less than 3 ng). Regardless, proceed to sequencing. Use kits recommended for very low-yield RNAs. Carry out a test sequencing run to validate presence of nascent mRNA. After confirmation, proceed to full sequencing.
After this step, cDNA can be stored at −80 °C. Aliquot 20 μL per tube and store at −80 °C; Avoid multiple freeze-thaw cycles.
Amplification times may vary because of the low RNA input. We usually use 10 μL of cDNA. When performing PCR, include a negative control by using water instead of cDNA.
Acknowledgments
We would like to thank the members of the Fondue-Mittendorf laboratory for critical reading of the manuscript. We would also like to thank the Markey Cancer Center’s Research Communications Office for manuscript editing (P30CA177558). Research reported in this publication was supported by the National Science Foundation grant, MCB 2016515 (Y. N. F.-M.) and the National Institutes of Environmental Health grants, R01 R01ES031846, R01ES034253 (Y. N. F.-M.).
References
- 1.Matveeva E, Maiorano J, Zhang Q et al. (2016) Involvement of PARP1 in the regulation of alternative splicing. Cell Discov 2:15046. 10.1038/celldisc.2015.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matveeva EA, Al-Tinawi QMH, Rouchka EC et al. (2019) Coupling of PARP1-mediated chromatin structural changes to transcriptional RNA polymerase II elongation and cotranscriptional splicing. Epigenetics Chromatin 12(1):15. 10.1186/s13072-019-0261-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drexler HL, Choquet K, Merens HE et al. (2021) Revealing nascent RNA processing dynamics with nano-COP. Nat Protoc 16(3):1343–1375. 10.1038/s41596-020-00469-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuchs G, Voichek Y, Rabani M et al. (2015) Simultaneous measurement of genome-wide transcription elongation speeds and rates of RNA polymerase II transition into active elongation with 4sUDRB-seq. Nat Protoc 10(4): 605–618. 10.1038/nprot.2015.035 [DOI] [PubMed] [Google Scholar]