

Abstract
Objectives
The objectives of this study included developing and validating a stability-indicating high-performance liquid chromatographic (HPLC) method with ultraviolet (UV) detection for the determination of buprenorphine in a buccal solution for veterinary use, and applying that method to determine the stability of a 3 mg/ml buprenorphine preparation in room temperature and refrigerated storage conditions. This preparation, intended for buccal administration in feline patients, plays an important role in pain management in cats.
Methods
A stability-indicating HPLC method was developed and validated for system suitability, accuracy, repeatability, intermediate precision, specificity, linearity and robustness based on US Pharmacopeia (USP) General Chapter <1225>. The method was then applied to the study of potency changes over 90 days in a buccal buprenorphine solution stored at two temperatures.
Results
All HPLC-UV method data met acceptable criteria for the quantification of buprenorphine in a buccal solution formulation. The buprenorphine concentrations found in each stability sample remained within the 90–110% of label claim throughout the 90 days of study. All stability test bottles of the buprenorphine buccal solution retained their original appearance. For the room temperature bottles, some white particulate matter was noted in the threads of the container bottles starting at day 21. The pH of the preparations during the course of the study was in the range of 3.57–4.06 and 4.01–4.16 for the room temperature and refrigerated samples, respectively.
Conclusions and relevance
Pharmacists have compounded a concentrated 3 mg/ml buccal solution to use easily in the home care or outpatient setting for treatment of feline pain. Prior to this investigation, pharmacists empirically assigned beyond-use dates to this formulation based on standards in USP General Chapter <795> Pharmaceutical Compounding – Nonsterile Preparations. This study of a 3 mg/ml buprenorphine buccal solution indicates stability through 90 days.
Introduction
Buprenorphine is a semi-synthetic opioid indicated for the general management of pain in humans. The commercially available products that are approved by the US Food and Drug Administration (FDA) include a bupenorphine hydrochloride 0.3 mg/ml injectable formulation for human use. 1 Veterinarians have administered this injectable formulation by the oral transmucosal, or buccal, route for sedation and analgesic effects in the management of pain associated with tissue inflammation and joint injuries in cats, dogs and other veterinary patients. 2 Buprenorphine is one of the most commonly used analgesics in cats, and provides a viable alternative to non-steroidal anti-inflammatory drugs, some of which may show elevated toxicity in cats owing to their deficiency in uridine diphosphate-glucuronosyltransferase.3,4 Specifically, the buccal administration of this drug is well tolerated by cats, and adverse effects such as salivation and vomiting are shown to be minimal. 5 Cat owners prefer buccal administration over injection and oral dosing, with the added benefit of avoidance of first-pass metabolism experienced with gastrointestinal absorption.6,7
According to a recent review of the studies using buprenorphine in cats, ‘increased attention has been given to pain management in cats, and it is now generally regarded as a mandatory component of clinical veterinary care in this species’. 8 Several investigations of the pharmacokinetic and pharmacodynamic characteristics of the buprenorphine oral transmucosal route of administration have detailed the drug and the buccal formulation’s place in feline analgesia and antinociception.3,7,9 In summary, buccal administration of buprenorphine has demonstrated good bioavailability (~32%), and has a disposition similar to intravenous administration when corrected for bioavailability, with both routes affecting thermal threshold.7,9 The alkaline nature of the cat’s mouth (pH 8.7–9.0) favors the buccal absorption of buprenorphine (pKa 8.2), as it shifts the drug into it unionized form. 3 The oral transmucosal route of administration has been accepted as a simple, non-invasive and pain-free technique. 3
The FDA-approved solution of buprenorphine for human use is not sufficiently concentrated to preclude feline patients from swallowing unreasonable volumes of the solution to achieve therapeutic effects. As a result, pharmacists have compounded a concentrated 3 mg/ml buccal solution for easy use with cats in the home care or outpatient setting. Without an established beyond-use date (BUD) for this formulation, pharmacists have empirically assigned BUDs to these formulations based on the standards in US Pharmacopeia (USP) General Chapter <795>. 10
According to USP General Chapter <795>, individuals compounding drug formulations are responsible for producing high-quality preparations for human and veterinary patients. These preparations must be of acceptable strength, quality and purity, and should be labeled in accordance with good compounding practices. Labeling indications related to BUD of a compounded preparation should be based on relevant scientific data. This includes stability investigations or other published information to ensure that compounded preparations maintain expected strength, quality, purity and other characteristics at least until the assigned and labeled BUD. 10
To our knowledge, there is currently no published information regarding validated stability-indicating methods to evaluate a compounded buprenorphine formulation, or any long-term stability information pertaining to compounded buprenorphine formulations. The research reported here aimed to develop and validate a stability-indicating high-performance liquid chromatographic (HPLC) method with ultraviolet (UV) detection for the determination of buprenorphine in buccal veterinary solution. Furthermore, the objective of this research was to apply that method to evaluate, based on appearance, pH and active pharmaceutical ingredient (API) concentration (potency), the 90-day stability of this 3 mg/ml buprenorphine preparation in room temperature and refrigerated storage conditions.
Materials and methods
Equipment and chromatographic conditions
Shimadzu Ultrafast Liquid Chromatograph systems with UV and photodiode array (PDA) detection set at 280 nm were used for all chromatographic measurements. Each system was equipped with column oven, in-line degasser and auto-sampler. Isocratic reverse-phase separation was completed on a Thermo Hypersil BDS C8 (50 × 2.1 mm ID, 5 micron) column (Thermo Fisher), which was maintained at 40°C. The mobile phase for the HPLC method was 10 mM ammonium acetate in HPLC-grade water and HPLC-grade acetonitrile (20:80), delivered at 0.250 ml/min. All chemicals were of HPLC grade or higher (Fisher Scientific). Additional chemicals included dextrose (Fisher Scientific), citric acid (Macron) and sodium citrate (Macron). All sample injection volumes were 10 μl.
Preparation of buprenorphine buccal solution samples
The buprenorphine HCl reference standard was supplied by USP. The buprenorphine buccal solution was prepared by a licensed pharmacist according to the formula detailed in Figure 1. Preparations were packaged in amber glass dropper bottles. Four bottles were stored at 2.4 ± 0.8°C in a refrigerator, and four bottles on the laboratory bench at an ambient temperature of 19.8 ± 0.6°C.
Figure 1.

Recipe for buprenorphine buccal solution, veterinary
Development and validation of stability-indicating HPLC method
A stability-indicating HPLC method was developed and validated for system suitability, accuracy, repeatability, intermediate precision, specificity, linearity and robustness. All tests were conducted in accordance with guidelines found in USP General Chapter <1225> Validation of Compendial Procedures. 11
System suitability and intermediate precision
This experiment involved the preparation of a placebo mixture containing all the inactive ingredients in the formulation. This placebo solution was used as a vehicle to make a 300 μg/ml sample that was injected five times. The 300 μg/ml concentration reflects a 10-fold dilution from the product concentration (3 mg/ml), and was used to bring the HPLC signal within the dynamic range of the instrument. This procedure was repeated on six different days, and the percent relative SD (%RSD) for each day, as well as across days, was calculated. These calculations reflected the intermediate precision. Additionally, column performance parameters, including theoretical plates, API resolution and tailing factor, were calculated to evaluate system suitability. Resolution, the degree of separation between two peaks on a chromatogram, can be calculated using equation (1): 12
| (1) |
where Rs is resolution, tR is time of retention and W is the base peak width. For the API peak, resolution was calculated relative to the peak from the formulation that elutes immediately before it. The concept of theoretical plates provides an indication of column efficiency, and is calculated using equation (2): 12
| (2) |
where N is the number of theoretical plates, tR is time of retention and W is the API peak width. Finally, peak symmetry was assessed using the tailing factor, calculated using equation (3), where ‘a’ and ‘b’ are the widths of the front and back half of the API peak, respectively: 12
| (3) |
Accuracy and repeatability
To assess accuracy, a percent recovery experiment was performed, which compared the response between an API-spiked standard and from an API-spiked placebo. This experiment was completed at three concentration levels (225 μg/ml, 300 μg/ml and 375 μg/ml) and the data were collected in triplicate. These concentrations reflect 75%, 100% and 125% of the assay concentration. The data from the API-spiked placebo at the three concentrations were also used to assess assay repeatability.
Specificity
The two stress conditions were used to force degradation, and show assay specificity for buprenorphine determination in the presence of drug and formulation degradants. To force chemical degradation through oxidation, a 380 μM solution of sodium hypochlorite (NaClO) was used to treat a 3 mg/ml buprenorphine solution in placebo. To expedite the reaction, the experimental sample was also heated in a water bath at a constant 50°C. Periodic aliquots of 100 μl each were removed to assess buprenorphine peak area. With each sample removed, the reaction with NaClO was quenched using 10 μl of sodium sulfite (Na2SO3). So as not to affect drug concentration by dilution, the same volume of Na2SO3 was added to the control sample with each aliquot removed. These samples were followed for 4 h and until >10% degradation had occurred. To force thermal degradation, a 3 mg/ml buprenorphine sample prepared in placebo was heated continuously in a 100°C water bath. Periodic aliquots of 100 μl were removed and assayed for buprenorphine peak area over a 24 h period and until >10% degradation had occurred.
Linearity and range
The buprenorphine reference standard was dissolved in methanol to achieve concentrations of 75 μg/ml, 150 μg/ml, 225 μg/ml, 300 μg/ml and 375 μg/ml. At each concentration, the samples were prepared in triplicate and injected in triplicate for a total of nine data points for each concentration.
Robustness
These experiments challenged the method, with slight variations to verify that the reproducibility and column performance were still acceptable. For challenge 1, the mobile phase buffer pH was adjusted from 7.2–9.4 using sodium hydroxide. For challenge 2, the mobile phase flow rate was adjusted from 0.25–0.75 ml/min. For challenge 3, the column temperature was decreased to 20°C from 40°C. For each challenge, a 300 μg/ml buprenorphine sample was injected five times, and data to evaluate %RSD, resolution from placebo peaks, theoretical plate number and tailing factor were collected.
Stability investigation
Calibration and sampling
For each sampling day, a placebo was used as the diluent for a 3 mg/ml buprenorphine HCl stock solution. This solution was further diluted in methanol to prepare a five-point calibration curve. Each calibration standard was then filtered through a 0.22 micron polytetrafluoroethylene (PTFE) syringe filter, and was injected in triplicate.
For determination of buprenorphine concentration in each preparation, 100 μl solution was removed from each prescription bottle and diluted in 900 μl methanol. For each time point, triplicate samples were taken from each preparation. These samples were vortexed and filtered through a 0.22 micron PTFE syringe filter. A sample from each placebo was also taken in this manner and diluted 10-fold in methanol at each time point. Concentrations of buprenorphine in each study sample were calculated using the linear regression equation associated with the calibration samples applicable to that day. Additionally, the temperature of the laboratory and refrigerator were recorded on each sampling day to ensure consistency of storage conditions.
Results
Development and validation of stability-indicating HPLC method
All HPLC-UV method data met acceptable criteria based on validation characteristics described in USP General Chapter <1225> for the quantification of buprenorphine in a buccal solution formulation. 11
System suitability and intermediate precision
The purpose of this experiment was to ensure adequate resolution of the API peak from degradants and inactive ingredients. Additionally, repeatability was assessed, as well as column performance with regard to theoretical plates and tailing factor. The average resolution was 6.48. Baseline resolution is defined as ⩾1.5, indicating that the API peak is well separated from components of the placebo. 12 The average tailing factor was 1.25. The average column efficiency was at 2100 theoretical plates. The %RSD for the system suitability samples was 0.16%. For intermediate precision (6 days), the total %RSD was 3.70%.
Accuracy and repeatability
The purpose of this experiment was to compare the instrument response between the API in a spiked standard with that dissolved in the placebo formulation. The average recovery was 99.80%, 99.61% and 98.85% at the 225 μg/ml, 300 μg/ml and 375 μg/ml levels, respectively. These data were within ± 2% of the intended concentration. Repeatability of the API-spiked placebo samples was 0.139%, 0.138% and 0.110% RSD at the 225 μg/l, 300 μg/ml and 375 μg/ml levels, respectively.
Specificity
The NaClO-treated preparation exceeded 10% degradation of the API peak with 6 h of exposure. As the reaction with NaClO is very rapid, measured degradation was noted with the initial experimental sample. The reaction was followed until the API peak area was reduced by 17% from the time zero API peak area. The heat-treated preparation exceeded 10% degradation after 24 h of exposure. These degradation profiles are shown in Figure 2. For each specificity experiment, the API peak was greater than baseline resolved (resolution ⩾1.5) from neighboring degradant peaks. The API peaks were verified as spectrally pure using the HPLC system equipped with PDA.
Figure 2.
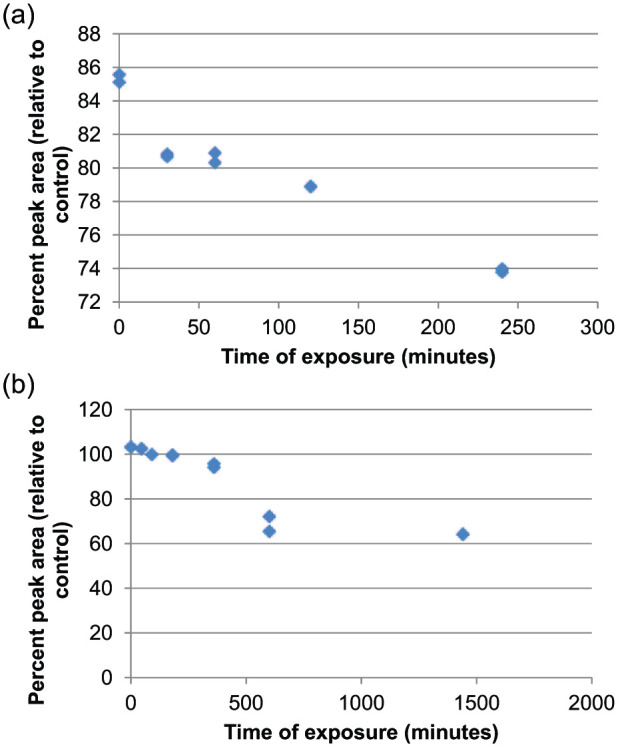
Degradation profile of buprenorphine treated with (a) sodium hypochlorite and (b) heat
Linearity and range
The linear range of the assay was 75–375 μg/ml, thoroughly bracketing the assay concentration of 300 μg/ml. Acceptable linearity, as designated by the correlation coefficient (R2), was demonstrated within the intended calibration range for the assay. These data yielded an R2 of 0.9996. Additionally, the data showed a random distribution of residuals.
Robustness
Despite the chromatographic changes induced by the three challenges, data from these conditions showed method robustness. These challenges included increased mobile phase pH (7.2–9.4), increased the flow rate from 0.25–0.75 ml/min and decreased the column temperature from 40–20°C. The mobile phase pH adjustment caused a decrease in buprenorphine retention on the column. However, the characteristics of the separation did not change dramatically with the change in buffer pH. The results were sufficiently reproducible with a %RSD of 0.080. As expected, the retention of buprenorphine was reduced when the flow rate increased. The column performance also increased accordingly. The %RSD of 0.020 indicates high reproducibility under high flow rate conditions. When the column temperature was decreased, buprenorphine retention was prolonged owing to the lower viscosity of the mobile phase at lower temperatures. The peak was broadened at the higher retention time, but still highly reproducible at a %RSD of 0.221.
Stability investigation
Stability data for buprenorphine buccal solution 3 mg/ml are shown in Tables 1 and 2. The concentrations of buprenorphine in each bottle shown in the results have been corrected for dilution into the linear range of the assay, as well as corrected for the hydrochloride salt used to prepare the calibration curve. Throughout the study, the chromatographic retention time of the API peak was consistent, at approximately 5.8 mins, and the chromatograms of the products and reference standard were consistent in appearance. The bulk samples in the bottle retained their original appearance; however, at day 21, some white particulate matter was noted on the threads of the room temperature bottles. The pH values of the refrigerated samples ranged from 4.01–4.16; however, the pH of the room temperature samples was more varied, with a range of 3.57–4.06. The trend in the refrigerated sample pH was very consistent; however, the room temperature samples trended downward (below pH 4), starting at day 30.
Table 1.
Stability data for buprenorphine buccal solution 3 mg/ml stored in amber glass dropper bottles at room temperature (19.8 ± 0.6°C) conditions for 90 days
| Specification | Initial | 8 h | 24 h | 48 h | 7 days | 15 days | 21 days | 30 days | 60 days | 75 days | 90 days |
|---|---|---|---|---|---|---|---|---|---|---|---|
| % of label (bottle A) | 99.7 | 100.0 | 102.0 | 98.4 | 93.6 | 98.9 | 98.8 | 100.0 | 99.4 | 101.9 | 96.6 |
| % of label (bottle B) | 96.5 | 98.9 | 100.3 | 99.5 | 95.4 | 102.4 | 100.1 | 100.0 | 103.4 | 98.7 | 94.9 |
| % of label (bottle C) | 101.2 | 99.6 | 100.3 | 102.6 | 95.8 | 100.3 | 100.1 | 102.4 | 98.5 | 102.0 | 91.5 |
Table 2.
Stability data for buprenorphine buccal solution 3 mg/ml stored in amber glass dropper bottles at refrigerated temperature (2.4 ± 0.8°C) conditions for 90 days
| Specification | Initial | 8 h | 24 h | 48 h | 7 days | 15 days | 21 days | 30 days | 60 days | 75 days | 90 days |
|---|---|---|---|---|---|---|---|---|---|---|---|
| % of label (bottle A) | 96.8 | 100.8 | 99.8 | 99.9 | 96.2 | 97.9 | 102.4 | 99.0 | 99.4 | 97.7 | 94.9 |
| % of label (bottle B) | 97.3 | 97.1 | 95.4 | 98.6 | 92.7 | 97.7 | 102.6 | 99.4 | 97.5 | 98.8 | 95.2 |
| % of label (bottle C) | 96.2 | 97.9 | 97.2 | 99.3 | 93.3 | 97.9 | 101.8 | 99.3 | 98.6 | 100.0 | 96.6 |
Discussion
This stability-indicating HPLC method is simple, fast and isocratic. The method produces highly accurate and precise results of quantification of buprenorphine in this formulation, and the method is minimally affected by small changes in method parameters (robust). Additionally, this method can separate the buprenorphine API from degradant peaks associated with this formulation when stress conditions are applied. The linear range (75–375 μg/ml) is highly appropriate and convenient for the study of a 3 mg/ml solution, requiring only a simple 10-fold dilution for sample preparation.
Assay results for buprenorphine were based on the chromatographic peak for buprenorphine HCl using a UV absorbance of 280 nm. Initially, the buprenorphine content of all tested bottles was in the range of 2.806–3.074 mg/ml, and was within 90–110% of the labeled potency (2.7–3.3 mg/ml). The buprenorphine concentrations found in each study sample remained within the 90–110% of label claim throughout the study, and were still within this range at 90 days.
The appearance of the chromatograms did not differ between days zero and 90 for either the samples or the calibration standards. All bottles of the buprenorphine buccal solution retained the original appearance of a slightly viscous, colorless, clear liquid. For the room temperature bottles, some white particulate matter was noted in the threads of the container bottles starting at day 21, but this did not affect API concentration. Finally, the pH range for the refrigerated samples was very narrow, while the room temperature samples showed a downward trend for this parameter, likely due to the breakdown of the citrate buffer system in the vehicle over time.
Conclusions
The stability-indicating HPLC method developed and used to assess the stability of the buprenorphine buccal formulation in this study is considered fully validated based on characteristics described in USP General Chapter <1225>. 11 This method has the advantage of being an isocratic run with a relatively short run time, utilizing a common C8 column. The stability evaluation, including potency measurements and physical inspections, of a 3 mg/ml buprenorphine buccal solution indicates that this product is stable under refrigeration through 90 days when stored in amber glass dropper bottles. These data also demonstrated that while the potency of the preparation remains within range (90–110%) of the label amount through 90 days under room temperature conditions, the pH of the formulation is more varied when stored at this condition.
Acknowledgments
We would like to thank ProCompounding Pharmacy in Johnson City, Tennessee, for preparation of the test samples, and East Tennessee State University Bill Gatton College of Pharmacy for its ongoing support of student research.
Footnotes
The authors do not have any potential conflicts of interest to declare.
Funding: Previous funding received from, but not related to this project, National Institutes of Health and Hamilton Company. This study was supported by a research grant from The United States Pharmacopeial Convention, Inc.
Accepted: 24 December 2014
References
- 1. American Reagent Inc. Buprenorphine (package insert). New York: American Reagent Inc, 2009. [Google Scholar]
- 2. Mathews KA. Pain management and general approach to management. Manag Pain 2000; 30: 729–755. [DOI] [PubMed] [Google Scholar]
- 3. Bortolami E, Slingsby L, Love EJ. Comparison of two formulations of buprenorphine in cats administered by the oral transmucosal route. J Feline Med Surg 2012; 14: 534–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shrestha B, Reed JM, Starks PT, et al. Evolution of a major drug metabolizing enzyme defect in the domestic cat and other felidae: phylogenetic timing and the role of hypercarnivory. PLoS ONE 2011; 6: e18046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Giordano T, Steagall PV, Ferreira TH, et al. Postoperative analgesic effects of intravenous, intramuscular, subcutaneous or oral transmucosal buprenorphine administered to cats undergoing ovariohysterectomy. Vet Anaesth Analg 2010; 4: 357–366. [DOI] [PubMed] [Google Scholar]
- 6. Robertson SA, Taylor PM, Sear JW. Systemic uptake of buprenorphine by cats after oral mucosal administration. Vet Rec 2003; 152: 675–678. [DOI] [PubMed] [Google Scholar]
- 7. Hedges AR, Pypendop BH, Shilo-Benjamini Y, et al. Pharmacokinetics of buprenorphine following intravenous and buccal administration in cats, and effects on thermal threshold. J Vet Pharmacol Ther 2013; 37: 252–259. [DOI] [PubMed] [Google Scholar]
- 8. Steagall PVM, Monteiro-Steagall BP, Taylor PM. A review of the studies using buprenorphine in cats. J Vet Intern Med 2014: 28: 762–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hedges AR, Pypendop BH, Shilo Y, et al. Impact of the blood sampling site on time-concentration drug profiles following intravenous or buccal drug administration. J Vet Pharmacol Ther 2013; 37: 145–150. [DOI] [PubMed] [Google Scholar]
- 10. US Pharmacopeia. United States Pharmacopeia and National Formulary (USP 37 – NF 32). Chapter 795 pharmaceutical compounding. Rockville, MD: US Pharmacopeia, 2014. [Google Scholar]
- 11. US Pharmacopeia. United States Pharmacopeia and National Formulary (USP 37 – NF 32). Chapter 1225 validation of compendial procedures. Rockville, MD: US Pharmacopeia, 2014. [Google Scholar]
- 12. Hansen SH, Pedersen-Bjergaard S, Rasmussen KE. Introduction to pharmaceutical chemical analysis. 1st ed. Chichester: John Wiley & Sons, 2012, pp 127–140. [Google Scholar]