

Abstract
The present review contains a representative sampling of mechanistic studies, which have appeared in the literature in the last 5 years, on 1,3-dipolar cycloaddition reactions, using DFT calculations. Attention is focused on the mechanistic insights into 1,3-dipoles of propargyl/allenyl type and allyl type such as aza-ylides, nitrile oxides and azomethyne ylides and nitrones, respectively. The important role played by various metal–chiral–ligand complexes and the use of chiral eductors in promoting the site-, regio-, diastereo- and enatioselectivity of the reaction are also outlined.
Keywords: DFT calculations, dipolar cycloadditions, aza-ylides, nitrile oxides, azomethine ylides, nitrones
1. Introduction
1.1. Computational Methods
In recent years, theoretical and computational studies have increasingly accompanied experimental data, providing support in the conformational analysis of molecules, biomolecules and their bioactive synthetic analogues and in the explanation of the mechanisms of organic reactions.
The conformational analysis of organic molecules is based on the principle that molecules are not rigid and their kinetic energy at room temperature is large enough to let all atoms exhibit permanent molecular movement. This fact means that the absolute positions of atoms in a molecule and of the molecule as a whole are not fixed and that the relative location of substituents on a single bond is different at each moment. So, every molecule, with one or more single bonds, exists in many different rotamers or conformers. Low-energy conformers make the major contribution to the overall population. A transformation from one geometry to another is primarily related to changes in the torsional angles of single bonds. A complete overview of the conformational potential surface of molecules can be gained by theoretical techniques, and numerous methods for conformational analysis, which are able to identify all minima on the potential energy surface, have been developed. The number of minima increases with the number of rotatable bonds. Thus, the detection of all minima becomes difficult and time consuming. The most used techniques for conformational analyses are systematic search procedures, performed by systematically varying each of the torsion angles to generate all possible conformations, and molecular dynamics, which reproduces the time-dependent motional behavior of a molecule [1].
Another very important application of theoretical studies, which has found widespread application in recent decades, is the clarification of various mechanisms of organic reactions, locating reactants, intermediates, transition states and products along the reaction pathways.
In order to locate the different geometries of the molecules or the different points along a reaction coordinate, different methods can be used. In computational chemistry, quantum mechanical methods are very important tools.
They are based on the calculation of the interaction energies between a nucleus–nucleus, electron–nucleus and electron–electron for all atoms of the studied molecules and on the solution of the corresponding Schrödinger Equation (1).
| HΨ = EΨ | (1) |
Quantum mechanical methods allow for calculating the energy and electronic structure of molecular systems in different spatial configurations, to analyze the breaking and formation of bonds and to study the chemical reactivity. Nevertheless, they have a disadvantage in their high computational cost.
Nowadays, a quantum mechanical approach that can locate widespread diffusion, is density functional theory (DFT) [1], which is based on the resolution of the Kohn–Sham equations. Calculation times are shorter with respect to other quantum mechanical methods, but the accuracy of the results is preserved.
DFT calculations, on the basis of the Hohenberg–Kohn theorem [2], consider the electronic energy of the molecular ground state to be completely determined by electron density (ρ). In fact, the wave function, depending on one spin and three spatial coordinates for every electron of the studied systems, is not intuitive for compounds with more than one electron. Therefore, an important target is the possibility of finding a physical observable that permits the a priori construction of the Hamiltonian operator. The dependence of the Hamiltonian on the positions and atomic numbers of nuclei and on the total numbers of electrons suggests ρ as a useful research observable since, when integrated over the entire space, it provides the total number of electrons.
The correspondence between the electron density (ρ) of a system and its energy represented the basis of DFT theory and it is of crucial importance considering a comparison with the wave function.
In fact, while the complexity of the wave function increases with the number of electrons, ρ, which only depends on the three spatial coordinates, it presents the same number of variables independently from the system’s dimensions. On the basis of this approach, the electron density, which determines the external potential, furnishes the Hamiltonian, from which the wave function can be obtained. Finally, once these parameters are determined, the energy of the system can be computed.
However, this direction does not award any simplification over the Molecular Orbital (MO) theory, since the final step still requires solving the Schrödinger equation, which is very difficult because of the electron–electron interaction term of the Hamiltonian. In 1965, Kohn and Sham [3] realized that it was possible to solve the problem by considering a non-interacting system of electrons and evaluating the kinetic energy, T. Because of this approximation, the calculated T is lightly undervalued, but the difference with respect to the real value of the energy is small and is included in the Exchange-Correlation term (Exc). So, in general, the DFT energy results in
| EDFT = T(ρ) + Ene(ρ) + J(ρ) + Exc(ρ) |
T(ρ) is the kinetic energy in the hypothesis that electrons do not interact each other;
Ene(ρ) is nucleus–electron attraction energy;
J is Coulomb energy between electrons;
Exc is, as said above, exchange and correlation term.
Between the different DFT methods, the B3LYP [4] hybrid functional (a hybrid method obtained by combining the five functionals Becke, Slater, HF exchange, LYP and VWN5 correlation) gave the most accurate results for a large number of compounds and in particular for organic molecules, finding wide application by computational organic chemists.
In the last few years, a significant number of new density functionals, for applications of Kohn–Sham density functional theory, in chemistry and physics were developed. For example, the Thrular’s hybrid metafunctionals M06 and M06-2X are widely used for their broad applicability, in particular in transition metal chemistry and in the study of excited-states.
Moreover, it is worthwhile pointing out that during the calculations, molecules can be considered in the gas phase and so treated as isolated, non-interacting species with enormous facilities for theoretical modeling. However, this approach is not accurate enough for water-soluble biomolecules. In fact, the solvent phase deeply influences the results because of the hydrogen bond interactions and the charge polarization differences from the gas and the water phase. When the solvent is considered, the first point is the number of solvent molecules to add to the system. To overcome this problem, the so-called implicit models or continuum solvation models have been developed [1], in which the solvent is replaced by a continuous electric field. One family of these methods is called the Self-Consistent Reaction Field (SCRF) that considered the solvent as a continuum of uniform dielectric constant (ε). Perhaps the most popular SCRF method is the Tomasi’s Polarized Continuum Model (PCM) [5], from which also a conductor-like modification was described: the C-PCM method [6].
Despite the accuracy of QM methods, they cannot be used to study some biochemical systems, such as enzymes, because of their dimensions. Nevertheless, the use of methods that are less time-demanding (i.e., MM methods) to investigate the breaking and forming of bonds during a reaction is not feasible. To overcome these limitations, hybrid quantum mechanics/molecular mechanics (QM/MM) simulations were developed. These approaches consist of the treatment of the region, in which the chemical process takes place, with a QM method, while the larger part of the system is modeled by a molecular mechanics force field [7].
In the last five years, DFT calculations were applied by organic chemists to support hypotheses on different reactions mechanisms. A very interesting field of application was the investigation of the synchronous or asynchronous technique of bonds formation in concerted mechanisms with particular regard to the 1,3-dipolar cycloadditions (1,3-DC) [8]. A very significant support to the rationalization of the various reaction pathways of pericyclic reactions is offered by the Frontiers Molecular Orbital (FMO) theory, which is based on the idea that a good approximation of reactivity can be obtained not considering all the orbitals of the system but rather only the highest occupied orbital of one reactant (HOMO) and the lowest unfilled (lowest unoccupied orbital, LUMO) of the other one.
Another very useful tool to interpret results and rationalize the mechanism of pericyclic reactions is the Houk/Bickelhaupt activation strain model [9]. With this approach, activation barriers, that determine reaction rates, can be obtained. In detail, in the case of bimolecular processes, the sum of the energies needed to distort the addends to reach the same geometry as in the TSs, and the interaction energies between the two distorted reactants, furnish the activation energies. The changes of these values during the reaction clarify the factors that control the reactivity.
In many of the reviewed works in the following sections, these important principles of physical organic chemistry are used to interpret results.
1.2. 1,3-Dipolar Cycloaddition
The 1,3-dipolar cycloadditions reactions can be considered the most productive heterocyclic synthesis that allow, using a single step, the introduction of three or four stereogenic centers in a stereospecific manner. The addends of this type of reactions are dipolarophiles, i.e., compounds with π bonds and 1,3-dipoles that can be distinguished in linear propargyl/allenyl types or allyl types.
The concerted cycloaddition shows a regioselectivity and stereoselectivity that can be appropriately rationalized with the frontier molecular orbital theory, studying the interactions between the LUMO and HOMO of dipole and dipolarophile in the endo and exo transition states. Moreover, the regio, diastereo and enantioselectivity of the process can also depend on the presence of metals in the reaction environment. In fact, metal ions, acting as Lewis’s acids, can alter not only the orbital coefficients of the reacting atoms but also the energy of the frontier orbitals of the reacting species [10].
All of the information reported above highlights the importance of computational studies aimed to predict or clarify the mechanistic and stereochemical outcomes of these reactions.
With the development of computational methodologies, in the last few decades, the growing interest of organic chemists for their application in the study of 1,3-dipolar cycloaddition reactions has been demonstrated by the increasing number of publications in this area [11,12,13,14,15,16].
In this review, we are going to show the recent applications (2018–2023) of DFT methods to mechanistic speculation on reaction routes of 1,3-CDs classified on the basis of the different involved dipoles, i.e., 1,3-dipoles of propargyl/allenyl type (aza-ylides and nitrile oxides) and allyl type (azomethine ylides and nitrones) that appeared in the literature in the last few years.
2. Computational Mechanistic Studies of 1,3-Dipoles
2.1. 1,3-Dipoles of Propargyl-Allenyl Type
Dipoles of the propargyl-allenyl type present a linear structure in which a triple bond is present in canonical form.
![]()
The main dipoles belonging to this category are: aza-ylides (I), diazoalkanes (II), nitrile oxides (III), and nitrilimines (IV).
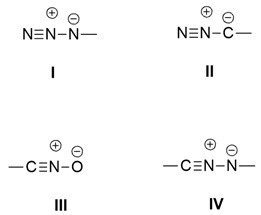
In the following sections, only I and III will be considered.
2.1.1. Aza-Ylides
Aza-ylides represent a very important tool for the synthesis of complex heterocyclic compounds and are involved as special frameworks in the so-called “click chemistry”. When in 2001 [17], Sharpless described the ideal conditions for click reactions, also the 1,3-dipolar cycloadditions were listed as candidates for application of this new branch.
In 2002, the first copper-catalyzed azide–alkyne cycloadditions were reported [18]. They were conducted in mild reaction conditions, with high yields, and the formation of only one regioisomer (i.e., 1,4-disubstituted 1,2,3-triazole derivatives). In this way, organic azides gained increased attention in this synthetic field and obtained an enormous success as 1,3-dipoles for the preparation of compounds with applications in life sciences.
In 2018, Ben El Ayouchia [19] investigated and explained the uncatalyzed and copper(I)-catalyzed [3 + 2] cycloaddition of methyl azide with propyne using DFT calculations at the B3LYP/6-31G(d) level. In the case of the uncatalyzed route, two regioisomeric paths (1,4- and 1,5-approaches) were studied, showing similar high activation energies of 18.84 (TS1) and 18.51 (TS2) kcal/mol, respectively (Figure 1). These values give an explanation to the lack of regioselectivity of the coupling conducted in the absence of catalysts. The asynchronicity was evaluated and shown to be higher for the 1,5-regioisomer process with respect to the 1,4-one. In general, the formation of triazoles is highly exothermic, and calculations showed an asynchronous one-step mechanism with a very non-polar character.
Figure 1.

Three-dimensional (3D) plots of the TSs of the uncatalyzed route leading to 1,4- and 1,5-regioisomer, respectively.
Instead, in the case of the catalyzed reaction, the metal is described through the LANL2DZ basis set. Firstly, the terminal alkyne binds to copper(I) as a π-ligand with a Cu6–N1 distance of 2.10 Å and an increase in acidity of its terminal alkyne proton. The second step consists of the binding of the distal nitrogen of the azide to the C-2 carbon of the acetylide with a barrier in water of 8.99 kcal/mol. In the corresponding TS, the C4-N3 bond length is 1.90 Å, while the Cu6–N1 becomes 2.02 Å. This is the rate-limiting step of the reaction for the Cu(I)-catalyzed process. From this passage, a six-membered Cupracycle is obtained (Figure 2). It is quite stable because of the absence of ring strain determined by the presence of two copper atoms and an sp2 hybridized carbon atom. Finally, the ring-closure process occurs with a barrier of 16.12 kcal/mol and a length of the C5–N1 forming a bond of 2.22 Å. As clearly evidenced, the coordination of the copper to alkyne changes the mechanism from a non-polar one-step to a polar stepwise with the preferred obtainment of a 1,4-cycloadduct.
Figure 2.
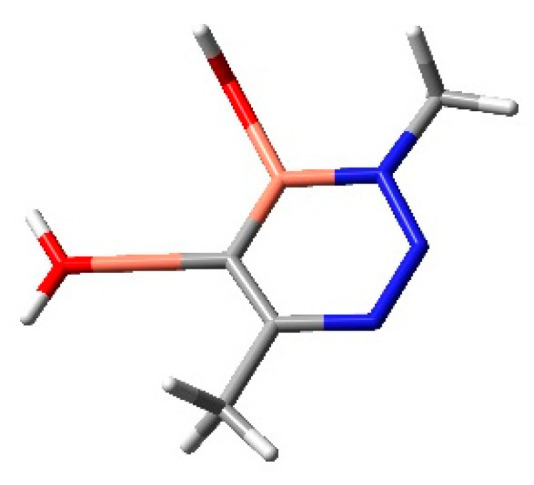
Six-membered cupracycle, intermediate of the reaction.
In the same field, Hamlin et al. [20] investigated the cycloadditions of methyl azide with 2-butyne in comparison with cycloalkynes (cycloheptyne, cyclooctyne, cyclononyne). Calculations were performed at the M06-2X/6-311++G(d)//M06-2X/6-31+G(d) level. The ΔG≠ values are 16.5–22.0 kcal/mol lower in energy for cyclic adducts with respect to 2-butyne, which gives a less exergonic reaction.
This study demonstrated that the higher reactivity of cyclic alkynes, in comparison with acyclic ones, can be attributed to three different factors. Surely, the first one is a reduction in strain or distortion energy, which is historically used to explain the acceleration of cycloaddition rate. However, the authors highlighted that it is accompanied by a smaller gap between HOMO and LUMO and a major orbital overlap, which is able to help the stabilization of orbital interactions. The smaller HOMO–LUMO gap is attributable to a stabilization the cycloalkyne π-LUMO, while the higher orbital overlap is probably due to a polarization of both the π-HOMO and π-LUMO lobes on the external π-face, pointing to the azide frontier molecular orbitals.
A series of Cu complexes with N-heterocyclic carbene were prepared by Lin et al. [21] and used for the copper-catalyzed azide–alkyne cycloaddition reaction. The activities of the Cu complexes that resulted were related to the nature of the counterion X− (I > Br > Cl∼BF4 > PF6) and to the rank of the trans effect. Experimental evidence allow for hypothesizing that firstly, the NHC ligand dissociated from the Cu and deprotonated phenylacetylene, giving (NHC-H)+ and phenylacetylide. Then, phenylacetylide and Cu bonded, forming (NHC-H)+[(PhC≡C−) CuX]−.
DFT calculations, performed at the M06/Def2-SVP level and using the C-PCM model for a solvent, suggest a mechanism characterized by the obtainment of acetylide copper halide as a consequence of NHC ligand dissociation and phenylacetylene deprotonation. After cycloadditions of methyl azide with mononuclear copper acetylide give a six-membered metallacycle, the reaction with the second copper catalyst occurs, forming an intermediate. This step is followed by ring contraction, leading to a dinuclear intermediate with higher stability.
In a second step, the six-membered metallacycle, achieved by the cycloadditions of methyl azide with mononuclear copper acetylide, reacted with the second copper catalyst, (NHC)CuX, to form the dinuclear intermediate, which, undergoing a quick ring contraction, form another more stable dinuclear intermediate. Finally, triazole is acquired after the NHC−H+ protonation. Moreover, calculations confirm that the NHC dissociation should occur in the first phase and not in the X− dissociation.
Yu et al. [22] studied cycloadditions involving methyl azide and various allenes (propadiene, ketenimine, ketene, carbodiimide, isocyanic acid, and carbon dioxide), investigating reactivity, site- and regioselectivity through DFT calculations using a BP86 functional and TZ2P basis set. The formation of two possible regiospecific cycloadducts, the 1,5- or the 1,4-adduct, was taken into consideration and, in all cases, the formation of 1,5-adducts (Scheme 1A) was kinetically and thermodynamically favored over that of 1,4-adducts (Scheme 1B).
Scheme 1.

Possible products ((A) = 1,5-adducts; (B) = 1,4 adducts) of the 1,3-dipolar cycloaddition of methylazide with linear allenes (X, Y = CH2, NH, O).
In the case of asymmetric heteroallenes, methyl azide prefers to attack the most electropositive terminal atom. Only in the case of ketene is the barrier for the attack to CO a little bit lower than that for the attack to CC (19.2 vs. 20.0 kcal/mol). With the increasing of the heteroatoms number, the process is less reactive (passing from CCC to CCN or CCO, from CCO to NCO or OCO, and from CCN to NCN). Cyclic allenes were also considered as cycloadducts. In these cases, the higher predistortion of cyclic allenes determines higher reactivity, connected with lower activation energies for the cycloaddition, because of a smaller HOMO–LUMO gap and thus the presence of more stabilizing orbital interactions.
Ben El Ayouchia et al. [23] investigated, through a combination of experimental and molecular electron density theory (MEDT) studies, the [3 + 2] cycloaddition reaction between methyl azide and propyne. Experimentally, the reaction, catalyzed by Ag(I), is fast at room temperature and the final product, 1,4-dimethyl-1,2,3-triazole, is obtained in good yields and easily isolated without any purification (Scheme 2).
Scheme 2.

Reaction conditions: (a) Ag(I), AgCl/H2O.
Calculations were performed at both the B3LYP/6-31G(d,p) (LANL2DZ for Ag) and the ωB97XD/6-311G(d,p) (LANL2DZ for Ag) levels, choosing the water and chloride anion as ligands.
In the case of the uncatalyzed reaction, the cycloaddition occurs with high barrier and poor regioselectivity, giving both 1,4- and 1,5-dimethyl-1,2,3-triazoles. Instead, considering the Ag-catalyzed reaction, the 1,4-adduct is estimated to be regioselectively obtained, as experimentally verified.
Three different possible catalytic species are supposed (Scheme 3 and Scheme 4).
Scheme 3.

Ag(I)-catalyzed azide-alkyne cycloaddition involving catalytic species 1.
Scheme 4.

Ag(I)-catalyzed azide–alkyne cycloaddition involving catalytic species 6 (X = Cl, OH2).
The reaction involving intermediate 1 is characterized by a single step, as shown in Scheme 3. The other two species, 6 (X = Cl, H2O), follow a two-step mechanism that has no experimental impact because of the endothermic nature of the corresponding intermediate complexes 8 (Scheme 4).
In both cases, the rate-determining step is the nucleophilic attack of the N1 nitrogen atom of azide to the substituted C atom of propyne without energetic differences between chloride or water ligands.
This dinuclear route presents lower barrier values (about 4 kcal/mol) with respect to the mononuclear pathway. It is noteworthy that a similar stepwise mechanism was found using the two different functionals (B3LYP and ωB97XD), confirming the accuracy of B3LYP for the study of these cycloadditions.
Kim et al. [24] performed a [3 + 2] cycloaddition of alkynes, cyanoalkynes, thioalkynes, and ynamides with differently substituted azides in the presence of nickel as a catalyst (Scheme 5). The products were obtained with significant regio- and chemoselectivity. In the case of cyanoalkynes, the 1,4,5-trisubstituted triazoles with the cyano group at position 4 of the ring are preferred. Instead, the thioalkynes and the ynamides showed an inverse regioselectivity leading to 1,4,5-trisubstituted triazoles with thiol and amide groups at position 5, respectively.
Scheme 5.

Reaction conditions: (a) Cp2Ni (10 mol %), Xantphos (10 mol %), Cs2CO3 (1 eqv), DCM, r.t., 24 h.
In order to clarify the reaction mechanism, a computational study was performed at the M06/6-31G**/cc-pVTZ(-f) level.
The computed mechanism, reported in Scheme 6, allowed for hypothesizing the formation of a nickelcyclopropene intermediate obtained by the oxidative addition of the alkyne substrate to the Ni(0)–Xantphos catalyst. Then, this intermediate gave the coupling with azide determining the regio- and chemoselectivity.
Scheme 6.

Computed reaction mechanism of nickel-catalyzed cycloaddition.
In detail, a Xantphos ligand forms a 14-electron Ni(0)–d10 complex 10 that, in turn, gives an exergonic oxidative addition of the alkyne. In this way, Ni(II)-cyclopropene intermediate 11 is afforded. Then, the interaction with azide allows the obtainment of 12. Finally, the cycloaddition occurs, giving 13, overcoming a barrier of 24.8 kcal/mol.
This cyclization step is the rate-determining one of this nickel-catalyzed reaction on which regioselectivity depends.
Chen et al. [25] obtained bicyclic tetrazoles from the anionic [3 + 2] dipolar cycloaddition between lithiated trimethylsilyldiazomethane and α-azido ketones. DFT calculations were performed at the M06/6-31+G level, highlighting that the first step of the reaction is the chelation of the nitrogen-based dipole and dipolarophile by a lithium ion (Scheme 7).
Scheme 7.

Dipolar cycloaddition between lithiated trimethylsilyldiazomethane and α-azido ketones.
The steric hindrance of groups bearing by the α-azido ketones affect the selectivity and efficiency of the cycloadditions in competition with the formation of triazines due to the C−H insertion of alkylidene carbene.
The mechanism (Figure 3) starts from 14, which is derived from lithiated trimethylsilyldiazomethane and α-azido ketones. It rearranges through a barrier of 8.0 kcal/mol to IN1. This intermediate undergoes the anionic [3 + 2] dipolar cycloaddition, passing TS2 (9.4 kcal/mol) and leading to cycloadduct IN2. Then, IN2 rearranges to the most stable aromatic intermediate IN3 that is protonated, furnishing tetrazoles 15.
Figure 3.

Energy profile of reaction leading to 15.
Navarro et al. [26] synthesized 1,4-(disubstituted)-5-triazenyl-1,2,3-triazoles using a ligand-free domino copper(I)-catalyzed azide−alkyne−azide process, involving chelating aryl azides with polar groups in ortho to the azide moiety (i.e., P(O)(NEt2)2) and terminal alkynes (i.e., phenylacetylene 17) (Scheme 8).
Scheme 8.

Reaction conditions: (a) CuSO4·5H2O/5 Na+ L-Asc. 1 eq, DMF (1 mL), t = 42 min, 25 °C, air.
From this model reaction, fully substituted 1,2,3-triazole 19, chemo- and regioselectively containing a triazenyl moiety, is obtained. Experimentally, a competition with a reaction route that generates the conventional click 4-(disubstituted)-triazole 20 was revealed. In order to clarify the process, DFT calculations were performed at the M06/6-311+G(d,p)-SDD/SMD(DMF)//M06/6-31+G(d)-SDD/SMD (DMF) level. Modeling data showed that chelation of the copper cation of the Cu-triazolide intermediate 18 by the ortho-substituted azide is crucial to avoid competition with the formation of conventional triazole 20.
The mechanism involving the formation of 19 presents four steps and thus four different transition states. The first one (TS1) corresponds to the nucleophilic attack of Cu-triazolide 18 to the azide 16. Then, the cis/trans isomerization of the triazene moiety (TS2), the Cu-oxygen decoordination of one P=O (TS3), and the protonation of the Cu-triazene (TS4) occurred (Figure 4). TS1 is the rate-determining step of the process and is 0.5 kcal/mol lower in energy with respect to the TS that from 18 leads to 20.
Figure 4.

Computed pathway for the formation of 19 and 20.
Fang et al. [27] studied the copper(I)-catalyzed azide alkyne cycloaddition (CuAAC) using as the catalyst a supported carbon nanotube (CNT) Au4Cu4 cluster. The Au4Cu4/CNT system heterogeneously and efficiently catalyzes the CuAAC reaction of terminal alkynes without alkyne deprotonation to a σ,π-alkynyl intermediate, following the cycle reported in Figure 5.
Figure 5.

Catalytic cycle of CuAAC reaction of terminal alkynes.
DFT results showed that HC≡CPh was activated by π-complexation with Au4Cu4 forming the bimetallic σ, π-alkynyl intermediate that is the real catalyst of the reaction. The terminal H atom of the alkyne is not stripped during catalysis. This type of catalyst allows also the reaction involving internal alkynes. On the contrary, the Au11/CNT and Cu11/CNT nanocatalysts are not active for the last type of alkynes, highlighting the synergistic effects of Au and Cu in Au4Cu4.
Zu and Kinjo [28] investigated the regioselective cycloaddition involving 1,2-diboraallene 25, showing cumulated C=B and B=B double bonds, and azide, without catalysis and using mild conditions, leading to diboratriazole 26.
Compound 25 reacted with 2,6-diisopropylphenyl azide, giving cycloadduct 26 (Scheme 9). The same reaction, conducted with other azides (i.e., trimethylsilyl azide, 1-adamantyl azide, and phenyl azide), does not provide the same product, highlighting the importance of the azide electronic and steric factors for the reaction trend.
Scheme 9.

Synthesis of compounds 26 and 27.
Cycloadduct 26 presents low solubility and so it was successfully isolated. Nevertheless, it spontaneously releases N2, giving 27.
The electronic structure of 26 was elucidated calculating frontier orbitals through DFT methods at the B3LYP/6-311G(d,p) level. The HOMO orbital was dominated by the π-bonding orbital over the C13–B2–B3 moiety (see numbering in Scheme 9), while HOMO-1 included the σ-bonding of the B2N3 ring. The HOMO-2 orbital coincided with π-bonding orbitals B3–N4 and B2–N2, which were polarized toward the nitrogen. The LUMO orbital corresponds to the p orbitals on C13 and B3 and also includes the π-type orbital over the B-N moiety in cyclic (alkyl)(amino)carbene.
Moreover, the authors investigated the reaction mechanism leading to 26 (Figure 6).
Figure 6.

Reaction mechanism leading to 26.
The phenyl azide attacks 1,2-diboraallene from the CH2 side. The initial coordination of two nitrogen atoms of azide with the two B atoms affords the five-membered ring Int1 without overcoming any barrier. Passing through the low barrier TS1, of only 2.1 kcal/mol, the elimination of one PCH3 group occurs, giving 2.
Singh et al. reported [29] 1,2,3-triazole scaffolds, obtained through Copper(I)-catalyzed alkyne−azide cycloaddition (CuAAC) as a chemosensor probe (Scheme 10), which is able to recognize heavy metal ions, such as Pb (II) and Hg (II).
Scheme 10.

Synthesis of 1,2,3-triazoles through CuAAC.
The structures of the ligand 30 and the complexes with Pb(II) and Hg(II) were optimized through DFT calculations at the B3LYP/6-311++G(d,p) level and B3LYP/LANL2DZ for metals.
In the complex, the metal ion is coordinated to one of the N atoms of the 1,2,3-triazole ring. The FMO analysis showed that the HOMO is mostly delocalized over the 4-tertbutylcatechol moiety, while the LUMO orbital is delocalized over one of the arms bonded to the 1,2,3-triazole. The energy difference between HOMO and LUMO is lower for the complex (1.11 eV) with respect to the free ligand (5.203 eV). These 1,2,3-triazoles, synthesized via the CuAAC methodology, can be efficiently used for their ion-sensing properties.
In 2022, Chiavegatti Neto et al. investigated the different steps of metal-free [3 + 2] cycloadditions with enolates/enols and azides using DFT calculations [30].
Experimentally, the reaction was conducted starting from 2-butylidenemalononitrile DBU and PhN3 in DMSO-d6 at 50 °C (Scheme 11). After the obtainment of both Z and E isomers of 31, PhN3 was added, leading to Z-32 and E-32. Calculations were performed at the M06-2X/6-31+G(d,p) level starting from 2-propylidenemalononitrile, which was achieved from the Knoevenagel condensation between propanal and malononitrile. The following deprotonation gives intermediates Z-31 and E-31 that react with phenyl azide, passing the two barriers E-TS1 and Z-TS1, the first one being slightly lower. Experimental data show that the trans isomers prevail. The irreversible cycloaddition leading to E-32 and Z-32 ends the final elimination, furnishing compound 33. The reaction energy profile suggests that the elimination is the rate-determining step of the process with a higher energy with respect to the cycloaddition.
Scheme 11.

Synthesis of compound 33.
On the basis of the growing interest toward economic viability and sustainability, attention was focused on the use of copper catalysts for coupling reactions. Gholivand et al. [31] reported the cinnamaldehyde-derived Schiff base ligand N,N′-bis (trans-cinnamaldehyde) ethylenediimine (C20H20N2, L) used to obtain Cu (I) complexes (i.e., [CuC20H20N2)PPh3Cl] (C1) and [Cu(C20H20N2)PPh3Br] (C2) and applied to the azide–alkyne click reaction (AAC) as catalysts (Scheme 12).
Scheme 12.

Cu(I)-catalyzed AAC reactions, leading to 34 a–h.
Experimentally, the best reaction conditions were shown to be 15 mol % catalyst, 45 °C, 30 min and water as the solvent. The structure and the reactivity of C1 and C2 were studied through quantum calculations using moduleDMol3 and the LDA/PWC level. The minimum energy conformations were located, and the FMO analysis was performed. Complex C2 is more reactive than C1, and adducts are obtained faster using C1 rather than C2 as the catalyst, except in case a and b (Scheme 12).
Song et al. [32] used DFT calculations, starting from 1,2,3,4-tetrazole a Mn-catalyzed click reaction using phenylacetylene and obtaining 1,5-disubstituted 1,2,3-triazole 35 with complete control of the regioselectivity (Scheme 13). The authors proposed, on the basis of their results, a very interesting manganese-copper/zinc cooperative mechanism.
Scheme 13.

Synthesis of 1,5-disubstituted 1,2,3-triazole 35.
Optimizations were performed using the M06/6-311++G** basis set for all other atoms and the LANL2DZ basis set for metals.
The mechanistic analysis highlighted three different steps for the process: (1) the opening of the tetrazole; (2) a variation of the Mn coordination site; and (3) the 1,3-cycloaddition assisted by ZnCl2. This last step is the regioselectivity-determining one of the reactions.
The reaction (Figure 7) starts with tetrazole ring opening giving complex IN2. Cycloaddition is not possible at this level because of the steric hindrance caused by the presence of pyridine on the N1 atom. The isomerization of IN2 to IN2_iso occurs. At this point, ZnCl2, obtained in conjunction with the catalyst, coordinates to the N4 atom of IN2-iso, giving IN2B-iso. Then, the 1,3-cycloaddition leading to the formation of C1–N1 and C2–N3 bonds in the presence of ZnCl2 occurs, passing a barrier of 27.4 kcal/mol (TS2). Finally, the reaction furnishes the 1,5-disubstituted 1,2,3-triazole 35 and Mn(Por) catalyst regenerated. The authors considered also the possibility of a pathway without ZnCl2 participation; however, it was shown to be kinetically disfavored.
Figure 7.

Calculated free energy profiles for the click reaction.
Fukuura and Yumara [33] investigated, through DFT calculations, the 1,3-dipolar cycloaddition reaction involving phenylacetylene and phenyl azide on the surface of carbon nanotubes, showing diameters in the range 10–14 Å. The authors used QM/MM ONIOM calculations, treating guest molecules with the B97D functional and host nanotubes with UFFs (universal force fields). The regioselectivity of the reaction was studied, paying attention to the formation of the 1,4- and 1,5-triazoles.
The nanotube support favors, both from the kinetic and thermodynamic point of view, the formation of the 1,4-triazoles as cycloadducts. Considering the 1,4-route, reactants are planar, because they are small with respect to the cavity of tubes. In this case, the nanotubes allow lower ΔE≠ without any dependence on their diameter values.
Conversely, in the 1,5-approach, the phenyls of cycloadducts overlap each other, giving a stacking interaction. This structure in thin tubes determines repulsive orbital interactions between the two rings, causing an increase in the activation energies with a decrease in the tube diameter.
2.1.2. Nitrile Oxides
The interest in the chemistry of nitrile oxides is primarily due, when they react with alkenes as dipolarophiles, to the synthesis of isoxazoline derivatives, which are regarded as masked b-hydroxycarbonyl compounds that can be further converted into synthetically useful building blocks [34].
Bucci et al. [35] reported the synthesis of tert-Butyl 1-(5-Benzyl-3a,5,6,6a-tetrahydro-4H-pyrrolo[3,4-d]-isoxazol-3-yl)-2S-phenylethyl)carbamate 38a,b through a nitrile oxide [1,3]-dipolar cycloaddition between N-tert-Butoxycarbonyl [1-Chloro-1-(hydroxyimino)-3-phenylpro-pan-2-yl)]carbamate 36 and N-benzyl-3-pyrroline 37 and base (Scheme 14).
Scheme 14.

Nitrile oxide [1,3]-dipolar cycloaddition leading to 38a,b.
From this reaction, two diastereoisomers can be formed, such as the Δ2-isoxazoline derivatives 38a and 38b.
In order to optimize the yield and control diastereoselectivity, different reaction conditions were tested. The use of more polar solvents allows for better yields when a weak organic base, with a pKb value in the range 5–3, is used. Also, inorganic bases and MeCN as the solvent give interesting results.
By-products can be formed consequently to the rate of formation of nitrile oxide.
The diastereoisomeric ratio is influenced by the choice of the organic or inorganic base, moving from a 1:3 ratio in favor of adduct 38b to a 1:1 ratio, respectively.
A complete conformational search of 38a and 38b was performed through MM calculations. The most stable geometries were optimized using DFT methods at the CPCMHCTC/6-11+G(d,p)//HCTH/6-31+G(d) level and MeCN as the solvent and C-PCM method. From the two conformations, the transition states leading to 3a and 3b were located. The reaction was shown to be under kinetic control, and the predicted ratio for 38a and 38b is 28:72, being a value very similar to that that experimentally determined.
The transition state TS-3b is stabilized by a T-shaped CH/π interaction involving the two aromatic rings. Conversely, in TS-3a, an interaction between the two phenyls is substituted by a CH/π interaction between the Boc group and the pyrroline benzyl with a slightly lower stabilization.
The authors conclude that the use of an organic base avoided π interactions between the aryls of pyrroline and nitrile oxide, influencing the diastereoselection of the cycloaddition.
The first cycloaddition of nitrile oxide 39 to the graphene sheets 40 was described both theoretically and experimentally by Uceta et al. [36]. The authors performed DFT studies of the interaction between graphene 40 and nitrile oxide 39, highlighting the practicability of 1,3-dipolar cycloadditions leading to the isoxazoline ring (Scheme 15). Calculations were performed at the (U)M06-2X level and using the 6-31+G(d,p) basis set.
Scheme 15.

Cycloaddition of graphene 40 and nitrile oxide 39.
Modeling data showed that the DE between HOMO and LUMO is not high, and the reaction can occur. The reaction of graphene with 39 is a normal electron demand cycloaddition, involving the HOMO of nitrile oxide and the LUMO of graphene.
The aromatic ring of compound 39 can approach graphene in two ways, i.e., to the armchair side (r1) or at the zigzag side (r2). Moreover, the aromatic moiety can be placed ahead (c1), behind (c2), or above (c3) with respect the graphene sheet, giving six possible “approximations”.
The approach r2 is kinetically and thermodynamically preferred because of the CH–π stabilizing interaction between 39 and graphene 40. The preferred approaches pass through the same TS that presents the aromatic group parallel to the graphene, minimizing π–π interactions and leading to three adducts. The favorite ones were those with the lower steric interactions with graphene (r2c1 and r2c3). Nevertheless, data highlight that nitrile oxides are less reactive than nitrile imines, whose cycloaddition to the graphene sheets was previously reported in the literature [37].
The 1,3-dipolar rearrangement involving acetonitrile oxide 42 and (1S,2R,4S)-2-cyano-7-oxabicyclo[2.2.1]hept-5-en-2-yl acetate derivatives 43a–c was studied, and a BET (bonding evolution theory) analysis was conducted [38]. Optimizations were performed at the ωB97X-D/6-311G(d) level of calculations. For this reaction, four different reaction channels can be investigated. In fact, nitrile oxide 42 reacts with the oxanorbornenic compounds 43a–c through syn or anti-attacks and para and meta regioisomeric approaches of nitrile oxide on 7-oxanorbornenic derivatives (Scheme 16).
Scheme 16.

Different regioisomeric and diastereofacial routes of reaction between 42 and 43 a–c.
The para routes are preferred with respect to the meta ones because of the most favorable electrophile–nucleophile interaction involving the O1 of 1 and the C5 of 43a–c. This lowers the activation barriers along the para channels. Moreover, a syn attack presents TSs as more stable than those of the anti way. In fact, the syn route benefits from an interaction between the partial positive charge on the N atom of 42 and the partial negative charge on the bridge oxygen of oxanorbornenic derivative. In addition, a steric hindrance, due to the proximity of the methyl group of 42 and that of acetate, disfavors the anti attack. In conclusion, the syn isomers are preferred both from a kinetic and a thermodynamic point of view.
The final BET analysis on the syn reaction channel showed that the formation of the C−C bond occurs before the formation of the O−C one through a usual sharing model. The topological changes along the reaction pathway take place in a highly synchronous way.
In order to control hyperglycemia that causes type 2 diabetes, glycogen phosphorylase (GP) inhibitors were prepared [39]. Spiro-oxathiazoles were demonstrated to be potent GP inhibitors and were obtained through a one-step 1,3-dipolar cycloaddition between an aryl nitrile oxide and a glucono-thionolactone.
As reported in Scheme 17, hemiacetal 48 was treated firstly with CCl4 and HMPT and then with potassium O-ethyldithiocarbonate at −40 °C, leading to dithiocarbonate 49. The following methanolysis gives 50, which in turn reacts with tert-butyl-sulfinyl chloride, affording β- thiosulfinate 51 as a diastereoisomeric mixture.
Scheme 17.

Synthesis of 1S-glucopyranosylidene-spiro-oxathiazole 54.
The subsequent thermal elimination conducted in toluene under reflux gives thionolactone 53 and D-gluconolactone 52 as a by-product. D-glucono-δ-thionolactone 53 was a dipolarophile of the 1,3-dipolar cycloaddition, synthesizing 54 with excellent regio- and stereoselectivity. The authors found that the rate-determining step of the reaction was the step from 51 to 53. So, a DFT mechanistic study of the thermolysis was performed at the WB97XD2/6- 311G(d,p) level and considering a solvent (toluene) with the PCM method and SMD solvation model. Calculations evidenced that the thermal elimination step occurs, passing a five-membered transition state and involving the anomeric proton. This mechanism benefits from an anomeric effect and furnishes thionolactone 53 and tert-butyl sulfenic acid.
Holman et al. [40] obtained substituted isoxazoline exploiting an electrochemical method. This technique shows short reaction times and results to be green because they are characterized by minimal waste generation and the absence of toxic or expensive oxidizing reagents. The model reaction is reported in Scheme 18.
Scheme 18.

1,3-dipoalr cycloadditions leading to isoxazolines.
The chlorination of oxime 55 is electrochemically achieved by the oxidation of chloride anions to chlorinating electrophilic form. The in situ formation of nitrile oxide is followed by an electrochemically enabled regio- and diastereoselective reaction with electron-deficient alkenes, giving the desired products. For this reaction, both aromatic and alkyl aldoximes can be used.
The mechanism of the reaction was studied through DFT calculations at the M06-2X/Def2TZVP/SMD(MeCN) level. Two different pathways, reported in Scheme 19, were investigated: (A) stepwise radical-mediated and (B) a concerted [3 + 2] cycloaddition.
Scheme 19.

Different proposed mechanisms for the [3 + 2] cycloaddition between nitrile oxide and alkenes.
All modeling data are consistent with the A pathway (radical mechanism), whose barriers were 2.5 times lower in energy than those of the B pathway. Also, the determined regioselectivity and the formation of the 3,5-isoxazoline confirms the radical mechanism reported in Figure 8.
Figure 8.

Free energy profile of the calculated open-shell radical mechanism leading to isoxazolines 57.
The 1,3-dipolar cycloaddition of nitrile oxides with cyclodienes leading to cycloalkene-fused isoxazolines then converted into dialkenylated heterocycles through ring opening and cross-metathesis was studied at the M06-2X/6-311+G(d,p) level of theory (Scheme 20) [41].
Scheme 20.

The 1,3-dipolar cycloadditions of 42, 59 with cyclic dienes 60, 62, 64, 65–67.
Calculations allowed for locating the different TSs, giving the adduct with the O atom (named a) or the C-atom (named a′) of the isoxazoline closer to the C-sp2 of the cycloalkene ring. The determined relative activation free energies are in very good agreement with the product ratios experimentally obtained. In fact, in case of 60, the TS of route a is more stable at about 2.7 kcal/mol, and the datum is confirmed by the experimentally found regioselectivity. The diene 62 showed lower barriers and consequently higher reactivity. Moreover, the product from via a is kinetically favored, while that from via a’ is thermodynamically preferred. Conversely, the reactions with 64–67 present very high barriers, and also experimentally, products are not yielded. Using an activation strain model, the authors reveal that the distortion energies of the nitrile oxide determine the regioselectivity.
Table 1, reported below, summarizes the main findings (i.e., reagents, reaction conditions, catalysts, and level of calculations) of the papers focused on 1,3-DC involving 1,3-dipoles of propargyl-allenyl type.
Table 1.
Summary of the key findings of the reviewed studies involving 1,3-dipoles of propargyl-allenyl type (reagents, reaction conditions, catalysts, and computational methods).
| 1,3-Dipoles of Propargyl-Allenyl Type | |||
|---|---|---|---|
| Aza-Ylides | |||
| Reagents | Reaction Conditions | Catalyst | Level of Calculations |
| methylazide + propine | Δ, catalyst | Cu (I) | B3LYP/6-31G(d) and LANL2DZ for Cu |
| methylazide + 2-butine or cycloalkyne | Δ | / | M062X/6-311++G(d) |
| methyl azide and various allenes | Bu4NF/CsF, CH3CN | / | PB86/TZ2P |
| methyl azide and propyne | r.t., catalyst | Ag(I) | B3LYP/6-31G(d) and LANL2DZ for Ag |
| differently substituted azides + alkynes, cyanoalkynes, thioalkynes, and ynamides | Cp2Ni (10 mol %), Xantphos (10 mol %), Cs2CO3 (1 eqv), DCM, r.t., 24 h | Ni | M06/6-31G(d,p) and cc-pVTZ for Ni |
| lithiated trimethylsilyldiazomethane and α-azido ketones | LTMSD, −78 °C, 1h then r.t. | / | M06/6-31+G |
| arylazide + phenylacetylene | CuSO4·5H2O/5 Na+ L-Asc. 1 eq, DMF (1mL), t = 42 min, 25 °C, air | Cu | M06/6-311+G(d,p) SDD |
| benzylazide+terminal alkynes | 50 °C, catalyst, EtOH | Cu(I), Au4Cu4/CNT | PBE/ECP6MWB and def2-TZVPP for Cu, def2-SVP for other |
| 1,2-diboraallene+2,6-diisopropylphenyl azide | Toluene/n-hexane/r.t. | / | B3LYP/6-311G(d,p) |
| alkyne+(azidomethyl)benzene | (a) Cs2CO3, DMF, r.t.; (b) THF/TEA (1:1), 55–60 °C, CuBr(PPh3)3, 5.5 h, | Cu(I) | B3LYP/6-311++G(d,p) level and B3LYP/LANL2DZ for metals |
| 2-butylidenemalononitrile + phenylazide | DBU, DMSO-d6 at 50 °C | / | M06-2X/6-31+G(d,p) |
| azide + alkyne | 15 mol % catalyst, 45 °C, 30 min and water | CuC20H20N2)PPh3Cl and Cu(C20H20N2)PPh3Br | LDA/PWC |
| 1,2,3,4-tetrazole + phenylacetylene | Mn(TPP)Cl, Zn, C6H6, 100 °C, 24 h | manganese-copper/zinc | M06/6-311++G** for all other atoms and LANL2DZ for metals |
| phenylacetylene + phenyl azide | carbon nanotubes | / | QM/MM ONIOM B97D/UFFs |
| Nitrile Oxides | |||
| Reagents | Reaction conditions | Catalyst | Level of calculations |
| N-tert-Butoxycarbonyl [1-Chloro-1-(hydroxyimino)-3-phenylpro-pan-2-yl)]carbamate + N-benzyl-3-pyrroline | Base/r.t. | / | CPCMHCTC/6-11+G(d,p)//HCTH/6-31+G(d) |
| nitrile oxide + graphene sheets | MW | / | (U)M06-2X/6-31+G(d,p) |
| acetonitrile oxide +(1S,2R,4S)-2-cyano-7-oxabicyclo[2.2.1]hept-5-en-2-yl acetate derivatives | r.t. | / | ωB97X-D/6-311G(d) |
| oxime + t-but acrylate | Et4NCl/CH3CN | / | M06-2X/Def2TZVP |
| nitrile oxides + cyclodienes | MeCN, 25 °C or 70 °C | / | M06-2X/6-311+G(d,p) |
2.2. 1,3-Dipoles of Allyl Type
Allyl-type 1,3-dipoles are bent and contain only one double bond in a canonical form.
Some of these are here shown (V–VIII).
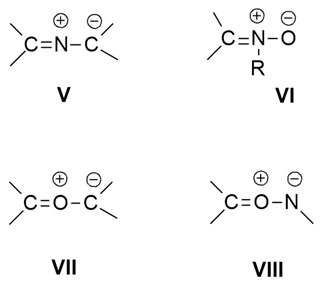
In the following sections, the cycloaddition reactions involving the azomethine ylides (V) and nitrones (VI) will be discussed.
2.2.1. Azomethyne Ylides
Azomethine ylides have been employed in many elegant applications for the preparation of a pyrrolidine nucleus and for the productions of natural products, drugs, and agrochemical compounds [42].
The organocatalytic reaction of ortho hydroxy imine 68, with nitro alkanes 69 in the presence of N,N′-bis[3,5-bis(trifluoromethyl)phenyl]-thiourea as catalyst, has been used to prepare a series of tetrasubstituted pyrrolidines 70 in high yields, high enantiomeric excess and excellent exo/endo selectivity (Scheme 21) [43]. The 1,3-dipolar cycloaddition process occurs because of the ortho hydroxy group present in phenyl ring of 68, that, via an Intramolecular hydrogen bond, increases the acidity of the proton in the α-position of the imine group and makes the formation of the azomethine ylide possible even if the starting imine contains only one activating group.
Scheme 21.

Organocatalytic synthesis of tetrasubstituted pyrrolidines.
A mechanistic study of the model reaction between 1-phenyl-2-nitroethene 69 with imine 68 and in presence of the catalyst reported in Scheme 21 was performed using DFT methods at the M06-2X/6-31g(d,p) level. Calculations reveal the ability of the hydroxy group to activate the dipolar cycloaddition reaction. The formation of two different hydrogen bonds was shown to be essential for reactivity, i.e., there was an intramolecular hydrogen bond of the azomethine ylide in equilibrium with the starting imine and an intermolecular one with the catalyst.
In detail, during the study, the authors considered for the reactants three different coordination points with the catalyst taking into account both the endo and exo approaches and considering different models (Takemoto, Papai and Zhong). In all the cases, the pre-association complex intermediates and the corresponding TSs were located. In Takemoto’s model, the nitroalkene is directly coordinated to the catalyst thiourea, while in Papai’s one, the same moiety is connected to the azomethine ylide. Finally, in Zhong’s model, an additional hydrogen bond of the aryl group of the catalyst with the azomethine ylide is detected (Figure 9).
Figure 9.

Pre-association complex intermediates for Takemoto, Papai and Zhong models.
Zhong’s model was shown to be the favorite one with a barrier of only 14.48 kcal/mol to pass from the intermediate to the product.
Selva et al. [44] successfully developed a two-step procedure leading to bicyclic pyrrolidines 75 in moderate to good yields (31–70%). They described as key intermediates non-stabilized azomethine ylides 73, obtained by the condensation of allyl ammine 71 with aldehydes 72 followed by the addition of maleimides 74, in toluene at 150 °C (Scheme 22).
Scheme 22.

Synthesis of bicyclic pyrrolidines 75.
The reaction occurs with high diasteroselection affording, as major adducts, the endo-2,5-trans derivatives 75 (Scheme 22). However, an inversion of diastereoselectivity was observed with 2-pyridinyl and 2-thienyl substituents, where the major products were the exo-2,5-trans adducts 76 (44–47% yield, respectively) (Scheme 23).
Scheme 23.

Synthesis of exo-2,5-trans adducts 76.
The above methodology was applied to the diastereoselective synthesis of tricyclic thrombin inhibitor 81 in 47% yield, starting from allyl amine 71, benzaldehyde 72 and N-(4-fluorobenzyl)maleimide 77 according to Scheme 24.
Scheme 24.

Synthesis of thrombin inhibitor 81.
In order to rationalize the observed diastereoselectivity, a complete DFT study was performed through optimizations, employing the B3LYP functional in conjunction with Grimme’s dispersion correction and the def2SVP basis set. The authors considered the different possible conformers of the ylide able to stabilize the negative charge at either the allylic or the benzylic position. The formation of the ylide was shown to be the rate-limiting step of the process.
Studying the different endo and exo approaches, the TSs were located and the S-ylide formed the fastest with respect to the most stable W-ylide (Scheme 25).
Scheme 25.

Located TSs for endo and exo routes, starting from S-ylide and W-ylide.
For the cycloaddition reaction, the endo route has lower barriers with respect to the exo one. Considering the benzaldehyde-derived ylides, a difference of 4.7 kcal/mol between the rate-limiting step and the isomerization barrier between the S-ylide and the W-ylide was detected, determining an equilibrium between both conformers. Consequently, endo-2,5-trans and endo-2,5-cis are both obtained.
In the case of 2-formylpyridine ylides, the barriers of the S-ylide formation, that is the rate-limiting step, and S-ylide isomerization to W-ylide are competitive with a difference of only 1.4 kcal/mol. Consequently, the isomerization is negligible, and the obtained products (endo-2,5-trans and exo-2,5-trans) are both derived from S-ylide.
The enantioselective 1,3-dipolar cycloaddition of azomethine ylide 83, produced by iminoester 82, with acrylonitriles or methacrylonitrile 83 in the presence of Cu(CH3CN)4BF4 as a catalyst and the chiral phosphine−urea bifunctional ligand (L) has been achieved, leading to a series of highly substituted chiral cyano pyrrolidines 84 in high yields and with excellent diastereo- and enantioslectivity (up to 99:1 d.r., 99% ee) (Scheme 26). Using this protocol, the antitumor (S,S,S,S)-ETP69 was successfully synthesized with a 38% yield and with an enantiomeric purity greater than 99% using as the key compound the pyrrolidine 85 [45].
Scheme 26.

Synthesis of antitumor compound (S,S,S,S)-ETP69.
In order to investigate the enantio- and diastereoselectivity of this asymmetric cycloaddition, calculations were performed using DFT methods at the M11/6–311+G(d,p) level and the SDD basis set for Cu atoms. The solvent effects were determined through single-point calculations with the SMD solvation model, considering diethyl ether as a solvent.
The free energy profiles for an asymmetric 1,3-dipolar cycloaddition of azomethine ylides using the synthesized phosphine–urea bifunctional ligand L are considered.
Firstly, iminoester coordinates to Cu, giving 86. At this point, there is a nucleophilic addition of the α-carbon atom of the iminoester to the terminal carbon of acrylonitrile 83. The subsequent cycloaddition rapidly gives the 91-SSS. The nucleophilic addition is the enantioselectivity-determining step. Diastereoselectivity is determined by the lower barrier, leading to 91-SSS with respect to the other stereoisomers. In fact, the corresponding transition state is stabilized by the formation of two different hydrogen bonds of the cyano group with the urea moiety (Figure 10). The authors finally concluded that both metal catalysis and organocatalysis are important for this reaction. Moreover, calculations reveal that the distortion energy has a determinant role in the enantioselectivity trend because of the steric effect between the phosphine ligand and the dipole.
Figure 10.

Energy profile leading to 91-SSS.
The asymmetric 1,3-dipolar cycloaddition using glycine imino ester 92, beta-fluoroalkyl alkenyl arylsulfones (-CF3, -CF2CF3, -CHF2, -CF2Cl, and CH2F) 93 and the (S)-tol-BINAP-Cu(II) complex, has been accomplished to synthesize the tetrasubstituted pyrrolidine carboxylates 94 with the simultaneous creation of four adjacent stereogenic centers [46] (Scheme 27).
Scheme 27.

Synthesis of tetrasubstituted pyrrolidine carboxylates 94.
The preferred diastereo- and enantioselectivity were rationalized through calculations at the M06-L/def2-TZVPP level. The mechanistic course of the reaction shown in Figure 11 is characterized by a stepwise nucleophilic addition in a head–head and/or tail–tail manner.
Figure 11.

Catalytic cycle leading to compound exo-94.
Firstly, the coordination of 92b to the (S)-tol-BINAP-CuII complex allows deprotonation, producing the azomethine ylide I. The subsequent enantioselective Michael addition to the C=C double bond of the imonoester formed the zwitterionic intermediate III-exo. Finally, an intramolecular Mannich-type addition of III-exo to the iminium moiety on the Si-face and a proton transfer give compound exo-94.
A stereo- and regioselective synthesis of cyclopropa[a]pyrrolizine derivatives, from moderate to excellent yields (33–95%), has been accomplished by Filatov et al. according to the reaction of azomethine ylide 98, obtained from ninhydrin 96 and L-Prolin 97, with different 1,2 substituted 3,3-disubstituted and 1,2,3-trisubstitued cycloprepenes (Scheme 28) [47]. Global electrophilicity indexes, the natural bond orbital (NBO) charges, FMO coefficients, Fukui function and free-energy profiles have been used to explain the 1,5-regio- and the endo- stereoselectivity of the cycloaddition process.
Scheme 28.

Stable azomethine ylide 98 from ninhydrin 96 and L-prolin 97.
Optimizations were carried out with DFT/HF methods using the M11 hybrid exchange-correlation functional and cc-pVDZ basis set. Solvent (THF) was taken into consideration with the polarizable continuum model (PCM). The regioselectivity was investigated considering the two possible 1,4-endo and 1,5-endo routes. The cycloaddition has a one-step mechanism, and the 1,5-pathway is kinetically favored, leading to the 1,5-regioisomers as preferred (Figure 12).
Figure 12.

Free energy barriers leading to possible 1,4-endo and 1,5-endo compounds.
The dipolaroid-like nature of the TSs suggests that this regioselective process is asynchronous and controlled by azomethine ylide 98. Finally, the authors rationalize the stereoselection of the cycloaddition, considering both the endo and exo route and showing the lower barriers of the endo-via. The endo TSs are stabilized by an interaction between the N atom of 98 and syn-H atom of Csp3 of cyclopropene. In fact, the s-orbital in the HOMO of cyclopropene interacts with the nitrogen π-orbital of the ylide LUMO.
Caleffi et al. [48] have found that the L1-CuOTf·PhMe complex is able to efficiently promote an enantioselective 1,3-dipolar cycloaddition between imino esters 99 and electron-deficient alkenes 100, leading to exo adducts 101 as major compounds (Scheme 29).
Scheme 29.

Enantioselective 1,3-dipolar cycloaddition using L1-CuOTf·PhMe complex and leading to exo adducts.
On the contrary, employing L2-AgSbF6-complex,a, a diastereo-divergent process occurs with the formation of endo-cycloadducts as major cycloadducts (Scheme 30).
Scheme 30.

Enantioselective 1,3-dipolar cycloaddition using L2- AgSbF6-complex and leading to endo adducts.
These different results have been explained with the aim of DFT calculations at the B3LYP/6-31G(d) level and PCM method with toluene as the solvent.
The possible Cu(I) and Ag(I) complexes were studied. Because of the chelating character of the L1 and L2 chiral ligands, Cu and Ag atoms have a distorted tetrahedral coordination with the P atoms of the ligand and the N and O atoms of the iminoester. When dipolarophile attends the dipole, in the case of INT1, the coordination does not change. However, INT1′ presents the replacement of one of the metal-P bonding interactions with a metal–oxygen interaction.
Considering INT1, the coordination sphere of the metal is complete. Thus, the distal disposition between the cyano or carboxamido group with respect to the ester of the dipole determined the exo selectivity. On the contrary, the formation of INT1′ and metal-O/metal-N bonding interaction is the driving force toward the obtainment of endo-products (Figure 13).
Figure 13.

Catalytic cycle involving Cu(I) and Ag(I) complexes (A:M = Cu, X = CO2Me; B:M = Cu, X = CONMe2; C:M = Ag, X = CN; D:M = Ag, X = CO2Me).
The reaction of the azomethine ylide 106 produced in situ by N-ethylglycine (50 equiv) and paraformaldehyde (400 equiv) at 120 °C for 15 min with Gd3N@Ih-C80 (Scheme 31) furnishes two ethyl pyrrolidino adducts 107 and 108 in a regioselective manner [49].
Scheme 31.

Synthesis of ethyl pyrrolidino adducts 107 and 108 using Gd3N@Ih-C80.
The authors tried to elucidate the structures of the two cycloadducts using different techniques. X-ray diffraction study allowed for understanding that minor-bis-108 has a C2-symmetric [6,6][6,6]-geometry on bond 53–54. It is noteworthy that the Gd3N cluster is strictly planar and much less strained with respect to pristine Gd3N@C80, which presents the N atom out of the plane. The evaluation of the stability of adduct minor-bis-2 under thermal conditions highlighted its isomerization to kinetic major-bis-1, perhaps through rearrangement. The inverse thermal conversion (from 107 to 108) was not observed. The structure of 107 was determined through DFT calculations and optimizations at the BP86-D2/TZP level of theory. The major-bis-adduct-107 has an asymmetric [6,6][6,6]-geometry with a second addition site on bond 57–58. The trimetallic nitride template moiety is planar only in minor-108, while in the other case, the pyramidalization was detected. Finally, the isomerization from minor-108 to major-107 was studied. Calculations revealed that the reaction proceeds through a thermodynamically favored way that involves the formation of the [6,6][6,6]-bis-adduct on 54–52 (Figure 14). Therefore, the isomerization observed in this study is a [6,6] to [6,6] process.
Figure 14.
Isomerization pathway from minor 108 to major 107.
Fulleropyrrolidines 2R and 2S containing a stereocenter at carbon 2 of pyrrolidine ring, as potential HIV-1 protease inhibitors, have been, recently, synthesized by Alonso et al. [50]. These compounds have been accomplished in a diastereoselective fashion (ratio 111:112 5:1) by the 1,3-dipolar cycloaddition reaction of the azomethine ylide 110, produced by the enantiopure formyl steroid 109 and N-methylglycine, to [60]fullerene, according to Prato conditions (Scheme 32).
Scheme 32.

Reaction conditions: C60, N-methylglycine, toluene, reflux, Ar.
Firstly, using a combination of semiempirical and DFT methods, a conformational study was performed. In particular, the 1,3-dipole preferred the S-trans conformation. The evaluation of the nucleophilic character of reacting carbons of the dipole excludes that it influences the diastereoisomeric ratio. Optimizations at the B3LYP/6-31+G(d,p)/C-PCM = toluene//OLYP/6-31G(d):PM6 level allowed locating TSs, leading to the final cycloadducts. A two-step mechanism was detected with the first one established as rate-determining (Figure 15).
Figure 15.

Energy profile of the 1,3-dipolar cycloaddition.
The computed ratio is in agreement with the experimental one (76:24 vs. 68:13). In TS1_112, the dipole is distorted with respect to the initial geometry in comparison with TS1_111. This effect is due to the steric hindrance determined by the methyl at C20, which influences diastereoselection. In conclusion, the attack of [60]fullerene on the Re face of the 1,3-dipole in the S-trans conformation is responsible for the final regioselectivity. Moreover, compound 111 is also thermodynamically favored.
Following a previous paper [51], Rivilla et al. have designed and synthesized consensus tetratricopeptide repeat proteins (CTPRs) as biocatalysts and used them to form nitroproline esters [52]. This process involves the formation of the azomethine ylide intermediate 113, obtained from starting N-Benzylideneglycinate via enolization, promoted by CTPRs, and a subsequent reaction with nitrostyrene 114. In particular, wild-type CTPR1a and CTPR3a catalyze the formation of the four pyrrolidine stereoisomers 115–116 (Scheme 33) with 40% yield in an equimolar ratio, while CTPR1_wt and CTPR3_wt catalyze the formation of only 115-exo and 115-endo with 20% yield in a 1:1 ratio. The formation of the four pyrrolidine isomers is rationalized to the in situ formation of azomethine ylides 113a and 113b.
Scheme 33.

Synthesis of nitroproline esters using consensus tetratricopeptide repeat proteins (CTPRs) as biocatalysts.
Starting imine 109 has two possible conformations in equilibrium through the rotation around the N−Cα bond: i.e., the homo-s-trans and homo-s-cis geometries that give dipoles 113a or 113b, respectively. This occurs through the proton transfer/abstraction of one Cα-H bond due to acid/base catalysis involving independent or connected active sites of CTPR proteins.
The mechanism leading to endo- and exo-115a adducts is concerted but asynchronous, while endo’ and exo’ were derived from a double suprafacial reaction. Also, the different stability and flexibility between CTPR and CTPRa series proteins can influence the course of the reaction.
The authors performed QM/MM calculations, using CTPR1a and the ONIOM method at the M06-2X/6-311+G(2d,2p):dreiding//B3LYP/6-31G-(d,p):dreiding level. The high level includes imine 109, nitroalkene 114, and five residues (E2, K13, Y23, Y24, Q25), while the rest of the protein is treated at low level of calculations.
Firstly, imine (homo-s-trans) and the dyad (E22/K26) form a complex Ca showing a H-bond involving the charged amino group of K26 and the carboxylate of E22 (Figure 16). Moreover, in 109, the carbonyl O atoms coordinated to the protonated N atom of the amino moiety of K26, and a H atom of the methylene weakly interacts with the carboxylate moiety of the E22. Passing TS1A (about 13 kcal/mol), 109 is converted into enolate (IntA). Easily, without any significant barrier, enol E is obtained. From E, a prototropy reaction furnishes 113a. The second way (B), leading to 113b, involved 109 in the homo-s-cis geometry that, interacting with CTPR1a, gives the complex Cb, which is less stable than Ca by 4.4 kcal/mol. Starting from Cb, iminium cation INTB is obtained by the protonation of 109 by an ammonium group of K26. This step is endoergoic. Finally, passing TS2B, azomethine ylide 113b is formed.
Figure 16.

Energy profile leading to 113a (A) and 113b (B) starting from imine 1a in the homo-s-trans geometry and near the dyad E22/ K26 of CTPR1a.
The two reaction profiles are almost isoenergetic, and so the formation of the two possible azomethine ylides 113a and 113b is competitive.
Finally, the cycloaddition between 115a and b with dipolarophile 114 is considered (Figure 17). Starting both from 113a or 113b, the formations of endo or exo compounds are in competition (TS3 vs. TS4 = 3.5 vs. 6.8 kcal/mol; TS5 vs. TS6 = 5.2 vs. 10.3 kcal/mol) and so a mixture of cycloadducts is obtained, although the endo products are favored. The reinitiating of the cycle is guaranteed by the recovery of the catalytic couple E22/K26. This is allowed by the weakening, in the cycloadducts, of two interactions (i.e., the one between the NH group of the cycloadduct and the N atom of the K26 residue, and the H bond present in turn between the N atom of K26 and the neutral carboxy group of E22).
Figure 17.

Energy profile of the formation of adducts endo/exo-115 (A) and endo′/exo′-115 (B) starting from 113a and 113b, respectively.
Yamazaki et al. [53] have recently reported the synthesis of various substituted pyrrolidines, in regio- and diastereoselective fashion, by the 1,3-dipolar cycloaddition of azomethine ylides, under mild condition, using iridium complex as the catalyst. The synthesis occurs in one step, involving N-benzoylprolinemethylester 116, chlorodicarbonylbis(triphenylphosphine)iridium(I) complex and 1,1,3,3-tetramethyldisiloxane. As described in Scheme 34, the azomethine ylides 118 involved in the process is obtained through a partial reduction of the amide group by Vaska’s complex and tetramethyldisiloxane, which is followed by the silanoate elimination of 117. Finally, the dipole reacts with methyl cinnamate 119 or N-enoyl oxazolidinone 120 as dipolarophiles to afford the corresponding pyrrolidine derivatives 121 and 122, respectively, in 64% yields.
Scheme 34.

Synthesis of compounds 121 and 122.
In order to clarify the different selectivity observed for the two reactions, DFT calculations were performed at the BP86/TZ2P level.
The diastereoselectivity is influenced by strain factors or interaction energy in the case of methyl cinnamate 119 or oxazolidinone 120, respectively. In both reactions, four TSs can be located, but the lower barrier was shown to be different: TS1 for methyl cinnamate and TS2 for N-enoyl oxazolidinone (TS2) (Figure 18). Using the activation strain model (ASM), the ΔE≠ was distinguished into two contributions: the strain energy or ΔE≠strain, determined by deformation of reactants, and the interaction energy (ΔE≠int) between deformed reagents. When methyl cinnamate is the dipolarophile, the selectivity of the favorite TS is ruled by ΔE≠strain, while in the TS preferred for N-enoyl oxazolidinone is ΔE≠int.
Figure 18.

Three-dimensional (3D) plots of the preferred TSs when dipolarophile is methyl cinnamate (TS1) or N-enoyl oxazolidinone (TS2).
The same author, in a previous paper [54], correlated the degree of asynchronicity of Diels–Alder reactions with strain energy value. In general, an asynchronous TS, in which the pyramidalization of the C atoms is minimized, shows a less destabilizing Estrain; this is the case of TS1. When an oxazolidinone is part of a dipolarophile, the Eint value prevails on Estrain as in TS2. Moreover, the molecular electrostatic potential analysis (MEP) highlighted that TS2 has the forming C−C bond.
Cu(I) complexed with a monodentate, triple-homoaxial chiral, phosphoramidite ligand (L) was employed by Chang et al. [55] in the synthesis of enantioenriched pyrrolidines 125 and 126, obtained by the 1,3-dipolar cycloaddition of azomethine ylides, produced by 123, with heteroaryl alkenyl derivatives 124 not possessing strong electron-withdrawing substituents (Scheme 35).
Scheme 35.

Synthesis of pyrrolidines 125 and 126.
The exclusive diastereoselectivity and the excellent enantioselectivity are explained by the uncommon ability of the complex Cu(I)-L to activate both the dipole and the dipolarophile involved.
The stereochemical outcome of the reaction leading to 126b (Figure 19) was clarified through DFT calculations using the M06-L method and the SDD basis set for Cu and 6-31G(d) for the other atoms. Two different regioselectivity routes are possible as reported in Figure 19.
Figure 19.

Regioisomeric routes for the synthesis of enantioenriched pyrrolidines 126.
The Cu(I)-L complex prefers the coordination mode in which the 4-Cl-C6H4 group does not suffer steric repulsion with the amido moiety.
Mechanistic investigation highlighted that the reaction occurs in two steps. Passing a first barrier (TS1), a zwitterionic intermediate (INT2) is obtained with the formation of the C1−C3 bond between the ylide and the alkene. The second step regards the formation of a new C−C bond determining cyclization. The cycloaddition is the rate-determining step of the process (Figure 20). The endo_126b, obtained by the attack from the upper side of the ylide, is the main product. The formation of its enantiomer 126b_ent is disfavored by the nonbonding interactions, present in TS2, between H4 of naphthol and benzo[d]oxazole moiety. All the other considered ways present higher barriers, and the formation of the other products is excluded. The completion of the catalytic cycle with the regeneration of L occurs thanks to the exchange between the product and another molecule of azomethine ylide. Computational data are in very good agreement with experimental results, showing that the proposed phosphoramidite ligand (rigid, sterically bulky and with a triple-axial chirality) is very suitable in providing a chiral environment around the cycloaddition.
Figure 20.

Energy profile of the reaction leading to 126b (black dotted line) or 126b_ent (red dotted line).
The azomethine ylides 129, obtained from the tetracyclic ketone 11H-benzo[4,5]imidazo[1,2-a]indol-11-one 127 and L-proline 128, have been utilized by Filatov et al. [56] to synthesize several spiro-heterocyclic compounds when they react with cyclopropenes 130 or maleimide 131, which are differently substituted (Scheme 36).
Scheme 36.

Synthesis of spiro heterocyclic compounds 132 and 133.
With the support of DFT calculations, performed at the M11/cc-pVDZ level using the PCM method to treat the solvent (1,4-dioxane), the mechanism was investigated, considering the azomethine ylide 129 that reacts with 1,2-diphenyl-3-vinylcyclopropene.
Compound 127 and L-proline 128 give azomethine ylide 129 in two possible conformations: S-shaped (129_A) and W-shaped (129_B). Their precursors are diastereomeric oxazolidin-5-ones, INT1_A and INT1_B, which are obtained by the condensation of 127 and 128. Then, the 1,3-dipolar cycloreversion, leading to 1,3-dipoles (129_A, 129_B), occurs in two steps passing through endo TSs. The first one is the rate-determining step, and the barriers of the two routes highlighted that AY-1 and AY-2 are obtained in an equimolar ratio.
The endo pathway, starting from ylide 129_A, is preferred with respect to the endo route from 129_B by about 0.9 kcal/mol. Cycloadduct 132 is thermodynamically favored, and it is present in greater quantities in the final mixture (Scheme 37).
Scheme 37.

Mechanistic route leading to compound 132.
Chiral unnatural amino acids 138 in enantiomeric pure form, characterized by the presence of 3-spiropyrrolidine oxindole skeletal, have been obtained in moderate to good yield (40–86%) and with high diasteroeselectivity (>20:1), by a three-component reaction, involving as a key intermediate the azomethine ylides 136 [57]. Thus, the reaction of various amino acids 134, with different isatins 135 and Belokon’s chiral Ni(II) complex A, at 50 °C for 24 h in ethanol, furnishes after a usual work-up of the desired cycloadducts 138 via the initial cycloadducts 137 (Scheme 38).
Scheme 38.

Reaction of amino acids 134 with isatins 135 and Belokon’s chiral Ni(II) complex leading to cycloadducts 138.
The regioselectivity of the reaction involving (S)-proline and sarcosine was investigated through DFT calculations at PBE0-D3BJ/def2SVP/CPCM(EtOH) level. The selected functional (PBE0) proved to be accurate for studying the organic reactions. Considering the CN bond, the obtained azomethine ylide has two possible geometries: Z or E. The approach to the Ni complex can differently occur: (i) in a tail-to-tail (tt) way, directing the bulkier moieties toward each other, or in a tail-to-head manner (th) with the same groups in the opposite direction; (ii) attacking the alkene C atom of the complex from the top and bottom side; (iii) determining the configuration of C3 of isatin.
The mechanistic investigation locates 16 different TSs for Pro and sarcosine.
In detail, the (S)-1 Ni-complex forms with azomethine ylide a π-complex (PI) with the groups of the N atom of ylide with endo orientation (Figure 21). Starting from this complex, the pericyclic reaction occurs, passing a transition state (TS) characterized by a planar configuration of ylide. In both cases, ttZSR_TS, leading to adduct (P) with ttZSR configuration, was shown to be preferred. Moreover, from the product, the N inversion is possible. Experimental results confirmed calculations for sarcosine, while when Pro is an aminoacid, a cycloadduct with the thZSR configuration is achieved. In the case of sarcosine, the reaction is under kinetic control and a ttZSR-adduct is formed. The retro-cycloaddition has a too high barrier to happen. Instead, in the case of proline, the retro-reaction is possible as experimentally verified. Calculations also showed that passing a barrier of 28.7 kcal/mol, the rearrangement from ttZSR to thZSR starting from product iP can occur at room temperature. The thZSR-iP adduct is thermodynamically preferred over the possible regioisomeric P and iP products. So, in this last case, the outcome of the reaction is influenced by both thermodynamic and kinetic control.
Figure 21.

Located structures for the preferred way (ttZSR) involving sarcosine.
Radwan et al. [58] have studied the metal-free 1,3-dipolar cycloaddition of azomethine ylide 141, derived from 3-formylchromone 140, with glycine ester 139, and arylidenes 142a–e or phenylmaleimide 145 (NPM) in the presence of AcONa in toluene at room temperature. The cycloaddition process, in the case of arylidenes 142a–e as dipolarophiles, leads to a mixture of endo (143a–e) and exo’ (144a–e) adducts in excellent yield (92–88%) and with an endo product as the major diastereomer. On the contrary, using NPM 145 as the dipolarophile, the endo cycloadduct 146 was the only obtained product (86% yield) (Scheme 39).
Scheme 39.

The 1,3-dipolar cycloaddition of azomethine ylide 141 with glycine ester 139 leading to endo adduct.
Interestingly, when the reaction of 139, 140 and 142a–e was conducted at reflux temperature (110 °C), only cycloadducts 144a–e were obtained. These results can be easily rationalized considering that the kinetic cycloadducts 143a–e at high temperature are transformed, via the in situ retro-1,3-dipolar cycloaddition and recyclization, in the thermodynamically exo’ derivatives 144a–e (Scheme 40).
Scheme 40.

The 1,3-dipolar cycloaddition of azomethine ylide 141 with glycine ester 139 leading to exo adduct.
The mechanism of the reaction was investigated using DFT calculations in order to clarify the role of AcO2H in this cycloaddition reaction. Optimizations were performed at the wB97xd/6-31G(d,p) level of theory.
The first studied step was the formation of the syn-/anti- dipoles, focusing attention on acetic acid.
In the first possible pathway, starting from (E)-imine (blue dotted lines in Figure 22), the protonation of the N atom, determined by AcOH, occurs in a concerted way with respect to the deprotonation of the α-hydrogen atom of the imine (TS1). Instead, the second possible route (black dotted lines) is characterized by a two-step mechanism with AcOH addition/elimination (TS2 and TS3).
Figure 22.

Energy profile of the 1,3-DC of azomethine ylide 141 with glycine ester 139 leading to endo and exo adducts.
The H-bonds in TS1 allow a more stable trifurcated structure with the obtainment of the syn-dipole preferred from a kinetic point of view. Transition state TS4, characterized by strong H-bonds that give a bifurcated structure and lower steric hindrance, is the favorite way to achieve the anti-dipole.
Then, the mechanistic study was extended to the [3 + 2] cycloaddition with dipolarophile 142a: starting from a dipole in syn or anti conformation, passing two low barriers TS7 and TS8, and leading to endo- and exo’-cycloadducts 143a and 144a, respectively. These data are in agreement with experimental results that show the endo-adduct, kinetically favored, as the major product.
The barriers highlighted the possibility of retro-cycloaddition at room temperature starting from the endo 143a followed by the isomerization of a syn-dipole into an anti-dipole. Instead, exo’-adduct 144a is thermodynamically preferred. In general, IRC analysis showed for the process an asynchronous concerted mechanism.
Finally, the computational study proceeded considering the [3 + 2] cycloaddition of glycine imino ester 139 with NPM (N-phenylmaleimide) in the presence or not of the AgOAc catalyst. Calculations were carried out at the same level as above, and the Lanl2dz basis set was used for the Ag atom. This reaction is experimentally fast and, without metal, gives a reaction that is not reversible, leading to highly stable cycloadduct 146. On the contrary, in the presence of Ag as the catalyst, a mixture of decomposition products is obtained (see Scheme 41).
Scheme 41.

Sansano reaction [59].
The barriers for the formation of the syn-dipole and following the reaction with NPM without a metal catalyst are analogous to those above described. The preferred cycloadduct endo-143a is more stable, and so the reaction is fast and irreversible.
The introduction of the catalyst AgOAc shows low barriers leading to Ag-azomethine ylide (7.4 kcal/mol) and cycloadduct (4.9 kcal/mol). However, the complexation of Ag with the starting imine is more stable than the complexation of metal with a final cycloadduct, rationalizing the experimental obtainment of decomposition products.
2.2.2. Nitrones
The cycloaddition of nitrones to alkenes represents a fast and elegant way to prepare isoxazolidines, which are very important compounds/intermediates in organic chemistry [60]. These cycloadducts have found wide applications as synthons in total synthesis by their conversion into 1,3-aminoalcohols and alkaloids. Furthermore, the isoxazolidine system is a mimetic of the ribose unit, and it has recently been exploited in the synthesis of modified nucleosides having antiviral and antitumoral activity [61,62].
Fused bicyclic tetrahydroisoxazoles, without the use of catalysts or additives, were synthesized by Yao et al. [63] by the 1,3-dipolar cycloaddition of nitrones 149 with oxa(aza)bicyclic alkenes 150 (Scheme 42). Final compounds were obtained, as a diastereoisomeric mixture, in very good yields (76–99%). This reaction is an important example of efficient [3 + 2] cycloaddition, conducted in mild conditions, to obtain highly functionalized compounds (Scheme 42).
Scheme 42.

Synthesis of fused bicyclic tetrahydroisoxazoles.
The mechanism of the reaction, reported in Scheme 43, was studied through optimizations at the B3LYP/6-311G(d,p) level and using the PCM solvation model and toluene as the solvent.
Scheme 43.

Endo and exo pathways of the 1,3-DC leading to fused bicyclic tetrahydroisoxazoles.
The endo and exo routes, that give 153 and 154 and 151 and 152, respectively, were taken into consideration. All the possible TSs were located. The endo pathway is disfavored, and the barriers are very close to 50 kcal/mol. Considering the exo pathway, with a lower barrier that is close to 30 kcal/mol, the obtainment of 151 is preferred over 152 by about 3 kcal/mol, which is in very good agreement with the experimental determined diastereoselectivity.
The 1,3-dipolar cycloaddition reactions of N-methyl-C-ethoxycarbonylnitrone 155 with allyl-heptaisobutyl-POSS 156b, under microwave irradiation, produce two cycloadducts: 157a and 158a with a high control of regio- and stereoselectivity in a 5.7:1 ratio, respectively. Differently, the reaction of 155 with 156a leads also to the formation of adduct 159 with inversion of the regioselectivity, although in moderate yield (10%), but high stereochemical control (only trans adduct) (Scheme 44) [64].
Scheme 44.

The 1,3-dipolar cycloaddition reactions of N-methyl-C-ethoxycarbonylnitrone 155 with allyl-heptaisobutyl-POSS 156.
The experimental results, in particular regio- and stereoselectivity, were rationalized by DFT calculations at the B3LYP/6-31G(d) level, investigating the reaction of POSS-substituted olefin 156a,b with nitrone 155. The different TSs, deriving from 155 in the E or Z configuration, and the exo or endo approach of the dipolarophiles were considered with the POSS and -CO2Et moiety in cis or trans configuration.
Figure 23 contains the 3D plots of the TSs leading to 157a and 158a. The favorite TS 157a_E/Z furnished trans adduct 157a with 95% yield, while TS 158a_E/Z barriers are higher in energy and gave a 3.1% yield of cis derivative 158a. The barrier relative to the formation of 159 is higher (>30 kcal/mol), and only 2% yield of this adduct is obtained. The formation of the new C–C and C–O bonds occurs through a concerted but slightly asynchronous approach.
Figure 23.

TSs of reaction between 155 and 156a leading to stereoisomers 157a and 158a.
The 1,3-dipolar cycloaddition of 6-alkylidenepenicillanates 160, in toluene at 80 °C for 24 h, with nitrones 161 was employed by Alves et al. to synthesize different chiral spiroisoxazolidine-b-lactams 162 (major) and 163 (minor) (Scheme 45) [65].
Scheme 45.

Synthesis of chiral spiro-b-lactams from 6-alkylidenepenicillanates.
The reaction proved to be stereo- and regioselective, giving rise to 162 as the main adduct with a yield varying between 26 and 70%.
A complete conformational study of the spiroisoxazolidine-β-lactams 162 and 163 was carried out at the B3LYP/6-31G(d) level. The endo compound 162 was shown to be more stable than the exo one 163 with a butterfly-like geometry characterized by an open shape conformation.
Other DFT studies were performed at the M062X/6-311G(d,p) level on the cycloaddition reaction of C,N-disubstituted nitrones 163 with disubstituted 4-methylene-1,3-oxazol-5(4H)-one 164. The authors highlight that the dipole, contrary to the mechanism reported by Mekheimer et al. [66], adds chemo selectively to the olefinic bond, forming the corresponding spiro cycloadduct 165, rather than to the methylene carbon and the carbonyl oxygen in a [4 + 3] cycloaddition, leading to 166. Furthermore, it was found that the presence of electron-withdrawing groups in the dipole reduces the energy barriers, while it is increased in the presence of electron-donating groups [67] (Scheme 46).
Scheme 46.

Synthesis of compound 166.
The lack of regioselectivity in 1,3-DC of methylenecyclopropane 170, compared to the high regioselectivity of other 1,1-disubstituted alkenes, has been computationally investigated through DFT calculations, to rationalize the experimental finding [68].
Considering the cycloaddition involving nitrone 167 and isobutene 168 (Scheme 47), only the 5,5-dimethyl-substituted isoxazolidine 169 is experimentally obtained with the two methyls on position 5. The TS leading to this cycloadduct is lower in energy with respect to that leading to the other possible regioisomer (the 4,4-dimethyl-substituted isoxazolidine). The calculated kinetic ratio is rationalized through the charge distribution and the orientation of the dipole moment of alkene.
Scheme 47.

Computed 1,3-dipolar cycloadditions involving different 1,1-disubstituted alkenes.
The reaction with methylenecyclopropane 170, experimentally determined, leads to the formation of a regioisomeric mixture of 171 and 172 in a 65:35 ratio, respectively, with the 5-spiro-fused isoxazolidine 171 as the major product. Instead, the reaction carried out with 173 provides only the methylenecyclobutane 174. The calculated barriers gave explanation to these results. For 173, μ is oriented toward the cyclopropane. The two alkenes (170 and 173) present a different reactivity determined by electrostatic and kinetic aspects. In the reaction involving 170, a lower EDG effect of the substituents is detected, and this results in a lack of the regioselectivity. Whereas, in the case of 173, the charge distribution of the alkene leads to furnishing the 5-spiro-fused product 174.
The study was also extended to isopropylidenecyclopropane 175 and isopropylidenecyclobutane 177. The 1,3-dipolar cycloaddition between 175 and nitrone 167 led only to the 5,5-dimethylisoxazolidine 176, in which the barrier of the other orientation was the highest. Instead, the difference of the regioisomeric barriers of the reaction between 177 and nitrone is much lower with 5,5-dimethylisoxazolidine 178, which is slightly kinetically favored. Finally, the reaction of nitrones with cyclobutylidenecyclopropane 179 was studied. Experimentally, only cycloadduct 180 bearing cyclopropane on C4 was obtained. Calculations showed that this product is both kinetically and thermodynamically favored.
In 2020, the 1,3-DC of adamantine aldo and ketonitrones with maleimides leading to isoxazolidines appeared in the literature [69]. From an experimental point of view, the reaction was conducted in toluene at 110 °C. In the case of 185, a diastereomeric mixture was obtained, while in the case of 181, only one isomer is achieved (Scheme 48). However, the authors did not specify whether this isomer was the exo or endo one and if its formation was due to thermodynamic or kinetic factors.
Scheme 48.

Synthesis of isoxazolidines through 1,3-DC of adamantine aldo and ketonitrones.
Considering the importance of adamantane and isoxazolidine moieties in the field of medicinal chemistry, the stereo selective reaction of keto- 181 and aldonitrone 185 containing an adamantine moiety with maleimides 182 was computationally investigated by Umar et al. [70] using DFT methods at the M06-2X/6-311++G(d,p) level and the polarizable continuum solvent (PCM) model. Both reactions proceed in a concerted, pericyclic manner. In detail, the reaction of 181 with maleimide 182 proceeds through two TSs, leading to the endo- or exo-stereoisomer with activation barrier values of 2.8 and 4.5 kcal/mol at room temperature, respectively (Figure 24).
Figure 24.

Free energy profile of reaction of 181 with maleimide in experimental conditions (T = 110 °C).
Calculations were repeated at experimental conditions and in particular considering T = 110 °C. The endo adduct is thermodynamically preferred, while the two TSs are very close in energy. Since only an isomer was experimentally detected, the less stable exo one is probably converted into the endo one.
The reaction of aldonitrone 185 with maleimide leads to trans and cis-stereoisomers with activation barriers of 10.3 and 8.0 kcal/mol, respectively (Figure 25). The reaction is exergonic, and from the energetic point of view, the stereoisomers are thermodynamically similar and kinetically different. The reaction is under kinetic control, and the cis adduct is preferentially obtained.
Figure 25.

Free energy profile of reaction of 185 with maleimide in experimental conditions (T = 110 °C).
Table 2 reports and summarizes the key findings of the reviewed studies involving 1,3-dipoles of allyl type.
Table 2.
Summary of the key findings of the reviewed studies of azomethyne ylides and nitrones (reagents, reaction conditions, catalysts, and level of calculations).
| 1,3-Dipoles of Allyl Type | |||
|---|---|---|---|
| Azomethyne Ylides | |||
| Reagents | Reaction Conditions | Catalyst | Level of Calculations |
| ortho hydroxy imine + nitro alkanes | p-xylene, r.t., cat | N,N′−bis[3,5−bis(trifluoromethyl)phenyl]−thiourea | M06-2X/6-31g(d,p) |
| allyl ammine + aldehydes | PhMe, r.t/ maleimide, 150 °C, PhMe | / | B3LYP/def2SVP |
| azomethine ylide+ acrylonitriles | Cu(CH3CN)4BF4 and chiral phosphine−urea bifunctional ligand | M11/6–311+G(d,p) | |
| glycine imino ester + beta-fluoroalkyl alkenyl arylsulfones | THF, Et3N | BINAP-Cu(II) | M06-L/def2-TZVPP |
| ninhydrin + L-Prolin | MeOH, r.t. | / | M11 / cc-pVDZ |
| imino esters + electron deficient alkenes | Et3N, PhMe | L1-CuOTf·PhMe or L2-AgSbF6 | B3LYP/6-31G(d) |
| N-ethylglycine (50equiv) + paraformaldehyde | o-ClC6H4, 120 °C | Gd3N@Ih-C80 | BP86-D2/TZP |
| azomethine ylide+[60]fullerene | PhMe, Δ, Ar | / | B3LYP/6-31+G(d,p)/C-PCM = toluene//OLYP/6-31G(d):PM6 |
| azomethine ylide +nitrostyrene | CTPRs, THF, r.t. | / | ONIOM M06-2X/6-311+G(2d,2p):dreiding//B3LYP/6-31G-(d,p):dreiding |
| azomethine ylide + methyl cinnamate or N-enoyl oxazolidinone | [Ir], TMDS, PhMe, r.t. | iridium complex | BP86/TZ2P |
| azomethine ylide + alkenyl derivatives | THF, r.t. | Cu(I) | M06-L/6-31G(d), SDD for Cu |
| azomethine ylide + cyclopropenesor maleimide | 1,4-dioxane, 100 °C, N2 | / | M11/cc-pVDZ |
| amino acids + isatins | EtOH, 50 °C | Ni(II) | PBE0-D3BJ/def2SVP |
| azomethine ylide + arylidenes or phenylmaleimide | AcONa, PhMe, r.t. or 110 °C | / | wB97xd/6-31G(d,p) |
| glycine imino ester + N-phenylmaleimide | AcONa, TEA, EtOH, reflux | AgOAc | wB97xd/6-31G(d,p)/ Lanl2dz for Ag |
| Nitrones | |||
| nitrones + oxa(aza)bicyclic alkenes | PhMe, 60 °C | / | B3LYP/6-311G(d,p) |
| N-methyl-C-ethoxycarbonylnitrone + allyl-heptaisobutyl-POSS | 100W, PhMe, 110 °C | / | B3LYP/6-31G(d) |
| nitrone +6-alkylidenepenicillanates | PhMe, 80 °C | / | B3LYP/6-31G(d) level |
| C,N-disubstituted nitrones + disubstituted 4-methylene-1,3-oxazol-5(4H)-one | PhMe, reflux | / | M062X/6-311G(d,p) |
| adamantine aldo and ketonitrones + maleimides | PhMe, reflux | / | M06-2X/6-311++G(d,p) |
3. Conclusions
The classical 1,3-dipolar cycloaddition (1,3-DC) is considered one of the most exploited strategies for the synthesis of five-membered heterocycles, allowing the introduction of several stereogenic centers in a stereospecific manner and in a single step. The mechanistic aspects of this reaction and in particular the regio-, diastereo- and enantioselectivity are rationalized and predicted using quantum mechanical calculations. The DFT calculations have proven to be particularly effective to explain the outcome of the cycloaddition and predict the products obtained during the reaction.
In conclusion, in this review, we have highlighted the importance of DFT calculations, focusing our observation on the mechanistic insights on aza-ylides, nitrile oxides, azomethine ylides and nitrones as dipoles which have appeared in the literature in the last 5 years. Moreover, a careful analysis of the different computational approaches allows us to conclude that although several new functionals have been introduced, such as the Thrular’s hybrid metafunctionals, to date, B3LYP remains the most used for the study of this type of organic reaction by the scientific community. However, the growing interest toward new catalysts, in particular enzymatic but also metallic, opens the way to the testing of new future combinations of computational methods, which are able to predict the selectivity of 1,3-DC reactions.
Author Contributions
Conceptualization, writing—original draft preparation, writing—review and editing, M.A.C. and L.L. All authors have read and agreed to the published version of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding Statement
The authors thank the Universities of Catania and Milano-Bicocca for partial financial support. M.A.C., thanks MIUR (NextGenerationEu, Bando 2022 PNRR Prot. P20224MN78).
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Cramer C.J. Essentials of Computational Chemistry: Theories and Models. John Wiley & Sons; Chichester, UK: 2004. [Google Scholar]
- 2.Hohenberg H., Kohn W. Inhomogeneous electron gas. Phys. Rev. 1964;136:864–871. doi: 10.1103/PhysRev.136.B864. [DOI] [Google Scholar]
- 3.Sham L.J., Kohn W. One-particle properties of an inhomogeneous interacting electron gas. Phys. Rev. 1966;145:561–567. doi: 10.1103/PhysRev.145.561. [DOI] [Google Scholar]
- 4.Lee C., Yang W., Parr R.G. Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- 5.Barone V., Cossi M., Tomasi J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 1998;19:404–417. doi: 10.1002/(SICI)1096-987X(199803)19:4<404::AID-JCC3>3.0.CO;2-W. [DOI] [Google Scholar]
- 6.Barone V., Cossi M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Mode. J. Phys. Chem. A. 1998;102:1995–2001. doi: 10.1021/jp9716997. [DOI] [Google Scholar]
- 7.Groenhof G. Introduction to QM/MM simulations. Methods Mol. Biol. 2013;924:43–66. doi: 10.1007/978-1-62703-017-5_3. [DOI] [PubMed] [Google Scholar]
- 8.Molteni G., Ponti A. Is DFT Accurate Enough to Calculate Regioselectivity? The case of 1,3-dipolar cycloaddition of azide to alkynes and alkenes. ChemPhysChem. 2023;24:e202300114. doi: 10.1002/cphc.202300114. [DOI] [PubMed] [Google Scholar]
- 9.Bickelhaupt F.M., Houk K.N. Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem. 2017;56:10070–10086. doi: 10.1002/anie.201701486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanemasa S. Metal-assisted stereocontrol of 1,3-dipolar cycloaddition reactions. Synlett. 2002;2002:1371–1387. doi: 10.1055/s-2002-33506. [DOI] [Google Scholar]
- 11.Roca-López D., Tejero T., Caramella P., Merino P. Cycloadditions: An alternative to forbidden [4π + 4π] processes. the case of nitrone dimerization. Org. Biomol. Chem. 2014;14:517–525. doi: 10.1039/C3OB42014K. [DOI] [PubMed] [Google Scholar]
- 12.Yu Z.-X., Caramella P., Houk K.N. Dimerizations of Nitrile Oxides to Furoxans Are Stepwise via Dinitrosoalkene Diradicals: A Density Functional Theory Study. J. Am. Chem. Soc. 2003;125:15420–15425. doi: 10.1021/ja037325a. [DOI] [PubMed] [Google Scholar]
- 13.Domingo L.R., Aurell M.J., Jalal R., Esseffar M. A DFT study of the role of Lewis acid catalysts in the mechanism of the 1,3-dipolar cycloaddition of nitrile imines towards electron-deficient acryloyl derivatives. Comp. Theor. Chem. 2012;986:6–13. doi: 10.1016/j.comptc.2012.01.035. [DOI] [Google Scholar]
- 14.Domingo L.R., Picher M.T., Arroyo P., Saez J.A. 1,3-Dipolar Cycloadditions of Electrophilically Activated Benzonitrile N-Oxides. Polar Cycloaddition versus Oxime Formation. J. Org. Chem. 2006;71:9319–9330. doi: 10.1021/jo0613986. [DOI] [PubMed] [Google Scholar]
- 15.Ugur I., Agopcan C.S., Dedeoglu B., Aviyente V., Hawthorne M.F., Liu P., Liu F., Houk K.N., Jimenez-Oses G.J. 1,3-Dipolar Cycloaddition Reactions of Low-Valent Rhodium and Iridium Complexes with Arylnitrile N-Oxides. Org. Chem. 2017;82:5096–5101. doi: 10.1021/acs.joc.7b00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schoenebeck F., Ess D.H., Jones G.O., Houk K.N. Reactivity and Regioselectivity in 1,3-Dipolar Cycloadditions of Azides to Strained Alkynes and Alkenes: A Computational Study. J. Am. Chem. Soc. 2009;131:8121–8133. doi: 10.1021/ja9003624. [DOI] [PubMed] [Google Scholar]
- 17.Kolb H.C., Finn M.G., Sharpless K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 18.Tornøe C.W., Christensen C., Meldal M.J. Peptidotriazoles on solid phase:[1,2,3]-triazoles by regiospecific copper (I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 19.Ben El Ayouchia H., Bahsis L., Anane H., Domingo L.R., Stiriba S.-E. Understanding the mechanism and regioselectivity of the copper(I) catalyzed [3 + 2] cycloaddition reaction between azide and alkyne: A systematic DFT study. RSC Adv. 2018;8:7670–7678. doi: 10.1039/C7RA10653J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamlin T.A., Levandowski B.J., Narsaria A.K., Houk K.N., Bickelhaupt F.M. Structural Distortion of Cycloalkynes Influences Cycloaddition Rates both by Strain and Interaction Energies. Chem. Eur. J. 2019;25:6342–6348. doi: 10.1002/chem.201900295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin Y.-C., Chen Y.-J., Shih T.-Y., Chen Y.H., Lai Y.-C., Chiang M.Y., Senadi G.C., Chen H.-Y., Chen H.-Y. Mechanistic Study in Click Reactions by Using (N-Heterocyclic carbene)Copper(I) Complexes: Anionic Effects. Organometallics. 2019;38:223–230. doi: 10.1021/acs.organomet.8b00565. [DOI] [Google Scholar]
- 22.Yu S., Vermeeren P., van Dommelen K., Bickelhaupt F.M., Hamlin T.A. Understanding the 1,3-dipolar cycloadditions of allenes. Chem.-Eur. J. 2020;26:11529–11539. doi: 10.1002/chem.202000857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ben El Ayouchia H., Bahsis L., Fichtali I., Domingo L.R., Ríos-Gutiérrez M., Julve M., Stiriba S.-E. Deciphering the mechanism of silver catalysis of “Click” chemistry in water by combining experimental and MEDT studies. Catalyst. 2020;10:956. doi: 10.3390/catal10090956. [DOI] [Google Scholar]
- 24.Kim W.G., Baek S.-Y., Jeong S.Y., Nam D., Jeon J.H., Choe W., Baik M.-H., Hong S.Y. Chemo- and regioselective click reactions through nickel-catalyzed azide–alkyne cycloaddition. Org. Biomol. Chem. 2020;18:3374–3381. doi: 10.1039/D0OB00579G. [DOI] [PubMed] [Google Scholar]
- 25.Chen F.-J., Lin Y., Xu M., Xia Y., Wink D.J., Lee D. C-H Insertion by alkylidene carbenes to form 1,2,3-triazines and anionic [3 + 2] dipolar cycloadditions to form tetrazoles: Crucial roles of stereoelectronic and steric effects. Org. Lett. 2020;22:718–723. doi: 10.1021/acs.orglett.9b04548. [DOI] [PubMed] [Google Scholar]
- 26.Navarro Y., García López J., Iglesias M.J., López Ortiz F. Chelation-Assisted Interrupted Copper(I)-Catalyzed Azide–Alkyne–Azide Domino Reactions: Synthesis of Fully Substituted 5-Triazenyl-1,2,3-triazoles. Org. Lett. 2021;23:334–339. doi: 10.1021/acs.orglett.0c03838. [DOI] [PubMed] [Google Scholar]
- 27.Fang Y., Bao K., Zhang P., Sheng H., Yun Y., Hu S.-X., Astruc D., Zhunsight M. Into the mechanism of the CuAAC reaction by capturing the crucial Au4Cu4–π-alkyne intermediate. J. Am. Chem. Soc. 2021;143:1768–1772. doi: 10.1021/jacs.0c12498. [DOI] [PubMed] [Google Scholar]
- 28.Zhu L., Kinjo R. An inorganic Huisgen reaction between a 1,2-diboraallene and an azide to access a diboratriazole. Angew. Chem. Int. Ed. 2022;61:e202207631. doi: 10.1002/anie.202207631. [DOI] [PubMed] [Google Scholar]
- 29.Singh G., George N., Singh R., Singh G., Kaur J.D., Kaur G., Singh H., Singh J. CuAAC-Derived selective fluorescent probe as a recognition agent for Pb(II) and Hg(II): DFT and docking studies. ACS Omega. 2022;7:39159–39168. doi: 10.1021/acsomega.2c05050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiavegatti Neto A., Soares K.C., da Silva Santos M., Jardini Aímola T., Ferreira A.G., Jardim G.A.M., Tormena C.F., Weber Paixão M., Barbosa Ferreira M.A. Mechanistic investigation of enolate/stabilized vinylogous carbanion-mediated organocatalytic azide (3 + 2) cycloaddition reactions for the synthesis of 1,2,3-triazoles. Org. Biomol. Chem. 2022;20:6019–6026. doi: 10.1039/D2OB00391K. [DOI] [PubMed] [Google Scholar]
- 31.Gholivand K., Faraghi M., Malekshah R.E., Jafari M., Akbarzadeh A.R., Latifi R., Fadaei-Tirani F., Fallah N., Farshadfar K. New Cu (I) complexes as catalyst for “click” reaction: An experimental and computational study. Appl. Organomet. Chem. 2023;37:e6924. doi: 10.1002/aoc.6924. [DOI] [Google Scholar]
- 32.Song G., Rong W., Liu Y., Li J. Mechanisms and origin of regioselectivity for manganese-catalyzed denitrogenative annulation and click reactions. Org. Chem. Front. 2023;10:3642–3653. doi: 10.1039/D3QO00406F. [DOI] [Google Scholar]
- 33.Fukuura S., Yumura T. Roles of carbon nanotube confinement in modulating regioselectivity of 1,3-dipolar cycloadditions. J. Phys. Chem. A. 2023;127:6962–6973. doi: 10.1021/acs.jpca.3c04214. [DOI] [PubMed] [Google Scholar]
- 34.Chiacchio M.A., Lanza G., Chiacchio U., Giofrè S.V., Romeo R., Iannazzo D., Legnani L. Oxazole-based compounds as anticancer agents. Curr. Med. Chem. 2019;26:7337–7371. doi: 10.2174/0929867326666181203130402. [DOI] [PubMed] [Google Scholar]
- 35.Bucci R., Giofré S., Clerici F., Contini A., Pinto A., Erba E., Soave R., Pellegrino S., Gelmi M.L. Tetrahydro-4H-(pyrrolo[3,4-d]isoxazol-3-yl)methanamine: A bicyclic diamino scaffold stabilizing parallel turn conformations. J. Org. Chem. 2018;83:11493–11501. doi: 10.1021/acs.joc.8b01299. [DOI] [PubMed] [Google Scholar]
- 36.Uceta E., Vizuete M., Carrillo J.R., Barrejón M., Fierro J.L.J., Prieto M.P., Langa F. Cycloaddition of nitrile oxides to graphene: A theoretical and experimental approach. Chem. Eur. J. 2019;25:14644–14650. doi: 10.1002/chem.201903105. [DOI] [PubMed] [Google Scholar]
- 37.Barrejón M., Gómez-Escalonilla M.J., Fierro J.L.G., Prieto P., Carrillo J.R., Rodríguez A.M., Abellán G.M., López-Escalante C., Gabas M., López-Navarrete J.T., et al. Modulation of the exfoliated graphene work function through cycloaddition of nitrile imines. Phys. Chem. Chem. Phys. 2016;18:29582–29590. doi: 10.1039/C6CP05285A. [DOI] [PubMed] [Google Scholar]
- 38.Adjieufack A.I., Nana C.N., Ketcha-Mbadcam J., Ndassa I.M., Andrés J., Oliva M., Safont V.S. Deciphering the curly arrow representation and electron flow for the 1,3-dipolar rearrangement between acetonitrile oxide and (1S,2R,4S)-2-cyano-7-oxabicyclo[2.2.1]hept-5-en-2-yl acetate derivatives. ACS Omega. 2020;5:22215–22225. doi: 10.1021/acsomega.0c02371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goyard D., Kónya B., Czifrák K., Larini P., Demontrond F., Leroy J., Balzarin S., Tournier M., Tousch D., Petit P., et al. Glucose-based spiro-oxathiazoles as in vivo anti-hyperglycemic agents through glycogen phosphorylase inhibition. Org. Biomol. Chem. 2020;18:931–940. doi: 10.1039/C9OB01190K. [DOI] [PubMed] [Google Scholar]
- 40.Holman S.D.L., Wills A.G., Fazakerley N.J., Poole D.L., Coe D.M., Berlouis L.A., Reid M. Electrochemical synthesis of isoxazolines: Method and mechanism. Chem. Eur. J. 2022;28:e202103728. doi: 10.1002/chem.202103728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kiss L., Escorihuela J. A computational study of 1,3-dipolar cycloadditions of nitrile oxides with dienes. Tetrahedron. 2023;139:133435. doi: 10.1016/j.tet.2023.133435. [DOI] [Google Scholar]
- 42.Dubey S., Pal A., Roy S., Sasmal S., Tamrakar A., Jana R., Das T. Recent advances in the (3 + 2) cycloaddition of azomethine ylide. New J. Chem. 2023;47:8997–9034. doi: 10.1039/D3NJ01018J. [DOI] [Google Scholar]
- 43.Esteban F., Cieślik W., Arpa E.M., Guerrero-Corella A., Díaz-Tendero S., Perles J., Fernández-Salas J.A., Fraile A., Alemán J. Intramolecular Hydrogen Bond Activation: Thiourea-organocatalyzed enantioselective 1,3-dipolar cycloaddition of salicylaldehyde-derived azomethine ylides with nitroalkenes. ACS Catal. 2018;8:1884–1890. doi: 10.1021/acscatal.7b03553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Selva V., Selva E., Merino P., Najera C., Sansano J.M. Sequential metal-free thermal 1,3-dipolar cycloaddition of unactivated azomethine ylides. Org. Lett. 2018;20:3522–3526. doi: 10.1021/acs.orglett.8b01292. [DOI] [PubMed] [Google Scholar]
- 45.Xiong Y., Du Z., Chen H., Yang Z., Tan Q., Zhang C., Zhu L., Lan Y., Zhang M. Well-designed phosphine–urea ligand for highly diastereo- and enantioselective 1,3-dipolar cycloaddition of methacrylonitrile: A combined experimental and theoretical study. J. Am. Chem. Soc. 2019;141:961–971. doi: 10.1021/jacs.8b10939. [DOI] [PubMed] [Google Scholar]
- 46.Cheng F., Kalita S.J., Zhao Z.-N., Yang X., Zhao Y., Schneider U., Shibata N., Huang Y.-Y. Diastereodivergent asymmetric 1,3-dipolar cycloaddition of azomethine ylides and β-fluoroalkyl vinylsulfones: Low copper(II) catalyst loading and theoretical studies. Angew. Chem. Int. Ed. 2019;58:16637–16643. doi: 10.1002/anie.201908227. [DOI] [PubMed] [Google Scholar]
- 47.Filatov A.S., Wang S., Khoroshilova O.V., Lozovskiy S.V., Larina A.G., Boitsov V.M., Stepakov A.V. Stereo- and regioselective 1,3-dipolar cycloaddition of the stable ninhydrin-derived azomethine ylide to cyclopropenes: Trapping of unstable cyclopropene dipolarophiles. J. Org. Chem. 2019;84:7017–7036. doi: 10.1021/acs.joc.9b00753. [DOI] [PubMed] [Google Scholar]
- 48.Caleffi G.S., Larrañaga O., Ferrándiz-Saperas M., Costa P.R.R., Nájera C., de Cózar A., Cossío F.P., Sansano J.M. Switching diastereoselectivity in catalytic enantioselective (3 + 2) cycloadditions of azomethine ylides promoted by metal salts and privileged segphos-derived ligands. J. Org. Chem. 2019;84:10593–10605. doi: 10.1021/acs.joc.9b00267. [DOI] [PubMed] [Google Scholar]
- 49.Semivrazhskaya O., Romero-Rivera A., Aroua S., Troyanov S.I., Garcia-Borràs M., Stevenson S., Osuna S., Yamakoshi Y. Structures of Gd3N@C80 Prato bis-ddducts: Crystal structure, thermal isomerization, and computational study. J. Am. Chem. Soc. 2019;141:10988–10993. doi: 10.1021/jacs.9b05603. [DOI] [PubMed] [Google Scholar]
- 50.Alonso D., Hernández-Castillo D., Almagro L., González-Alemán R., Molero D., Herranz M.A., Medina-Páez E., Coro J., Martínez-Álvarez R., Suárez M., et al. Diastereoselective synthesis of steroid–[60]fullerene hybrids and theoretical underpinning. J. Org. Chem. 2020;85:2426–2437. doi: 10.1021/acs.joc.9b03121. [DOI] [PubMed] [Google Scholar]
- 51.Rivilla I., de Cózar A., Schäfer T., Hernandez F.J., Bittner A.M., Eleta-Lopez A., Aboudzadeh A., Santos J.I., Miranda J.I., Cossio F.P. Catalysis of a 1,3-dipolar reaction by distorted DNA incorporating a heterobimetallic platinum(II) and copper(II) complex. Chem. Sci. 2017;8:7038–7046. doi: 10.1039/C7SC02311A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rivilla I., Odriozola-Gimeno M., Aires A., Gimeno A., Jiménez-Barbero J., Torrent-Sucarrat M., Cortajarena A.L., Cossío F.P. Discovering Biomolecules with Huisgenase activity: Designed repeat proteins as biocatalysts for (3 + 2) cycloadditions. J. Am. Chem. Soc. 2020;142:762–776. doi: 10.1021/jacs.9b06823. [DOI] [PubMed] [Google Scholar]
- 53.Yamazaki K., Gabriel P., Di Carmine G., Pedroni J., Farizyan M., Hamlin T.A., Dixon D.J. General pyrrolidine synthesis via iridium-catalyzed reductive azomethine ylide generation from tertiary amides and lactams. Adv. Syn. Catal. 2021;11:7489–7497. doi: 10.1021/acscatal.1c01589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vermeeren P., Hamlin T.A., Fernández I., Bickelhaupt F.M. Origin of rate enhancement and asynchronicity in iminium catalyzed Diels−Alder reactions. Chem. Sci. 2020;11:8105–8112. doi: 10.1039/D0SC02901G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang X., Yang Y., Shen C., Xue K.-S., Wang Z.-F., Cong H., Tao H.Y., Chung L.W., Wang C.-J. β-Substituted alkenyl heteroarenes as dipolarophiles in the Cu(I)-catalyzed asymmetric 1,3-dipolarc cycloaddition of azomethine ylides empowered by a dual activation strategy: Stereoselectivity and mechanistic insight. J. Am. Chem. Soc. 2021;143:3519–3535. doi: 10.1021/jacs.0c12911. [DOI] [PubMed] [Google Scholar]
- 56.Filatov A.S., Pronina J.A., Selivanov S.I., Shmakov S.V., Uspenski A.A., Boitsov V.M., Stepakov A.V. 1,11H-Benzo[4,5]imidazo[1,2-a]indol-11-one as a new precursor of azomethine ylides: 1,3-Dipolar cycloaddition reactions with cyclopropenes and maleimides. Int. J. Mol. Sci. 2022;23:13202. doi: 10.3390/ijms232113202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gugkaeva Z.T., Panova M.V., Smol’yakov A.F., Medvedev M.G., Tsaloev A.T., Godovikov I.A., Maleev V.I., Larionov V.A. Asymmetric metal-templated route to amino acids with 3-spiropyrrolidine oxindole core via a 1,3-dipolar addition of azomethine ylides to a chiral dehydroalanine Ni(II) complex. Adv. Syn. Catal. 2022;364:2395–2402. doi: 10.1002/adsc.202200446. [DOI] [Google Scholar]
- 58.Radwan M.F., Elboray E.E., Dardeer H.M., Kobayashi Y., Furuta T., Hamada S., Dohi T., Aly M.F. 1,3-Dipolar cycloaddition of 3-chromonyl-substituted glycine imino esters with arylidenes and in situ diastereodivergent via retrocycloaddition. Chem. Asian J. 2023;18:e202300215. doi: 10.1002/asia.202300215. [DOI] [PubMed] [Google Scholar]
- 59.Selva E., Castello L.M., Mancebo-Aracil J., Selva V., Najera C., Foubelo F., Sansano J.M. Synthesis of pharmacophores containing a prolinate core using a multicomponent 1,3-dipolar cycloaddition of azomethine ylides. Tetrahedron. 2017;73:6840–6846. doi: 10.1016/j.tet.2017.10.030. [DOI] [Google Scholar]
- 60.Chiacchio M.A., Legnani L., Chiacchio U. Recent advances on the synthesis of isoxazolidines. In: Faisca Phillips A.M., editor. Chapter 7 in Synthetic Approaches to Nonaromatic Nitrogen Heterocycles. Volume 2. John Wiley & Sons; Hoboken, NJ, USA: 2020. pp. 161–177. [Google Scholar]
- 61.Chiacchio U., Padwa A., Romeo G. Cycloaddition methodology: A useful entry towards biologically active heterocycles. Curr. Org. Chem. 2009;13:422–447. doi: 10.2174/138527209787582268. [DOI] [Google Scholar]
- 62.Romeo G., Chiacchio U., Corsaro A., Merino P. Chemical synthesis of heterocyclic sugar nucleoside analogues. Chem. Rev. 2010;110:3337–3370. doi: 10.1021/cr800464r. [DOI] [PubMed] [Google Scholar]
- 63.Yao Y., Yang W., Lin Q., Yang W., Li H., Wang L., Gu F., Yang D. 1,3-Dipolar cycloaddition of nitrones to oxa(aza)bicyclic alkenes. Org. Chem. Front. 2019;6:3360–3364. doi: 10.1039/C9QO00660E. [DOI] [Google Scholar]
- 64.Legnani L., Iannazzo D., Pistone A., Celesti C., Giofrè S., Romeo R., Di Pietro A., Visalli G., Fresta M., Bottino P., et al. Functionalized polyhedral oligosilsesquioxane (POSS) based composites for bone tissue engineering: Synthesis, computational and biological studies. RSC Adv. 2020;10:11325–11334. doi: 10.1039/D0RA01636E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alves A.J.S., Pinho e Melo T.M.V.D. Synthesis of novel chiral spiroisoxazolidine-β-lactams from 6-alkylidenepenicillanates: A 1,3-dipolar cycloaddition approach. Eur. J. Org. Chem. 2020;39:6259–6269. doi: 10.1002/ejoc.202001085. [DOI] [Google Scholar]
- 66.Mekheimer R.A., Al-Zaydi K., Ibrahim M.A.A., Al-Shamary A., Sadek K. Regio and stereoselective 1,3-dipolar cycloaddition reactions of C-aryl (or hetaryl)-N-phenylnitrones to monosubstituted ylidene malononitriles and 4-benzylidene-2-phenyloxazol-5(4H)-one. Z. Naturforsch B J. Chem. Sci. 2017;72:317e326. doi: 10.1515/znb-2016-0263. [DOI] [Google Scholar]
- 67.Pipim G.B., Opoku E., Tia R., Adei E. Peri-, chemo-, regio-, stereo- and enantio-selectivities of 1,3-dipolar cycloaddition reaction of C, N-disubstituted nitrones with disubstituted 4-methylene-1,3-oxazol-5(4H)- one: A quantum mechanical study. J. Mol. Graph. Model. 2020;97:107542. doi: 10.1016/j.jmgm.2020.107542. [DOI] [PubMed] [Google Scholar]
- 68.Briccolani-Bandini L., Pagliai M., Cordero F.M., Brandi A., Cardini G. Cyclopropylidene effect in the 1, 3-dipolar cycloaddition of nitrones to alkylidene cyclopropanes: A computational rationalization. J. Phys. Chem. A. 2021;125:3892–3899. doi: 10.1021/acs.jpca.1c02204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Molchanov A.P., Lukina V.M., Efremova M.M., Muryleva A.A., Slita A.V., Zarubaev V.V. The 1,3-dipolar cycloaddition of adamantine-derived nitrones with maleimides. Synth. Commun. 2020;50:1367–1374. doi: 10.1080/00397911.2020.1738494. [DOI] [Google Scholar]
- 70.Umar A.R., Tia R., Adei E. The 1,3-dipolar cycloaddition of adamantine-derived nitrones with maleimides: A computational study. Comput. Theor. Chem. 2021;1195:113099. doi: 10.1016/j.comptc.2020.113099. [DOI] [Google Scholar]