

Abstract
Repetitive implant-related infections (IRIs) are devastating complications in orthopedic surgery, threatening implant survival and even the life of the host. Biofilms conceal bacterial-associated antigens (BAAs) and result in a "cold tumor"-like immune silent microenvironment, allowing the persistence of IRIs. To address this challenge, an iron-based covalent organic framed nanoadjuvant doped with curcumin and platinum (CFCP) was designed in the present study to achieve efficient treatment of IRIs by inducing a systemic immune response. Specifically, enhanced sonodynamic therapy (SDT) from CFCP combined with iron ion metabolic interference increased the release of bacterial-associated double-stranded DNA (dsDNA). Immunogenic dsDNA promoted dendritic cell (DC) maturation through activation of the stimulator of interferon gene (STING) and amplified the immune stimulation of neutrophils via interferon-β (IFN-β). At the same time, enhanced BAA presentation aroused humoral immunity in B and T cells, creating long-term resistance to repetitive infections. Encouragingly, CFCP served as neoadjuvant immunotherapy for sustained antibacterial protection on implants and was expected to guide clinical IRI treatment and relapse prevention.
Keywords: Implant-related infections, Systemic immunotherapy, cGAS-STING pathway, Interferon, Neutrophil activation
Graphical abstract
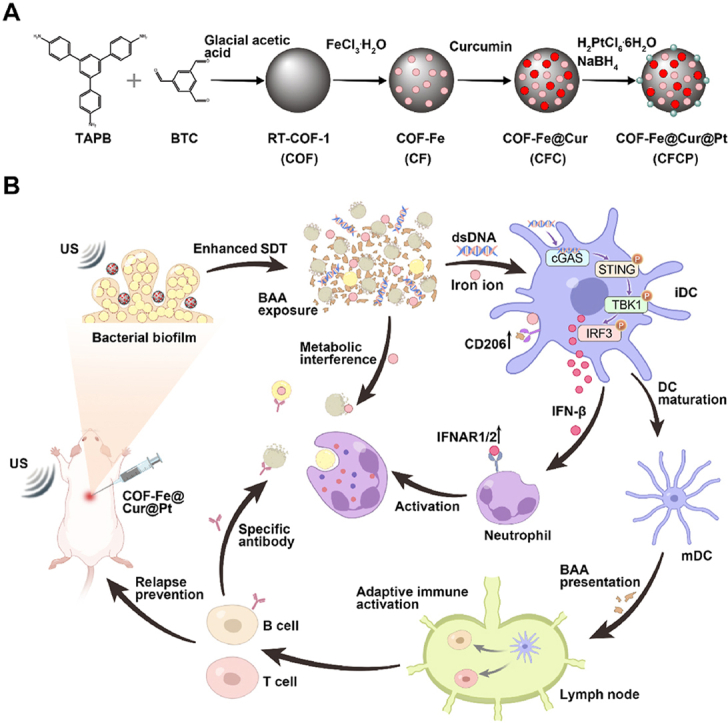
Scheme. (A) Synthesis procedure of CFCP. (B) CFCP promotes the exposure of bacterial-associated antigens (BAAs) through enhanced sonodynamic therapy (SDT) and metabolic interference, which in turn activates the cGAS-STING pathway in dendritic cells (DCs). DC-mediated interferon secretion and antigen presentation evoke systemic antimicrobial immune responses, effectively treating implant-associated infections (IRIs) and preventing their relapse and reinfection.
Highlights
-
•
Nanoadjuvant motivates bacterial-associated antigen exposure through enhanced sonodynamic therapy and metabolic interference.
-
•
STING activation evokes systemic immune responses for the treatment of implant-related infections (IRIs).
-
•
Nanoadjuvant-mediated neoadjuvant immunotherapy prevents relapse and reinfection of IRIs.
1. Introduction
Implant surgery has become a common clinical procedure worldwide, benefiting numerous patients suffering from heart, kidney, eye, bone and joint, and dental diseases [1]. However, postoperative complications, particularly implant-related infections (IRIs), reduce the survival of implants and even endanger the host's life [2]. Despite the powerful phagocytic and bactericidal capabilities of the innate immune system, cunning pathogens adhere and aggregate under the protection of implants to form bacterial biofilms with physical barrier properties and antigen concealment, making infection eradication difficult [1].
In recent years, nanomedicine has shown great application promise in oncological, infectious, inflammatory, and autoimmune diseases [[3], [4], [5], [6], [7], [8]]. The unique size and physical properties (e.g. light, sound, and heat) of nanomaterials provide them significant advantages in the treatment of bacterial biofilm infections [9]. In particular, the development of nanomaterials with both bactericidal and immunomodulatory activities for the immune microenvironment of infection is becoming a popular research direction [10]. We previously developed a nanomaterial for the treatment of IRIs exploiting the inherent bactericidal properties of copper ions and the immunomodulatory activity of luteolin on macrophages [11]. However, the therapeutic efficacy of unilateral innate immunomodulatory strategies is stretched in the face of the challenge of complex, chronic, and recurrent infections [12]. Therefore, there is a need to develop novel nanotherapeutic platforms to enhance systemic immune responses against repetitive implant-related biofilm infections.
Dendritic cells (DCs) are specialized antigen-presenting cells (APCs) that play a pivotal role in initiating specific systemic immune responses [13,14]. However, the antigen-hiding properties of biofilms in IRIs make it difficult for DCs to recognize, phagocytose, and present bacterial-associated antigens (BAAs). How to break the vicious cycle of infection-immune silencing-infection persistence is key to evoking systemic immunotherapy. Cyclic GMP-AMP synthase (cGAS) is a natural immune sensor that activates the stimulator of interferon genes (STING) by recognizing dsDNA derived from bacteria or tumor cells. STING activation forms aggregates that recruit and phosphorylate downstream TANK binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3) to initiate the interferon immune response [15,16]. In oncology, researchers transform dead tumor cells from non-immunogenic to immunogenic, thereby stimulating the body's anti-tumor immune response [17]. In this process, tumor-associated antigens activate the cGAS-STING pathway in DCs to catalyze their maturation and antigen presentation. However, it remains unclear whether increasing BAA exposure has a similar ignition effect in low-immunogenic IRIs.
In this work, we developed an iron-based covalent organic framed nanoadjuvant doped with curcumin and platinum (CFCP) to achieve efficient treatment of IRI by inducing a systemic immune response. As a natural sonosensitiser, curcumin generated singlet oxygen (1O2) with high energy under ultrasound (US) irradiation [18]. The loaded single-atom platinum (Pt) exerted catalase enzyme activity (CAT) to catalyze oxygen (O2) production and enhanced the sonodynamic therapy (SDT) of curcumin [19]. CFCP-mediated SDT disrupted biofilm structure in IRIs and hastened bacterial death in synergistic with the metabolic interfering activity of iron ions. Subsequently, immunogenic dsDNA released from disrupted bacterial biofilms upregulated the STING pathway to induce DC maturation while activating the innate antibacterial immune response of neutrophils. Further, DCs increased BAA presentation to T and B cells, thereby motivating the adaptive immunity characterized by specific antibody secretion and memory immunity. Encouragingly, CFCP has been demonstrated to be effective as neoadjuvant immunotherapy in preventing IRI relapse and reinfection and is expected to be further explored and applied clinically.
2. Results and discussion
2.1. Synthesis and characteristics of CFCP
The covalent organic frame (COF), iron-based COF (CF), CF loaded with curcumin (CFC), and CFC doped with Pt (CFCP) were synthesized sequentially according to the schematic diagram in Scheme A. As the observation of transmission electron microscopy (TEM) showed, CF, CFC, and CFCP exhibited spherical structures with regular morphology and uniform size (Fig. 1A). The average hydrated particle sizes of CF, CFC, and CFCP were 153.1 nm, 142.6 nm, and 156.8 nm, respectively (Fig. 1B). The zeta potential of CF, CFC, and CFCP was 0.20 ± 0.55 mV, −12.20 ± 2.56 mV, and −21.89 ± 1.88 mV, respectively (Fig. S1). The increase of zeta potential suggested good dispersion and stability of CFCP. Energy dispersion spectrum (EDS) analysis showed that C, N, Fe, and Pt elements were dominant and uniformly distributed in CFCP (Fig. 1C). In addition, Fe-N stretching vibration peak was detected near 1000 cm−1 in the Fourier transform infrared spectroscopy (FTIR) of CF, CFC, and CFCP (Fig. 1D), which was consistent with the literature reports [20]. The X-ray diffraction (XRD) spectra of CFCP revealed more diffraction peaks than CF and CFC, which could be attributed to the loading of curcumin and Pt (Fig. S2). High-resolution X-ray photoelectron spectroscopy (XPS) measurements demonstrated that Fe, O, N, C, and Pt were present in CFCP and Fe was predominantly divalent and trivalent (Fig. 1E and F).
Fig. 1.

Characterization of CFCP. (A) TEM images of CF, CFC, and CFCP. (B) Average hydrated particle size of CF, CFC, and CFCP measured by DLS. (C) The elemental distribution of CFCP measured by EDS mapping analysis. The colors corresponding to the elements C, Fe, Pt, and N are red, yellow, purple, and green, respectively. (D) FTIR spectra of CF, CFC, and CFCP. (E) XPS spectrum of CFCP and (F) high-resolution spectrum of Fe 2p within them. (G) Time-dependent H2O2 depletion by CFCP. (H) O2 production by the reaction of 100 μg/ml CFCP with 20 mM H2O2. (I) ESR spectra of different samples with TEMP as the radical trap. (J to L) UV–vis spectra of DPBF in 20 mM H2O2 after co-incubation with US, CFC+US, and CFCP+US for 2, 4, 6, and 8 min. (M) Depletion rates of DPBF in various reaction systems.
The Ultraviolet–visible spectroscopy (UV–vis) spectra of CF, CFC, and CFCP were measured further and their characteristic absorption peak was detected at 410 nm (Fig. S3). No significant decrease in the UV–vis spectral peaks of CFCP was observed over four days in different solution systems including water, phosphate buffer saline (PBS), Dulbeccos Modified EagleMedium (DMEM), and serum, indicating its good stability (Fig. S4). In addition, the UV–vis spectra of curcumin at different concentrations were detected (Fig. S5A). Curcumin concentration showed a significant positive correlation with the absorbance at 434 nm (Fig. S5B). The loading efficiency of curcumin in CFC and CFCP was 23.3 ± 2.26 % and 20.6 ± 1.75 %, respectively (Fig. S6). At pH 7.4, 6, and 5, CFCP released 17.9 ± 0.7 %, 46.8 ± 1.1 %, and 52.8 ± 1.9 % of curcumin over 24 h, respectively (Fig. S7).
Next, we investigated the enhanced SDT effect of CFCP. The CAT activity of single-atom Pt was first evaluated. The results showed that CFCP continuously consumed hydrogen peroxide (H2O2) during the reaction (Fig. 1G). Meanwhile, the O2 concentration in the CFCP group increased with the reaction time and gradually entered a plateau after 5 h (Fig. 1H). In addition, the SDT activity of CFCP was investigated using electron spin resonance (ESR) spectroscopy. As shown in Fig. 1I, more 1O2 was produced by CFCP+US, confirming the Pt-enhanced SDT effect. 1,3-Diphenylisobenzofuran (DPBF) was used as another probe to determine the efficiency of CFCP in catalyzing the generation of 1O2. The consumption of DPBF was significantly accelerated in the CFCP+US group compared to the US and CFC+US, suggesting a stronger SDT effect (Fig. 1J-M). Assays using methylene blue (MB) as a reactive oxygen species (ROS) probe led to similar conclusions (Fig. S8).
The biocompatibility of CFCP was determined before biological experiments. Cell Counting Kit-8 (CCK-8) assay showed that the toxic side effect of CFCP on the proliferative activity of DCs and neutrophils was negligible when the concentration was up to 100 μg/ml (Figs. S9A and B). In addition, the therapeutic combination of CFCP+US did not produce significant hemolysis at a concentration of 100 μg/ml (Figs. S9C and D). Therefore, 100 μg/ml was chosen as the action concentration of CFCP in subsequent in vitro experiments.
2.2. CFCP+US motivates BAA exposure through enhanced SDT
Bacterial biofilms protect BAAs from recognition by the immune system, resulting in immune silencing and chronicity of infection [21]. We have demonstrated that CFCP exhibits enhanced SDT under US irradiation, which is expected to disrupt biofilm structure and motivate BAA exposure. With this in mind, we explored the performance of CFCP+US in eliminating biofilm infections in vitro.
Staphylococcus aureus (S. aureus) and Escherichia coli (E. coli) were cultured in vitro and their biofilms were obtained. The prepared biofilms were treated with different conditions and evaluated for their therapeutic effects. The biofilms without any treatment were used as a control. Reconstructed images of confocal laser scanning microscopy (CLSM) revealed that the control group had the most intact biofilm structure and almost no bacterial death. After CFCP+US treatment, the severely damaged biofilm became loose and a large number of bacteria died (Fig. 2A). In contrast, US, CFC, CFC+US, and CFCP were less effective in killing biofilm infections. Scanning electron microscopy (SEM) similarly showed that CFCP+US-treated biofilms lost their structural integrity, and the surviving bacteria within them were drastically reduced (Fig. S10A). Destruction of biofilm by CFCP+US caused a reduction in biomass as evidenced by crystalline violet staining (Fig. 2B and C). Quantitative results from the spread plate method (SPM) demonstrated that CFCP+US decreased bacterial survival within S. aureus and E. coli biofilms from 100.0 ± 6.0 % and 100 ± 2.9 % to 14.8 ± 2.3 % and 18.9 ± 2.0 %, respectively (Figs. S10B and C).
Fig. 2.

CFCP+US motivates BAA exposure through the enhanced SDT. (A) Three-dimensional structure of bacterial biofilms reconstructed by CLSM. Green for live bacteria, red for dead bacteria. (B) Representative images of the anti-biofilm effect in different treatment groups assessed using crystalline violet staining. (C) The crystalline violet dye bound to the biofilms was dissolved and the absorbance was measured at 595 nm. (D) Flow cytometry results of bacterial membrane potential in different treated biofilms. The y-axis (red axis) represents normal membrane potential, while the x-axis (green axis) represents depolarized membrane potential. Dots in purple boxes represent bacteria with depolarized membrane potential, and the numbers refer to their proportions. (E) Morphology of bacteria in biofilm from different treatment groups as observed under TEM. The dotted box on the right refers to the meaning of the different colored arrows. Scale bars, 200 nm. (F) Flow cytometry results of ROS levels in S. aureus within biofilms measured by DCFH-DA. (G) Relative ROS levels in the S. aureus biofilms as measured by a fluorescent microplate reader. (H) The absorbance at 450 nm for ONPG hydrolysis assay to assess the membrane permeability of S. aureus in different treated biofilms. (I) The intracellular iron concentration of S. aureus in biofilms from different treatment groups. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, and ns means no significance).
The mechanisms of CFCP+US killing biofilm infections were further explored. The membrane potential regulates a wide range of bacterial physiology and behaviors, including pH homeostasis, membrane transport, motility, antibiotic resistance, cell division, electrical communication, and environmental sensing [22,23]. Depolarization of the membrane potential is accompanied by an increase in the permeability of the cytoplasmic membrane, leading to the leakage of cellular contents [24]. In Fig. 2D, we examined the membrane potentials of bacteria from different treatment groups using flow cytometry and circle-gated the bacteria with depolarized membrane potentials. The proportion of S. aureus and E. coli with depolarized membrane potentials in the CFCP+US group increased sharply from 1.25 % to 90.30 % and from 2.08 % to 89.18 %, respectively. Rapid depolarization of membrane potential indicated the severe destruction of bacterial membranes, which was verified by the detection of TEM. Specifically, the continuity of the bacterial membrane was lost and the membrane boundary became blurred after CFCP+US treatment, accompanied by the efflux of intracellular material such as cytoplasm and DNA (Fig. 2E). We further found that the ROS levels were significantly elevated in CFCP+US-treated biofilms and bacteria, which attributed to the enhanced SDT and was a potential mechanism for biofilm infection elimination (Fig. 2F and G, and Figs. S11A and B). In addition, CFCP+US increased the permeability of the bacterial membrane, facilitating greater access of iron ions released by CFCP to the bacteria (Fig. 2H and I, and Figs. S11C and D). Iron overload has been demonstrated to exert a bactericidal effect by interfering with bacterial metabolism or inducing ferroptosis [25].
2.3. Iron-mediated metabolic interference hastens bacterial biofilm death
To determine the comprehensive mechanisms by which CFCP+US eliminated biofilm infections, RNA-seq transcriptomics was employed to detect the differential gene expression of S. aureus. Compared with the control group, 447 genes were down-regulated and 480 genes were up-regulated in CFCP+US-treated S. aureus (Fig. S12). According to the enrichment analysis of the Kyoto Encyclopedia of Genes and Genomes (KEGG), the energy metabolism pathways were significantly disrupted in the CFCP+US group of S. aureus, as evidenced by down-regulation of the ABC transporters, glycolysis/gluconeogenesis, pentose phosphate pathway, and fructose and mannose metabolism (Fig. 3A). As an essential family of membrane transport proteins, ABC transporters utilize the energy of ATP hydrolysis to transport nutrients such as glucose across membranes. In addition, ABC transporters are involved in the development of bacterial drug resistance and the secretion of virulence factors [26]. It was reasonably inferred that the ABC transporters were key functional targets for the bactericidal action of CFCP+US. The Gene Ontology (GO) functional enrichment demonstrated that CFCP+US interfered with the processes of substance transport and energy metabolism in S. aureus, which were essential for the maintenance of normal bacterial growth and proliferative activity (Fig. 3B) [27]. Overall, the interference of CFCP+US with bacterial metabolic processes was significant. For further validation of the transcriptomic results, the representative differentially expressed genes related to ABC transporters, glycolysis, and anti-oxidative stress functions were screened and respectively presented in the form of a hierarchical clustering and heat map (Fig. 3C–E).
Fig. 3.

Iron ions released by CFCP+US interfere with bacterial metabolism in S. aureus biofilms. (A) KEGG enrichment of downregulated genes in S. aureus biofilms after CFCP+US treatment. (B) GO term of downregulated pathway related to biological processes, cellular components, and molecular function in S. aureus biofilms treated with CFCP+US. (C) Heat map of differentially expressed genes involved in ABC transport system. (D) Heat map of differentially expressed genes involved in glycolysis process. (E) Heat map of differentially expressed genes involved in antioxidant defense system. Yellow for relatively high expression genes and purple for relatively low expression genes. (F to H) qPCR validation results for expression levels of representative genes in the ABC transport system (natA and UvrB), glycolysis process (FBA1 and FBA2), and antioxidant defense system (Usp and yloU). (I to L) Levels of glucose, pyruvate, NAD+/NADH, and ATP in biofilms of S. aureus with different treatments. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, and ns means no significance).
Iron is a key element for bacterial growth [28]. Through co-evolution with the hosts, bacteria have mastered specific mechanisms for accessing host iron sources [29]. However, it has been shown that iron overload within bacteria is detrimental to their maintenance of normal growth and metabolism [30,31]. CFCP+US was proven to increase the inward flow and accumulation of iron ions in bacteria. To investigate the role played by iron in CFCP+US interference with bacterial metabolism, we selected deferoxamine (DFO) as a chelator to antagonize the effect of iron [32]. qPCR results showed that CFCP+US down-regulated the expression of genes related to ABC transporter, glycolysis, and anti-oxidative stress functions, validating the transcriptomic results (Fig. 3F–H, and Fig. S13). This down-regulatory effect was significantly attenuated by DFO, indicating that it was mainly iron that interfered with bacterial metabolism. We further determined the glycolytic process of S. aureus at the metabolite level. As shown in Fig. 3I, the glucose content was reduced in CFCP+US-treated S. aureus, probably as a result of the down-regulation of the ABC transporter. Additionally, the decrease of pyruvate (Fig. 3J), the increase of the NAD+/NADH ratio (Fig. 3K), and the downregulation of the ATP level (Fig. 3L) demonstrated that CFCP+US inhibited the glycolytic process in S. aureus [33]. As expected, such inhibition effects were largely alleviated by DFO. Similar conclusions were drawn from the tests on E. coli (Fig. S14). The above results indicated that CFCP+US severely interfered with bacterial metabolic processes, which would hasten bacterial biofilm death and BAA exposure.
2.4. Bacterial-associated dsDNA induces DC maturation through STING activation
CFCP+US has been shown to promote the degradation of biofilm matrices and the destruction of bacterial membranes, which is expected to increase the release of immunogenic substances represented by dsDNA. As the fluorescent image revealed, the content of residual dsDNA in CFCP+US-treated biofilm was sharply reduced (Fig. S15A). The quantitative assay results suggested that, after CFCP+US treatment, the dsDNA in the medium supernatant of S. aureus and E. coli was elevated from 172.6 ± 31.3 ng/ml and 162.4 ± 25.6 ng/ml to 947.9 ± 35.1 ng/ml and 546.7 ± 37.3 ng/ml, respectively (Figs. S15B and C). In contrast, dsDNA efflux was less significant in the other treatment groups. Whether CFCP-driven dsDNA release could regulate immune stimulation and antigen presentation of DCs aroused our great research interest. Therefore, the S. aureus biofilm supernatant was collected after different treatments and configured as the biofilm-conditioned mediums (BCMs) for DC cultivation (Fig. 4A). Flow cytometry was employed to assess the expression of the surface co-stimulatory molecules CD80 and CD86 in DCs cultured in different BCMs [34]. It was found that the proportion of CD80+CD86+ DCs in the CFCP+US group increased from 16.70 % to 32.24 % and decreased to 20.37 % after offsetting the effect of dsDNA using Deoxyribonuclease I (DNase I) (Fig. 4B). Similar findings were obtained on bone marrow-derived dendritic cells (BMDCs) (Fig. S16). Subsequently, the activation of the cGAS-STING pathway and its role in DC maturation was verified. The western blot (WB) results revealed that the phosphorylation process of TBK1 and IRF3 was upregulated in the CFCP+US group and downregulated after the intervention with DNase I (Fig. 4C, and Fig. S17). We inferred that dsDNA as a bacterial immunogenic substance activated the cGAS-STING signaling pathway to promote DC maturation. Immunofluorescence staining of STING in DCs supported this conclusion (Fig. S18). Correspondingly, DCs in the CFCP+US group synthesized and secreted more immunomodulatory factor IFN-β. (Fig. 4D, and Fig. S19). In addition, gene expression levels of inflammatory cytokines and chemokines increased as DC matured (Fig. 4E). In particular, DCs in the CFCP+US group secreted more tumor necrosis factor-α (TNF-α) and interleukin 6 (IL-6) (Fig. 4F), which would stimulate and amplify the innate immune response [35].
Fig. 4.

Bacterial-associated dsDNA promotes DC maturation through activating STING. (A) Preparation of BCMs (biofilm-conditioned mediums) and DCMs (DC-conditioned mediums). (B) The proportion of mature DCs (CD86+CD80+) measured by flow cytometry. DNase I was used to eliminate dsDNA in BCMs. (C) Representative WB bands of proteins and their phosphorylated forms associated with the cGAS-STING pathway in different treated DCs. (D) Concentration of IFN-β secreted by DCs into mediums after co-culture with different BCMs. (E) Relative expression levels of genes related to immune activation in DCs treated with different BCMs. (F) Concentration of TNF-α and IL-6 secreted by DCs into medium after co-culture with different BCMs. (G) CD206 expression levels in DCs assessed by immunofluorescence staining. DFO is used to chelate iron ions in BCMs. Green for CD206, red for cytoskeleton, and blue for nucleus. (H) Relative fluorescence intensity of CD206 expressed by DCs in different groups. (I) The expression levels of genes associated with antigen presentation in DCs after co-culture with BCMs. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, and ns means no significance).
The mannose receptor (CD206) is an important pattern recognition and endocytosis receptor in the innate immune system involved in microorganism recognition, endocytosis, antigen presentation, and immune homeostasis maintenance [36]. Iron ions have been reported to upregulate CD206 expression [37]. The results of immunofluorescence staining showed that DCs in the CFCP+US group expressed more CD206 and were inhibited by DFO (Fig. 4G and H). Flow cytometry analysis further validated the upregulation effect of iron ions on CD206 (Fig. S20). Benefiting from the activation of the cGAS-STING pathway and the immunomodulatory effects of iron ions, the expression of genes associated with antigen presentation and T-cell migration was upregulated in CFCP+US group DCs (Fig. 4I).
In summary, CFCP+US prompted the release of bacterial-associated dsDNA, which subsequently promoted the maturation of DCs by activating the cGAS-STING pathway. Mature DCs expressed more immunomodulatory factors such as IFN-β, inflammatory factors, and chemokines. In addition, iron ions released by CFCP+US synergistically enhanced the antigen-presenting ability of DCs by increasing the expression of CD206.
2.5. IFN-β secreted by DCs mediates neutrophil immune activation
Neutrophils are the first line of innate immune system defense against bacterial infections [38]. In turn, bacterial biofilms suppress neutrophil immunological activity [39]. We demonstrated that the CFCP+US-driven bacterial-associated dsDNA promoted the secretion of IFN-β by activating the STING pathway in DCs. IFN-β has been reported to enhance the immune response of neutrophils by regulating their differentiation, activation, and migration [40]. Therefore, the culture medium of DCs from different treatment groups was collected and configured as DC conditioned mediums (DCMs) for neutrophil cultivation and identification.
We first examined the effects of different DCMs on the IFN-1 pathway in neutrophils. As shown in Fig. 5A, the genes related to the IFN-1 pathway was generally up-regulated in the CFCP+US group. Furthermore, DCMs of the CFCP+US group increased the expression of IFN-1 receptor (IFNAR1) in neutrophils (Fig. S21). Flow cytometry results showed that the percentage of activated neutrophils (CD182+CD54+) treated with DCM of the CFCP+US group increased from 39.60 % to 57.73 % (Fig. 5B). Meanwhile, the expression of intercellular adhesion factor and pro-inflammatory cytokines was significantly enhanced in neutrophils of CFCP+US group (Fig. 5C). To clarify whether IFN-β exerted specific immune activation on neutrophils, we co-cultured antibody of IFNAR1 (anti-IFNAR1) with neutrophils to block the binding of IFN-β in DCMs to IFNAR1. Subsequently, the immune activation of neutrophils was examined using flow cytometry and qPCR. Compared with the CFCP+US group, the proportion of CD54+CD182+ neutrophils decreased from 59.9 ± 2.6 % to 48.3 ± 2.8 % after antagonizing IFN-β (Figs. S22A and B). qPCR results indicated that anti-IFNAR1 reversed the up-regulation of genes related to neutrophil activation (Fig. S22C). These results suggested that IFN-β played an important role in the immune activation of neutrophils by DCs.
Fig. 5.

IFN-β secreted by DCs enhances the antibacterial immune response of neutrophils. (A) Expression levels of genes related to the IFN-1 pathway in neutrophils after co-culture with different DCMs. (B) Representative flow cytometry plots of activated neutrophils (CD54+CD182+) in different DCM co-culture groups. Dots in purple boxes represent the activated neutrophils, and the numbers refer to their proportions. (C) Expression levels of chemotaxis and inflammation-related genes in neutrophils from different groups. (D) Representative flow cytograms of ROS in neutrophils of different DCM co-culture groups. (E) Assessment of NE and cit-H3 expression in different groups of neutrophils using immunofluorescence staining. Green for NE, red for cit-H3, and blue for nucleus. (F) The fluorescence intensity of NE and cit-H3 in neutrophils co-cultured with different DCMs. (G) Concentration of NE and MPO secreted by neutrophils into mediums after co-culture with different DCMs. (H) CLSM images after co-culturing neutrophils from different DCM co-culture groups with fluorescent bacteria. Green for GFP-S. aureus, red for the neutrophil cytoskeleton, and blue for the nucleus. (I) DC-secreted IFN-β activates neutrophils and enhances their NETs, degranulation, and phagocytosis. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, and ns means no significance).
Neutrophil extracellular traps (NETs), degranulation, and phagocytosis are the main means neutrophils perform their antibacterial functions [41]. NETs are reticular fibrous structures released by neutrophils into the extracellular space, with a DNA backbone containing antimicrobial peptides and hydrolases [42]. As a potent weapon for NET release and sterilization, ROS was increased in neutrophils in the CFCP+US group (Fig. 5D). Meanwhile, a distinct NET reticulum structure was observed in the CFCP+US group, with high expression of citrullinated histone H3 (cit-H3) and neutrophil elastase (NE) (Fig. 5E and F). In addition, neutrophils in the CFCP+US group secreted more NE and myeloperoxidase (MPO), suggesting an enhanced degranulation process (Fig. 5G). Finally, we co-cultured neutrophils treated with different DCMs with bacteria and assessed their phagocytosis and bacterial killing activity. Compared to the control group, neutrophils and their NETs in the CFCP+US group captured more bacteria. For the CFCP+US+DNase I group, the phagocytosis of neutrophils was inhibited (Fig. 5H). Furthermore, in the bacterial co-culture system of the CFCP+US group, the survival rates of S. aureus and E. coli were reduced from 85.8 ± 3.4 % and 87.1 ± 4.0 % to 31.3 ± 2.3 % and 24.7 ± 9.1 %, respectively (Fig. S23). In contrast, the ability of DCM-treated neutrophils in the CFCP+US+DNase I group to kill bacteria was significantly attenuated.
Overall, the outcome of antimicrobial therapy of CFCP+US triggered the host immune response, further increasing the antimicrobial efficiency of the treatment process. Specifically, bacterial-associated dsDNA released by CFCP+US therapy activated the STING pathway in DCs and promoted their maturation. Subsequently, IFN-β secreted by DCs activated neutrophils and enhanced their innate antibacterial functions, including NETs, degranulation, and phagocytosis (Fig. 5I).
2.6. CFCP+US boosts cure of primary IRIs by evoking systemic immune response
Considering the excellent antimicrobial and immune-activating properties of CFCP+US in vitro, we further adopted a mouse model of primary IRIs to verify its therapeutic efficacy in vivo. The illustration of model construction, treatment implementation, and therapeutic efficacy evaluation was shown in Fig. 6A. Vancomycin, a routine antibiotic currently used for the prophylaxis and treatment of IRIs in clinical practice, was used as a positive control in this study [43].
Fig. 6.

CFCP+US treatment activates systemic immune responses against primary IRIs in vivo. (A) Schematic diagram of the construction and intervention of the murine primary IRI model. (B) Curves of the individual infection areas at different time points for each treatment group and the representative photograph of wounds on day 10. (C) Curves of the average infection area in different treatment groups. (D) Representative SEM images of subcutaneous implants of mice with different treatments on day 10. Yellow for S. aureus. (E) Giemsa staining results of peri-implant tissues on day 10 from different treatment groups. (F and G) Bacterial counts of implants and peri-implant tissues on day 10 as determined by SPM. (H and I) Quantification of STING and IFN-β in peri-implant tissues on day 7 with different treatments by immunohistochemical staining. (J to M) Representative flow cytometry plots of mature DCs (CD80+CD86+) by gating on CD11c+ MHC II+ cells, activated neutrophils (CD54+CD182+) by gating on CD11b+Ly6G+ cells, CD4+ T cells (CD4+) by gating on CD3+ T cells, and plasmablasts (CD138+CD19+) by gating on CD45+ cells. DCs and neutrophils were analyzed from locally infected tissues on day 7. T and B cells were analyzed from IDLNs on day 7. (N) Quantitative analysis of the corresponding immune cell population. (O) IgM and (P) IgG antibody levels in the serum of mice on day 7 from different treatment groups. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, and ns means no significance).
Throughout the observation period, the body weights of mice increased smoothly in all groups (Fig. S24). In addition, CFCP+US-treated mice showed the slightest infection signs. Specifically, the infected area in the control group continued to increase with observation time. Vancomycin-treated mice showed signs of infection control on day 5, as evidenced by a gradual decrease in the infected area. In contrast, the CFCP-treated mice showed no reduction in the area of infection, although some delay in the spread rate was achieved compared to control. Encouragingly, the infected area of the mice in the CFCP+US group consistently decreased after the first treatment on postoperative day 3. At day 10, few signs of infection were observed in CFCP+US-treated mice (Fig. 6B and C). Microbiological and histological analysis on day 10 revealed that CFCP+US eliminated biofilm structures adhering to the implant while reducing the bacterial load on the implants and in the surrounding tissues (Fig. 6D–G).
The anti-infective immune responses of mice in different treatment groups were evaluated on day 7. The enhanced expression of STING and IFN-β in the wound tissues indicated the upregulation of the STING pathway (Fig. 6H and I). Flow cytometry results showed activation of multiple immune cells in locally infected tissues and infection-draining lymph nodes (IDLNs) (Fig. 6J–N). Specifically, the proportion of mature DCs (CD80+CD86+) was increased in the infected tissues of CFCP+US treated mice, which would enhance the recognition and presentation of BAAs. The high expression of Ly6G and MPO indicated that more neutrophils were recruited to the infected area in the CFCP+US group (Fig. S25). Subsequently, neutrophils were activated (CD54+CD182+) to perform their innate immune function against bacterial infections. T cells are activated through cell-to-cell interactions when the T cell antigen receptor complex encounters peptide antigen presented by APCs [44]. More CD4+ T cells were detected in the CFCP+US-treated mice. For bacterial-associated infections, an increase in the infiltration of CD4+ T cells predicts stronger humoral immunity, while inhibiting CD4+ T cell priming limits biofilm eradication [45]. Notably, there was a small decrease in the proportion of CD8+ T cells, which may be due to the activation of more CD4+ T cells. Overall, the antimicrobial humoral immune response dominated by CD4+ T cells was enhanced. With the assistance of CD4+ T cells, antigen-sensitized B cells differentiate into plasma cells and memory B cells [46]. Plasma cells secrete a large number of specific antibodies that circulate to the infection site and exert antibacterial effects [47]. Plasma cells (CD138+CD19+) differentiation was enhanced in CFCP+US treated mice, suggesting the initiation of the antigen-specific humoral immune response. The increase of serum IgM and IgG verified the potentiating effect of CFCP+US on the antibody response (Fig. 6O and P) [48]. In addition, there was no abnormal elevation of Fe within the blood of mice in the CFCP and CFCP+US treatment groups compared with the control group (Fig. S26). No significant differences were observed in the indices of blood routine, liver function, and kidney function in mice of all treatment groups (Fig. S27).
To clarify whether CFCP+US directly affected the phenotype of immune cells surrounding the implant, we constructed a mouse model of aseptic subcutaneous implants and treated it with CFCP+US on days 3 and 5. Subsequently, we collected peri-implant tissues on day 7 and examined the phenotypes of DCs and neutrophils therein using flow cytometry. As shown in Fig. S28, there was no significant difference in the proportion of CD80+CD86+ DCs and CD54+CD182+ neutrophils in the CFCP+US and control groups. The above results validated in vivo that CFCP+US activated the systemic immune response of mice and boosted the cure of primary IRIs.
2.7. CFCP+US prevents IRI relapse and reinfection as neoadjuvant immunotherapy
In the mouse model of primary IRIs, we demonstrated that the therapeutic effect of CFCP+US was superior to that of vancomycin. However, single antibiotic therapy often fails when IRIs relapse or develop into chronic infections. Revision surgery remains the clinical gold standard for the treatment of chronic or repetitive IRIs, including immediate revision (stage I revision) and delayed revision (stage II revision) [49]. Stage I revision refers to the immediate placement of a new implant after removing the infected one. For stage II revision, regular antibiotic therapy will be administrated until the infection is well controlled before the placement of a new implant. However, both revision surgeries are challenged by high rates of relapse and reinfection due to incomplete debridement, antibiotic resistance, bacterial deposits, and immunosuppression [50]. Neoadjuvant immunotherapy has shown prospects in activating immune response before surgery to prevent postoperative relapse of infection or tumor relapse [51]. Therefore, an IRI revision model was established to explore the potential of CFCP+US as a neoadjuvant immunotherapy to address the relapse issue after revision surgery.
Specifically, titanium plates were implanted subcutaneously into mice and infected with S. aureus on day 0. Mice were randomized into four groups: Van (I), Van (II), CFCP, and CFCP+US. On day 9, mice in each group were treated preoperatively with vancomycin, vancomycin, CFCP, and CFCP+US, respectively. On day 11, mice in the Van (I), CFCP, and CFCP+US groups were subjected to stage I revision surgery, which consisted of as thorough a debridement as possible and immediate implantation of a new titanium plate. For mice in the Van (II) group, debridement on day 11 was followed by three days of vancomycin treatment and replacement with new titanium plates by day 14 (Fig. 7A). The relapse of infection in the mice was closely monitored and recorded during this period. Infection assessment showed a higher relapse rate and a larger infection area in the Van (I) and CFCP groups, while almost no relapse happened in the Van (II) and CFCP+US groups (Fig. 7B and C). Bacterial counts of the implants and surrounding tissues on day 20 similarly demonstrated that Van (II) and CFCP+US were effective in preventing the recurrence of IRIs after revision surgery (Fig. 7D and E). In addition, serum IgM and IgG antibody levels were significantly increased in the CFCP+US treatment group, which may be key to preventing the relapse of infection (Fig. 7F and G). In summary, CFCP+US achieved a comparable effect in preventing infection relapse after revision surgery with stage II revision surgery but reducing antibiotic use and surgical frequency.
Fig. 7.

CFCP+US-mediate neoadjuvant immunotherapy prevents infection relapse and re-infection after IRI revision surgery. (A) Schematic diagram of the construction and intervention of the murine relapse and re-infection IRI model. (B) Curves of individual infection areas in the relapse IRI model and representative photographs of wounds on day 20. (C) Infection relapse rates in different treatment groups. (D and E) Bacterial counts of implants and peri-implant tissues on day 20 in the relapse IRI model. (F) IgM and (G) IgG antibody levels in the serum from different treated mice on day 20. (H) Curves of individual infection areas in the re-infection IRI model and representative photographs of wounds on day 30. (I) The curves of the average infection areas in different treatment groups. (J and K) Bacterial counts of implants and peri-implant tissues on day 30 of the re-infection IRI model. (L) Immunofluorescence staining images for CD20 (B cells) and CD3/CD4 (T cells) in the spleen on day 30. (M) Representative flow cytometry plots of memory B cells (IgG+ IgD−) in IDLNs by gating on B220+ cells on day 30. (N) Quantitative analysis result of memory B cell population. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, and ns means no significance).
However, even if no natural relapse of infection occurs after revision surgery, the chances of re-infection by exogenous bacteria are multiplied [52]. With this in mind, we selected non-relapse mice in each group and subjected them to a new bacterial challenge on day 21 to assess their defenses to reinfection. The results showed that mice in the Van (I), Van (II), and CFCP groups failed to show satisfactory reinfection resistance. Although stage II revision surgery based on antibiotic treatment was effective in preventing the natural infection relapse postoperative, it did not protect against reinfection caused by exogenous bacterial challenges. In contrast, the reinfection area of mice that underwent neoadjuvant treatment with CFCP+US was significantly reduced (Fig. 7H and I). Meanwhile, the bacterial load of the implants and surrounding tissues on day 30 from the CFCP+US group was minimal, validating its greatest resistance to reinfection (Fig. 7J and K). SEM examination of implants supported this conclusion (Fig. S29A). Notably, the weakest inflammatory response was observed in the peri-implant tissues of CFCP+US-treated mice compared to the other treatment groups (Figs. S29B and C). Further, we examined immune activation in mice with different treatments. CFCP+US-treated mice showed an active immune response of B and T cells with enhanced expression of CD20 and CD3/CD4 (Fig. 7L). Meanwhile, the proportion of memory B cells was significantly increased in the CFCP+US group, suggesting activated memory humoral immune response (Fig. 7M and N). Additionally, histological analysis of the major organs (heart, liver, spleen, lung, and kidney) demonstrated that the CFCP+US treatment had a high biosafety profile (Fig. S30). These results demonstrated that the application of CFCP+US as neoadjuvant immunotherapy in preventing infection relapse and reinfection after revision surgery for IRIs was effective.
3. Conclusions
In summary, we developed a nanoadjuvant with enhanced SDT effects to treat repetitive IRIs by evoking the systemic antimicrobial immune response. Specifically, the 1O2 generated by CFCP under US irradiation coupled with interference of iron ions on bacterial metabolism increased BAA exposure. The dsDNA released from destroyed biofilm and bacteria promoted the maturation of DCs by activating the cGAS-STING pathway. IFN-β secreted by DCs enhanced the innate antimicrobial immune response of neutrophils to eliminate bacterial infections through NETs, degranulation, and phagocytosis. In addition, iron ions increased the expression of CD206 on DCs, which further promoted the presentation of BAAs. DC-presented BAA continuously activated B and T cells and hastened the curing of IRIs by secreting bacteria-specific antibodies and enhancing the immune memory effect. It was encouraging that CFCP+US was not only effective in treating primary IRI, but also as a neoadjuvant immunotherapy to prevent recurrence of infection and reinfection after revision surgery.
4. Materials and methods
4.1. Materials
Methanol, Acetone, 1,3,5-Tris(4-aminophenyl) benzene (TAPB, CAS: 118727-34-7), Benzene-1,3,5-tricarbaldehyde (BTC, CAS: 3163-76-6), Glacial acetic acid (CAS: 64-19-7), Acetonitrile (CAS: 75-05-8), Chloroplatinic acid hexahydrate (H2PtCl6.6H2O, CAS: 16941-12-1), Sodium borohydride (NaBH4, CAS:16940-66-2), Iron (III) chloride hexahydrate (FeCl3.6H2O, CAS: 10025-77-1). All the materials were purchased from Aladdin Reagent Co. (Shanghai, China).
4.2. Synthesis of CFCP
Preparation of COF. 30 ml of methanol was stirred vigorously with 30 ml of ethanol at room temperature (RT) for 10 min. Subsequently, 205 mg of TAPB and 95 mg of BTC were added and sonicated at low temperatures to aid dissolution. After the complete dissolution of TAPB and BTC, 6 ml of glacial acetic acid was added and stirred vigorously for 15 min. The product obtained was washed with methanol, acetone, and DI-water in turn by centrifugation (13000 rpm, 20 min) and dried under vacuum to obtain COF for later use.
Synthesis of CF. 10 mg of COF was dispersed in 5 ml of DI water, followed by adding 0.1 ml of 20 mg/ml of aqueous FeCl3.6H2O. After stirring for 24 h at RT, the product obtained was washed by centrifugation with DI-water (13000 rpm, 20 min) and dried under vacuum to obtain CF for later use.
Loading of curcumin. 10 mg of CF was dispersed in 5 ml of DI-water, followed by the addition of 1 ml of 1 mg/ml of ethanolic solution of curcumin. After stirring for 24 h at RT, the product obtained was washed by centrifugation with DI-water (13000 rpm, 20 min) and dried under vacuum to obtain CFC for later use.
Preparation of CFCP. 10 mg of CFC was dispersed in 5 ml of DI-water. Subsequently, 1 ml of 20 mM aqueous H2PtCl6 was added and stirred at a low temperature for 1 h. Then 10 ml of 4 mg/ml of NaBH4 was added and the reaction was carried out at low temperature for 3 h. The product obtained was washed by centrifugation with DI-water (13000 rpm, 20 min) and dried under vacuum to give CFCP.
4.3. Characterization of CFCP
A TEM (JEOL-1400 PLUS, Japan) equipped with an EDS was used to observe the morphology and elemental distribution of the nanoparticles (NPs) at 120 KV. DLS (ZS-100, Horiba) was used to collect the hydrodynamic diameter and zeta potential data of various NPs. FTIR spectra were recorded by a VERTEX70V FTIR spectrometer (Bruker, Germany). XRD patterns were scanned from 10° to 80° on a Rigaku automated X-ray diffractometer (Smartlab, Rigaku Co. Ltd., Japan). XPS (Thermo Scientific K-Alpha, USA) analysis was performed to determine the elemental types and valence states. The stability of CFCP was analyzed based on UV–visible absorption spectra (PerkinElmer Lambda 750, USA) in different solution systems. O2 was detected using a portable dissolved oxygen meter (HACH, USA). US therapeutic apparatus (Nu-Tek, China) was used as the irradiation source for implementing sonodynamic therapy (1 MHZ frequency, 1 W/cm2 power, and 50 % duty cycle).
Loading efficiency and release characteristics of curcumin. A series of standard solutions of curcumin in methanol were prepared at mass concentrations of 2, 4, 6, and 8 μg/ml. The absorbance of different standard solutions was determined using UV–vis spectrophotometer (PerkinElmer Lambda 750, USA). The regression equation was calculated between mass concentration and absorbance at maximum absorption wavelength. To calculate the loading efficiency of curcumin, 1 mg of CFCP was completely dissolved in 10 ml of methanol and the absorbance of the supernatant was determined using UV–vis spectrophotometer (PerkinElmer Lambda 750, USA). The amount of curcumin and loading efficiency were calculated from the standard curve. To test the release characteristics of curcumin, 1 mg of CFCP was dissolved in 10 ml of PBS buffer of different pH values and then pipetted into a dialysis bag of 3500 u cut-off molecular mass. The dialysis bag was immersed in 90 ml of PBS buffer of the corresponding pH. 1 ml of supernatant was taken out at regular intervals and replenished with 1 ml of PBS buffer of the corresponding pH. The absorbance of the supernatant at different time points was measured and the cumulative release rate of curcumin was calculated from the standard curve.
Evaluation of SDT in vitro. DPBF (MedChemExpress, US) was used as an indicator to detect the generation of 1O2. Briefly, DMSO (20 μL) containing DPBF (10 mM) was added to 2 ml of PBS, CFC (100 μg/ml), or CFCP (100 μg/ml) in the presence of H2O2 (20 mM). After being transferred into a cuvette, the various diverse solutions were exposed to US irradiation, followed by the record with UV–vis spectrophotometer (PerkinElmer Lambda 750, USA) every 2 min. In addition, the 1O2 generation was detected using 4-Amino-2,2,6,6-tetramethylpiperidine (TEMP) as a trapping agent for ESR spectroscopy measurements. Various groups were set as follows: US irradiation, CFC+US, and CFCP+US. After corresponding treatments, the solution (180 μL) in each group was mixed with TEMP solution (20 μL, 1 M) and transferred to a quartz capillary tube. The ESR signals were recorded at RT using an ESR spectrometer (Bruker EMXplus, Germany).
4.4. In vitro biocompatibility assessment
The cytotoxicity of CFCP to DCs and neutrophils was assessed using the CCK-8 kits (Beyotime, China). Briefly, cells were inoculated into 96-well plates (1 × 104 per well) and cultured overnight. Different concentrations of CFCP (0, 20, 30, 50, 100, and 200 μg/ml) were added to the culture medium, and incubation was continued for 24 h. Subsequently, cells were washed with PBS to remove the excess NPs and incubated with a DMEM medium containing 10 % CCK-8 reagent for 30 min. Finally, the absorbance of each culture system was measured at 450 nm using a microplate reader (BioTek ELX800, USA).
The hemolysis assay was used to assess the hemocompatibility of CFCP. Fresh blood from mice was collected, centrifuged, and resuspended in a PBS solution containing heparin. After the different treatments, the blood cell suspension was incubated at 37 °C for 2 h. The supernatant was collected and the absorbance at 540 nm was measured using a microplate reader (BioTek ELX800, USA).
4.5. In vitro anti-biofilm infection evaluation
Monoclonal strains of S. aureus (ATCC 43300) and E. coli (ATCC 35218) were picked from blood agar plates and incubated in Tryptic Soy Broth (TSB, Solarbio, China) at 37 °C for 8 h. Bacterial suspensions were diluted to 1 × 106 CFU/ml and inoculated in 24-well plates at 1 ml per well. After incubation at 37 °C for 24 h, the biofilms were obtained and divided into six groups: Control (no treatment), US (8 min of US irradiation), CFC (100 μg/ml of CFC), CFC+US (100 μg/ml of CFC + 8 min of US irradiation)), CFCP (100 μg/ml of CFCP), and CFCP+US (100 μg/ml of CFCP + 8 min of US irradiation). The biofilms were treated accordingly and incubated at 37 °C for another 6 h.
The anti-biofilm effect of CFCP+US was systematically evaluated, including CLSM, SEM, crystalline violet staining, and SPM. Specifically, after incubation with LIVE/DEAD BacLight Bacterial Viability Kits (Invitrogen, USA) for 30 min, the biofilm structure was observed and reconstructed using CLSM (ZEISS LSM 710, Germany). For SEM, biofilms were fixed in 2.5 % glutaraldehyde (Servicebio, China) overnight, dehydrated with different concentrations of ethanol solution (50 %, 60 %, 70 %, 80 %, 90 %, 95 %, and 100 % for 10 min each), and freeze-dried for 4 h. The samples were sprayed with gold and finally observed using SEM (JEOL JSM-6310LV, Japan). In addition, biofilms were fixed with 99 % methanol and stained with 0.1 % crystalline violet dye (Beyotime, China) for 10 min. The stained biofilm was dissolved with 33 % acetic acid and the absorbance at 595 nm was measured using a microplate reader (BioTek ELX800, USA). To detect the bacterial load in bacterial biofilms, the biofilms were diluted in a gradient and spread evenly on blood agar plates. Individual colonies were counted after the plates were incubated overnight in a 37 °C incubator (Thermo, USA).
The anti-biofilm mechanisms of CFCP+US were further explored. To detect the bacterial membrane potential and ROS levels, single bacterial suspensions were obtained by resuspending biofilms from different treatment groups. The bacteria were incubated with the BacLight™ Bacterial Membrane Potential Kit (Invitrogen, USA) and the ROS probe DCFH-DA (Beyotime, China) for 30 min, respectively. The stained bacteria were washed with PBS and examined using a flow cytometer (Beckman Coulter, USA). In addition, single bacterial suspensions were prepared and fixed with 1 % osmium tetroxide for 2 h. The bacteria were dehydrated in a gradient concentration of ethanol and embedded in acetone and resin. 60 nm thick sections were prepared, stained with 2 % uranyl acetate, and observed using TEM (JEOL 1200Ex, Japan) at 80 KV. The ONPG hydrolysis test was used to assess the permeability of bacterial membranes. Specifically, the biofilm suspension was mixed 1:1 with the prepared ONPG solution (0.75 M in NaH2PO4 buffer, pH = 7) an incubated at 37 °C for 1 h. The absorbance of the reaction solution at 420 nm was then measured using a microplate reader (Bio-TekELx800, USA). Finally, iron quantification kits (Merck, USA) and BCA kits (Beyotime, China) were used to determine the iron content and protein content within biofilm bacteria respectively. And their ratios were considered standardized intra-bacterial iron concentrations.
4.6. Transcriptomic analysis and validation of bacterial biofilms
S. aureus biofilms were collected after different treatments and immediately stored in liquid nitrogen. The samples were detected for transcriptomic analysis with the mass spectrometry platform of OE Biotech (Shanghai, China). The data obtained were further analyzed using OE Cloud (OE Biotech) software.
qPCR was used to validate the expression levels of genes that were significantly regulated in the transcriptomics analysis results. Briefly, total S. aureus RNA was extracted from biofilms using a bacterial RNA extraction kit (Vazyme, China). cDNA was synthesized using a reverse transcription kit (EZBioscience, USA) after the determination of RNA concentration. cDNA obtained was subsequently amplified using SYBR Green Master Mix (EZBioscience, USA) and a LightCycler 480 (Roche, USA). The 16s RNA was selected as the housekeeping gene and the primers used were listed in Table S1.
Finally, the glucose metabolism of S. aureus in biofilms was measured using the Glucose Assay Kit (Beyotime, China), Pyruvate Assay Kit (Solarbio, China), ATP Assay Kit (Beyotime, China), and NAD+/NADH Assay Kit (Beyotime, China), respectively.
4.7. In vitro DC maturation and function assay
DC 2.4 cells (CL-0545) and BMDCs (CP-M151A) were purchased from Pricella (Wuhan, China) and cultured in a constant temperature incubator at 37 °C with 5 % CO2. The BCMs was prepared to culture DCs. Briefly, supernatants of S. aureus biofilms from different treatment groups were collected, centrifuged (12000 g, 10 min), and filtered using 0.22 μm filters (Merck, Germany). The dsDNA concentration in the supernatant was determined using a dsDNA quantification kit (Yeason, China). Subsequently, the filtered biofilm supernatant was mixed 1:2 with DMEM medium (Gibco, US), followed by the addition of 10 % fetal bovine serum (FBS, Gibco, US) and 1 % penicillin/streptomycin (Gibco, US). When necessary, 50 μg/ml of DNase I (Merck, Germany) or 100 μM of DFO (Merck, Germany) were used to eliminate dsDNA or Fe in BCMs. Notably, DCs cultured in BCMs prepared with untreated biofilm supernatant were used as a control.
For WB, RIPA cell lysate (EpiZyme, China) was used to extract DC protein. The protein concentration was determined using the BCA kit (EpiZyme, China) and adjusted to 1 mg/ml for sample loading. After electrophoresis using the Bio-Rad system, the proteins on the gel were transferred to the PVDF membrane (EpiZyme, China). The membrane was blocked with 5 % BSA solution (EpiZyme, China) at RT for 30 min and incubated with the primary antibody at 4 °C overnight. The primary antibody was recovered and the membrane was rinsed, followed by incubation with the corresponding HRP-conjugated secondary antibody at RT for 1 h. Finally, protein bands were detected using chemiluminescent reagents (EpiZyme, China) and the imaging system (ImageQuant 800, Amersham), and their relative expression levels were quantified using ImageJ software. The WB antibodies used in this study were shown in Table S2.
For flow cytometry, DCs from different groups were collected and co-incubated with fluorescein-labeled flow cytometry antibody on ice for 30 min. After recovering the antibodies and washing the cells, the samples were examined using a flow cytometer (Beckman Coulter, USA), and the data were analyzed using Flowjo V10. The antibodies used in this test were shown in Table S3.
For qPCR, total cellular RNA of different treated DCs was extracted using an RNA extraction kit (EZBioscience, USA). Subsequently, the RNA concentrations were measured with a Nanodrop 2000 (Thermo, USA) and the cDNA was synthesized using a reverse transcription kit (EZBioscience, USA). Finally, qPCR was carried out using SYBR Green Master Mix (EZBioscience, USA) and a LightCycler 480 (Roche, USA). The gene primers used in this test were shown in Table S1.
For ELISA, the DC medium was collected and centrifuged (12000 g, 10 min) to obtain the supernatant. Subsequently, ELISA kits (Dakewe Biotech, China) were used to determine the corresponding cytokine levels in the medium supernatant.
For immunofluorescent staining, DCs were fixed in 4 % paraformaldehyde for 10 min and blocked with Immunostaining Blocking Solution (Beyotime, China) at RT for 30 min. Subsequently, cells were co-incubated with Anti-Mannose Receptor/CD206 Rabbit pAb (GB113497, Servicebio, China) at 4 °C overnight. After removing the primary antibody, cells were incubated with Alexa Fluor® 488-conjugated Goat Anti-Rabbit IgG (H+L) (GB25303, Servicebio, China) at RT for 1 h. Actin-Tracker Red-Rhodamine (Beyotime, China) and DAPI (Beyotime, China) were used to label the cytoskeleton and nuclei, respectively. Finally, samples were observed with a CLSM (ZEISS LSM 710, Germany) and the fluorescence intensity was determined with a fluorescent microplate reader (BioTek, USA).
4.8. In vitro neutrophil activation and function assay
Neutrophils were isolated from mouse bone marrow using the EasySep™ Mouse Neutrophil Enrichment Kit (STEMCELL Technologies, Canada) according to the manufacturer's instructions. The DCMs were prepared for neutrophil culture. Briefly, medium supernatants from different groups of DCs were collected, filtered, and mixed 1:2 with DMEM medium (Gibco, US), followed by the supplementation of 10 % FBS (Gibco, US) and 1 % penicillin/streptomycin (Gibco, US). The cell culture conditions were as described previously. For IFN-β antagonism experiments, neutrophils were pre-incubated with 2 μg/ml of anti-IFNAR1 antibody (Cat. abs111649, absin, China) for 2 h before culturing with DCMs.
After 4 h of incubation, the gene expression levels related to IFN-1 pathway and inflammation-associated cytokines in neutrophils was examined using the qPCR assay. The proportion of activated neutrophils was detected through flow cytometry after co-incubation with APC-labeled anti-CD54 and PE/Cy7 labeled anti-CD182 antibodies. ROS levels within neutrophils were measured using flow cytometry after co-incubation with the ROS probe DCFH-DA (Beyotime, China) for 30 min. To assess the ability to form NETs, neutrophils were co-incubated with anti-cit-H3 and anti-NE primary antibodies overnight, labeled with fluorescent secondary antibodies, and examined under a CLSM (ZEISS LSM 710, Germany). A fluorescent microplate reader (BioTek, USA) was used to quantify their fluorescent intensity. The content of NE and MPO in the cell culture medium was measured by ELISA kits (Cloud-Clone Corp., USA) to assess the ability of neutrophils to degranulate. In addition, neutrophils after different treatments were co-incubated with GFP-S. aureus (Cell: Bacteria = 5:1) for 2 h and then washed with PBS and vancomycin solution to remove the bacteria that had not been phagocytosed. The cytoskeleton and nucleus were labeled with Actin-Tracker Red-Rhodamine (Beyotime, China) and DAPI (Beyotime, China), respectively. Finally, the phagocytosis of bacteria by neutrophils was observed using CLSM (ZEISS LSM 710, Germany). To assess their bactericidal capacity, neutrophils were lysed using 0.1 % Triton-X100 and mixed with the culture supernatant. The mixed bacterial solution was diluted and spread to blood agar plates. Single colonies were counted after incubation at 37 °C overnight.
4.9. Primary murine subcutaneous IRI model
48 male ICR mice (8–10 weeks old) were selected to establish the model of primary IRIs. Briefly, mice were anesthetized with 1 % pentobarbital (10 μL/g), shaved on the back, and disinfected with 75 % ethanol. Sterile titanium pieces (8 mm diameter) were implanted subcutaneously through a transverse dorsal incision. A 100 μL of S. aureus bacterial suspension (1 × 106 CFU/ml) was injected around the titanium pieces and the incision was then sutured. Afterward, mice were randomly divided into four groups (12 mice each) and treated with local injections on days 3 and 5 postoperative: Control group (100 μL of saline), Van group (2 mg/kg of vancomycin), CFCP group (2 mg/kg of CFCP), CFCP+US group (2 mg/kg of CFCP + 8 min of US irradiation). The wound infection area was measured using calipers regularly. On postoperative days 7 and 10, partial mice from each group were executed and their blood, implants, peri-implant tissues, and IDLNs were collected.
Implants, peri-implant tissues, and blood collected on day 10 were used primarily for treatment efficacy and biosafety assessment. The implants were fixed, dehydrated, lyophilized, and finally observed for residual bacteria using SEM (JEOL JSM-6310LV, Japan). Equal weights of peri-implant tissues were homogenized with a tissue homogenizer (KZ-III-FP, Servicebio), while the implants were sonicated and vortexed in sterile PBS (sonication for 5 min, vortex for 30 s, 3 cycles). The bacterial load in the tissue homogenate and implant rinse were quantified using SPM. In addition, the blood was used for Fe, blood routine, and blood biochemistry tests.
Peri-implant tissues, IDLNs, and blood collected on Day 7 were used primarily for immunological testing. Partial peri-implant tissues were fixed, dehydrated, embedded, sectioned, and stained to assess the activation of the immune response. Primary antibodies of STING (Cat.ab288157, Abcam), IFN-β (Cat.PRS4243, Merck), Ly6G (Cat.GB11229, Servicebio), and MPO (Cat.GB11224, Servicebio) were used in this study. Flow cytometry was used to detect the activation of immune cells in peri-implant tissues and IDLNs. Specifically, freshly collected tissues and IDLNs was cut into small pieces and digested with a mixture of enzymatic solutions at 37 °C for 30 min, including 1 mg/ml of collagenase I (Merck, Germany), 250 μg/ml of collagenase IV (Gibco, US), and 50 μg/ml of deoxyribonuclease (Merck, Germany). Single-cell suspensions were obtained after filtration through 70 μm filters (Biosharp, China) and the erythrocytes were removed using 1 ml of ACK lysis buffer (Biosharp, China). After washing with PBS, the samples were co-incubated with anti-CD16/32 antibodies on ice for 15 min to block the Fc receptor. The blocked immune cells were incubated with fluorescein-labeled flow cytometry antibodies on ice for 30 min. The target cells were gated using flow cytometry for analysis according to the strategies shown in Fig. S31. Detailed information of flow cytometry antibodies was listed in Table S3. In addition, antibody levels (IgM and IgG) in the blood were detected using ELISA kits (Beyotime, China) according to manufacturer's instructions.
4.10. Relapse murine subcutaneous IRI model
48 male ICR mice (8–10 weeks old) were used to establish the model of relapse IRIs. Titanium pieces were implanted subcutaneously and infected with bacteria, as described previously. Mice were randomly divided into four groups and treated with local injection on day 9: Van (I) group (2 mg/kg of vancomycin), Van (II) group (2 mg/kg of vancomycin), CFCP group (2 mg/kg of CFCP), and CFCP+US group (2 mg/kg of CFCP + 8 min of US irradiation). After removing the infected titanium pieces on day 11, the necrotic tissue was excised and the wound was irrigated in a saline-hydrogen peroxide-saline-vancomycin sequence. In the Van (I), CFCP, and CFCP+US groups, new titanium pieces were implanted immediately after debridement and the incision was closed. For the Van (II) group, the wound was treated with 2 mg/kg of vancomycin (once a day for three days) before implanting the new titanium piece. On day 20, some of the mice were sacrificed and the titanium pieces as well as the surrounding tissues were collected for infection assessment through SPM. Meanwhile, blood levels of IgG and IgM were detected using ELISA kits (Beyotime, China). Subsequently, mice with no visible recurrence of infection were given a new injection of S. aureus suspension (1 × 105 CFU/ml) on day 21. The area of infection was measured and recorded in the same manner. On day 30, the remaining mice were sacrificed and the required samples were collected for further testing and analysis, including titanium pieces, surrounding tissues, IDLNs, and main organs (heart, liver, spleen, lung, and kidney). Specifically, SPM and SEM were used to detect the bacterial load in titanium pieces and surrounding tissues. Peri-implant tissue homogenates were obtained and the levels of inflammatory factors (TNF-α and IL-1β) were measured using ELISA kits (Dakewe Biotech, China). Flow cytometry was used to assess the activation of memory B cells in IDLNs. In addition, histological analysis of the major organs was used to assess the in vivo biocompatibility of CFCP.
4.11. Statistical analysis
All data were obtained from replicated independent experiments and expressed as mean ± standard deviation. The data obtained were analyzed and plotted using Microsoft Office 2016 and GraphPad Prism 8 software. One-way ANOVA and Student's t-test were used to determine statistical significance. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. p < 0.05 were considered statistically significant.
Ethics approval and consent to participate
Research involving animals was approved by the Ethics Committee of the First Affiliated Hospital of USTC (approval number: 2023 N(A)-0126).
Conflict of interest
The authors declare no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Dongdong Xu: Writing – original draft, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Jun Hu: Methodology, Investigation, Formal analysis, Data curation. Jiawei Mei: Methodology, Investigation, Formal analysis, Data curation. Jun Zhou: Methodology. Zhengxi Wang: Data curation. Xudong Zhang: Software. Quan Liu: Methodology. Zheng Su: Supervision, Resources, Data curation. Wanbo Zhu: Writing – review & editing, Supervision, Formal analysis. Hongjian Liu: Supervision, Resources, Formal analysis, Conceptualization. Chen Zhu: Writing – review & editing, Resources, Project administration, Funding acquisition.
Footnotes
Peer review under responsibility of KeAi Communications Co., Ltd.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioactmat.2024.01.020.
Contributor Information
Zheng Su, Email: suz924@ustc.edu.cn.
Wanbo Zhu, Email: zhuwanbo@sjtu.edu.cn.
Hongjian Liu, Email: fccliuhj@zzu.edu.cn.
Chen Zhu, Email: zhuchena@ustc.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Arciola C.R., Campoccia D., Montanaro L. Implant infections: adhesion, biofilm formation and immune evasion. Nat. Rev. Microbiol. 2018;16:397–409. doi: 10.1038/s41579-018-0019-y. [DOI] [PubMed] [Google Scholar]
- 2.Lebeaux D., Ghigo J.M., Beloin C. Biofilm-related infections: bridging the gap between clinical management and fundamental aspects of recalcitrance toward antibiotics. Microbiol. Mol. Biol. Rev. : MMBR (Microbiol. Mol. Biol. Rev.) 2014;78:510–543. doi: 10.1128/MMBR.00013-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Song L., Lu L., Pu Y., Yin H., Zhang K. Nanomaterials-based tumor microenvironment modulation for magnifying sonodynamic therapy. Acc. Mater. Res. 2022;3:971–985. [Google Scholar]
- 4.Wang D., Zhang M., Zhang Y., Qiu G., Chen J., Zhu X., Kong C., Lu X., Liang X., Duan L., Fang C., Liu J., Zhang K., Luo T. Intraparticle double‐scattering‐decoded sonogenetics for augmenting immune checkpoint blockade and CAR‐T therapy. Adv. Sci. 2022;9 doi: 10.1002/advs.202203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang X., Zhao Q., Yang J., Wang T., Chen F., Zhang K. Tumor microenvironment-triggered intratumoral in-situ biosynthesis of inorganic nanomaterials for precise tumor diagnostics. Coord. Chem. Rev. 2023;484 [Google Scholar]
- 6.Zhang Y., Du D., Fang C., Yu X., Fang Y., Liu X., Ou D., Yin H., Liu H., Wang T., Lu L., Li X., Zhang K. Fundamental Research; 2022. Epigenetics Disruptions Enabled by Porphyrin-Derived Metal-Organic Frameworks Disarm Resistances to Sonocatalytic ROS Anti-tumor Actions. [Google Scholar]
- 7.Zhang Y., Fang C., Zhang W., Zhang K. Emerging pyroptosis-engineered nanobiotechnologies regulate cancers and inflammatory diseases: a double-edged sword. Matter. 2022;5:3740–3774. [Google Scholar]
- 8.Zhou J., Fang C., Rong C., Luo T., Liu J., Zhang K. Reactive oxygen species-sensitive materials: a promising strategy for regulating inflammation and favoring tissue regeneration. Smart Materials in Medicine. 2023;4:427–446. [Google Scholar]
- 9.Wang Z., Liu X., Duan Y., Huang Y. Infection microenvironment-related antibacterial nanotherapeutic strategies. Biomaterials. 2022;280 doi: 10.1016/j.biomaterials.2021.121249. [DOI] [PubMed] [Google Scholar]
- 10.Azharuddin M., Zhu G.H., Sengupta A., Hinkula J., Slater N.K.H., Patra H.K. Nano toolbox in immune modulation and nanovaccines. Trends Biotechnol. 2022;40:1195–1212. doi: 10.1016/j.tibtech.2022.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu D., Zhu W., Ding C., Mei J., Zhou J., Cheng T., Guo G., Zhang X. Self-homeostasis immunoregulatory strategy for implant-related infections through remodeling redox balance. ACS Nano. 2023;17:4574–4590. doi: 10.1021/acsnano.2c10660. [DOI] [PubMed] [Google Scholar]
- 12.Raafat D., Otto M., Reppschläger K., Iqbal J., Holtfreter S. Fighting Staphylococcus aureus biofilms with monoclonal antibodies. Trends Microbiol. 2019;27:303–322. doi: 10.1016/j.tim.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Del Prete A., Salvi V., Soriani A., Laffranchi M., Sozio F., Bosisio D., Sozzani S. Dendritic cell subsets in cancer immunity and tumor antigen sensing. Cell. Mol. Immunol. 2023;20:432–447. doi: 10.1038/s41423-023-00990-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sabado R.L., Balan S., Bhardwaj N. Dendritic cell-based immunotherapy. Cell Res. 2017;27:74–95. doi: 10.1038/cr.2016.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao L., Tian H., Fang M., Xu Z., Tang D., Chen J., Yin J., Xiao H., Shang K., Han H., Li X. Activating cGAS-STING pathway with ROS-responsive nanoparticles delivering a hybrid prodrug for enhanced chemo-immunotherapy. Biomaterials. 2022;290 doi: 10.1016/j.biomaterials.2022.121856. [DOI] [PubMed] [Google Scholar]
- 16.Lv M., Chen M., Zhang R., Zhang W., Wang C., Zhang Y., Wei X., Guan Y., Liu J., Feng K., Jing M., Wang X., Liu Y.C., Mei Q., Han W., Jiang Z. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res. 2020;30:966–979. doi: 10.1038/s41422-020-00395-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Z., Chu Z., Yang J., Qian H., Xu J., Chen B., Tian T., Chen H., Xu Y., Wang F. Immunogenic cell death augmented by manganese zinc sulfide nanoparticles for metastatic melanoma immunotherapy. ACS Nano. 2022;16:15471–15483. doi: 10.1021/acsnano.2c08013. [DOI] [PubMed] [Google Scholar]
- 18.Li Y., Huang C., Xu Y. Colon cancer exosome-derived biomimetic nanoplatform for curcumin-mediated sonodynamic therapy and calcium overload. Front. Bioeng. Biotechnol. 2022;10 doi: 10.3389/fbioe.2022.1069676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin A., Sun Z., Xu X., Zhao S., Li J., Sun H., Wang Q., Jiang Q., Wei H., Shi D. Self-cascade uricase/catalase mimics alleviate acute gout. Nano Lett. 2022;22:508–516. doi: 10.1021/acs.nanolett.1c04454. [DOI] [PubMed] [Google Scholar]
- 20.Wang D., Lin L., Li T., Meng M., Hao K., Guo Z., Chen J., Tian H., Chen X. Etching bulk covalent organic frameworks into nanoparticles of uniform and controllable size by the molecular exchange etching method for sonodynamic and immune combination antitumor therapy. Adv. Mater. 2022;34 doi: 10.1002/adma.202205924. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y., Shi L., Su L., van der Mei H.C., Jutte P.C., Ren Y., Busscher H.J. Nanotechnology-based antimicrobials and delivery systems for biofilm-infection control. Chem. Soc. Rev. 2019;48:428–446. doi: 10.1039/c7cs00807d. [DOI] [PubMed] [Google Scholar]
- 22.Mikuláss K.R., Nagy K., Bogos B., Szegletes Z., Kovács E., Farkas A., Váró G., Kondorosi É., Kereszt A. Antimicrobial nodule-specific cysteine-rich peptides disturb the integrity of bacterial outer and inner membranes and cause loss of membrane potential. Ann. Clin. Microbiol. Antimicrob. 2016;15:43. doi: 10.1186/s12941-016-0159-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benarroch J.M., Asally M. The microbiologist's guide to membrane potential dynamics. Trends Microbiol. 2020;28:304–314. doi: 10.1016/j.tim.2019.12.008. [DOI] [PubMed] [Google Scholar]
- 24.Miyata S.T., Kitaoka M., Brooks T.M., McAuley S.B., Pukatzki S. Vibrio cholerae requires the type VI secretion system virulence factor VasX to kill Dictyostelium discoideum. Infect. Immun. 2011;79:2941–2949. doi: 10.1128/IAI.01266-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dai W., Shu R., Yang F., Li B., Johnson H.M., Yu S., Yang H., Chan Y.K., Yang W., Bai D., Deng Y. Deerfield Beach, Fla.; 2023. Engineered Bio-Heterojunction Confers Extra- and Intracellular Bacterial Ferroptosis and Hunger-Triggered Cell Protection for Diabetic Wound Repair, Advanced Materials. [DOI] [PubMed] [Google Scholar]
- 26.Locher K.P. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat. Struct. Mol. Biol. 2016;23:487–493. doi: 10.1038/nsmb.3216. [DOI] [PubMed] [Google Scholar]
- 27.Kaila V.R.I., Wikström M. Architecture of bacterial respiratory chains. Nat. Rev. Microbiol. 2021;19:319–330. doi: 10.1038/s41579-020-00486-4. [DOI] [PubMed] [Google Scholar]
- 28.Golonka R., Yeoh B.S., Vijay-Kumar M. The iron tug-of-war between bacterial siderophores and innate immunity. J. Innate Immun. 2019;11:249–262. doi: 10.1159/000494627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ellermann M., Arthur J.C. Siderophore-mediated iron acquisition and modulation of host-bacterial interactions. Free Radic. Biol. Med. 2017;105:68–78. doi: 10.1016/j.freeradbiomed.2016.10.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin L., Chen H., Zhao R., Zhu M., Nie G. Nanomedicine targets iron metabolism for cancer therapy. Cancer Sci. 2022;113:828–837. doi: 10.1111/cas.15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harrison A.V., Lorenzo F.R., McClain D.A. Iron and the pathophysiology of diabetes. Annu. Rev. Physiol. 2023;85:339–362. doi: 10.1146/annurev-physiol-022522-102832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sandoval-Acuña C., Torrealba N., Tomkova V., Jadhav S.B., Blazkova K., Merta L., Lettlova S., Adamcová M.K., Rosel D., Brábek J., Neuzil J., Stursa J., Werner L., Truksa J. Targeting mitochondrial iron metabolism suppresses tumor growth and metastasis by inducing mitochondrial dysfunction and mitophagy. Cancer Res. 2021;81:2289–2303. doi: 10.1158/0008-5472.CAN-20-1628. [DOI] [PubMed] [Google Scholar]
- 33.Chandel N.S. Glycolysis, Cold Spring Harbor perspectives in biology. 2021;13 doi: 10.1101/cshperspect.a040535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dilioglou S., Cruse J.M., Lewis R.E. Function of CD80 and CD86 on monocyte- and stem cell-derived dendritic cells. Exp. Mol. Pathol. 2003;75:217–227. doi: 10.1016/s0014-4800(03)00072-8. [DOI] [PubMed] [Google Scholar]
- 35.Hunter C.A., Jones S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015;16:448–457. doi: 10.1038/ni.3153. [DOI] [PubMed] [Google Scholar]
- 36.Layek B., Lipp L., Singh J. APC targeted micelle for enhanced intradermal delivery of hepatitis B DNA vaccine. J. Contr. Release : official journal of the Controlled Release Society. 2015;207:143–153. doi: 10.1016/j.jconrel.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 37.Camiolo G., Barbato A., Giallongo C., Vicario N., Romano A., Parrinello N.L., Parenti R., Sandoval J.C., García-Moreno D., Lazzarino G., Avola R., Palumbo G.A., Mulero V., Li Volti G., Tibullo D., Di Raimondo F. Iron regulates myeloma cell/macrophage interaction and drives resistance to bortezomib. Redox Biol. 2020;36 doi: 10.1016/j.redox.2020.101611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Németh T., Sperandio M., Mócsai A. Neutrophils as emerging therapeutic targets, Nature reviews. Drug discovery. 2020;19:253–275. doi: 10.1038/s41573-019-0054-z. [DOI] [PubMed] [Google Scholar]
- 39.Bonavia A., Miller L., Kellum J.A., Singbartl K. Hemoadsorption corrects hyperresistinemia and restores anti-bacterial neutrophil function. Intensive care medicine experimental. 2017;5:36. doi: 10.1186/s40635-017-0150-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng Y., Wu X., Zhang S., Deng C., Zhao L., Wang M., Wu Q., Yang H., Zhou J., Peng L., Luo X., Chen Y., Wang A., Xiao Q., Zhang W., Zhao Y., Zeng X., Fei Y. The potential roles of type I interferon activated neutrophils and neutrophil extracellular traps (NETs) in the pathogenesis of primary Sjögren's syndrome. Arthritis Res. Ther. 2022;24:170. doi: 10.1186/s13075-022-02860-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burn G.L., Foti A., Marsman G., Patel D.F., Zychlinsky A. The neutrophil. Immunity. 2021;54:1377–1391. doi: 10.1016/j.immuni.2021.06.006. [DOI] [PubMed] [Google Scholar]
- 42.Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018;18:134–147. doi: 10.1038/nri.2017.105. [DOI] [PubMed] [Google Scholar]
- 43.Steinmetz T., Eliakim-Raz N., Goldberg E., Leibovici L., Yahav D. Association of vancomycin serum concentrations with efficacy in patients with MRSA infections: a systematic review and meta-analysis. Clin. Microbiol. Infection : the official publication of the European Society of Clinical Microbiology and Infectious Diseases. 2015;21:665–673. doi: 10.1016/j.cmi.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 44.Acuto O., Cantrell D. T cell activation and the cytoskeleton. Annu. Rev. Immunol. 2000;18:165–184. doi: 10.1146/annurev.immunol.18.1.165. [DOI] [PubMed] [Google Scholar]
- 45.Mathiasen S.L., Gall-Mas L., Pateras I.S., Theodorou S.D.P., Namini M.R.J., Hansen M.B., Martin O.C.B., Vadivel C.K., Ntostoglou K., Butter D., Givskov M., Geisler C., Akbar A.N., Gorgoulis V.G., Frisan T., Ødum N., Krejsgaard T. Bacterial genotoxins induce T cell senescence. Cell Rep. 2021;35 doi: 10.1016/j.celrep.2021.109220. [DOI] [PubMed] [Google Scholar]
- 46.Crotty S. Follicular helper CD4 T cells (TFH) Annu. Rev. Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 47.Nutt S.L., Hodgkin P.D., Tarlinton D.M., Corcoran L.M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 2015;15:160–171. doi: 10.1038/nri3795. [DOI] [PubMed] [Google Scholar]
- 48.Sanges S., Wallet F., Blondiaux N., Theis D., Vérin I., Vachée A., Dessein R., Faure K., Viget N., Senneville E., Leroy O., Maury F., Just N., Poissy J., Mathieu D., Prévotat A., Chenivesse C., Scherpereel A., Smith G., Lopez B., Rosain J., Frémeaux-Bacchi V., Hachulla E., Hatron P.Y., Bahuaud M., Batteux F., Launay D., Labalette M., Lefèvre G. Diagnosis of primary antibody and complement deficiencies in young adults after a first invasive bacterial infection. Clin. Microbiol. Infection : the official publication of the European Society of Clinical Microbiology and Infectious Diseases. 2017;23 doi: 10.1016/j.cmi.2017.02.005. 576.e571-576.e575. [DOI] [PubMed] [Google Scholar]
- 49.Patel R. Periprosthetic joint infection. N. Engl. J. Med. 2023;388:251–262. doi: 10.1056/NEJMra2203477. [DOI] [PubMed] [Google Scholar]
- 50.Massin P., Delory T., Lhotellier L., Pasquier G., Roche O., Cazenave A., Estellat C., Jenny J.Y. Infection recurrence factors in one- and two-stage total knee prosthesis exchanges. Knee Surg. Sports Traumatol. Arthrosc. 2016;24:3131–3139. doi: 10.1007/s00167-015-3884-1. [DOI] [PubMed] [Google Scholar]
- 51.Topalian S.L., Taube J.M., Pardoll D.M. Neoadjuvant checkpoint blockade for cancer immunotherapy. Science. 2020:367. doi: 10.1126/science.aax0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kunutsor S.K., Whitehouse M.R., Blom A.W., Board T., Kay P., Wroblewski B.M., Zeller V., Chen S.Y., Hsieh P.H., Masri B.A., Herman A., Jenny J.Y., Schwarzkopf R., Whittaker J.P., Burston B., Huang R., Restrepo C., Parvizi J., Rudelli S., Honda E., Uip D.E., Bori G., Muñoz-Mahamud E., Darley E., Ribera A., Cañas E., Cabo J., Cordero-Ampuero J., Redó M.L.S., Strange S., Lenguerrand E., Gooberman-Hill R., Webb J., MacGowan A., Dieppe P., Wilson M., Beswick A.D. One- and two-stage surgical revision of peri-prosthetic joint infection of the hip: a pooled individual participant data analysis of 44 cohort studies. Eur. J. Epidemiol. 2018;33:933–946. doi: 10.1007/s10654-018-0377-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.