

Abstract
Virus-encoded circular RNA (circRNA) participates in the immune response to viral infection, affects the human immune system, and can be used as a target for precision therapy and tumor biomarker. The coronaviruses SARS-CoV-1 and SARS-CoV-2 (SARS-CoV-1/2) that have emerged in recent years are highly contagious and have high mortality rates. In coronaviruses, little is known about the circRNA encoded by the SARS-CoV-1/2. Therefore, this study explores whether SARS-CoV-1/2 encodes circRNA and characteristics and functions of circRNA. Based on RNA-seq data of SARS-CoV-1 and SARS-CoV-2 infections, we used circRNA identification tools (circRNA_finder, find_circ and CIRI2) to identify circRNAs. The number of circRNAs encoded by SARS-CoV-1 and SARS-CoV-2 was identified as 151 and 470, respectively. It can be found that SARS-CoV-2 shows more prominent circRNA encoding ability than SARS-CoV-1. Expression analysis showed that only a few circRNAs encoded by SARS-CoV-1/2 showed high expression levels, and the positive strand produced more abundant circRNAs. Then, based on the identified SARS-CoV-1/2-encoded circRNAs, we performed circRNA identification and characterization using the previously developed CirRNAPL. Finally, target gene prediction and functional enrichment analysis were performed. It was found that viral circRNA is closely related to cancer and has a potential role in regulating host cell functions. This study studied the characteristics and functions of viral circRNA encoded by coronavirus SARS-CoV-1/2, providing a valuable resource for further research on the function and molecular mechanism of coronavirus circRNA.
Keywords: SARS-CoV-1/2, identification, characterization, expression analysis, function
INTRODUCTION
Circular RNA (circRNA) was first found among the RNA of plant pathogenic viruses, which are called viroids [1]. With the in-depth study of the function of eukaryotic circRNAs, scholars are re-examining viruses to find the possibility of truly encoding circRNAs that are back-spliced from viral genes [2–4]. Recent circRNA analysis has found and verified that viruses can encode circRNA. Viral circRNAs are found in DNA viruses (such as herpes virus and papilloma virus) [5]. These circRNAs range from 220 to 457 bp, and they lack protein-coding ability, different from viroids, which also have a single-stranded circRNA structure [5, 6]. To detect viruses and host circRNAs in samples, several groups have applied RNase R for processing and have performed sequencing, revealing various single- and double-stranded viruses [7]. They identified multiple circRNAs from various viruses in samples of cervical cancer, liver cancer, herpes virus and Epstein Barr virus (EBV) [8]. Flemington et al. found that viral circRNAs are expressed between the latent and lytic cycles of EBV and span multiple cell lines [9]. With the deepening of research, the role of circRNAs in viral infections is increasingly recognized, and it has been found that circRNAs are involved in various viral infections (such as those involving hepatitis B virus and human papillomavirus) [10]. Viruses can hijack host circRNAs, leading to enhanced viral replication and pathogenesis, and can also counteract innate immune responses, playing a role in immune surveillance [11, 12]. Studying the influence of virus-encoded circRNAs in the host–virus interaction process can facilitate understanding of unknown physiological functions of circRNAs and their mechanisms in disease pathogenesis and promote new diagnosis, treatment and prognosis strategies for virus-related diseases [13–16].
As of 10 September 2022, the ongoing pandemic of a novel coronavirus, named SARS-CoV-2, which is a coronavirus with a positive single-stranded RNA genome, has led to infection of 605 467 311 people and resulted in 6 488 382 deaths [17, 18]. During 2002–2004, SARS-CoV-1 was circulating in 32 countries, infected more than 8000 people and killed more than 900 people [19]. The pathogens SARS-CoV-1 and SARS-CoV-2 belong to Coronaviridae and Betacoronavirus [20]. Both use ACE2 as their receptor, but they belong to two types. The high fatality rate caused by SARS-CoV-1/2 and the long-term epidemic indicate that coronaviruses are among the most threatening viruses to human life [21]. Despite effective vaccines and some treatment, infected individuals continue to die. Coronaviruses are the largest RNA viruses, with a genome range of 26–32 kb and a diameter of 125 nm. The full genome sequences of SARS-CoV-1 and SARS-CoV-2 are 29.7 kb and 29.9 kb in length, respectively, consisting of 11–14 open reading frames (ORFs) encoding 15–16 nonstructural proteins, four main structural proteins, including spike protein (S), envelope protein (E), membrane protein (M) nucleocapsid protein (N) and 5–8 helper proteins [22]. In addition to these typical ORFs, several virus-encoded noncoding RNAs have also been found in coronavirus infection. Kim et al. showed a high-resolution map of the SARS-CoV-2 transcriptome and found many transcripts encoding unknown ORFs [23]. Du et al. found a new circRNA molecule, circTNFAIP3, and found that overexpression of circTNFAIP3 promotes coronavirus replication by reducing host cell apoptosis [24]. Morales et al. found that three small RNAs encoded by SARS-CoV-1 are associated with infection-related lung pathology [25]. These studies indicate that the circRNA encoded by these viruses may play an important role in coronavirus infection. In particular, the team of Professor Wei Wensheng of Peking University has developed a circRNA vaccine for COVID-19 and its variants, which further confirms the important role of circRNA in virus therapy [26].
In recent years, virus-encoded circRNAs have been discovered and characterized in several cancer-related viruses, such as Orf virus [27] and Zika virus [28]. Some viral circRNAs are associated with cancer progression, such as HPV-circE7 [29], ebv-circRPMS1 [30] and ebv-circLMP2A [31]. Despite some achievements in targeting viral circRNAs, however, due to the lack of systematic annotation of the SARS-CoV-1/2 viral genome, there are relatively few reports on coronavirus circRNAs, and there are still many problems and challenges. Therefore, we identified thousands of circRNAs in SARS-CoV-1/2 from viral infection RNA-seq data. We also characterized the expression, sequence characteristics, genomic localization preferences and functions of these viral circRNAs (the flow chart is shown in Figure 1). We collected RNase R-treated and enriched circRNA-enriched SARS-CoV-1/2 RNA-Seq data, and used circRNA detection tools (circRNA_finder [32], find_circ [33] and CIRI2 [34]) that do not rely on genome annotation for circRNA identification. Those identified as circRNA by all three methods are considered high-confidence circRNA. Then, based on the high-confidence circRNAs, we analyzed and annotated these circRNAs to systematically understand the characteristics of circRNAs encoded by SARS-CoV-1/2. Based on our previous CirRNAPL algorithm [35], using high-confidence circRNA encoded by SARS-CoV-1/2 as test set for identification, we explored the characteristic differences between circRNA encoded by SARS-CoV-1/2 and human circRNA. Then, the interaction between SARS-CoV-1/2 circRNAs and miRNAs was predicted, and functional enrichment analysis based on target genes was conducted. Our systematic characterization of coronavirus circRNAs provides a valuable resource that can be used to further explore the action mechanism of circRNAs in coronaviruses.
Figure 1.

The flow chart of the research framework of this paper. (A) Dataset preparation. (B) CircRNA identification. (C) Advanced analysis.
RESULTS
Computational analysis of circRNAs encoded by SARS-CoV-1/2
CIRI2, find_circ and circRNA_finder were used to identify circRNAs encoded by SARS-CoV-1 and SARS-CoV-2. The specific number of circRNAs identified by the three methods is shown in Figure 2.
Figure 2.
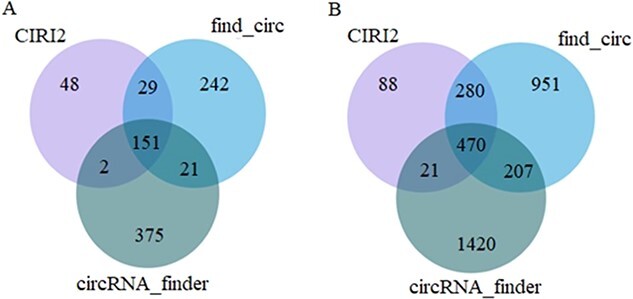
Number of circRNAs identified by three computational methods. (A) SARS-Cov-1; (B) SARS-Cov-2.
The number of circRNAs encoded by SARS-CoV-1 and SARS-CoV-2 identified by CIRI2, find_circ and circRNA_finder was 230 and 859, 443 and 1908, and 549 and 2118, respectively. Among them, at least two methods detected 52 and 508 circRNAs encoded by SARS-CoV-1 and SARS-CoV-2, and the number of circRNAs detected by the three methods is as follows: 151 and 470. SARS-CoV-2 shows a more prominent circRNA encoding ability than SARS-CoV-1. Those identified as circRNA by all three methods are considered high-confidence circRNAs. High-confidence circRNA was used in the following series of studies. Among them, the results of the three methods are presented in Additional file S1.
Sequence conservation of SARS-CoV-1/2 circRNA
The sequence conservation of viral circRNAs in the two coronaviruses was assessed using basic local alignment search tool (BLAST). Hits with identity ≥80%, coverage ≥80% and e-value ≤1E-5 were defined as homologues of viral circRNA. According to BLAST sequence alignment analysis, 118 circRNAs encoded by SARS-CoV-1 have sequence homology with SARS-CoV-2, and 352 circRNAs encoded by SARS-CoV-2 have sequence homology with SARS-CoV-1.
Expression of SARS-CoV-1/2 circRNA
To gain a deeper understanding of the characteristics of coronavirus circRNAs, high-confidence circRNAs were extracted, and the distribution junction-spanning reads of virus circRNA was statistically analyzed (Figure 3A and D), as were the relationship between the length of virus circRNA sequence and the number of circRNAs (Figure 3B and E) and the distribution characteristics of expression levels (Figure 3C and F).
Figure 3.

(A–C) Statistics of the circRNA of the SARS-Cov-1. (A) Quantity distribution of junction reads of circRNA. (B) Length distribution of viral circRNA. (C) Distribution of viral circRNA expression. (D–F). Statistics of the circRNA of the SARS-Cov-2. (D) Quantity distribution of junction reads of viral circRNA. (E) Length distribution of viral circRNA. (F) Distribution of viral circRNA expression.
For SARS-CoV-1, the number of junction-spanning reads for each sample circRNA ranges from 1 to 21, with 93 of 151 circRNAs having at least 2 and 17 having at least 5 (Figure 3A). The distribution characteristics of virus circRNA expression were analyzed based on the number of connection readings (Figure 3B and C). Among them, 55 circRNAs encoded by SARS-CoV-1 had expression levels less than −1, while 8 were greater than 1, with the majority located in a small number of SARS-CoV-1 circRNAs with high expression levels. Then, the distribution characteristics of the length and the number of circRNAs encoded by SARS-CoV-1 were calculated. CircRNAs identified from different samples with the same reverse splicing site were integrated into one circRNA, as shown in Figure 3(B). Statistical data showed that 84 of 151 circRNA encoded by SARS-CoV-1 have lengths between 200 and 1 kb, which is consistent with the characteristics of circRNA. Interestingly, the 22 circRNAs encoded by SARS-CoV-1 have almost a head-to-tail connection length of ≥10 kb in the viral genome.
For SARS-CoV-2, the number of junction-spanning reads for each sample’s circRNA ranges from 1 to 156 (Figure 3D), and the number of junction-spanning reads was significantly greater than that of SARS-CoV-1. Of 470 viral circRNAs, 264 have at least two junction-spanning reads, accounting for over 50% of the total. Among them, 50 SARS-CoV-2 circRNAs had at least five junction-spanning reads. The distribution characteristics of SARS-CoV-2 circRNA expression were analyzed based on the number of connection readings. Among them, 114 SARS-CoV-2 circRNAs had expression levels less than −1, while 55 were greater than 1. It can also be observed that the number of SARS-CoV-2 circRNAs with high expression levels is still in the minority. Then, the length and number of SARS-CoV-2 circRNAs were counted, as shown in Figure 3C. Statistical data show that the SARS-CoV-2 circRNA lengths of 292 viruses range from 200 to 1 kb, which is similar to the characteristics of SARS-CoV-1 and conforms to circRNA characteristics. Only 39 viruses produce circRNAs with lengths ≥10 kb.
Then, the location of the circRNA in the SARS-CoV-1 and SARS-CoV-2 genomes was analyzed, and the distribution characteristics of the positive (+) and negative (−) strands of the virus circRNAs were analyzed (Figure 4A and B). Based on the published SARS-CoV-1/2 reference genomes, a schematic diagram was drawn, and the distribution of the positive (+) and negative (−) strands in the virus genome was calculated based on the junction-spanning reads of the virus circRNAs.
Figure 4.

(A) Coverage curve of viral circRNAs on the SARS-Cov-1 reference genome. (B) Coverage curve of viral circRNAs on the SARS-Cov-2 reference genome. (C) Scatter diagram of circularization position of circRNA in SARS-Cov-1. (D) Scatter diagram of circularization position of circRNA in SARS-Cov-2.
Although in most cases the length of circRNAs is hundreds of bases, 11% (17 of 151) of SARS-CoV-1 and 10% (50 of 470) of SARS-CoV-2 remained after selecting circRNAs with at least 5 junction-spanning reads. When mapping the connection readings of virus circRNA to the positive (+) and negative (−) strands of SARS-Cov-1/2 genomes, circRNAs were found to be widely expressed from throughout the entire genome and highly expressed from certain regions of ORF1ab, M, ORF6/ORF7a/ORF7b and N/ORF8b. It was also observed that the number of circRNAs on the negative (−) chain was twice that on the positive (+) chain. As a positive-stranded, single-stranded RNA virus, SARS-CoV-1 uses full-length replication or discontinuous transcription to generate a negative-strand RNA genome or subgenomic negative-strand RNA as the template for synthesis of positive-strand RNA rather than encoding protein. Whether negative-strand viral circRNAs play a regulatory role in positive-strand RNA synthesis is currently uncertain. The viral circRNAs identified in SARS-CoV-2 show negative-strand bias. Taken together, the findings for SARS-CoV-1 and SARS-CoV-2 suggest that positive-strand viruses produce more abundant circRNAs than negative-strand.
CircRNAs interact with the 5′-terminal sequence and the 3′-terminal sequence, leading to cross-linking between the two ends of the genome or subgenome and resulting in circularization. Therefore, we continued to count and analyze the cyclization positions of circRNAs in SARS-CoV-1 and SARS-CoV-2, as shown in Figure 4(C) and (D). With the short-length gray circRNA near the diagonal as the background, the orange and red dots in the upper left corner indicate different circularized circRNAs in the figure. The two color distributions represent the genome length covered, that is, they represent ≥50 and ≥90% of the circRNAs, respectively. It was also found that the expression level of SARS-CoV-2 is more pronounced than that of SARS-CoV-1.
Sequence characteristic analysis of SARS-CoV-1/2 circRNA
After analyzing the conservation and homology of various SARS-CoV-1/2 circRNAs, we used CirRNAPL model for circRNA identification, and 307 sequences were predicted as circRNAs, achieving an accuracy rate of 0.59. Then, the differences in sequence characteristics between SARS-CoV-1/2 circRNAs and general human circRNAs were explored. To create a more intuitive graphical representation, the sequence features are dimensionally reduced using the feature dimensionality reduction algorithm t-SNE, and the scatterplots are visualized in two-dimensional space. Figure 5A–K shows the scatter plot of nine groups of features after t-SNE dimension reduction (purple points are SARS-CoV-1/2 circRNAs, blue points are human circRNAs).
Figure 5.

Sequential feature analysis. (A–K) Comparison of the characteristics of SARS-CoV-1/2 coronavirus circRNA and human circRNA. (L) Violin plot to visualize feature distribution. (M) The 20 most important features. (N) Summary plot for SHAP values. For each feature, one point corresponds to a single sample. The SHAP value along the x-axis represents the impact that feature had on the model’s output for that specific sample. Features in the higher position in the plot indicate the more important it is for the model. (O) SHAP dependence plots. These plots show the effect that a single feature has on the model predictions and the interaction effects across features. Each point corresponds to an individual sample, the value along the x-axis corresponds to the feature value.
Figure 5(A)–(K) shows that the 2- and 3-mer characteristic repeat points are the most, indicating that the differences between the two are less, which shows that the sequence frequency characteristics of k-mer of circRNA encoded by SARS-CoV-1-1/2 have no significant differences among human circRNAs. Second, triplet features have much fewer duplicate points, and the number of overlapping points of Mismatch and pc-psednc are also reduced. For graph, dac, dacc, dcc, nmbac and sc-psednc features, the point distribution of circRNA encoded by SARS-CoV-1-1/2 and human circRNA has boundaries, and there is a significant difference between the two. By comparison, the characteristic differences between the circRNA encoded by SARS-CoV-1-1/2 and human circRNA are quite significant, and the boundaries are relatively clear. When coronavirus encodes circRNA, it may cause changes in the structure and composition of circRNA, resulting in differences between the two. This can also provide a new direction for the functional research of circRNA encoded by SARS-Cov-1/2.
Violin plots were drawn to visualize the distribution of 2D horizontal vectors before t-SNE after dimensionality reduction for SARS-CoV-1/2 circRNAs and human circRNAs (Figure 5L). Significant differences exist in the distribution of characteristic data between SARS-CoV-1/2 circRNA and human circRNA, further demonstrating the characteristic difference between the two.
We leverage SHAP to analyze feature contributions and dependencies. The SHAP value represents the contribution of a feature to the model output change, reflects the influence of the feature in each sample, and can also show positive and negative effects. First, the absolute SHAP values of each feature in the circRNA were averaged, and the 20 most important features were calculated and described with a SHAP summary map, as shown in Figure 5(M). Among the first 13 features, graph71 occupies the first place, and graph occupies four places, followed by pc-psednc, dcc, dac, Mismatch and sc-psednc.
Then, summary plots were constructed to better understand the relationship between eigenvalues and model outputs, as well as the patterns of overall sample characteristics. Count the SHAP values for each feature for each sample and observe for outliers if present. Figure 5(N) shows a summary plot of the top 20 most important features. First, the graph71 feature is the most important. When the value of graph71 is larger, the model will have a negative impact on the model’s prediction of circRNA, and when the value is smaller, it will have a positive impact. There are similar situations in six features including graph89, pc-psednc31 and sc-psednc43. The opposite situation occurs on features such as pc-psednc43, dac9 and pc-psednc52, that is, a larger value of pc-psednc43 has a positive impact on the model prediction as a circRNA. The influence of multiple features such as dcc is relatively vague. graph71 (−0.06 to 0.06) and graph89 (−0.05 to −0.04) have large variations and dominate the behavior of the model.
Then, to understand how individual features affect the output of the model, we analyzed the SHAP value of the graph71 feature, comparing it with the feature value of all samples in the set data. To help reveal graph71 interactions, another feature was automatically selected for coloring and was not similarly dependent on graph71 (Figure 5O). It can be seen that graph71 has a turning point of about 0.00. For functional interactions, graph71 (−0.05, 0.00) and high-featured graph3, graph71 (0.00–0.05) and high-featured graph3 contribute to accurate model predictions, while low eigenvalues of graph3 show little effect. More functional interaction dependency graphs are available in Additional file S2.
Interactions between SARS-CoV-1/2 circRNAs and miRNAs
To further analyze whether coronavirus circRNA can act as a sponge for miRNAs in infected host cells, we used miRanda and TarPmiR to predict interaction. We implemented predictive analysis of circRNAs (length <1 kb and junction-spanning reads ≥2, combined analysis of circRNAs from SARS-CoV-1 and SARS-CoV-2 data) and host miRNAs from the comprehensive miRNA database miRBase. Our analysis predicted 489 miRNA–circRNA pairs and 72 unique human miRNAs. The results of the two methods are presented in Additional file S3.
To identify important miRNAs with strong interactions with circRNA encoded by SARS-CoV-1/2, we integrated the output of miRanda and TarPmiR to calculate the comprehensive quality score of interaction [36]. We focused on analyzing miRNAs with multiple connections to coronavirus circRNA. We found that hsa-miR-557 interacted with 8 circRNA encoded by SARS-CoV-2 and that hsa-miR-5088-5p, hsa-miR-103a-2-5p and hsa-miR-367-5p interacted with 21, 13 and 25 circRNAs encoded by SARS-CoV-1, respectively. More importantly, the eight circRNAs encoded by SARS-CoV-2 interacting with hsa-miR-6747-5p showed a very high mass fraction. Then, a literature search was conducted for several miRNAs. According to the literature [37], biogenesis of miR-5088-5p is upregulated by Fyn, promoting hypomethylation of its promoter, which is related to malignant tumors in breast cancer. Hsa-miR-103a-2-5p can affect the occurrence and development of liver cancer cells by inhibiting miR-34a [38]. It was also observed that hsa-miR-4745-5p is upregulated in colorectal cancer patients [39]. These results all indicate potential interactions between host miRNAs and viral circRNAs, and the role of some miRNAs in viral pathological regulation deserves further research.
Functional analysis of SARS-CoV-1/2 circRNAs and miRNAs
To further explore the function of circRNA encoded by SARS-CoV-2/1, we used three miRNA target databases, TargetScan, miRDB and miRTarBase, to retrieve the target genes of the 72 miRNAs identified. A total of 638 miRNA–mRNA interactions and 604 target genes were identified. Then, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed based on the target genes (as shown in Figure 6A and B). The results of the GO and KEGG analyses are presented in Additional file S4.
Figure 6.

GO and KEGG functional analysis of SARS-CoV-1/2 coronavirus circRNA. (A) GO. (B) KEGG.
Enrichment of the target genes revealed significantly enriched pathways and functions. The top 10 KEGG pathways correlate highly with mucoprotein synthesis, cancer pathway, thyroid signal, proteoglycan in cancer, prostate cancer and other pathways, indicating that the circRNA in coronavirus tumor samples is involved in viral infection and tumorigenesis. Biological process (BP) analysis showed that the target genes are involved in cellular nitrogen metabolism, ion binding, biosynthesis, cell protein modification, small molecule metabolism, gene expression, neurotrophic TRK receptor signaling pathway, catabolism, viral processes and symbiosis, including parasitic mutualism and other biological processes. For cellular component (CC) analysis, terms are mainly associated with organelle, protein complex, honeycomb components, cytoplasm, nucleoplasm, platelets α granular lumen, microtubule organizing center, lysosome lumen, vacuole and information node. MF analysis showed links to molecular function, nucleic acid binding transcription factor activity, protein binding transcription factor activity, enzyme binding, cytoskeleton protein binding, enzyme modulator activity, ribonucleic acid binding, transmembrane transport protein activity, protein binding and bridging, etc. GO-enriched terms provide clues on how viral circRNA plays a regulatory role in infected host cells. Viral circRNAs can regulate the cell cycle and protein structure and location through transcriptional inhibitory activity. In addition, the terminology of CCs, including nuclear envelope and nuclear spots, also showed enriched. Overall, the enriched functions are consistent with the role of viruses, indicating that some virus circRNAs act as host miRNA sponges to regulate infected host cells.
CONCLUSION
An increasing number of viral circRNAs have been found to play important roles in single- and double-stranded viruses. However, in coronaviruses, little is known about the virus-encoded circRNAs. This study conducted data mining on coronavirus infection-related RNA sequencing data and analyzed the circRNA encoded by SARS-CoV-1/2. The main contents of this study are as follows: (1) compared with human circRNA, circRNA encoded by SARS-CoV-1/2 is rarely conserved and may evolve faster than human circRNA. In terms of characteristics, the circRNAs encoded by SARS-CoV-1/2 differed in RCM, conservation score, graph structure and compositional features. Except for the less significant differences between RCMs, significant differences were found in the other three groups. This result indicates that the virus may change its structure and composition when encoding circRNA, which also provides a direction for future research. (2) A total of 621 highly reliable viral circRNAs were identified from two coronaviruses. By analyzing the reads across junctions, only a few SARS-CoV-1/2-encoded circRNAs showed high expression levels. SARS-CoV-1/2 recognizes the largest number of circRNAs encoded between 200 and 1 kb, which is consistent with the characteristics of human circRNAs. (3) The circRNA encoded by SARS-CoV-1/2 shows positive- and negative-strand bias, with the positive strand producing more abundant circRNA. (4) Target gene prediction and functional enrichment analysis of circRNA encoded by SARS-CoV-2 show strong correlation with viruses and cancer. Several targets were predicted through the interaction between SARS-CoV-2-encoded circRNA and miRNA. It has also been found that coronavirus circRNA may participate in many KEGG pathways related to the nervous system and cancer.
This study also has some limitations that need to be improved and addressed. (1) Identification of viral circRNAs from RNA-seq data sets of in vitro viral infections. However, viral circRNAs are highly tissue specific [40]. Therefore, the expression patterns of these viral circRNAs in other types of human cells or tissues in vivo require further study. (2) This study only identified the existence of viral circRNA through computational methods, and experimental verification of its existence requires further efforts. (3) Due to limited public database data, the number of viral circRNAs and SARS-CoV-1/2 detected in this study is still limited.
METHODS AND MATERIALS
Dataset collection
Gene Expression Omnibus (http://ww.ncbinlm.nih.gov/geo/) and Sequence Read Archive (https://ncbiinsights.ncbi.nlm.nih.gov/tag/sra/) at National Center of Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/) were searched with the keywords ‘virus’ and ‘SARS-CoV-1/2’. All datasets were manually checked, and datasets containing one of the following keywords were retained: ‘Total RNA’, ‘rRNA deleted’, ‘rRNA deleted’, ‘ribominus’, ‘RNase R’, ‘polyA RNA deleted’ and ‘non polyadenylated’. The NCBI SRA toolkit was downloaded to obtain a total RNA sequencing dataset (ID: GSE148729) for SARS-CoV-1/2-infected Calu-3 cells [41, 42].
The SARS-CoV-1/2 sequencing data downloaded from NCBI cannot be directly used because the sequencing data formats are not commonly used; the format SRA is a common data storage format for NCBI and uses binary compression. Fastq files are text files, and some software needs to be used to convert data types. In addition, there are a considerable number of low-quality bases and few splice sequences in the obtained sequencing data. To ensure the accuracy of subsequent analysis and the quality of the data, we performed quality control on the data and removed all these low-quality sequences. We assessed the quality of sequencing reads using FastQC v0.21.0 (http://www.bioinformatics.babraham.ac.ukjects/fastqc/). We used FASTQ v0.21.0 with default parameters to remove low-quality bases and PRINSEQ-lite v0.20.4 [43] with deep = 1 to reduce sequence repeats.
Computational identification of SARS-CoV-1/2 circRNA
Based on the RNA-seq data of SARS-CoV-1/2, we used circRNA_finder [32], find_circ [33] and CIRI2 [34] identify coronavirus circRNA. The circRNA_finder uses a STAR reader aligner to identify chimeric junction reads and then filters them by splicing the distance between the donor and receptor to be less than 100 000 bp. The find_circ first maps the read data to the reference genome through Bowtie2 and then discards the mapped read data. CIRI2 obtains read mapping information through local alignment with BWA-MEM. After processing the data through the above steps, BWA-MEM was first used to compare the clean reads with the reference genome of the virus.
Among them, the version of BWA is 0.7.17, parameter: -T 19. The version of STAR is 2.7.1a, using default parameters. The version of Bowtie2 is 2.1.0, parameter: -p16. Then three computational tools (CIRI2, find_circ and circRNA_finder) were used to identify viral circRNAs using parameters recommended in the manual.
Homology analysis of SARS-CoV-1/2 circRNA
Homology analysis for SARS-CoV-1/2 was performed using the Basic Local Alignment Search Tool (BLAST) (version 2.9.0+) sequence alignment algorithm [44] and HAlign [45–48]. Hits with homology ≥80%, coverage ≥80% and e-value ≤1E-5 were considered to be homologous with the queried virus circRNAs.
Analysis of expression of SARS-CoV-1/2 circRNA
After identifying coronavirus circRNAs, the host gene, length, transcriptional chain, number of junction-spanning reads and expression level of the circRNAs of the viruses were analyzed. When using three software programs (circRNA_finder, find_cic, CIRI2) to identify circRNAs, the number of reverse splicing sites (bsj reads) for each circRNA can be obtained. Then, the number of bsj reads per million mapped reads for that circRNA is calculated based on the number of bsj reads per million mapped reads for each circRNA [49]. The expression level of coronavirus circRNAs was measured by the logarithm of counts per million mapped reads (CPM), which is ln(CPM). The CPM calculation formula is shown in Equation (1):
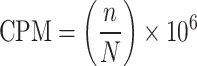 |
(1) |
where n and N represent the number of junction-spanning reads and the number of mapped reads, respectively.
We initially identified circRNAs encoded by SARS-CoV-1/2 with at least two splice site junctions at reverse splicing sites. After determining basic statistics, to control false positives, circRNAs with at least five connection readings were selected for further analysis.
Analysis of sequence characteristics of viral circRNAs
Based on the results of circRNA encoded by SARS-CoV-1/2 and our previous study [35], the identified and highly reliable 562 circRNA sequences were used as the test set, and the constructed CirRNAPL was used to identify the SARS-CoV-1/2 circRNA (as a machine learning algorithm, circRNA prediction is made based on RNA sequence data, and its evaluation index is accuracy). Then, we extracted the sequence features of SARS-CoV-1/2 using Pse-in-one (2-mer, 3-mer, dac, dacc, dcc, Mismatch, nmbac, pc-psednc, sc-psednc, triplet and graph) [50], performed feature analysis on the SARS-CoV-1/2 sequence data, and compared the difference in sequence features between SARS-CoV-1/2 circRNA and human circRNA. Use the t-SNE dimensionality reduction method to perform feature selection on each feature and perform two-dimensional visual feature difference analysis on important features, and draw a violin diagram to visualize the data distribution of important features. Using the SHapley Additive ExPlanations (SHAP) [51] analysis method, using the coronavirus circRNA sequence as a test set, based on the previous prediction model, the coronavirus feature interpretability analysis was carried out to explore the relationship between the feature and the model output. SHAP is a method for interpreting individual predictions [52–55]. The SHAP interpretation method calculates the Shapley value based on coalition game theory. The goal of SHAP is to explain the prediction of an instance by calculating the contribution of each feature to the prediction model.
Analysis of interaction between viral circRNA and miRNA
MiRNAs were obtained from the miRBase database (http://mirbase.org/). TarPmiR [56] and miRanda [57] software programs were used to predict circRNAs’ interactions with miRNAs. Viral circRNAs with a length of ≤1 kb and human miRNAs were collected from miRBase using miRanda v3.3a and TarPmiR to predict miRNA–circRNA interactions. MiRanda uses the free energy combination of miRNA and its target gene for miRNA target prediction: the higher the free energy is, the weaker the binding force is, and the binding strength is inversely proportional to the free energy. TarPmiR is a machine learning-based method that calculates more features. To control false positives, we conducted analysis based on the intersection of the recognition results of these two tools; we used strict parameter settings for target prediction, and the interaction predicted by the two methods was accepted for further analysis. For miRanda, two important parameters are set: Max Score ≥160 and Max Energy ≤−20 kcal mol−1. Two important parameters of TarPmiR are binding probability = 1 and energy ≤−20 kcal mol−1. miRanda has two evaluation metrics, maximum score and maximum energy, and TarPmiR is the output energy. The outputs of the two software programs are then combined to calculate a composite quality score. We performed min–max normalization on Max Score, Max Energy and Energy and scaled them between 0 and 1, whereby lower energy indicates more stable binding. Therefore, Max Energy and Energy are inverted before scaling. The composite quality score was defined as the average of the scaled outputs of miRanda and TarPmiR, with higher scores indicating higher binding quality.
Functional analysis of SARS-CoV-1/2 circRNA
Functional enrichment of circRNA source genes or potential target genes encoded by SARS-CoV-1/2 to explore their potential biological functions is important for the study of circRNA. First, we retrieved miRNA–mRNA interaction data from the miRNA target databases TargetScan [58], miRDB [59] and miRTarBase [60]. Then, based on the identified coronavirus circRNAs with high confidence, we performed GO (https://geneontology.org/) and KEGG (https://www.genome.jp/kegg/pathway.html) enrichment analyses of miRNA target genes using DAVID (https://david.ncifcrf.gov/) to further study circRNA functions. As a comprehensive database, DAVID not only organizes biological function annotation data such as gene or protein lists but also provides tools for analysis. GO analysis involves BP, MF and CC. Preliminary prediction of the function of SARS-CoV-1/2 virus circRNAs was also performed through KEGG enrichment analysis of related pathways. Among them, all KEGG pathways and GO terms with q-values <0.05 were considered significantly enriched.
Key Points
We identified thousands of circRNAs encoded by SARS-CoV-1 and SARS-CoV-2.
The expression of viral circRNA in SARS-CoV-1 and SARS-CoV-2 was analyzed.
Viral circRNA has different functions in SARS-CoV-1 and SARS-CoV-2.
Supplementary Material
Mengting Niu is a postdoctoral fellow at the University of Electronic Science and Technology of China and Shenzhen Polytechnic University. Her research interests include bioinformatics, data mining.
Chunyu Wang is a professor at Faculty of Computing, Harbin Institute of Technology. His research fields include computational biology and machine learning, especially on the structure and function prediction of biomolecules, artificial intelligence-assisted drug discovery.
Yaojia Chen is a PHD candidate at the Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu, China. Her research interests include machine learning and bioinformatics.
Quan Zou is is a professor in University of Electronic Science and Technology of China. His research interests include bioinformatics and machine.
Lei Xu is an Associate Professor at the school of Electronic and Communication Engineering, Shenzhen Polytechnic University. Her research interests are focused on bioinformatics, pattern recognition.
Contributor Information
Mengting Niu, School of Electronic and Communication Engineering, Shenzhen Polytechnic University, Shenzhen 518055, China; School of Life Science and Technology, University of Electronic Science and Technology of China, Chengdu, China.
Chunyu Wang, Faculty of Computing, Harbin Institute of Technology, Harbin, Heilongjiang 150000, China.
Yaojia Chen, Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, No.4 Block 2 North Jianshe Road, Chengdu 610054, China; Yangtze Delta Region Institute (Quzhou), University of Electronic Science and Technology of China, Quzhou, China.
Quan Zou, Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, No.4 Block 2 North Jianshe Road, Chengdu 610054, China; Yangtze Delta Region Institute (Quzhou), University of Electronic Science and Technology of China, Quzhou, China.
Lei Xu, School of Electronic and Communication Engineering, Shenzhen Polytechnic University, Shenzhen 518055, China.
FUNDING
The National Natural Science Foundation of China (62231013, 62201129, 62303328, 62302341, 62271329, 62372332); the National Key R&D Program of China (No. 2022ZD0117700); Research fund of Shenzhen Polytechnic University (No. 6022331007K, No. 6022310036K, No. 6023310037K); Key Field of Department of Education of Guangdong Province (No. 2022ZDZX2082); the Special Science Foundation of Quzhou (No. 2023D036).
DATA AVAILABILITY
All code and data generated or analyzed during this study are included in this published article, its additional file and publicly available repositories, which are available in the Zenodo repository (https://zenodo.org/record/8216215) and GitHub (https://github.com/nmt315320/viral.git).
AUTHORS’ CONTRIBUTIONS
QZ and LX designed the research; MT carried out the studies, participated in the sequence alignment and drafted the manuscript. YJC and CYW participated in the design of the study and performed the statistical analysis. LX conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
References
- 1. Sanger HL, Klotz G, Riesner D, et al. . Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci 1976;73(11):3852–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang J-t, Chen J-n, Gong L-p, et al. . Identification of virus-encoded circular RNA. Virology 2019;529:144–51. [DOI] [PubMed] [Google Scholar]
- 3. Ungerleider NA, Jain V, Wang Y, et al. . Comparative analysis of gammaherpesvirus circular RNA repertoires: conserved and unique viral circular RNAs. J Virol 2019;93(6):e01952–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tan KE, Lim YY. Viruses join the circular RNA world. FEBS J 2021;288(15):4488–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Toptan T, Abere B, Nalesnik MA, et al. . Circular DNA tumor viruses make circular RNAs. Proc Natl Acad Sci 2018;115(37):E8737–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yin S, Tian X, Zhang J, et al. . PCirc: random forest-based plant circRNA identification software. BMC Bioinform 2021;22(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen S, Zheng J, Zhang B, et al. . Identification and characterization of virus-encoded circular RNAs in host cells. Microb Genomics 2022;8(6):mgen000848. 10.1099/mgen.0.000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cameron JE, Yin Q, Fewell C, et al. . Epstein-Barr virus latent membrane protein 1 induces cellular MicroRNA miR-146a, a modulator of lymphocyte signaling pathways. J Virol 2008;82(4):1946–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ungerleider NA, Tibbetts SA, Renne R, Flemington EK. Gammaherpesvirus RNAs come full circle. MBio 2019;10(2):e00071–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mo Y, Liu Y, Lu A, et al. . Role of circRNAs in viral infection and their significance for diagnosis and treatment. Int J Mol Med 2021;47(5):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li X, Liu C-X, Xue W, et al. . Coordinated circRNA biogenesis and function with NF90/NF110 in viral infection. Mol Cell 2017;67(2):214–227.e7e7. [DOI] [PubMed] [Google Scholar]
- 12. Yao W, Pan J, Liu Z, et al. . The cellular and viral circRNAome induced by respiratory syncytial virus infection. MBio 2021;12(6):e03075–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yan L, Chen YG. Circular RNAs in immune response and viral infection. Trends Biochem Sci 2020;45(12):1022–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang Y, Zhang X, Shen Z, et al. . BmNPV circular RNA-encoded peptide VSP39 promotes viral replication. Int J Biol Macromol 2023;228:299–310. [DOI] [PubMed] [Google Scholar]
- 15. Tagawa T, Oh D, Dremel S, et al. . A virus-induced circular RNA maintains latent infection of Kaposi’s sarcoma herpesvirus. Proc Natl Acad Sci 2023;120(6):e2212864120. 10.1073/pnas.2212864120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li P, Tiwari P, Xu J, et al. . Sparse regularized joint projection model for identifying associations of non-coding RNAs and human diseases. Knowl-Based Syst 2022;258:110044. [Google Scholar]
- 17. Lamers MM, Haagmans BL. SARS-CoV-2 pathogenesis. Nat Rev Microbiol 2022;20(5):270–84. [DOI] [PubMed] [Google Scholar]
- 18. Krammer F. SARS-CoV-2 vaccines in development. Nature 2020;586(7830):516–27. [DOI] [PubMed] [Google Scholar]
- 19. Marchand-Senécal X, Kozak R, Mubareka S, et al. . Diagnosis and management of first case of COVID-19 in Canada: lessons applied from SARS-CoV-1. Clin Infect Dis 2020;71(16):2207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rouet R, Mazigi O, Walker GJ, et al. . Potent SARS-CoV-2 binding and neutralization through maturation of iconic SARS-CoV-1 antibodies. MAbs 2021; Taylor & Francis 2021;13(1):1922134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aherfi S, Pradines B, Devaux C, et al. . Drug repurposing against SARS-CoV-1, SARS-CoV-2 and MERS-CoV. Future Microbiol 2021;16(17):1341–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lata S, Akif M. Comparative protein structure network analysis on 3CLpro from SARS-CoV-1 and SARS-CoV-2. Proteins 2021;89(9):1216–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim D-H, Im H, Jee J-G, et al. . β-Arm flexibility of HU from Staphylococcus aureus dictates the DNA-binding and recognition mechanism. Acta Crystallogr D Biol Crystallogr 2014;70(12):3273–89. [DOI] [PubMed] [Google Scholar]
- 24. Du L, Wang X, Liu J, et al. . A previously undiscovered circular RNA, circTNFAIP3, and its role in coronavirus replication. MBio 2021;12(6):e02984–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Morales-Salazar I, Montes-Enríquez FP, Garduño-Albino CE, et al. . Synthesis of bis-furyl-pyrrolo[3,4-b]pyridin-5-onesviaUgi–Zhu reaction and in vitro activity assays against human SARS-CoV-2 and in silico studies on its main proteins. RSC Med Chem 2023;14(1):154–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qu L, Yi Z, Shen Y, et al. . Circular RNA vaccines against SARS-CoV-2 and emerging variants. Cell 2022;185(10):1728–1744.e16e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pang F, Zhang M, Yang X, et al. . Genome-wide analysis of circular RNAs in goat skin fibroblast cells in response to Orf virus infection. PeerJ 2019;7:e6267. 10.7717/peerj.6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Giulietti M, Righetti A, Cianfruglia L, et al. . To accelerate the Zika beat: candidate design for RNA interference-based therapy. Virus Res 2018;255:133–40. [DOI] [PubMed] [Google Scholar]
- 29. Zhao J, Lee EE, Kim J, et al. . Transforming activity of an oncoprotein-encoding circular RNA from human papillomavirus. Nat Commun 2019;10(1):2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu Q, Shuai M, Xia Y. Knockdown of EBV-encoded circRNA circRPMS1 suppresses nasopharyngeal carcinoma cell proliferation and metastasis through sponging multiple miRNAs. Cancer Manag Res 2019;Volume 11:8023–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lp G, Jn C, Dong M, et al. . Epstein–Barr virus-derived circular RNA LMP 2A induces stemness in EBV-associated gastric cancer. EMBO Rep 2020;21(10):e49689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fu X, Liu R, editors. CircRNAFinder: a tool for identifying circular RNAs using RNA-Seq data. In: Proceedings of the 6th International Conference on Bioinformatics and Computational Biology, BICOB; 2014, (Las Vegas, Nevada, USA, 2014).
- 33. Memczak S, Jens M, Elefsinioti A, et al. . Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013;495(7441):333–8. [DOI] [PubMed] [Google Scholar]
- 34. Gao Y, Zhang J, Zhao F. Circular RNA identification based on multiple seed matching. Brief Bioinform 2018;19(5):803–10. [DOI] [PubMed] [Google Scholar]
- 35. Niu M, Zhang J, Li Y, et al. . CirRNAPL: a web server for the identification of circRNA based on extreme learning machine. Comput Struct Biotechnol J 2020;18:834–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu B, Gao X, Zhang H. BioSeq-Analysis2.0: an updated platform for analyzing DNA, RNA and protein sequences at sequence level and residue level based on machine learning approaches. Nucleic Acids Res 2019;47(20):1280–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tsintarakis A, Papalouka C, Kontarini C, et al. . The intricate interplay between cancer stem cells and oncogenic miRNAs in breast cancer progression and metastasis. Life 2023;13(6):1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pervez MA, Khan DA, Gilani STA, et al. . Hepato-protective effects of delta-tocotrienol and alpha-tocopherol in patients with non-alcoholic fatty liver disease: regulation of circulating MicroRNA expression. Int J Mol Sci 2022;24(1):79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gungormez C, Gumushan Aktas H, Dilsiz N, Borazan E. Novel miRNAs as potential biomarkers in stage II colon cancer: microarray analysis. Mol Biol Rep 2019;46:4175–83. [DOI] [PubMed] [Google Scholar]
- 40. Cai Z, Fan Y, Zhang Z, et al. . VirusCircBase: a database of virus circular RNAs. Brief Bioinform 2021;22(2):2182–90. [DOI] [PubMed] [Google Scholar]
- 41. Wyler E, Mösbauer K, Franke V, et al. . Bulk and single-cell gene expression profiling of SARS-CoV-2 infected human cell lines identifies molecular targets for therapeutic intervention. bioRxiv2020.05. 05.079194. Search in. 2020: 2020.05. 05.079194. 10.1101/2020.05.05.079194. [DOI]
- 42. Zhang X, Chu H, Wen L, et al. . Competing endogenous RNA network profiling reveals novel host dependency factors required for MERS-CoV propagation. Emerg Microbes Infect 2020;9(1):733–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cantu VA, Sadural J, Edwards R. PRINSEQ++, a multi-threaded tool for fast and efficient quality control and preprocessing of sequencing datasets. PeerJ Preprints 2019;7:e27553v1. [Google Scholar]
- 44. Ye J, McGinnis S, Madden TL. BLAST: improvements for better sequence analysis. Nucleic Acids Res 2006;34:W6–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tang FR, Chao JN, Wei YM, et al. . HAlign 3: fast multiple alignment of ultra-large numbers of similar DNA/RNA sequences. Mol Biol Evol 2022;39(8):msac166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zou Q, Hu QH, Guo MZ, Wang GH. HAlign: fast multiple similar DNA/RNA sequence alignment based on the centre star strategy. Bioinformatics 2015;31(15):2475–81. [DOI] [PubMed] [Google Scholar]
- 47. Wan S, Zou Q. HAlign-II: efficient ultra-large multiple sequence alignment and phylogenetic tree reconstruction with distributed and parallel computing. Algorithms Mol Biol 2017;12:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang Y, Zhai Y, Ding Y, Zou Q. SBSM-Pro: support bio-sequence machine for proteins. arXiv preprint arXiv:2308.10275 2023. 2023.
- 49. Curry-Hyde A, Gray LG, Chen BJ, et al. . Cell type-specific circular RNA expression in human glial cells. Genomics 2020;112(6):5265–74. [DOI] [PubMed] [Google Scholar]
- 50. Liu B, Liu F, Wang X, et al. . Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res 2015;43(W1):W65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lundberg SM, Lee S-I. A unified approach to interpreting model predictions. Adv Neural Inf Process Syst 2017;30:4768–4777. [Google Scholar]
- 52. Yang H, Luo YM, Ma CY, et al. . A gender specific risk assessment of coronary heart disease based on physical examination data. NPJ Digit Med 2023;6(1):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ao C, Ye X, Sakurai T, et al. . m5U-SVM: identification of RNA 5-methyluridine modification sites based on multi-view features of physicochemical features and distributed representation. BMC Biol 2023;21(1):93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang R, Jiang Y, Jin J, et al. . DeepBIO: an automated and interpretable deep-learning platform for high-throughput biological sequence prediction, functional annotation and visualization analysis. Nucleic Acids Res 2023;51(7):3017–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jin J, Yu Y, Wang R, et al. . iDNA-ABF: multi-scale deep biological language learning model for the interpretable prediction of DNA methylations. Genome Biol 2022;23(1):1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ding J, Li X, Hu H. TarPmiR: a new approach for microRNA target site prediction. Bioinformatics 2016;32(18):2768–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Enright A, John B, Gaul U, et al. . MicroRNA targets in Drosophila. Genome Biol 2003;5:R1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shaker F, Nikravesh A, Arezumand R, Aghaee-Bakhtiari SH. Web-based tools for miRNA studies analysis. Comput Biol Med 2020;127:104060. 10.1016/j.compbiomed.2020.104060. [DOI] [PubMed] [Google Scholar]
- 59. Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res 2020;48(D1):D127–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Huang H-Y, Lin Y-C-D, Li J, et al. . miRTarBase 2020: updates to the experimentally validated microRNA–target interaction database. Nucleic Acids Res 2020;48(D1):D148–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All code and data generated or analyzed during this study are included in this published article, its additional file and publicly available repositories, which are available in the Zenodo repository (https://zenodo.org/record/8216215) and GitHub (https://github.com/nmt315320/viral.git).