

Abstract
Trypanosoma cruzi blood stage trypomastigotes are highly resistant to complement-mediated killing in normal serum. A previously described trypomastigote surface glycoprotein was shown to have binding affinity for human complement components C3b and C4b and restrict activation of the complement cascade, thus preventing lysis of the parasites. Insect stage epimastigotes do not produce detectable levels of this 160-kDa complement regulatory protein (CRP) and are highly sensitive to the lytic effects of complement. Epimastigotes were stably transfected with a T. cruzi expression vector carrying the trypomastigote CRP cDNA and produced fully functional recombinant CRP. The recombinant CRP had binding affinity for C3b, and the transfected epimastigotes were protected from complement-mediated lysis. These results demonstrate for the first time that a developmentally regulated gene of T. cruzi trypomastigotes can be expressed in noninfectious epimastigotes and that production of CRP by epimastigotes is sufficient to confer a virulence-associated trait. Furthermore, these studies demonstrate the critical role that trypomastigote CRP plays in the protection of parasites from the deleterious effects of complement, thus establishing the protein as a virulence factor of T. cruzi.
To establish infection and persist in a mammalian host, agents of chronic infections are particularly adept at avoiding intrinsic and acquired host immune effector mechanisms. One of the early lines of host defense is activation of the complement system, which can result in direct killing of microorganisms and their enhanced clearance by phagocytes. As a countermeasure, many pathogens, especially blood-dwelling organisms and those that disseminate from the initial site of infection, have evolved elaborate means to avoid the deleterious effects of complement activation.
Trypanosoma cruzi is a protozoan parasite and the causative agent of Chagas’ disease, a major public health concern in Latin America. During its life cycle, the parasite undergoes a series of developmentally regulated morphologic and physiologic changes to survive within insect and mammalian hosts. When the insect vector takes a bloodmeal on a parasitemic host, blood stage trypomastigotes are taken up and convert to epimastigotes in the insect digestive tract. Epimastigotes divide within the midgut of the insect and eventually convert to metacyclic trypomastigotes, which are passed in the feces. Metacyclic trypomastigotes enter the mammalian host at the bite wound site or through mucous membranes. Trypomastigotes enter cells and convert to the dividing amastigote stage. Shortly before the infected cell ruptures and releases parasites, amastigotes convert to trypomastigotes, which survive extracellularly in the bloodstream and disseminate to target tissues. People with untreated T. cruzi infections, which are lifelong, have low-level parasitemias and easily detectable antibodies to parasite antigens. Approximately 20 to 30% of chronically infected persons eventually develop severe sequelae, such as cardiac conduction defects and cardiomyopathy, or gastrointestinal dysfunction.
The extracellular survival and dissemination of blood stage trypomastigotes in a vertebrate host is likely enhanced by the capacity of trypomastigotes to resist complement-mediated killing (1, 10, 11). Trypomastigotes avoid lysis and clearance through the production of surface glycoproteins that interfere with complement activation (2, 3, 12). One such glycoprotein, the 160-kDa complement regulatory protein (CRP), functions to restrict activation of the alternative and classical complement pathways by binding complement components C3b and C4b, thus preventing assembly of proteolytically active C3 convertase (12). In contrast to blood stage trypomastigotes, insect stage epimastigotes are sensitive to the lytic effects of complement and do not produce detectable amounts of CRP (11–13). Conversion of epimastigotes to trypomastigotes is coincident with the expression of CRP on the cell surface and the acquisition of complement resistance.
The role of CRP as a virulence factor has been previously studied in vitro by using antibodies that block the CRP-C3b interaction. In these studies, anti-CRP antibodies which inhibited CRP-C3b binding were capable of supporting high levels of complement-mediated lysis of trypomastigotes (12, 13) and were protective when adoptively transferred to mice prior to a lethal T. cruzi challenge (2a). To further study the function of this protein and determine its role in the survival and persistence of the parasites in mammalian hosts, we recently isolated a cDNA encoding the full-length CRP (17). Recent advances in genetic manipulation of trypanosomes have made these organisms more amenable to genetic studies of virulence traits. In the present studies, insect stage epimastigotes were stably transfected with a plasmid encoding the trypomastigote-specific CRP. Transfected epimastigotes expressed the CRP transgene, and production of recombinant CRP was sufficient to convert epimastigotes from a complement-sensitive to a complement-resistant state. The results of these studies demonstrate for the first time that a trypomastigote-specific virulence trait can be produced by noninfectious epimastigotes and that expression of the CRP cDNA is sufficient to confer a complement-resistant phenotype.
MATERIALS AND METHODS
Media, buffers, and reagents.
All of the chemicals and reagents used were of molecular biology grade and were obtained from Sigma Chemical Co. (St. Louis, Mo.) or Boehringer Mannheim (Indianapolis, Ind.), unless otherwise indicated. Dulbecco’s minimal essential medium (GIBCO BRL, Gaithersburg, Md.) was supplemented as previously described (16). Guinea pig complement and rabbit complement were obtained from Accurate Chemical and Scientific Corp., Westbury, N.Y., and heat-inactivated complement (HIC) was prepared by incubation of complement at 56°C for 30 min. Lysis buffer contained 2% Triton X-114 (Pierce Chemicals, Rockford, Ill.) in 50 mM Tris (pH 7.4)–150 mM NaCl. Labeling medium was Dulbecco’s minimal essential medium, without cysteine and methionine (ICN Biochemicals, Costa Mesa, Calif.), buffered with 10 mM HEPES (pH 7.4) and supplemented with 10 μg of ovalbumin per ml and 2 mM glutamine. Tris-buffered saline (TBS) consisted of 50 mM Tris base (pH 7.5) and 150 mM NaCl. Blocking buffer was TBS containing 5% nonfat powdered milk. Transfer buffer was 50 mM Tris (pH 8.3)–380 mM glycine–0.1% sodium dodecyl sulfate [SDS]–20% methanol). Protease inhibitors (leupeptin, aprotinin, and E-64, all from Sigma Chemical Co.) were each added as indicated at a final concentration of 1 μg/ml.
Bacterial strains and plasmid preparation.
Escherichia coli SURE cells were used in transformations as recommended by the supplier (Stratagene, La Jolla, Calif.). Small-scale plasmid preparations were obtained by using Wizard minipreparation kits (Promega, Madison, Wis.). DNA was prepared for electroporation and automated sequencing by using Qiagen maxipreparation kits (Qiagen, Inc., Chatsworth, Calif.).
Parasites.
T. cruzi Y was used throughout these experiments (18). Epimastigotes were maintained in logarithmic growth phase at 28°C in supplemented LDNT as previously described (5). Tissue culture-derived trypomastigotes were recovered from the supernatant of infected monolayers of NIH 3T3 cells as previously described (20).
Construction of pTEX-CRP.
Plasmid pCRII containing the CRP-10 gene (17) was digested with BamHI and XhoI, and the 3,072-bp fragment containing the entire coding region and 53 bp of the upstream sequence was isolated by gel electrophoresis, purified by using Geneclean II (Bio 101, Inc., La Jolla, Calif.), and cloned into the BamHI- and XhoI-digested pTEX vector (gift of John Kelly, London School of Hygiene and Tropical Medicine, London, England) to yield pTEX-BX23 (see Fig. 1). A derivative of pTEX-BX23 lacking the 53-bp upstream, noncoding sequence was cloned and found to be unstable in E. coli, most likely due to readthrough from a prokaryotic promoter in the vector 5′ to the multiple cloning site. pTEX-BX23 was therefore modified such that the entire expression cassette from the SacII site to the KpnI site was flipped in orientation as follows. The plasmid was digested with SacII and KpnI, restriction overhangs were filled by treatment with T4 DNA polymerase and deoxynucleotides (19), and the DNA fragments were separated by gel electrophoresis and purified by Geneclean II. The purified vector was treated with calf intestine phosphatase as previously described (19) and ligated to the purified insert containing the expression cassette. Clones in the proper orientation were isolated. One clone, pTEX-BX24, was used to derive the final construct, pTEX-CRP, which is devoid of the 53-bp upstream sequence. The pTEX-CRP construct was obtained by amplification of the 5′ end of the CRP-10 gene with a 5′ primer starting at the putative translation initiation codon of CRP-10 (nucleotide 235) and incorporating a BamHI recognition sequence at the 5′ end (160-31; 5′-GAATTCGGATCCATGTCCCGTCATGTGTTTG-3′). The antisense primer (160-30; 5′-GGCGACGTTCTCACCCACAG-3′) covered an EcoRV recognition sequence at nucleotide 1377 in CRP-10. PCR was carried out for 30 cycles of 94°C for 1 min, 52°C for 2 min, and 72°C for 3 min and a 10-min extension at 72°C at the end of cycling. The 1.1-kb PCR product was gel purified and digested with BamHI and EcoRV. pTEX-BX24 was digested with BamHI and EcoRV, and the vector containing the 3′ end of the CRP gene was isolated and purified. This fragment was ligated to the BamHI- and EcoRV-digested PCR product to produce the final construct, pTEX-CRP. The CRP insert of the final construct was sequenced on both strands as previously described (17) to verify that no errors were introduced during the PCR amplification.
FIG. 1.

Construction of pTEX-BX23 and pTEX-CRP expression plasmids. (A) pTEX-BX23 was constructed by ligating a fragment containing the entire crp coding region and the 53-bp upstream (5′ UT) and 19-bp downstream (3′ UT) sequences into pTEX at the BamHI and XhoI sites. (B) pTEX-CRP was constructed by flipping the orientation of the SacII-KpnI fragment of pTEX-BX23 containing the crp gene and removing the 5′ UT crp sequence. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; NeoR, neomycin phosphotransferase gene.
Transfection of T. cruzi epimastigotes.
Plasmid DNA was isolated as described above and resuspended in buffer containing 10 mM Tris base (pH 7.5), and 1 mM EDTA. Epimastigotes were grown to a density of 1.5 × 107/ml in supplemented LDNT, washed once at room temperature in phosphate-buffer saline (PBS) (132 mM NaCl, 8 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4 [pH 7.0]), and resuspended at 108/ml in electroporation buffer (PBS, 0.5 mM magnesium acetate, 0.1 mM CaCl2). Cells (0.4 ml) were dispensed into disposable 0.2-mm gap cuvettes (Bio-Rad Laboratories, Richmond, Calif.), and 50 μl (150 μg) of plasmid DNA was added. The cells were electroporated by using a Bio-Rad Gene Pulser set at 300 V and 500 μF with four pulses in close succession. Samples were placed on ice for 10 min, and the contents were transferred to a flask containing 5 ml of supplemented LDNT. The parasites were incubated at 28°C, and after 48 h in culture, G418 was added to a final concentration of 50 μg/ml.
RT-PCR and Southern blotting of CRP in transfected epimastigotes.
Total cellular RNA was prepared from transfected epimastigotes by using Trizol reagent (GIBCO BRL) in accordance with the supplier’s direction. For cDNA synthesis, 5 μg of RNA was reverse transcribed by using the GIBCO BRL Superscript Preamplification System as directed by the manufacturer. The reverse transcription (RT) primer (160-18; 5′-TGTCTCGAGCACGTCAATCCACCATCAGG-3′) was used at a concentration of 0.5 μM and is the antisense sequence of CRP-10 at nucleotide 1767 (17). PCR amplification was carried out with 1/10 of the final volume of the cDNA reaction mixture, a 0.2 μM concentration of a sense oligomer derived from the last 25 nucleotides of the T. cruzi miniexon sequence (ME-2; 5′-ATCTGCAAGTTAACGCTATTATTGATACAGTTTCTGTA-3′), and a 0.2 μM concentration of an antisense oligomer beginning at nucleotide 1337 in CRP-10 (17) (160-20; 5′-AATTCGCTGCATGTCCGTCA-3′). The cycling conditions used were the same as those described above, except that annealing was carried out at 55°C. Products were electrophoresed in 1% agarose gels and visualized by ethidium bromide staining and UV illumination. DNA was transferred to a Nytran blotting membrane (Schleicher & Schuell), and the membrane was hybridized with a CRP-specific oligomer (160-9; 5′-CCTGCAGCAGTGAAGCCACT-3′). The probe (30 pmol) was labeled with digoxigenin-11-dUTP (Boehringer Mannheim) by incubation with 20 U of terminal transferase (United States Biochemical, Cleveland, Ohio) as directed by the supplier. The labeled probe was added to the membranes, and hybridization was carried out overnight at 60°C in 6× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) (19)–0.1% SDS–0.5% nonfat powdered milk. Membranes were washed, developed by using the Genius chemiluminescence system (Boehringer Mannheim), and exposed to Kodak X-Omat AR film for 10 min at room temperature.
Biosynthetic labeling and membrane protein preparation.
Epimastigotes were recovered from culture as described above, washed twice in PBS–1% glucose, and resuspended at 108/ml in labeling medium. [35S]methionine (Trans35S-label; ICN) was added at 50 μCi/ml, and the cells were incubated for 1 h at 28°C. After labeling, the cells were washed twice at 4°C in PBS–1% glucose. Trypomastigotes were labeled in the same manner, except that the labeling was carried out at 37°C. Membrane extracts were prepared as follows. After biosynthetic labeling, parasites were frozen and thawed, resuspended at 4 × 108/ml in deionized water containing protease inhibitors, and incubated at room temperature for 5 min. Membrane fragments were pelleted for 5 min at 5,000 × g. The pellet was solubilized in the original volume of 2% Triton X-114 in TBS with fresh protease inhibitors and incubated for 30 min on ice. Insoluble material was removed by microcentrifugation for 10 min at 4°C. To extract the detergent from the solubilized proteins, the supernatant was incubated at 37°C for 3 min and microcentrifuged for 3 min at room temperature, and then the aqueous phase was recovered. Nonidet P-40 (Pierce Chemicals) was added to the aqueous-phase proteins at a final concentration of 0.05% (vol/vol).
C3b preparation and affinity purification of CRP from T. cruzi trypomastigotes and transfected epimastigotes.
A human C3b affinity matrix was prepared as previously described (16). For C3b affinity chromatography of T. cruzi CRP, biosynthetically labeled membrane protein preparations from trypomastigotes and epimastigotes transfected with either the pTEX vector or pTEX-CRP were harvested as described above. Triton X-114 membrane protein extracts were prepared, and protein levels were quantitated by trichloroacetic acid precipitation of radiolabeled material. The same number of radioactive counts from each membrane preparation was adjusted to a 50-μl volume with TBS–0.05% Nonidet P-40 and incubated with 50 μl of C3b affinity beads which had been equilibrated in TBS–0.05% Nonidet P-40. Incubation was carried out for 1 h at room temperature with gentle shaking. The beads were washed three times in the same buffer, and bound protein was released by incubating the beads in SDS sample buffer and boiling them for 3 min. Samples were analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) through an 8.5% polyacrylamide gel followed by fluorography.
Western blotting.
Parasite membrane extracts were prepared as described above, and the protein concentration was measured with the Bio-Rad DC assay as directed by the manufacturer (Bio-Rad Laboratories). A 5-μg total-protein sample was subjected to SDS-PAGE as previously described (16). Proteins were transferred to nitrocellulose in transfer buffer at 45 V for 16 h at 4°C. The filter was blocked and incubated with mouse anti-recombinant CRP serum (17) or normal mouse serum, diluted 1:400 in blocking buffer, for 1 h. The filter was washed in TBS and incubated with alkaline phosphatase-conjugated goat anti-mouse immunoglobulin (Cappel-ICN, Costa Mesa, Calif.) diluted 1:15,000. After washing, the filter was incubated in 150 mM Tris-base (pH 9.5)–100 mM NaCl–5 mM MgCl2 for 5 min and developed with 5-bromo-4-chloro-3-indolylphosphate and nitroblue tetrazolium substrate (Promega).
Complement-mediated lysis.
Transfected epimastigotes were grown to a concentration of 5 × 106/ml, washed once in PBS–1% glucose, and resuspended at 107/ml in PBS–1% glucose. To measure complement-mediated lysis, a 100-μl volume of cells was mixed with 100 μl of rabbit or guinea pig complement or HIC. Cells were incubated for 1 h at 28°C, diluted 1:5 in PBS–1% glucose, and then counted in a hemocytometer. Percent survival after treatment was calculated as follows: percent survival = 100 × (number of motile parasite after treatment with complement/number of motile parasites after treatment with HIC).
RESULTS
Construction of pTEX-CRP expression vector.
The shuttle vector pTEX, which replicates in E. coli and T. cruzi (4), was used as a vehicle for expression of the trypomastigote CRP cDNA in epimastigotes. The vector contains a multiple cloning site flanked by the untranslated (UT) 5′ and 3′ regions of the T. cruzi glyceraldehyde 3-phosphate dehydrogenase gene and the neomycin phosphotransferase gene (neo) for selection of transfectants. The full-length CRP-10 cDNA, which had been previously cloned in pCRII, has three in-frame ATG codons upstream of the sequence encoding the mature protein (17). The third ATG, at nucleotide 235, was chosen as the putative translation initiation site because the predicted translation from this codon identifies a 24-amino-acid sequence that conforms to the eukaryotic signal sequence consensus and is consistent with the amino acid sequence identifying the start of the mature protein (17). This is not the case with the two upstream ATGs (17). In addition, the nucleotide sequence surrounding the ATG at nucleotide 235 is consistent with the Kozak consensus for translation initiation, although it has not been determined whether the Kozak consensus sequence is important in trypanosome translation initiation. A 3-kb fragment containing the entire CRP coding region plus 53 bp upstream of the putative start site was cloned into pTEX at the BamHI and XhoI sites (Fig. 1A). This construct, pTEX-BX23, when transfected into epimastigotes, produced G418-resistant cell lines; however, no CRP mRNA or protein could be detected in these cells (data not shown). This construct was modified so that the upstream sequence was removed and only the coding sequence from nucleotide 234 of CRP was cloned into pTEX at the BamHI and XhoI sites. Restriction digests of several of the E. coli transformants revealed gross rearrangements of the insert, indicating instability, possibly due to a prokaryotic promoter upstream of the T. cruzi expression cassette. To test this possibility and to derive a more stable construct, the entire expression cassette of pTEX from the SacII site through the KpnI site was inverted and recloned into the pTEX vector backbone as described in Materials and Methods (Fig. 1B). The CRP-10 fragment from the putative translational start at nucleotide 235 through nucleotide 3253 was cloned into this modified vector, and stable transformants were isolated. One isolate, pTEX-CRP, was verified by nucleic acid sequencing and used for epimastigote transfection.
Transfection of T. cruzi epimastigotes.
Wild-type epimastigotes were tested for sensitivity to G418, and it was found that at a concentration of 50 μg/ml, all of the parasites were dead at approximately 10 days. Control cells electroporated with no DNA and cultured in the presence of G418 were dead by 10 to 14 days postelectroporation. Parasites transfected with either the vector alone or pTEX-CRP yielded G418-resistant cell lines. After stable lines were established (approximately 4 weeks posttransfection), the G418 concentration was increased at 2-week intervals to 100, 200, and 500 μg/ml. No detectable differences in the growth rates of parasites were seen during the antibiotic increases (data not shown). In addition, no detectable differences in growth rates were observed between epimastigotes transfected with the pTEX vector alone and those transfected with the pTEX-CRP construct (data not shown). Two transfected cell lines, CRP 2.3 and CRP 2.14, were derived from separate transfections and maintained in supplemented LDNT medium with G418 at 500 μg/ml.
Detection of CRP-10 cDNA expression in T. cruzi epimastigotes by RT-PCR.
Total RNA was isolated from the transfected cell lines, and RT-PCR was performed to determine whether the CRP gene was being expressed in the transfected epimastigotes. The expected product of the RT-PCR was a 1,330-bp fragment covering the 5′ end of CRP. This fragment was detected by agarose gel electrophoresis and ethidium bromide staining of the RT-PCR product from CRP 2.3 and by Southern blotting using a CRP-specific probe (Fig. 2A). Similar results were obtained with RNA derived from the CRP 2.14 epimastigotes (data not shown). To verify that the signal observed was due to RT and amplification of the CRP transgene and not due to amplification of trace amounts of genomic CRP DNA in the RNA preparations, a mock RT-PCR was carried out on CRP 2-3 RNA with no addition of reverse transcriptase. No band was detected by ethidium bromide staining or Southern blotting (Fig. 2A), thus confirming that the transfected epimastigotes were transcribing the trypomastigote-specific CRP cDNA. Likewise, no band corresponding to CRP was detected in the RT-PCR product of epimastigotes transfected with the vector alone, again confirming that CRP expression is developmentally regulated and specific for the trypomastigote stage.
FIG. 2.

Ethidium bromide-stained gel and Southern blot of RT-PCR products derived from transfected T. cruzi epimastigote RNA. (A) Lanes 2 to 4 contain the RT-PCR products from the CRP 2.3 cell line without RT in the reaction mixture (lane 2), the vector-transfected cell line (lane 3), and the CRP 2.3 cell line (lane 4). Lanes 5 to 7 are an autoradiograph of a Southern blot of RT-PCR samples that are the same as those in lanes 2 to 4 but probed with a CRP-specific oligomer. Lane 1 contains ethidium bromide-stained DNA size markers. Sizes in base pairs are indicated at the left. (B) Schematic representation of the full-length CRP-10 cDNA (approximately 3,300 bp), illustrating the positions of the oligomers used in the RT-PCR and the Southern blot in panel A. The RT primer was the antisense sequence of the CRP-10 cDNA at nucleotide 1767. PCR amplification was carried out with a sense oligomer derived from the last 25 nucleotides of the T. cruzi mini-exon sequence and an antisense oligomer beginning at nucleotide 1337 of the CRP-10 cDNA (CRP-20), as described in Materials and Methods.
Western blot analysis of CRP-transfected T. cruzi epimastigotes.
Epimastigotes were grown to log phase and detergent solubilized, and membrane-enriched protein extracts were used in Western blots to detect recombinant CRP. Polyclonal antiserum raised against the full-length recombinant CRP expressed in E. coli was used to detect CRP (17). A protein migrating at approximately 100 kDa was detected in the protein extracts from the CRP 2.3 and CRP 2.14 cell lines but not in extracts prepared from the pTEX-transfected cell line (Fig. 3). The difference in apparent molecular mass between the epimastigote-derived recombinant CRP and the native trypomastigote protein is approximately 60 kDa and may be the result of protein degradation in the epimastigote preparations or differential glycosylation of the CRP protein produced in the two parasite stages.
FIG. 3.

Western blot of membrane protein extracts derived from transfected T. cruzi epimastigotes. Protein extracts were fractionated by SDS-PAGE, blotted onto nitrocellulose, and probed with mouse anti-CRP serum (1:400) (lanes 1 to 3) or normal mouse serum (NMS; 1:400) (lanes 4 to 6). Lanes contained 5 μg of protein derived from epimastigote cell lines transfected with CRP 2.3 (lanes 1 and 4), CRP 2.14 (lanes 2 and 5), or the pTEX vector (lanes 3 and 6). Molecular mass standards (kilodaltons) are shown on the left.
Epimastigote CRP has C3b-binding capacity and restricts complement activation.
To determine whether the epimastigote-derived recombinant CRP was functional, metabolically labeled membrane proteins from the pTEX-transfected and CRP 2.3 epimastigote cell lines were subjected to affinity chromatography on a human C3b affinity matrix. Radiolabeled material in the membrane extracts was quantitated by trichloroacetic acid precipitation, and equal amounts of radiolabeled protein from pTEX-transfected and CRP 2.3 cell membrane preparations were loaded onto the C3b matrix. Proteins specifically bound to the C3b matrix were eluted and separated by SDS-PAGE, which was followed by fluorography (Fig. 4). Similar to the Western blot results, epimastigotes transfected with the pTEX-CRP construct produced a C3b-binding protein which migrated at approximately 100 kDa and smaller proteins migrating in the 40- to 60-kDa range, possibly representing proteolytic degradation. Membrane preparations from CRP 2.14 cells gave similar results (not shown).
FIG. 4.
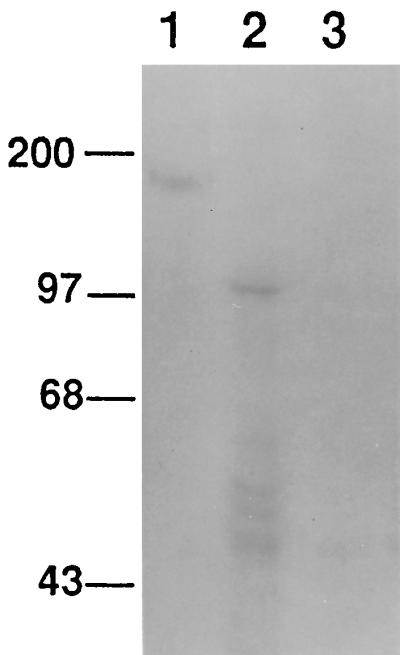
C3b affinity chromatography of membrane protein extracts derived from transfected T. cruzi epimastigotes. Epimastigotes were metabolically labeled with [35S]methionine, and detergent-solubilized membrane protein extracts were subjected to C3b affinity chromatography. Eluted proteins were fractionated by SDS-PAGE, and the gel was prepared for fluorography and exposed to X-ray film for 24 h. Lanes: 1, C3b-eluted protein from tissue culture-derived T. cruzi trypomastigotes; 2, transfected CRP 2.3 epimastigotes; 3, pTEX-transfected epimastigotes. Molecular mass standards (kilodaltons) are shown at the left.
To test the functional activity of the recombinant epimastigote CRP in intact cells, parasites were grown to log phase and treated with normal guinea pig serum (as a source of complement) or heat-inactivated serum at a final dilution of 1:2. As shown in Table 1, CRP 2.14 epimastigotes exhibited higher levels of resistance to complement-mediated lysis than did epimastigotes transfected with the vector alone. Decreasing the complement concentration by 50% increased the survival of CRP-transfected epimastigotes to 100%; however, 35 to 40% of the vector-transfected cells survived this treatment (data not shown). Complement-mediated lysis assays with CRP 2.3 cells produced comparable results (not shown).
TABLE 1.
Complement-mediated lysis of pTEX-transfected or CRP 2.14 T. cruzi epimastigotes
| Cell line | Treatmenta | Mean cell count ± SDb | % Survivalc |
|---|---|---|---|
| pTEX transfected | Complement | 1 ± 1 | |
| HIC | 52.3 ± 2.1 | 1.9 | |
| CRP 2.14 | Complement | 43.3 ± 4.5 | |
| HIC | 66 ± 8.1 | 65.6 |
Parasites were incubated for 60 min at 28°C in either fresh guinea pig complement or HIC at a final dilution of 1:2.
Values are the mean numbers of motile parasites counted in a hemocytometer at a magnification of ×400. Each value is the mean of quadruplicate samples. The results shown are representative of multiple experiments.
Percent survival = 100 × number of motile parasites treated with complement/number of motile parasites treated with HIC. Each treatment was carried out in duplicate, and duplicate counts were made of each sample. The results shown are representative of multiple individual experiments.
DISCUSSION
The results reported here demonstrate that protection of CRP-transfected epimastigotes from complement-mediated lysis is the result of expression of the CRP transgene and CRP production. This conclusion is most strongly supported by results of the complement-mediated lysis assays, in which transfection of epimastigotes with a plasmid encoding the CRP structural gene was sufficient to confer the complement-resistant phenotype. Additionally, it was confirmed that the protein produced by the transfected epimastigotes was cross-reactive with anti-CRP antibodies and was also capable of binding human C3b, demonstrating that the recombinant protein retained its native function. These results were in contrast to those obtained with vector-transfected epimastigotes, which remained complement sensitive and did not produce detectable levels of CRP.
The CRP isolated from membrane preparations of transfected epimastigotes differed in apparent molecular mass from the native trypomastigote-derived CRP (Fig. 3 and 4), and it has not been determined whether this decrease in size, from 160 to 100 kDa, represents proteolytic degradation of the CRP in the epimastigote membrane preparations or whether the protein produced is modified by epimastigotes. It is still clearly functional, however, based on its conferral of complement resistance on transfected cells and its binding affinity for C3b. We have previously found that the native CRP expressed in trypomastigotes undergoes rapid turnover at the cell surface, and when live parasites are incubated with C3b, CRP is rapidly released from the cell surface in a predominantly 100-kDa form (14). The release of the 100-kDa form by trypomastigotes is the result of proteolytic cleavage of the CRP fragment after it binds C3b, and its release may serve to protect the parasite by removing potentially opsonic C3b (14). It is reasonable to suspect that the CRP produced in epimastigotes has the same protease-sensitive site and that solubilization of endogenous proteases during membrane preparation may generate the observed 100-kDa form. Studies examining the effects of protease inhibitors on CRP release from transfected epimastigotes are under way.
Approximately 60 to 70% of the parasites were protected from lysis by complement under the assay conditions used. These results suggest that there is some heterogeneity in expression levels or some genetic rearrangement of the recombinant plasmid leading to altered expression of CRP in cells maintained in continuous culture. Rearrangement of vector sequences has previously been reported to occur in up to 15% of pTEX-transfected epimastigotes (4), and this phenomenon may have contributed to the phenotype nonuniformity observed in this study.
The analysis of potential virulence factors of protozoan pathogens has been limited by the lack of genetic systems for manipulation and expression of specific DNA sequences. The development of shuttle vectors with expression sequences for trypanosome genes has opened this avenue of investigation. Evidence is presented here that expression of the trypomastigote-specific CRP gene in insect stage epimastigotes is sufficient to confer a complement-resistant phenotype on these cells. The constitutive expression of developmentally regulated CRP in insect stage epimastigotes and conversion of these cells to a complement-resistant phenotype strongly argue that CRP functions to restrict complement activation and thus contributes to the virulence of trypomastigotes by allowing survival in the bloodstream. In this regard, CRP is among the first T. cruzi proteins to be established as a virulence factor.
The T. cruzi CRP gene family consists of at least 700 copies per genome (17, 21), and sequence analysis of RT-PCR products reveals that several different members of the gene family are transcribed in a clonal cell line (11a). The observation that the parasites maintain several hundred copies of CRP, with an undetermined number of them transcriptionally active, raises the question of whether all transcribed copies have the potential for translation and production of the functional protein. The ability to isolate multiple full-length CRP cDNAs and produce the functional protein in transfected epimastigotes allows us to further explore the extent to which clonal lines of the parasite express multiple variants of this gene and whether the variant proteins are functionally and antigenically distinguishable. Unfortunately, the large number of gene copies precludes the construction of T. cruzi strains deficient in CRP expression. Thus, expression in epimastigotes is an alternative means to analyze the function of this protein.
The beneficial role of complement-mediated clearance of trypomastigotes in T. cruzi infections has long been recognized (1, 10). In addition, an association between anti-T. cruzi antibodies that could support complement-mediated lysis of trypomastigotes and clearance of parasites in experimental infections and in human Chagas’ disease has been reported (6–9, 15). Previous studies have demonstrated that purified CRP interacts with C3b noncovalently such that formation of the C3 convertase is inhibited or the active convertase is destabilized (12, 16). The present studies demonstrate that CRP, as it is expressed in epimastigotes, is sufficient to block complement activation at the parasite surface. These results clearly establish the role of CRP in protecting the parasites against complement-mediated killing.
It has previously been shown that native CRP does not serve as a cofactor for the serum protease factor I and thus does not influence complement activation by degradation of C3b (12, 16). Inasmuch as binding of CRP to C3b appears to be responsible for its complement-inhibitory activity, it is possible that reagents such as specific immunoglobulins or peptides that block the CRP-C3b binding interaction may inhibit the complement regulatory activity and lead to more rapid clearance of trypomastigotes. Exploitation of CRP as a potential vaccine candidate will be facilitated by functional analysis of CRP and mutated derivatives expressed in epimastigotes. These studies will allow the determination of the C3b binding regions of the protein and the characterization of reagents which block this activity.
ACKNOWLEDGMENTS
This work was supported by Public Health Service award AI32719 from the National Institute of Allergy and Infectious Diseases and American Heart Association Established Investigator award 9640055.
I thank John Kelly for the pTEX vector and David Engman and Lisa Godsel for helpful advice with the epimastigote transfections. The technical assistance of Jane Schrimpf, Marika Szabo, and Alexia Mascilli is gratefully acknowledged.
Footnotes
Mailing address: Department of Molecular Genetics and Biochemistry, E 1240 Biomedical Science Tower, University of Pittsburgh School of Medicine, Pittsburgh, PA 15261. Phone: (412) 648-8848. Fax: (412) 624-1401. E-mail: kan1@vms.cis.pitt.edu.
REFERENCES
- 1.Budzko D B, Pizzimenti M C, Kierszenbaum F. Effects of complement depletion in experimental Chagas’ disease: immune lysis of virulent blood forms of Trypanosoma cruzi. Infect Immun. 1975;11:86–91. doi: 10.1128/iai.11.1.86-91.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fischer E, Ouassi M, Velge P, Cornette J, Kazatchkine M. gp58/68, a parasite component that contributes to the escape of the trypomastigote form of T. cruzi from damage by the human alternative complement pathway. Immunology. 1988;65:299–303. [PMC free article] [PubMed] [Google Scholar]
- 2a.Galvão, L., and K. Norris. Unpublished data.
- 3.Joiner K, Dias da Silva W, Rimoldi M, Hammer C, Sher A, Kipnis T. Biochemical characterization of a factor produced by trypomastigotes of Trypanosoma cruzi that accelerates the decay of complement C3 convertases. J Biol Chem. 1988;263:11327–11335. [PubMed] [Google Scholar]
- 4.Kelly J, Ward H, Miles M, Kendall G. A shuttle vector which facilitates the expression of transfected genes in Trypanosoma cruzi and Leishmania. Nucleic Acids Res. 1992;20:3963–3969. doi: 10.1093/nar/20.15.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirchhoff L, Hieny S, Shiver G, Snary D, Sher A. Cryptic epitope explains the failure of a monoclonal antibody to bind to certain isolates of Trypanosoma cruzi. J Immunol. 1984;133:2731–2735. [PubMed] [Google Scholar]
- 6.Krettli A. Protective antibodies in Trypanosoma cruzi infections: detection, functional activity and possible mechanisms of trypomastigote killing in vivo and in vitro. Mem Inst Oswaldo Cruz. 1984;79:59–65. [Google Scholar]
- 7.Krettli A, Brener Z. Protective effects of specific antibodies in Trypanosoma cruzi infections. J Immunol. 1976;116:755–760. [PubMed] [Google Scholar]
- 8.Krettli A, Brener Z. Resistance against Trypanosoma cruzi associated to anti-living trypomastigote antibodies. J Immunol. 1982;128:2009–2012. [PubMed] [Google Scholar]
- 9.Krettli A, Cancado J, Brener Z. Criterion of cure of human Chagas disease after specific chemotherapy: recent advances. Mem Inst Oswaldo Cruz. 1984;79:157–164. [Google Scholar]
- 10.Mota I, Umekita L. The effect of C3 depletion on the clearance of Trypanosoma cruzi induced by IgG antibodies. Immunol Lett. 1989;21:223–226. doi: 10.1016/0165-2478(89)90108-9. [DOI] [PubMed] [Google Scholar]
- 11.Noguiera N, Bianco C, Cohn Z. Studies on the selective lysis and purification of Trypanosoma cruzi. J Exp Med. 1975;142:224–229. doi: 10.1084/jem.142.1.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11a.Norris, K. Unpublished data.
- 12.Norris K, Bradt B, Cooper N, So M. Characterization of a Trypanosoma cruzi C3 binding protein with functional and genetic similarities to the complement regulatory protein, decay accelerating factor. J Immunol. 1991;147:2240–2247. [PubMed] [Google Scholar]
- 13.Norris K, Harth G, So M. Purification of a Trypanosoma cruzi membrane glycoprotein which elicits lytic antibodies. Infect Immun. 1989;57:2372–2377. doi: 10.1128/iai.57.8.2372-2377.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Norris K A. Ligand-binding renders the 160 kDa Trypanosoma cruzi complement regulatory protein susceptible to proteolytic cleavage. Microb Pathog. 1996;21:235–248. doi: 10.1006/mpat.1996.0058. [DOI] [PubMed] [Google Scholar]
- 15.Norris K A, Galvão L M C, Schrimpf J E, Cancado J R, Krettli A U. Humoral immune response to the Trypanosoma cruzi complement regulatory protein as an indicator of parasitologic clearance in human Chagas’ disease. Infect Immun. 1994;62:4072–4074. doi: 10.1128/iai.62.9.4072-4074.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Norris K A, Schrimpf J E. Biochemical analysis of the membrane and soluble forms of the complement regulatory protein of Trypanosoma cruzi. Infect Immun. 1994;62:236–243. doi: 10.1128/iai.62.1.236-243.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Norris K A, Schrimpf J E, Szabo M J. Identification of the gene family encoding the 160-kilodalton Trypanosoma cruzi complement regulatory protein. Infect Immun. 1997;65:349–357. doi: 10.1128/iai.65.2.349-357.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pereira da Silva L, Nussenzweig V. Sobre uma cepa de Trypanosoma cruzi altamente virulenta para o camundongo branco. Folio Clin Biol. 1953;20:191–195. [Google Scholar]
- 19.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 20.Sanderson C, Thomas J, Twomey C. The growth of Trypanosoma cruzi in human diploid cells for the production of trypomastigotes. Parasitology. 1980;80:153–162. doi: 10.1017/s0031182000000615. [DOI] [PubMed] [Google Scholar]
- 21.Van Voorhis W, Barrett L, Koelling R, Farr A. FL-160 proteins of Trypanosoma cruzi are expressed from a multigene family and contain two distinct epitopes that mimic nervous tissues. J Exp Med. 1993;178:681–694. doi: 10.1084/jem.178.2.681. [DOI] [PMC free article] [PubMed] [Google Scholar]