

Significance
This study describes the development of a second-generation eIF4A RNA helicase inhibitor (MG-002), which potently inhibits the synthesis of tumor- and metastasis-promoting proteins. We show that MG-002 is superior to first-generation eIF4A inhibitors in eliciting tumoricidal effects in aggressive triple-negative breast cancers, which otherwise would be refractory to standard therapies.
Keywords: eIF4A, mRNA translation, triple-negative breast cancer, lung metastasis
Abstract
In this study, we aimed to address the current limitations of therapies for macro-metastatic triple-negative breast cancer (TNBC) and provide a therapeutic lead that overcomes the high degree of heterogeneity associated with this disease. Specifically, we focused on well-documented but clinically underexploited cancer-fueling perturbations in mRNA translation as a potential therapeutic vulnerability. We therefore developed an orally bioavailable rocaglate-based molecule, MG-002, which hinders ribosome recruitment and scanning via unscheduled and non-productive RNA clamping by the eukaryotic translation initiation factor (eIF) 4A RNA helicase. We demonstrate that MG-002 potently inhibits mRNA translation and primary TNBC tumor growth without causing overt toxicity in mice. Importantly, given that metastatic spread is a major cause of mortality in TNBC, we show that MG-002 attenuates metastasis in pre-clinical models. We report on MG-002, a rocaglate that shows superior properties relative to existing eIF4A inhibitors in pre-clinical models. Our study also paves the way for future clinical trials exploring the potential of MG-002 in TNBC and other oncological indications.
Breast cancer is a heterogeneous disease that is classified into three main histological subtypes, which informs both patient outcome and treatment options for clinical management of the disease (1). These include estrogen/progesterone receptor (ER/PR)-positive (+) (~65% incidence), human epidermal growth factor receptor 2 (HER2)+ (~20% incidence), and triple-negative breast cancers (TNBC: ER-/PR-/HER2-) (~15% incidence). In comparison to other subtypes, TNBCs tend to be more aggressive and are more likely to be diagnosed in pre-menopausal women (2). Moreover, in contrast to ER+ and HER2+ breast cancers, effective targeted therapies for TNBC remain elusive, and chemotherapy remains the standard of care.
To date, chemotherapeutic regimens are largely ineffective for many patients (3), owing to the intra-tumoral heterogeneity of TNBC tumors (4). Indeed, following neo-adjuvant chemotherapy, patients diagnosed with TNBC show decreased 3-y survival rates and increased rate of recurrence and progression to metastatic disease as compared to women with non-TNBC disease (5). Increased genomic instability has informed pre-clinical studies to explore whether PARP and/or immune checkpoint inhibitors represent valid therapeutic strategies in TNBC. Unfortunately, both therapeutic modalities showed less than expected results in clinical trials (6, 7). It is therefore essential to identify clinically targetable vulnerabilities that are not affected by the inherent heterogeneity of TNBC malignancies.
Dysregulation of mRNA translation has been associated with tumor initiation, dissemination, and drug resistance (8, 9). Malignancy-associated perturbations in mRNA translation can arise as a consequence of hyperactivation of mitogen-activated protein kinase (MAPK) orphosphatidylinositol-3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathways (10)—both of which indirectly activate the eukaryotic initiation factor (eIF) 4F complex (8). Attempts to target the PI3K/Akt/mTOR pathway in women diagnosed with TNBC have failed (11–13), likely due to their inability to sufficiently suppress eIF4F complex assembly and/or function (14). This led us to focus on directly interfering with eIF4F function. The eIF4F complex consists of the eIF4E cap-binding protein, the DEAD-box RNA helicase eIF4A, and a scaffolding protein, eIF4G. eIF4F stimulates the recruitment of 40S ribosomes (and associated initiation factors) to the mRNA 5′ cap (15). Mammals encode three related eIF4A proteins: i) eIF4A1 (DDX2A), ii) eIF4A2 (DDX2B), and iii) eIF4A3 (DDX48). eIF4A1 and eIF4A2 share 90% amino acid identity, recycle through the eIF4F complex, and are implicated in translation (15). DDX48/eIF4A3, not involved in translation initiation, is a component of the exon junction complex and shares 66% amino acid identity with eIF4A1 (16). The requirement for eIF4A in translation is proportional to cap-proximal secondary structure (17), which imparts unequal responses to different mRNAs following changes in eIF4F levels and/or activity. For example, increased eIF4F activity preferentially increases mRNA translation of pro-tumorigenic factors, including cellular MYC (c-MYC) and cyclins (8, 9, 18, 19). In contrast, alterations in eIF4F levels and/or activity exert a minimal effect on translation of mRNAs required for normal cellular homeostasis (8). To this end, decreasing levels and/or activity of the eIF4F complex provides a sufficient therapeutic window to target the eIF4F complex in oncological indications (8).
Importantly, increased eIF4F levels are a common feature of cancer cells and are thus largely independent of intra-tumor heterogeneity (20). The high reliance of TNBC cells on eIF4F activity therefore positions this complex as an attractive target to overcome intra-tumor heterogeneity. Moreover, existing therapeutic approaches targeting the eIF4E subunit of the eIF4F complex induce cytostatic effects that are associated with induction of autophagy and metabolic dormancy (21). On the contrary, inhibition of eIF4A results in distinct perturbations in mRNA translation that are paralleled with the suppression of these compensatory mechanisms and cytotoxic effects (21). Motivated by this, we focused on the identification and characterization of inhibitors targeting the eIF4A helicase subunit of eIF4F (22).
Rocaglates are cyclopenta[b]benzofuran natural products produced by plants of the Aglaia genus (23). Several hundred natural and synthetic rocaglates have been isolated, synthesized, and studied (24). Most rocaglates target translation by acting as molecular staples that clamp eIF4A (and eIF4F) to polypurine-rich RNA sequences (24–26). This results in gain-of-function complexes that exert multiple effects on translation initiation. When eIF4A•Rocaglate• RNA (eIF4A•Roc•RNA) complexes form in mRNA 5′ leader regions they block ribosome scanning (25). In addition, eIF4F• Roc•RNA complexes are not competent for ribosome recruitment (24). Blocking of scanning also leads to 40S ribosome collisions, activating initiation ribosome–associated quality control and 40S ribosome degradation (27). Our previous work on rocaglates showed that in general the ability of a compound to induce RNA clamping correlates with inhibition of translation; however, there are notable exceptions. For example, the natural product silvestrol is not as potent as the synthetic rocaglate CR-1-31B at inducing clamping, yet both inhibit translation to similar extents (24). Unlike silvestrol, CR-1-31B has clear selectivity for blocking translation of mRNAs with polypurine-rich 5′ leader regions (24). Finally, rocaglamide A (RocA) also induces clamping of another DEAD-box RNA helicase, DDX3X, to poly (AG) RNA, albeit with significantly lower affinity than eIF4A1 (28).
There is significant interest in developing rocaglates as anti-cancer drugs, as exemplified by eFT226 (Zotatifin), administered intravenously, in a phase I/II clinical trial (NCT04092673) (29). Here, we present the functional characterization and biological activity of a next-generation rocaglate-derived synthetic compound, MG-002. More effective in suppressing translation than existing eIF4A inhibitors, MG-002 is orally bioavailable, and shows superior single-agent efficacy in primary and metastatic TNBC pre-clinical models.
Results
Design of MG-002.
Silvestrol and RocA are well-studied rocaglates (Fig. 1A). They have been useful in unraveling the mechanism of action for the rocaglate family (23, 25, 26). These compounds demonstrate selectivity toward transformed cells (23, 30) and anti-tumor activity in vivo (23, 30, 31). The complexity associated with silvestrol synthesis (32), and its rapid in vivo conversion to inactive silvestric acid following intravenous administration (33), prompted synthetic exploration of the rocaglamide core for simpler compounds. Features of rocaglates that interact with RNA (polypurine bases) are aryl rings A and B and the 8b-OH group (Fig. 1A, RocA structure, highlighted in blue). Anchoring rocaglates to eIF4A1 is achieved through stacking with ring C to eIF4A1 F163 and Q195 and hydrogen bonding between Q195 and the carbonyl of the C-2-N, N-dimethyl-carboxamide group (Fig. 1A, RocA structure, highlighted in green) (26). Introduction of a hydroxymate group at the C-2 position yielded a series of bioactive molecules with improved activity, among which CR-1-31B is the best characterized (34) (Fig. 1A). The improved potency of CR-1-31B compared to RocA (24) is likely related to the ability of the hydroxamate moiety to act as a hydrogen bond acceptor and/or as a bidentate chelating group, possibly stabilizing interactions with eIF4A1 Q195 and D194 (Fig. 1A). A second modification introduced to the rocaglate core that improved activity was the addition of a nitrile group at the B ring, potentially forming hydrogen bonding interactions with eIF4A1 N167 (Cmpd76, Fig. 1A) (35). Substitution of the A aryl ring for a pyridine ring lowered lipophilicity of the core while maintaining π-stacking with RNA (36), leading to the development of a clinical candidate, eFT226 (36). With these advances in potency and pharmacology, we designed MG-002 that incorporated all these improvements into a single molecule (Fig. 1A).
Fig. 1.
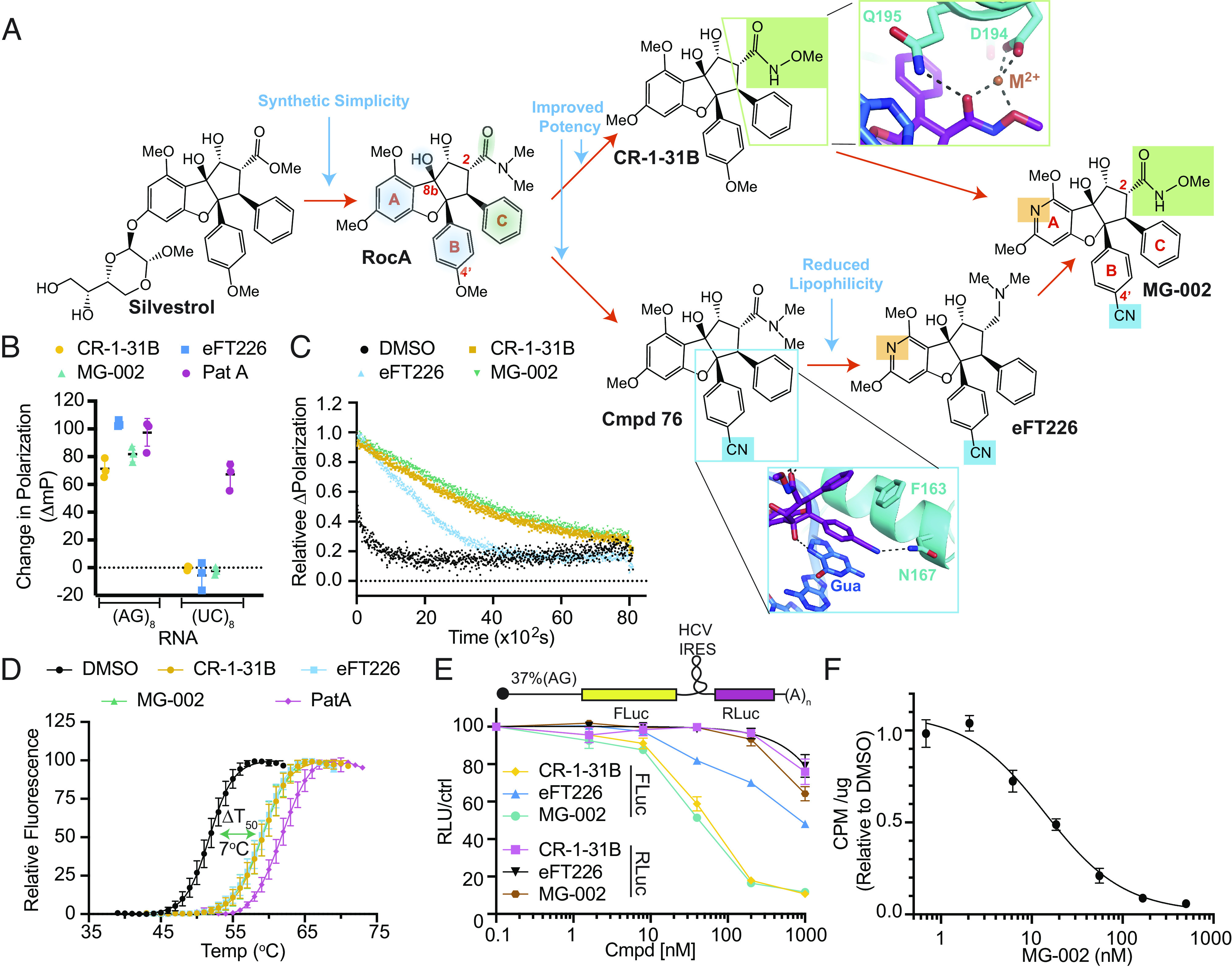
Design of an eIF4A inhibitor (MG-002) that potently inhibits cap-dependent mRNA translation. (A) Schematic outlining the rationale behind MG-002 development. RNA- (blue shading) and eIF4A1 (green shading)-interacting regions are highlighted on the RocA structure. Insets show close in views of a model of the MG-002 hydroxymate and nitrile groups, based on the RocA-bound crystal structure of eIF4A1 (26). (B) Assessing compound-induced clamping of eIF4A1 to FAM-labeled RNA of the indicated nucleotide composition by fluorescence polarization assay. The ΔmP (change in polarization) obtained with eIF4A1•AMPPNP •poly (NN)8 RNA was measured for the indicated compounds at 10 µM. The ΔmP obtained relative to dimethylsulfoxide (DMSO) is shown (n = 3 ±SD). (C) Relative dissociation of pre-formed eIF4A1•ATP•Cmpd•FAM-poly (AG)8 complexes measured as a function of time in the presence of 1,000-fold molar excess poly (AG)8 RNA. DMSO, t1/2 ~4.1 ± 1 min; CR-1-31B (10 µM), t1/2 ~59 ± 6.5 min; MG-002 (10 µM), t1/2 ~68 ± 2.8 min; eFT226 (10 µM), t1/2 ~ 27 ± 5.8 min. Error values were calculated from the 95% CI. (D) Differential scanning fluorimetry analysis of eIF4A1 (8 µM) in the presence of the indicated compounds (15 µM), 15 µM poly (AG)8, and 1 mM AMPPNP. The transition midpoint temperature shifts (ΔT50) are: CR-1-31B, 7.3 °C; eFT226, 7 °C; MG-002, 7.2 °C; PatA, 10 °C (n = 3 ±SD). (E) Inhibition of cap-dependent (FLuc) and independent (RLuc) translation was measured in response to the indicated compounds in Krebs-2 translation extracts programmed with the noted bicistronic mRNA. IC50s toward inhibition of FLuc synthesis from (CAG)-FF/HCV-IRES/Ren mRNA were: CR-1-31B, 54 ± 4 nM; eFT226, 813 ± 91 nM; MG-002, 43 ± 4 nM (n = 2 ±SD). (F) eHap1 cells were incubated in the presence of the indicated concentrations of compound for 1 h. During the last 15 min of incubation, 35S-Met was added followed by TCA precipitation and quantitation of 35S-Met incorporation into protein (n = 3 ±SD).
MG-002 Induces eIF4A1•RNA Clamping to Potently Inhibit Translation.
The RNA clamping activity of rocaglates can be measured using a fluorescence polarization (FP) assay (25). Like CR-1-31B, both eFT226 and MG-002 discriminated between poly (AG)8 and poly (UC)8 RNA templates (Fig. 1B). As noted, the unrelated natural product, pateamine A (PatA), which also induces RNA clamping, stabilized binding of eIF4A1 to both polypurine and polypyrimidine RNA templates (37). We then assessed the stabilizing effects of these compounds on eIF4A1•poly (AG)8•ATP complexes. The dissociation of pre-formed complexes was monitored following addition of excess unlabeled RNA. Complexes formed with CR-1-31B or MG-002, in the presence of ATP, had a ~two-fold longer half-life than those containing eFT226 (Fig. 1C). Stabilization of eIF4A1 by CR-1-31B, eFT226 and MG-002 was independently confirmed by differential scanning fluorimetry (DSF) (Fig. 1D), which monitors temperature-dependent protein unfolding, an event that is attenuated by target/ligand complex formation (38). A transition midpoint temperature shift (ΔT50) of 7 °C was noted, consistent with the ability of CR-1-31B, eFT226, and MG-002 to induce RNA clamping to eIF4A1. Relative to eIF4A1, the tested rocaglates only minimally influenced DDX3X:RNA clamping (SI Appendix, Fig. S1A).
CR-1-31B and MG-002 were similarly effective at inhibiting cap-dependent translation from a reporter harboring a 37% (AG)-rich 5′ leader region in vitro (Fig. 1E). eFT226 was the least active of the three tested compounds (Fig. 1E). MG-002, like CR-1-31B, more potently suppressed translation of mRNA reporters harboring (AG)-rich 5′ leader sequence relative to a reporter with a (UC)-rich 5′ leader sequence (SI Appendix, Fig. S1 B and C). As expected, all three compounds preferentially targeted cap-dependent versus HCV IRES-driven eIF4A-independent mRNA translation (39). Exposure (1 h) of eHAP1 cells (a nearly haploid leukemia cell line) to MG-002 robustly inhibited protein synthesis, as assessed by 35S-Met metabolic labeling (Fig. 1F) and polysome profiling (SI Appendix, Fig. S2). Taken together, these results indicate that MG-002 stabilizes eIF4A1-RNA complexes, preferentially inhibits mRNAs with polypurine leader regions, and is a potent inhibitor of translation in vitro and in cellula.
Biological Activity of MG-002.
We next examined the anti-tumorigenic properties of MG-002 toward TNBCs, which are known to express elevated eIF4A1 levels compared to other breast cancer subtypes (40). Exposure of MDA-MB-231 TNBC cells to MG-002 or CR-1-31B caused a profound delay in G2/M progression (Fig. 2A), consistent with previous reports regarding the effects of rocaglates on other cell lines (41–43). The consequences of prolonged exposure (2 to 4 d) to MG-002 were assessed using TNBC lines (4T1, MDA-MB-231) and a model of ER+/HER2+ breast cancer (BT474) to evaluate the generalizability of our findings (Fig. 2B). In all instances, cell viability was compromised with EC50 values for MG-002 ranging from approximately 1 to 10 nM, indicating potent cytotoxic activity in cell culture. We observed that MG-002 was superior to eFT226 in reducing breast cancer cell viability for the human breast cancer cell lines tested (Fig. 2B). When these same compounds were tested against non-transformed, immortalized cells (IMR-90, MRC-5) (Fig. 2C) or primary human umbilical vein endothelial cells (HUVEC) (Fig. 2D), only a modest cytostatic effect was observed as the number of viable cells never fell below 50%, even at high concentrations (3 μM). Reduction in c-MYC and cyclin D1 proteins, which are encoded by eIF4F-sensitive mRNAs (8), was observed in MDA-MB-231, 4T1, and MRC-5 cells exposed to MG-002 or CR-1-31B, but not in cells exposed to doxorubicin [DXR] (Fig. 2E). On the other hand, cleavage of PARP was observed when MDA-MB-231 or 4T1 cancer cells were exposed to MG-002, CR-1-31B, or DXR but not in MRC-5 cells treated with MG-002 or CR-1-31B (Fig. 2 C and E). In summary, these experiments demonstrated that all three rocaglates preferentially inhibit the proliferation of breast cancer and induce apoptosis, relative to non-transformed cells in culture. Moreover, MG-002 appeared to exhibit more potent anti-neoplastic activity than eFT226.
Fig. 2.
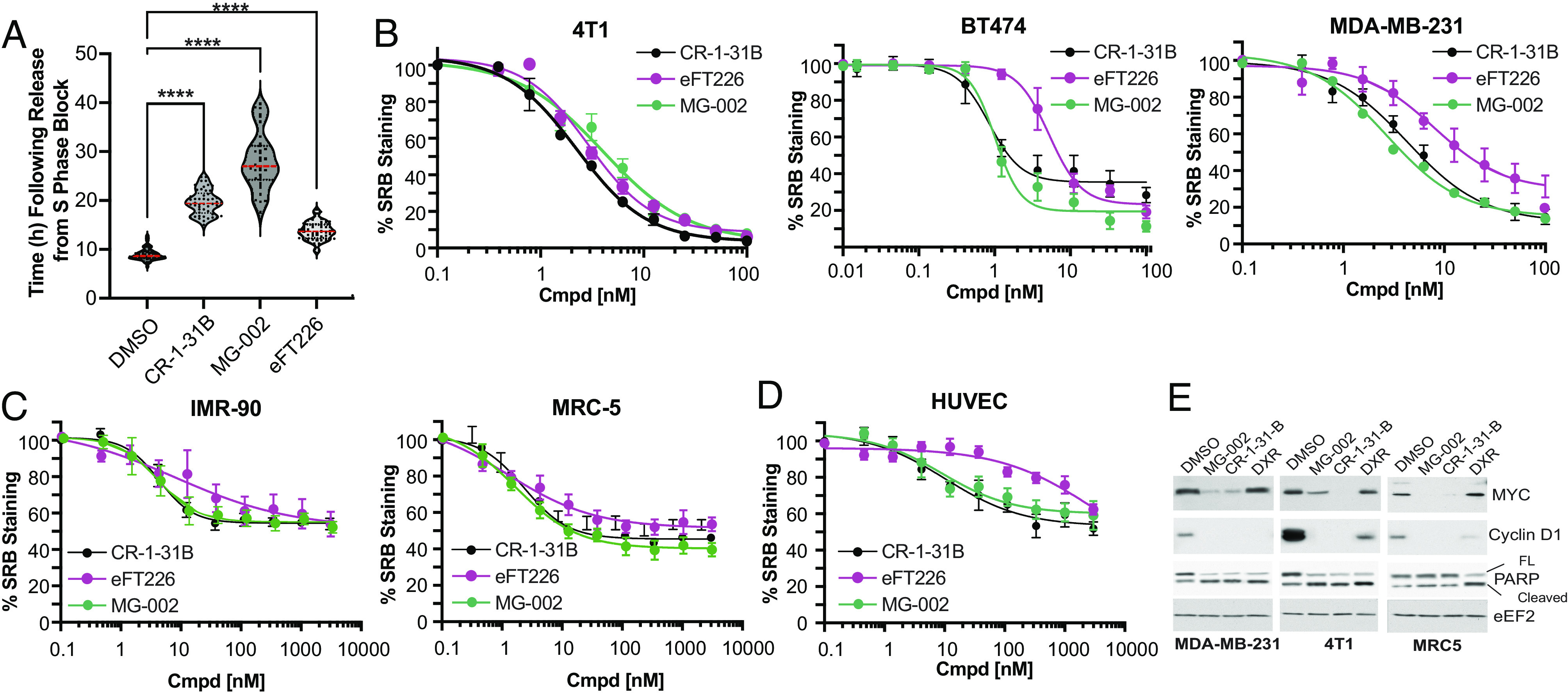
MG-002 significantly and selectively impairs breast cancer cell growth and viability. (A) Time of MDA-MB-231 cells to enter mitosis following release from DNA synthesis block. Release was performed in the presence of DMSO or 10 nM of the indicated compounds. The data shows the average of 50 cells ±SD. (B) Activity of CR-1-31B, eFT226, and MG-002 toward three TNBC cells lines (4T1, BT474, MDA-MB-231). Cells were exposed to the indicated concentrations of compound for 2 d (except for BT474 cells where exposure was for 4 d) and viability measured using the SRB assay. n = 3 ±SD. (C) Activity of CR-1-31B, eFT226, and MG-002 toward IMR-90 and MRC-5 immortalized and non-transformed cells. Cells were exposed to the indicated concentrations of compound for 2 d and stained with SRB. n = 3 ±SD. (D) Activity of CR-1-31B, eFT226, and MG-002 toward HUVEC cell line. Cells were exposed to the indicated concentrations of compound for 2 d and then stained with SRB. n = 3 ±SD. (E) Western blot analysis of the indicated proteins in cells exposed to MG-002 (100 nM), CR-1-31B (100 nM), or doxorubicin (DXR, 0.5 µg/mL) for 24 h.
MG-002 Cytotoxicity is eIF4A1 and eIF4A2 Dependent.
We have shown that rocaglates primarily exert their biological effects through eIF4A1 engagement (31, 44). However, RocA has been reported to induce RNA clamping of DDX3X, although the binding affinity is at least 30-fold lower when compared to the binding of eIF4A1 (28). To query whether MG-002 also exerts its cytotoxic effects through eIF4A1/2 engagement, we tested the haploid human cell line, eHap1, expressing either wild-type eIF4A1 or an eIF4A1F163L mutant, which is unable to bind the rocaglate compound, while eIF4A2 was deleted (eIF4A2−) to eliminate its ability to compensate for inactive eIF4A1 (37). We show that eIF4A1F163L/eIF4A2− eHap1 cells are significantly less sensitive to MG-002 compared to the parental control (Fig. 3A). These findings were consistent with our previous reports for other rocaglates (31, 37). We next engineered eIF4A1F163L/eIF4A2− eHap1 cells expressing wild-type eIF4A1, eIF4A2, eIF4A3, or DDX3X to further establish which of these proteins mediate the anti-proliferative effects of MG-002 (Fig. 3 B and C). eIF4A1F163L/eIF4A2− cells ectopically expressing eIF4A1 or eIF4A2 showed increased sensitivity to MG-002 (Fig. 3D). This sensitization was not observed with parental eHAP1 cells ectopically expressing eIF4A1 or eIF4A2 (Fig. 3E), suggesting that eIF4A1 is not limiting for target engagement in these parental cells. Sensitization was not observed in eIF4A1F163L/eIF4A2− cells ectopically expressing DDX3X (Fig. 3D) or eIF4A3 (SI Appendix, Fig. S3 A and B), suggesting that they are not primary targets of MG-002. Finally, ectopic expression of eIF4A1F163L failed to sensitize eIF4A1F163L/eIF4A2− cells to MG-002, demonstrating that wild-type eIF4A1 is required for the observed effects (eIF4A1:RNA clamping) (Fig. 3F). Taken together, these results indicate that MG-002 is primarily exerting its cytotoxic effects through eIF4A1/2 engagement.
Fig. 3.
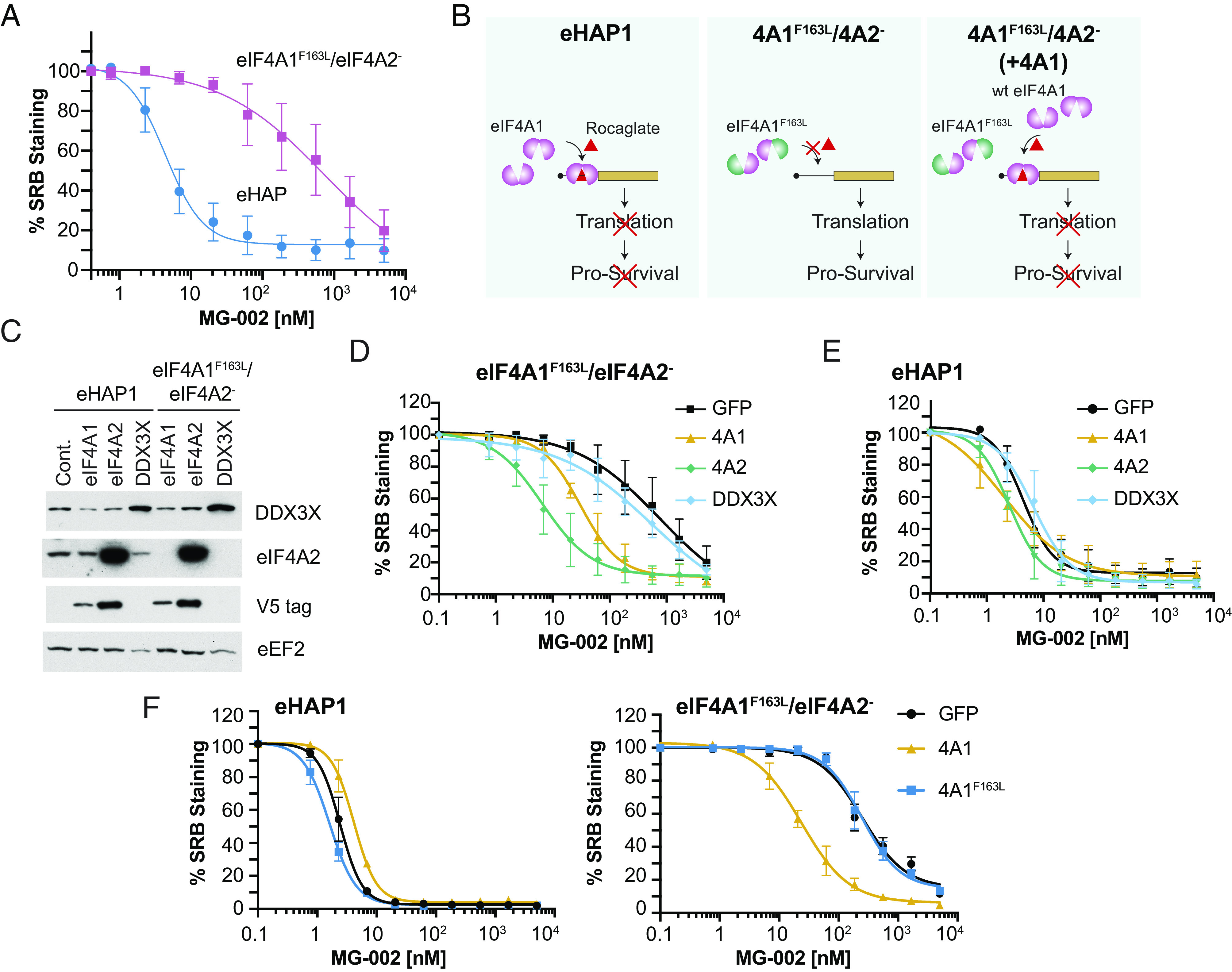
eIF4A is the molecular target for MG-002. (A) Activity of MG-002 toward parental eHAP1 or eIF4A1F163L/eIF4A2− cells. (B) Rocaglate-induced clamping of eIF4A1 to mRNAs leads to inhibition of translation and cell death. In contrast, eIF4A1F163L/eIF4A2− eHAP1 cells are significantly resistant to rocaglates as clamping of eIF4A1F163L to RNA is impaired. Overexpression of wt eIF4A1 in eIF4A1F163L/eIF4A2− cells restores rocaglate-induced translation inhibition and cell death. (C) Immunoblot of the indicated proteins from cell lines ectopically expressing V5 tagged eIF4A1, eIF4A2, or untagged DDX3X. (D) Cytotoxicity of eIF4A1F163L/eIF4A2− eHAP1 cells ectopically expressing GFP, eIF4A1, eIF4A2, or DDX3X following a 2-d exposure to the indicated MG-002 concentrations (n = 3 ±SD). (E) Response of eHAP1 cells ectopically expressing GFP, eIF4A1, eIF4A2, or DDX3X following a 2-d exposure to the indicated MG-002 concentrations (n = 3 ±SD). (F) Cytotoxicity of eHAP1 or eIF4A1F163L/eIF4A2− cells ectopically expressing eIF4A1F163L following a 2-d exposure to the indicated MG-002 concentrations (n = 3 ±SD).
MG-002 Is an Orally Bioavailable Inhibitor of Translation.
MG-002 shows similar mouse and human plasma protein binding as eFT226 (Table 1). MG-002 was found to be stable in simulated gastric fluid (SGF) (pH 1.2) (Table 1), prompting us to investigate its plasma concentration following drug delivery by oral gavage (PO). We found that PO delivery resulted in the sustained presence of MG-002 in the plasma, with a terminal half-life of ~10 h (Fig. 4A and Table 1). We next assessed the consequences of PO delivery on translation in vivo by analyzing liver polysomes post-compound delivery by gavage. Our results indicate strong suppression of translation in the liver following PO delivery of MG-002, as evidenced by a >three-fold decrease in polysome-to-monosome ratio 7 h post-delivery (Fig. 4 B and C). This was followed by partial translational recovery 24 h post-treatment (Fig. 4 B and C), which was consistent with the reduction in plasma concentration of MG-002 (Fig. 4A). MG-002 distribution to plasma, spleen, lungs, and liver was also quite extensive 4 h post-delivery PO (Fig. 4D). Importantly, MG-002 administration was well tolerated as noted by the lack of adverse effects on blood cells (Fig. 4E and SI Appendix, Fig. S4), organ (Fig. 4F), and body weight during administration (see below). Histopathological analysis of the heart, lung, spleen, kidney, and bone marrow following six treatments with MG-002 did not uncover any overt tissue damage (SI Appendix, Table S1). These encouraging results prompted us to undertake studies to assess the anti-cancer activity of MG-002 in pre-clinical TNBC mouse models.
Table 1.
Plasma protein binding, acid stability, and plasma half-life for MG-002
| Plasma protein binding (% bound) | SGF stability* | T1/2 (h) | ||
|---|---|---|---|---|
| Cmpd | Mouse | Human | ||
| MG-002 | 87 ± 0.5 | 73.2 ± 1.2 | 94.5 ± 0.7% | 9.97 ± 2.53† 4.11 ± 1.9‡ |
| eFT226 | 73.4 ± 1.1 | 79.4 ± 0.3 | ND | 10.9§ |
*Cmpd (50 µM) was incubated in SGF containing pepsin at pH 1.2 for 1 h at 37 °C.
†Delivered PO 5 mg/kg n = 3 ±SD.
‡Delivered IV 0.5 mg/kg n = 3 ±SD.
§Taken from ref. 36—delivered IV 1 mg/kg.
Fig. 4.

MG-002 accumulates in biological tissues and has minimal toxicities. (A) Average plasma concentration of MG-002 at the indicated time points following compound delivery by intravenous (IV) versus oral gavage (PO). (B) Polysomes isolated from livers of mice treated with vehicle or at the indicated time points following delivery of 0.5 mg/kg MG-002 PO. Polysome/monosome (P/M) ratio represents the average of two experiments. (C) Quantification of the P/M ratios shown in panel B. (D) Concentration of MG-002 in the indicated tissues 4 h after PO delivery of MG-002 (5 mg/kg) (n = 3 ±SD). (E) Total blood cell counts (total leukocytes, neutrophils, lymphocytes, monocytes, or eosinophils) from mice treated with vehicle or 0.5 mg/kg MG-002 PO on Mon/Wed/Fri for two consecutive weeks (n = 4 to 5 ±SD). (F) Tissue weights from mice treated with vehicle or 0.5 mg/kg MG-002 PO Mon/Wed/Fri for two consecutive weeks, after which time organs were harvested (n = 4 to 5 ±SD).
MG-002 Inhibits Tumor Cell Growth In Vivo Following PO Delivery.
We next sought to establish the effects of MG-002 on neoplastic growth and dissemination in vivo. 4T1-526 breast cancer cells represent an immunocompetent model of TNBC disease, which were selected in vivo for their propensity to metastasize to the lungs (45). MG-002 and eFT226 showed IC50 values of ~ 7 and 8 nM, respectively, for inhibition of mRNA translation in 4T1-526 cells, as assessed by puromycin incorporation in cell culture (SI Appendix, Fig. S5 A and B). Significant cell death was observed following 72h exposure of 4T1-526 cells to the above-determined IC50 doses of MG-002 and eFT226, with MG-002 being the more potent molecule (SI Appendix, Fig. S5C).
Following mammary fat pad injection, tumor-bearing mice were randomized into three groups when 4T1-526 tumors reached a ~100-mm3 volume. Cohorts were treated by PO with MG-002 (0.5 mg/kg), eFT226 (0.5 mg/kg), or vehicle control until the experimental endpoint (Fig. 5A). The results indicated that MG-002 more significantly impaired mammary tumor growth compared to eFT226 (Fig. 5A) without adversely affecting total body (Fig. 5B) or liver weights (Fig. 5C) in tumor-bearing animals. Immunohistochemical (IHC) analysis further revealed that MG-002 significantly reduced proliferation, increased apoptotic cell death, and reduced Myc protein levels, which were used as a surrogate marker for “eIF4F-sensitive” translation (Fig. 5 D–G). Despite some variability in Myc protein levels between individual tumors, elevated Myc levels positively correlated with increasing Ki67 positivity and inversely correlated with the percentage of cleaved Caspase 3 positive cells, as expected (Fig. 5H).
Fig. 5.
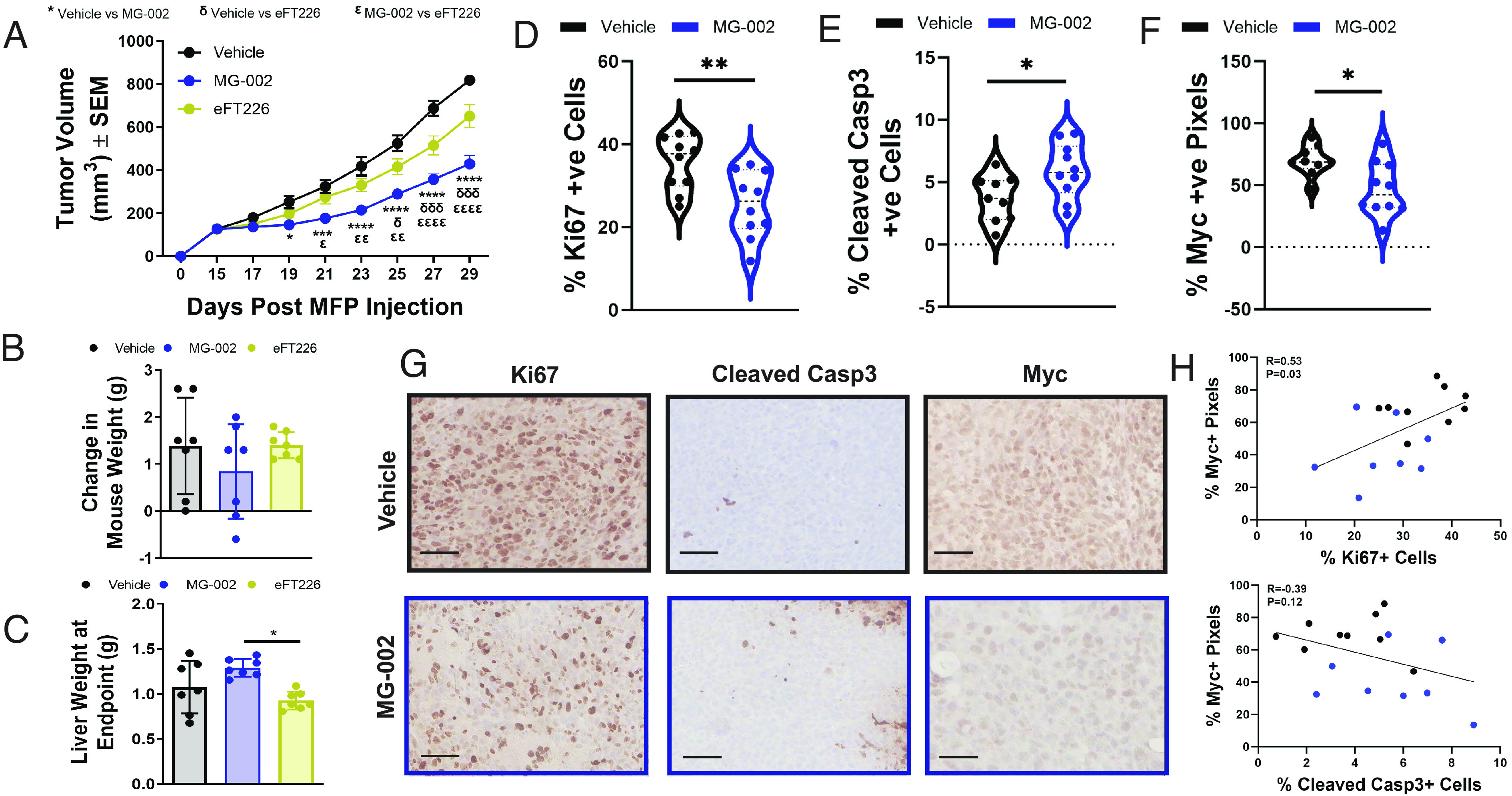
MG-002 inhibits primary TNBC tumor growth through reduced cell proliferation and increased apoptosis. (A) 4T1-526 mammary tumors were allowed to develop in the mammary fat pads of BALB/c mice and upon reaching ~100 mm3, the animals were randomized into three groups, which were treated by PO every 3 d with MG-002 (0.5 mg/kg), eFT226 (0.5 mg/kg) or vehicle control (n = 12 tumors/group). The data is shown as average tumor volume ± SEM. At the experimental endpoint, animals (n = 6 mice per group) were assessed for the (B) average change in mouse weight or (C) their liver weights. (D–F) IHC analysis of 4T1-526 tumors treated with 0.5 mg/kg MG-002 or vehicle control using (D) Ki67, (E) cleaved Caspase-3, and (F) Myc-specific antibodies. The data are shown as % positive cells ± SD and are representative of 9 to 10 tumors per group. (G) Representative IHC images for the graphs depicted in panel D–F. (scale bar: 100 microns.) (H) Pearson correlation examining the relationship between Myc positivity and either the % Ki67+ or % Cleaved Casp3+ cells (individual vehicle-treated tumors are represented by black dots; individual MG-002 treated tumors are represented by blue dots. For panel A, statistical analysis was performed with a two-way ANOVA (Tukey’s multiple comparisons test). For panels D–F, statistical analysis was performed using a two-tailed (unpaired) Student’s t test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
The differences in response to oral delivery of MG-002 and eFT226 motivated us to compare the in vivo response of both drugs following intraperitoneal or intravenous delivery (SI Appendix, Fig. S6 A–I). Here, we found that both MG-002 and eFT226 similarly delayed tumor growth (SI Appendix, Fig. S6 A and B). These experiments suggest that the inferior response of eFT226 versus MG-002 following PO delivery in vivo is likely to stem from the lesser bioavailability of eFT226 after oral application. We further validated the anti-neoplastic effects of intraperitoneal delivery of MG-002 against MDA-MB-231 tumors, representing a model of human TNBC (SI Appendix, Fig. S6C). Collectively, these results demonstrate that MG-002 is effective when administered by PO while being well tolerated. Moreover, we show that oral application of MG-002 results in more potent suppression of tumor growth than oral administration of eFT226.
Activity of MG-002 toward Metastatic TNBC Disease.
The ability of MG-002 to accumulate in lung tissue following PO delivery (Fig. 4C), a frequent site of breast cancer metastasis, prompted us to assess the efficacy of MG-002 toward metastatic lesions. We first investigated whether neo-adjuvant MG-002 treatment may prevent the formation of spontaneous lung metastases (Fig. 6 A and B). Here, tumor-bearing mice were treated with MG-002 or vehicle control (0.5 mg/kg PO every 3 d), and tumors were surgically resected at a 500-mm3 volume, after which drug treatment was terminated, and the animals were followed for the development of metastatic disease. We found that MG-002 was effective as a monotherapy at reducing the formation of spontaneous lung metastases (Fig. 6B). Next, we assessed the ability of MG-002 to target breast cancer metastases that had already colonized to the lungs (Fig. 6C). 4T1-526 breast cancer cells were injected into the tail vein to directly seed the lungs. After 10 d, treatment with MG-002 (0.5 mg/kg PO every 3 d) or vehicle was initiated over a 21-d period. We found that MG-002 was associated with a modest reduction in lung tumor burden, compared to vehicle control–treated mice (Fig. 6D). Thus, although promising, MG-002 is likely not sufficient as a monotherapy to treat individuals who present with macroscopic metastatic disease. Altogether, these data suggest that MG-002 as a monotherapy exerts anti-metastatic properties, primarily by reducing the likelihood that a patient will progress to metastatic disease.
Fig. 6.
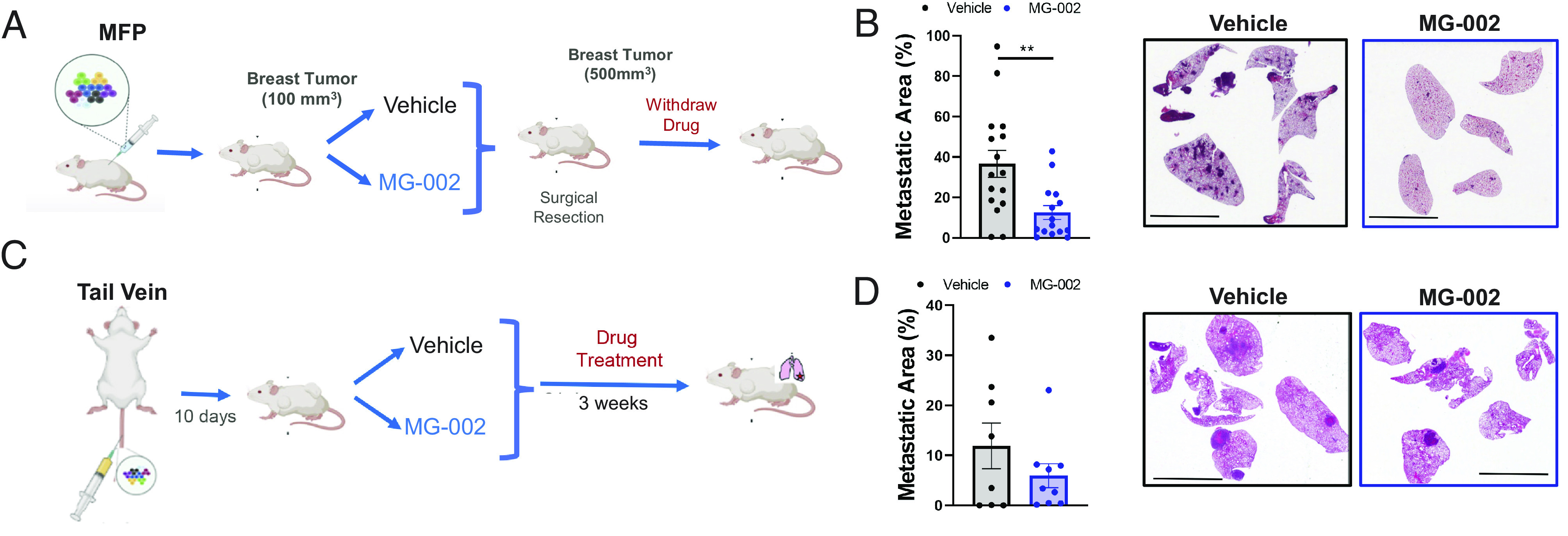
MG-002 monotherapy prevents the development of spontaneous breast cancer lung metastasis. (A) Diagram outlining the experimental schedule to assess the ability of MG-002 to prevent the formation of spontaneous lung metastases from the primary tumor. (B) Using the experimental protocol outlined in panel A, mice were either treated by PO with MG-002 (0.5 mg/kg) or vehicle control every 3 d until surgical resection. The metastatic area in the lungs was quantified from H&E-stained sections (15 to 16 mice/group) and the average metastatic burden is shown ±SD. (C) Schematic diagram outlining the experimental schedule to assess the ability of MG-002 to treat lung metastases following lung colonization. (D) Using the protocol outlined in panel C, mice were either treated by PO with MG-002 (0.5 mg/kg) or vehicle control every 3 d until the experimental endpoint. The metastatic area in the lungs was quantified from H&E-stained sections (8 to 9 mice/group) and the average metastatic burden is presented ±SD. For panels B and D, representative H&E sections are shown (scale bar, 6 mm.) Statistical analysis was performed using a two-tailed (unpaired) Student’s t test. *P < 0.05.
MG-002/DXR Combination Therapy Reduces Primary and Metastatic Disease Burden.
Aggressive breast cancers are likely to require combination therapies to exploit the therapeutic benefit of MG-002. We assessed this by combining MG-002 with DXR, a front-line chemotherapy that is already standard of care for TNBC (46). To this end, we contrasted the effects of MG-002/DXR combination versus single-agent treatments. Following mammary fat pad injection of 4T1-526 breast cancer cells, we started either monotherapy or combination treatment when tumors reached ~100 mm3. Tumors were equally responsive to DXR or MG-002, but significantly more sensitive to combination treatment (Fig. 7A). Combination treatment did not lead to significant weight loss in treated animals, although animals failed to gain weight during treatment (Fig. 7B). To better understand the basis for the improved anti-neoplastic activity of MG-002/DXR combination, we harvested breast tumors early during treatment. Herein, we selected a timepoint when we first observed reduced tumor volumes with MG-002 as a monotherapy and a further reduction with MG-002/DXR combination treatment (Fig. 7C). IHC analysis at this time point revealed no significant difference in Ki67 staining with either treatment (Fig. 7D). In contrast, MG-002 treatment increased the rate of apoptosis in breast tumors, which was even more pronounced when MG-002 and DXR were combined (Fig. 7E). Finally, treatment of mice already bearing lung metastases with MG-002/DXR combination therapy significantly reduced the lung tumor burden compared to animals treated with either drug as a monotherapy (Fig. 7F). Combined, these results support that MG-002 bolsters cytotoxic responses to DXR treatment toward primary TNBC tumors and TNBC metastatic lesions, exemplifying the potential benefits of utilizing MG-002 to increase the anti-neoplastic efficacy of clinically applied therapies.
Fig. 7.
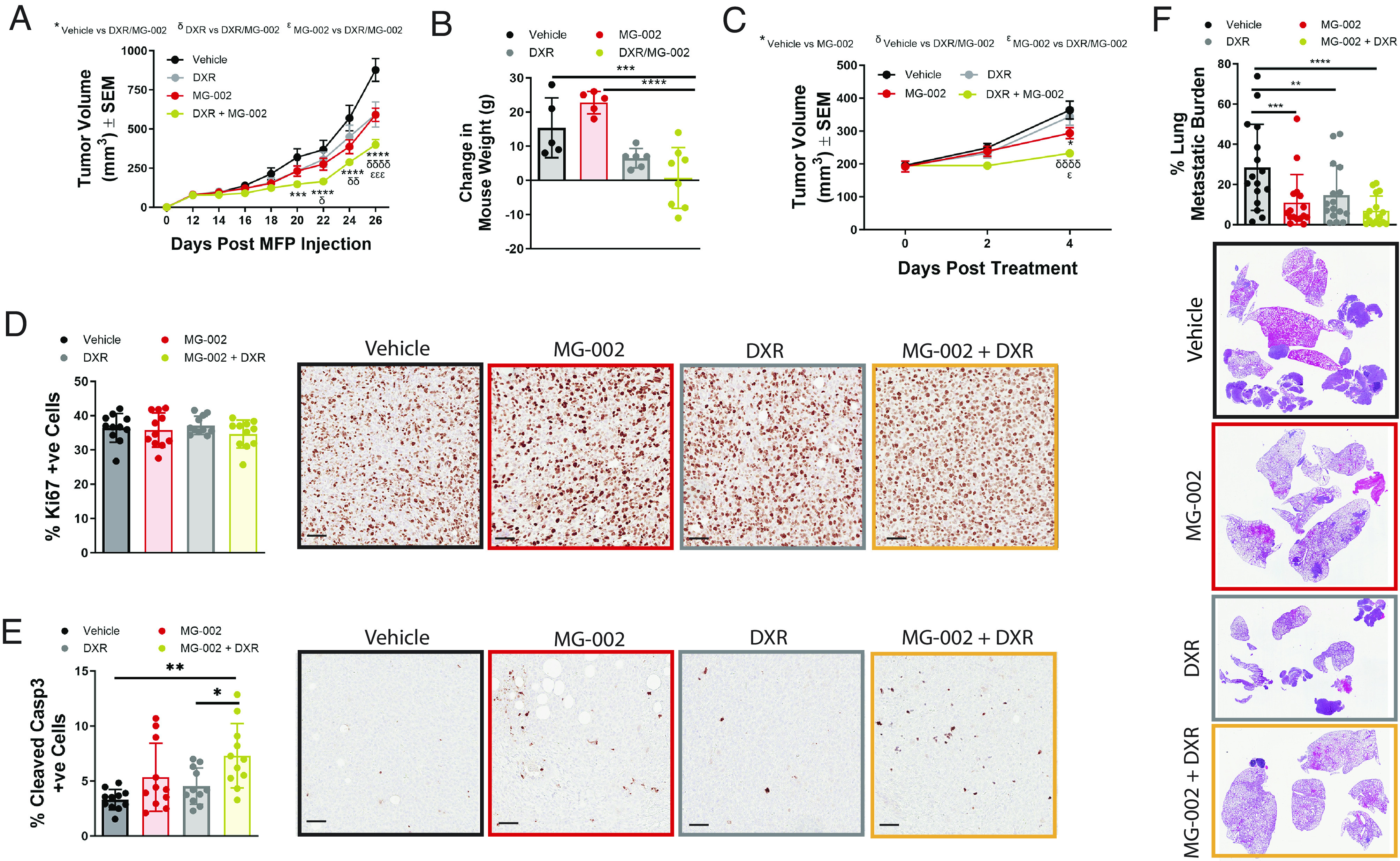
MG-002/DXR combination therapy reduces the growth of established primary TNBC breast tumors and lung metastases. (A) 4T1-526 mammary tumors were allowed to develop in the mammary fat pads of Balb/c mice and when tumors reached ~100 mm3, animals were randomized into four groups: 1) 0.5 mg/kg MG-002 PO; 2) 2.5 mg/kg DXR IP; 3) MG-002/DXR combination treatment using the same concentrations; and 4) vehicle control. Animals were treated every 3 d until the experimental endpoint (n = 10 to 14 tumors/group). The data are shown as average tumor volume ± SEM. (B) At the experimental endpoint, each animal in panel A was measured for the average change in weight from the beginning to the end of the study. (C) Tumors were treated as described in panel A, but mice were sacrificed 4 d after the start of drug treatment. (D and E) Tumors from the mice harvested in panel C were analyzed by immunohistochemistry using (D) Ki67 or (E) cleaved caspase-3 specific antibodies (n = 11 tumors per group). (F) Mice with established metastases (as outlined in Fig. 6C) were randomized into four groups and treated as outlined in panel A. The metastatic area in the lungs was quantified from H&E-stained sections (n = 16 mice/group), and the average metastatic burden is presented ±SD. Statistical analysis was performed with a two-way ANOVA (Tukey’s multiple comparisons test).
Discussion
Given that reprogramming of protein synthesis is an omnipresent feature of cancer cells (10), pharmacological targeting of the translation initiation machinery has great therapeutic potential. To date, most clinical trials in this space have focused on mTOR inhibitors, which in addition to modulating a plethora of cellular functions, suppress protein synthesis (47, 48), however, with marginal improvement on patient survival as shown in several large studies (49–51). This can at least in part be explained by the inability of mTOR inhibitors to efficiently hinder eIF4F complex assembly (14, 52–56). Thus, direct inhibitors of eIF4F activity may be superior in sustaining prolonged anti-neoplastic responses. Particularly, eIF4A is an attractive therapeutic target for treating TNBCs given that it is overexpressed in human ER–negative breast cancers compared to other breast cancer subtypes (40).
MG-002 reduces protein expression of c-MYC levels (Figs. 2E and 5F), a well-known oncogene (57–59). Genetic modeling in the mouse demonstrated that systemic c-MYC suppression results in profound anti-cancer activity, while being well tolerated (60). Indeed, exogenous administration of a dominant-negative MYC (Omomyc) is efficacious against TNBC and displays anti-metastatic properties in pre-clinical models (61). However, targeting c-MYC as an anti-cancer treatment has proven to be challenging (62, 63). The ability of MG-002 to reduce c-MYC levels by suppressing translation of its corresponding mRNA thus opens new therapeutic avenues to inhibit c-MYC.
We demonstrate that MG-002, like other rocaglates, clamps eIF4A onto RNA with poly (AG)-rich tracts. Cells encode multiple eIF4A paralogues including eIF4A1 and eIF4A2, both of which are substrates for rocaglates (64). Notwithstanding that eIF4A1 is the more abundant isoform (65, 66), our experiments using eIF4A1F163L/eIF4A2− eHAP1 cells engineered to express modified eIF4A paralogues revealed that overall levels of eIF4A1 and eIF4A2, but not DDX3X, in a tumor cell likely to contribute to MG-002 sensitivity. eIF4A1 is an essential gene (67), making it unlikely that cancer cells will develop resistance to MG-002 via eIF4A1 loss. However, mutations in drug targets are a common resistant mechanism to targeted cancer therapeutics. In a yeast-based screen for rocaglate-resistant alleles, we previously identified missense mutations in six amino acids of eIF4A1 that may confer resistance to rocaglates: T158, P159, F163, F192, Q195, and I199 in human eIF4A1 (44). From the crystal structure of eIF4A1 bound to RocA, two of these amino acids, F163 and Q195, make direct contacts with RocA (26). Mutations in eIF4A1 that confer resistance to MG-002 are unlikely to be pre-existing in a tumor cell population. Even if such mutations are acquired after exposure to MG-002, our studies suggest that all eIF4A1 and eIF4A2 alleles would have to be compromised to confer resistance to MG-002, which is unlikely to happen.
We observed clear differences in activity between MG-002 and eFT226. eFT226 is not as potent as MG-002 at inhibiting translation. Although eFT226 is slightly better at clamping eIF4A to poly (AG) RNA sequences, the resulting complexes show a half-life that is approximately 2.5-fold less than those formed in the presence of MG-002. This latter point may explain the lower potency of eFT226 in inhibiting protein synthesis. In general, MG-002 was also more effective than eFT226 toward TNBCs in cell culture. Last, when delivered by PO, eFT226 exerted less potent anti-neoplastic effects than MG-002.
We showed that suppression of the eIF4E cap-binding protein is effective at reducing breast cancer cell migration and invasion in vitro, decreasing both pulmonary colonization and metastasis growth (68). This correlated with a reduction in the translation of mRNAs encoding proteins of the metastatic cascade (68). Our results highlight the value of eIF4A as a therapeutic target in vivo and offer a strategy to directly inhibit mRNA translation by targeting eIF4A activity. Studies testing other rocaglates have shown that eIF4A inhibition increases the radiosensitivity of tumors (69) and also improves the response to targeted therapies, including MEK (70) and CDK4/6 inhibitors (71).
In summary, we report on the development of MG-002, an orally available rocaglate capable of reducing primary tumor growth, blocking metastatic seeding, and potentiating the effects of DXR on suppression of metastatic growth. Finally, MG-002 is likely to exert cytotoxic effects in other malignancies given promising pre-clinical studies showing that other eIF4A inhibitors elicit strong anti-neoplastic effects in models of leukemia and pancreatic cancer (72, 73).
Materials and Methods
Purification of Recombinant eIF4A1 Protein.
BL21 (DE3) codon+ Escherichia coli cells were transformed with pET15b-His6-eIF4A1, cultured at 37 °C until the OD600 reached 0.6, at which point induction was undertaken by the addition of 1 mM IPTG at 37 °C for 3 h. Recombinant His6-eIF4A1 was purified on a Ni2+-NTA agarose column and the eluent applied to a Q-Sepharose fast-flow matrix. The protein was eluted with a linear salt gradient (100 to 500 mM KCl), dialyzed against Buffer A (20 mM Tris-HCl [pH 7.5], 10% glycerol, 0.1 mM ethylenediaminetetraacetic acid (EDTA) overnight at 4 °C, and stored in aliquots at −80 °C.
FP Assay.
FP assays were performed as described (24). Briefly, 1.5 µM recombinant eIF4A1 protein was added to 10 nM FAM-labeled RNA in binding buffer (14.4 mM HEPES-KOH [pH 8], 108 mM NaCl, 1 mM MgCl2, 14.4% glycerol, 0.1% DMSO, and 2 mM DTT) and 1mM AMPPMP in the presence or absence of indicated compound. Following assembly, binding reactions were incubated for 30 min at room temperature (RT) in the dark, after which polarization values were determined using a microplate reader. In experiments monitoring the dissociation of the eIF4A1•Roc•FAM-poly (AG)8 complexes, complexes were formed in the presence of 1mM ATP, and after the initial 30-min incubation, reactions were supplemented with 1,000-fold molar excess unlabeled poly (AG)8 RNA and polarization measurements performed. The relative dissociation was measured as a function of time. The half-lives of complexes were calculated using the “one phase decay” method on Graph Pad.
DSF.
Experiments were performed as reported (38). Briefly, 8 µM of recombinant eIF4A1 was incubated with compound (15 µM) or DMSO in DSF buffer (20 mM HEPES-KOH [pH 7.5], 70 mM KCl, 2 mM DTT, 1 mM Mg(OAc)2, 1 mM AMPPNP, 7.5X Sypro Orange, and 15 µM poly (AG)8 RNA. The samples were heated and read from 37 to 75 °C by PCR using a 1 °C/min ramp rate.
In Vitro Translations.
In vitro translations were performed using 4 ng/µL of reporter mRNA and the indicated compound concentrations in Krebs-2 extracts at 30 °C for 1 h, as previously described (74). FLuc and RLuc luciferase activities were assessed on a Berthold Lumat LB 9507 luminometer (Berthold Technologies). IC50s were determined using a non-linear regression model on GraphPad Prism 8.4.0.
Polysome Profiles.
Polysomes from livers were isolated from mice that had received 0.5 mg/kg MG-002 PO. At 7 and 24 h, animals were sacrificed, and the livers excised and washed in cold phosphate-buffered saline (PBS) containing 100 µg/mL cycloheximide. Tissue was homogenized in 3 volumes of lysis buffer (40 mM HEPES-KOH [7.5], 100 mM KCl, 5 mM MgCl2, and 100 µg/mL cycloheximide). Homogenates were centrifuged for 10 min at 1,200 × g at 4 °C. Triton X-100 and sodium deoxycholate were added to the supernatant to a final concentration of 0.5%. Samples were loaded on 10 to 50% sucrose gradients and centrifuged in an SW40 rotor at 35,000 rpm for 2.5 h. Gradients were collected using a Brandel tube piercer and delivering 60% sucrose through the bottom of the centrifuge tube. Recording of the data was performed using InstaCal Version 5.70 and TracerDaq Version 1.9.0.0 (Measurement Computing Corporation).
35S-Met Labeling.
To measure protein synthesis, 60,000 cells/well were seeded in a 24-well plate. The following day, the medium was removed, and cells were washed with PBS and incubated in methionine-free Dulbecco’s Modified Eagle’s (DMEM) supplemented with 10% dialyzed serum for 1 h. For the last 15 min, cells were labeled with 35S-methionine. Medium was removed, and cells were washed in PBS and lysed in radioimmunoprecipitation assay buffer (RIPA) buffer (20 mM Tris-HCl [pH 7.5], 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 20 mM ß-glycerophosphate, 10 mM NaF, 1 mM phenylmethylsulfonyl fluoride (PMSF), 4 µg/mL aprotinin, 2 µg/mL leupeptin, and 2 µg/mL pepstatin) for 20 min with shaking at 4 °C. The protein was applied to Whatman 3M paper, precipitated with TCA, washed, and radioactivity quantitated by scintillation counting.
Animal Studies.
Balb/c and SCID-beige female mice (6 to 8 wk old) were purchased from Charles River Laboratories. 4T1-526 (50,000 cells) or MDA-MB-231 (1 × 106 cells) was injected into the mammary fat pads of Balb/c or SCID-Beige mice, respectively. For the experimental metastasis assays, 1 × 105 4T1-526 cells were injected into the tail vein of animals. All mice had ad libitum access to food and water and were housed on a 12 h light day cycle, mean temperature 22.5 ± 1.5 °C. MG-002 and eFT226 were injected either intraperitoneally, intravenously, or by PO (0.5 mg/kg every 2 to 3 d).
Statistics.
An unpaired two-tailed Student’s t test was used for the following: Figs. 4A, 5 D, E, and F, and 6 B and D and SI Appendix, Fig. S6C; a one-way ANOVA (Dunnett’s multiple comparisons test) was used for the following: Fig. 2A and SI Appendix, Fig. S5A; a two-way ANOVA (Tukey’s multiple comparisons test) was used for the following: Figs. 5 A and C and 7 A–C, E, and F and SI Appendix, Fig. S6 A, B, E, G, and H.
Study Approval.
Animal studies were approved by the Animal Resource Centre at McGill University in accordance with guidelines from the Canadian Council of Animal Care.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We would like to thank Patrick Sénéchal for excellent technical assistance. Funding for this work was from Canadian Institutes of Health Research grants to J.P. (FDN-148366, PJT-183610, PJT-186015) and J.U.-S. (PJT-168832). It was also supported by a LeadAction Breast Cancer Grant from IR1CoR to A.K., P.M.S., I.T., J.U.-S., and J.P. Finally, P.J. (Doctoral Studentship) and I.T. (Chercheur Boursier, Senior) are supported by the Fonds de Recherche du Québec-Santé. P.M.S. is a William Dawson Scholar of McGill University.
Author contributions
R.C., T.M.S., A.K., M.P., J.G.T., S.H., P.M.S., I.T., J.U.-S., and J.P. designed research; R.C., Y.K.I., S.K.N., M.M.-K., P.J., V.S., M.G.A., and F.R. performed research; P.M.S. and J.P. contributed new reagents/analytic tools; R.C., Y.K.I., S.K.N., M.M.-K., P.J., M.G.A., F.R., J.U.-S., and J.P. analyzed data; and R.C., T.M.S., A.K., M.P., J.G.T., S.H., P.M.S., I.T., J.U.-S., and J.P. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Regina Cencic, Email: regina.cencic@mcgill.ca.
Josie Ursini-Siegel, Email: giuseppina.ursini-siegel@mcgill.ca.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Parker J. S., et al. , Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 27, 1160–1167 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zagami P., Carey L. A., Triple negative breast cancer: Pitfalls and progress. NPJ Breast Cancer 8, 95 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mayer E. L., Burstein H. J., Chemotherapy for triple-negative breast cancer: Is more better? J. Clin. Oncol. 34, 3369–3371 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Bianchini G., Balko J. M., Mayer I. A., Sanders M. E., Gianni L., Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 13, 674–690 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liedtke C., et al. , Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 26, 1275–1281 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Loibl S., et al. , Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): A randomised, phase 3 trial. Lancet Oncol. 19, 497–509 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Cortes J., et al. , Pembrolizumab plus chemotherapy in advanced triple-negative breast cancer. N. Engl. J. Med. 387, 217–226 (2022). [DOI] [PubMed] [Google Scholar]
- 8.Bhat M., et al. , Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 14, 261–278 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Robichaud N., Sonenberg N., Ruggero D., Schneider R. J., Translational control in cancer. Cold Spring Harb. Perspect. Biol. 11, a032896 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y., et al. , A pan-cancer proteogenomic atlas of PI3K/AKT/mTOR pathway alterations. Cancer Cell 31, 820–832.e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xing Y., et al. , Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 21, 78 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savas P., et al. , Alpelisib monotherapy for PI3K-Altered, pretreated advanced breast cancer: A phase II study. Cancer Discov. 12, 2058–2073 (2022). [DOI] [PubMed] [Google Scholar]
- 13.Lee J. S., et al. , Phase I clinical trial of the combination of eribulin and everolimus in patients with metastatic triple-negative breast cancer. Breast Cancer Res. 21, 119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alain T., et al. , eIF4E/4E-BP ratio predicts the efficacy of mTOR targeted therapies. Cancer Res. 72, 6468–6476 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Pelletier J., Sonenberg N., The organizing principles of eukaryotic ribosome recruitment. Annu. Rev. Biochem. 88, 307–335 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Ferraiuolo M. A., et al. , A nuclear translation-like factor eIF4AIII is recruited to the mRNA during splicing and functions in nonsense-mediated decay. Proc. Natl. Acad. Sci. U.S.A. 101, 4118–4123 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Svitkin Y. V., et al. , The requirement for eukaryotic initiation factor 4A (elF4A) in translation is in direct proportion to the degree of mRNA 5’ secondary structure. RNA 7, 382–394 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Truitt M. L., Ruggero D., New frontiers in translational control of the cancer genome. Nat. Rev. Cancer 16, 288–304 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blagden S. P., Willis A. E., The biological and therapeutic relevance of mRNA translation in cancer. Nat. Rev. Clin. Oncol. 8, 280–291 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Ramon Y. C. S., et al. , Beyond molecular tumor heterogeneity: Protein synthesis takes control. Oncogene 37, 2490–2501 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gandin V., et al. , nanoCAGE reveals 5’ UTR features that define specific modes of translation of functionally related MTOR-sensitive mRNAs. Genome Res. 26, 636–648 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chu J., Pelletier J., Targeting the eIF4A RNA helicase as an anti-neoplastic approach. Biochim. Biophys. Acta 1849, 781–791 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Bordeleau M. E., et al. , Therapeutic suppression of translation initiation modulates chemosensitivity in a mouse lymphoma model. J. Clin. Invest. 118, 2651–2660 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chu J., et al. , Rocaglates induce gain-of-function alterations to eIF4A and eIF4F. Cell Rep. 30, 2481–2488.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iwasaki S., Floor S. N., Ingolia N. T., Rocaglates convert DEAD-box protein eIF4A into a sequence-selective translational repressor. Nature 534, 558–561 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwasaki S., et al. , The translation inhibitor rocaglamide targets a bimolecular cavity between eIF4A and polypurine RNA. Mol. Cell 73, 738–748.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garshott D. M., et al. , iRQC, a surveillance pathway for 40S ribosomal quality control during mRNA translation initiation. Cell Rep. 36, 109642 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen M., et al. , Dual targeting of DDX3 and eIF4A by the translation inhibitor rocaglamide A. Cell Chem. Biol. 28, 475–486.e8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao N., et al. , Targeting EIF4A triggers an interferon response to synergize with chemotherapy and suppress triple-negative breast cancer. J. Clin. Invest. 133, e172503 (2023), 10.1172/JCI172503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cencic R., et al. , Antitumor activity and mechanism of action of the cyclopenta[b]benzofuran, silvestrol. PLoS One 4, e5223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chu J., et al. , CRISPR-Mediated drug-target validation reveals selective pharmacological inhibition of the RNA helicase, eIF4A. Cell Rep. 15, 2340–2347 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerard B., Cencic R., Pelletier J., Porco J. A. Jr., Enantioselective synthesis of the complex rocaglate (-)-silvestrol. Angew. Chem. Int. Ed. Engl. 46, 7831–7834 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Saradhi U. V., et al. , Characterization of silvestrol pharmacokinetics in mice using liquid chromatography-tandem mass spectrometry. AAPS J. 13, 347–356 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodrigo C. M., Cencic R., Roche S. P., Pelletier J., Porco J. A., Synthesis of rocaglamide hydroxamates and related compounds as eukaryotic translation inhibitors: Synthetic and biological studies. J. Med. Chem. 55, 558–562 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu T., et al. , Synthetic silvestrol analogues as potent and selective protein synthesis inhibitors. J. Med. Chem. 55, 8859–8878 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Ernst J. T., et al. , Design of development candidate eFT226, a first in class inhibitor of eukaryotic initiation factor 4A RNA helicase. J. Med. Chem. 63, 5879–5955 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Naineni S. K., et al. , Functional mimicry revealed by the crystal structure of an eIF4A:RNA complex bound to the interfacial inhibitor, desmethyl pateamine A. Cell Chem. Biol. 28, 825–834.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Niesen F. H., Berglund H., Vedadi M., The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Fletcher S. P., Ali I. K., Kaminski A., Digard P., Jackson R. J., The influence of viral coding sequences on pestivirus IRES activity reveals further parallels with translation initiation in prokaryotes. RNA 8, 1558–1571 (2002). [PMC free article] [PubMed] [Google Scholar]
- 40.Modelska A., et al. , The malignant phenotype in breast cancer is driven by eIF4A1-mediated changes in the translational landscape. Cell Death Dis. 6, e1603 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rubio C. A., et al. , Transcriptome-wide characterization of the eIF4A signature highlights plasticity in translation regulation. Genome Biol. 15, 476 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hausott B., Greger H., Marian B., Flavaglines: A group of efficient growth inhibitors block cell cycle progression and induce apoptosis in colorectal cancer cells. Int. J. Cancer 109, 933–940 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Mi Q., et al. , Silvestrol regulates G2/M checkpoint genes independent of p53 activity. Anticancer Res. 26, 3349–3356 (2006). [PubMed] [Google Scholar]
- 44.Sadlish H., et al. , Evidence for a functionally relevant rocaglamide binding site on the eIF4A-RNA complex. ACS Chem. Biol. 8, 1519–1527 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rose A. A., et al. , ADAM10 releases a soluble form of the GPNMB/Osteoactivin extracellular domain with angiogenic properties. PLoS One 5, e12093 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carey L. A., et al. , The triple negative paradox: Primary tumor chemosensitivity of breast cancer subtypes. Clin. Cancer Res. 13, 2329–2334 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Burnett P. E., Barrow R. K., Cohen N. A., Snyder S. H., Sabatini D. M., RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. U.S.A. 95, 1432–1437 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pause A., et al. , Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5’-cap function. Nature 371, 762–767 (1994). [DOI] [PubMed] [Google Scholar]
- 49.Baselga J., et al. , Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 366, 520–529 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kornblum N., et al. , Randomized phase II trial of fulvestrant plus everolimus or placebo in postmenopausal women with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer resistant to aromatase inhibitor therapy: Results of PrE0102. J. Clin. Oncol. 36, 1556–1563 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moore H. C. F., et al. , A randomized trial of fulvestrant, everolimus, and anastrozole for the front-line treatment of patients with advanced hormone receptor-positive breast cancer, SWOG S1222. Clin. Cancer Res. 28, 611–617 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cope C. L., et al. , Adaptation to mTOR kinase inhibitors by amplification of eIF4E to maintain cap-dependent translation. J. Cell Sci. 127, 788–800 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Ilic N., Utermark T., Widlund H. R., Roberts T. M., PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc. Natl. Acad. Sci. U.S.A. 108, E699–E708 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mallya S., et al. , Resistance to mTOR kinase inhibitors in lymphoma cells lacking 4EBP1. PLoS One 9, e88865 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martineau Y., et al. , Pancreatic tumours escape from translational control through 4E-BP1 loss. Oncogene 33, 1367–1374 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Satheesha S., et al. , Response to mTOR inhibition: Activity of eIF4E predicts sensitivity in cell lines and acquired changes in eIF4E regulation in breast cancer. Mol. Cancer 10, 19 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dang C. V., A time for MYC: Metabolism and therapy. Cold Spring Harb. Symp. Quant. Biol. 81, 79–83 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Dhanasekaran R., et al. , The MYC oncogene—The grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 19, 23–36 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lourenco C., et al. , MYC protein interactors in gene transcription and cancer. Nat. Rev. Cancer 21, 579–591 (2021). [DOI] [PubMed] [Google Scholar]
- 60.Soucek L., et al. , Modelling Myc inhibition as a cancer therapy. Nature 455, 679–683 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Masso-Valles D., et al. , MYC inhibition halts metastatic breast cancer progression by blocking growth, invasion, and seeding. Cancer Res. Commun. 2, 110–130 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dang C. V., Reddy E. P., Shokat K. M., Soucek L., Drugging the “undruggable” cancer targets. Nat. Rev. Cancer 17, 502–508 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Duffy M. J., Crown J., Drugging “undruggable” genes for cancer treatment: Are we making progress? Int. J. Cancer 148, 8–17 (2021). [DOI] [PubMed] [Google Scholar]
- 64.Chu J., et al. , Amidino-rocaglates: A potent class of eIF4A inhibitors. Cell Chem. Biol. 26, 1586–1593.e3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Galicia-Vazquez G., Cencic R., Robert F., Agenor A. Q., Pelletier J., A cellular response linking eIF4AI activity to eIF4AII transcription. RNA 18, 1373–1384 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kulak N. A., Pichler G., Paron I., Nagaraj N., Mann M., Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 11, 319–324 (2014). [DOI] [PubMed] [Google Scholar]
- 67.Senechal P., et al. , Assessing eukaryotic initiation factor 4F subunit essentiality by CRISPR-induced gene ablation in the mouse. Cell Mol. Life Sci. 78, 6709–6719 (2021), 10.1007/s00018-021-03940-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nasr Z., Robert F., Porco J. A. Jr., Muller W. J., Pelletier J., eIF4F suppression in breast cancer affects maintenance and progression. Oncogene 32, 861–871 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lehman S. L., et al. , Inhibition of the translation initiation factor eIF4A enhances tumor cell radiosensitivity. Mol. Cancer Ther. 21, 1406–1414 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Malka-Mahieu H., et al. , Synergistic effects of eIF4A and MEK inhibitors on proliferation of NRAS-mutant melanoma cell lines. Cell Cycle 15, 2405–2409 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kong T., et al. , eIF4A inhibitors suppress cell-cycle feedback response and acquired resistance to CDK4/6 inhibition in cancer. Mol. Cancer Ther. 18, 2158–2170 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nishida Y., et al. , Inhibition of translation initiation factor eIF4a inactivates heat shock factor 1 (HSF1) and exerts anti-leukemia activity in AML. Leukemia 35, 2469–2481 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fooks K., et al. , EIF4A inhibition targets bioenergetic homeostasis in AML MOLM-14 cells in vitro and in vivo and synergizes with cytarabine and venetoclax. J. Exp. Clin. Cancer Res. 41, 340 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Novac O., Guenier A. S., Pelletier J., Inhibitors of protein synthesis identified by a high throughput multiplexed translation screen. Nucleic Acids Res. 32, 902–915 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.