

Significance
It is well recognized that copper is critical for the immune system, and copper dyshomeostasis is associated with inflammation in several diseases. However, the underlying mechanism remains incompletely understood. Here, we demonstrate that cytosolic copper can enhance the proinflammatory signaling. Distinct from its conventional role as a direct bactericidal agent in the phagosomes, copper interacts with pattern-recognition receptor alpha-kinase 1 (ALPK1) and regulates the downstream proinflammatory cytokines production during infection. Thus, copper mediates a previously unrecognized immune signaling activation mechanism by enhancing ALPK1 kinase. These findings unveil the cross talk between copper homeostasis and the innate immune pathway and provide a broad perspective on the role of metal ions in the detrimental immune activation during the development of related diseases.
Keywords: copper, metalloprotein, ALPK1, innate immunity, bacterial infection
Abstract
Copper is an essential trace element for the human body, and its requirement for optimistic immune functions has been recognized for decades. How copper is involved in the innate immune pathway, however, remains to be clarified. Here, we report that copper serves as a signal molecule to regulate the kinase activity of alpha-kinase 1 (ALPK1), a cytosolic pattern-recognition receptor (PRR), and therefore promotes host cell defense against bacterial infection. We show that in response to infection, host cells actively accumulate copper in the cytosol, and the accumulated cytosolic copper enhances host cell defense against evading pathogens, including intracellular and, unexpectedly, extracellular bacteria. Subsequently, we demonstrate that copper activates the innate immune pathway of host cells in an ALPK1-dependent manner. Further mechanistic studies reveal that copper binds to ALPK1 directly and is essential for the kinase activity of this cytosolic PRR. Moreover, the binding of copper to ALPK1 enhances the sensitivity of ALPK1 to the bacterial metabolite ADP-heptose and eventually prompts host cells to elicit an enhanced immune response during bacterial infection. Finally, using a zebrafish in vivo model, we show that a copper-treated host shows an increased production of proinflammatory cytokines, enhanced recruitment of phagosome cells, and promoted bacterial clearance. Our findings uncover a previously unrecognized role of copper in the modulation of host innate immune response against bacterial pathogens and advance our knowledge on the cross talk between cytosolic copper homeostasis and immune system.
Copper is an essential trace element for life from bacteria to humans and is involved in many cellular processes (1–5). In addition, it has been recognized for decades that copper plays a critical role in the host–bacterium interface (6–11). Copper deficiency often results in the susceptibility of hosts to bacterial infections, and an increased copper level in the sites of infection has been detected in response to bacterial infections (12). Furthermore, excess copper has been associated with harmful inflammation in several disease conditions (13, 14). However, the precise roles played by copper in the regulation of host immune response, as well as in inflammation, remain largely unknown.
The innate immune system is the body’s first line of defense against pathogenic microbial invasion. Host epithelial cells express pattern-recognition receptors (PRRs) to sense pathogen-associated molecular patterns (PAMPs) from invading microbes and to trigger downstream signaling cascades, which ultimately induce the expression of various cytokines and antimicrobial peptides, contributing to host defense against pathogens (15). alpha-kinase 1 (ALPK1) has recently been identified as a new cytosolic PRR, recognizing ADP-heptose from gram-negative and some gram-positive bacteria (16, 17). Upon detection of ADP-heptose by the N-terminal domain (NTD) of ALPK1, the kinase activity of C-terminal kinase domain (CKD) of ALPK1 is promoted, leading to the phosphorylation of TIFA and the formation of TIFAsomes with various associated proteins, including classical NF-κB regulators TAB2 and TRAF2/TFAF6 (16–18). Once formed, TIFAsomes promote the phosphorylation and nuclear translocation of p65, eventually culminating in the production of NF-κB-dependent proinflammatory cytokines (16). The central role of ALPK1 in controlling host innate immune response against various bacterial pathogens, such as Shigella flexneri, Salmonella enterica, Helicobacter pylori, and Campylobacter jejuni, has been well documented (16, 17, 19–21). Nevertheless, how host cells regulate ALPK1 activity, as well as orchestrate the ALPK1-mediated downstream innate immune signaling, remains largely unaddressed.
Here, we report that copper plays a critical role in the regulation of host innate immune response against bacterial infection via sensitizing ALPK1. In response to bacterial infection, copper was uptaken from extracellular space or mobilized from mitochondria into the cytosol. The accumulated cytosolic copper efficiently promoted the resistance of host cells to infection by either intracellular or, surprisingly, extracellular pathogens. Mechanistic study revealed that copper bound to ALPK1 and increased the kinase activity of this PRR. As a result, host cells elicited an enhanced innate immune signaling to ADP-heptose stimulation or bacterial infection. In a zebrafish in vivo model, we observed a copper–ALPK1 axis-dependent production of proinflammatory cytokines and more phagocyte recruitment to the infection sites during bacterial infection. We discussed in detail how copper is involved in the ALPK1–NF-κB immune pathway against bacterial pathogens.
Results
Accumulated Copper in the Host Cell Cytosol during Bacterial Infection Enhances Host Cell Defense against Evading Pathogens, Including Extracellular Bacteria.
Evidence accumulated for several decades suggests a role of copper in the host–pathogen interface, but how copper is involved in the regulation of host defense against bacterial infection remains elusive (6, 7, 11). To address this issue, we first visualized copper in host cells in response to pathogen challenge by Coppersensor-3 (CS3), a specific fluorescence probe for imaging labile copper in mammalian cells (22) (Fig. 1A). Analysis of fluorescence signals revealed the steadily elevated levels of cytosolic copper in host cells upon bacterial infection (Fig. 1 B and C). We observed that bacterial challenge induced obvious copper accumulation after 3 h, whereas prolonged infection (16 h) resulted in a more significantly increased cytosolic copper level in host cells (Fig. 1 B and C). Meanwhile, addition of excess copper (40 μM) into the culture medium diminished the difference in fluorescence signals between the control group and infection group (SI Appendix, Fig. S1 A and B). Consistently, ICP-MS assay also showed that infection increases cytosolic copper levels of host cells (SI Appendix, Fig. S2).
Fig. 1.
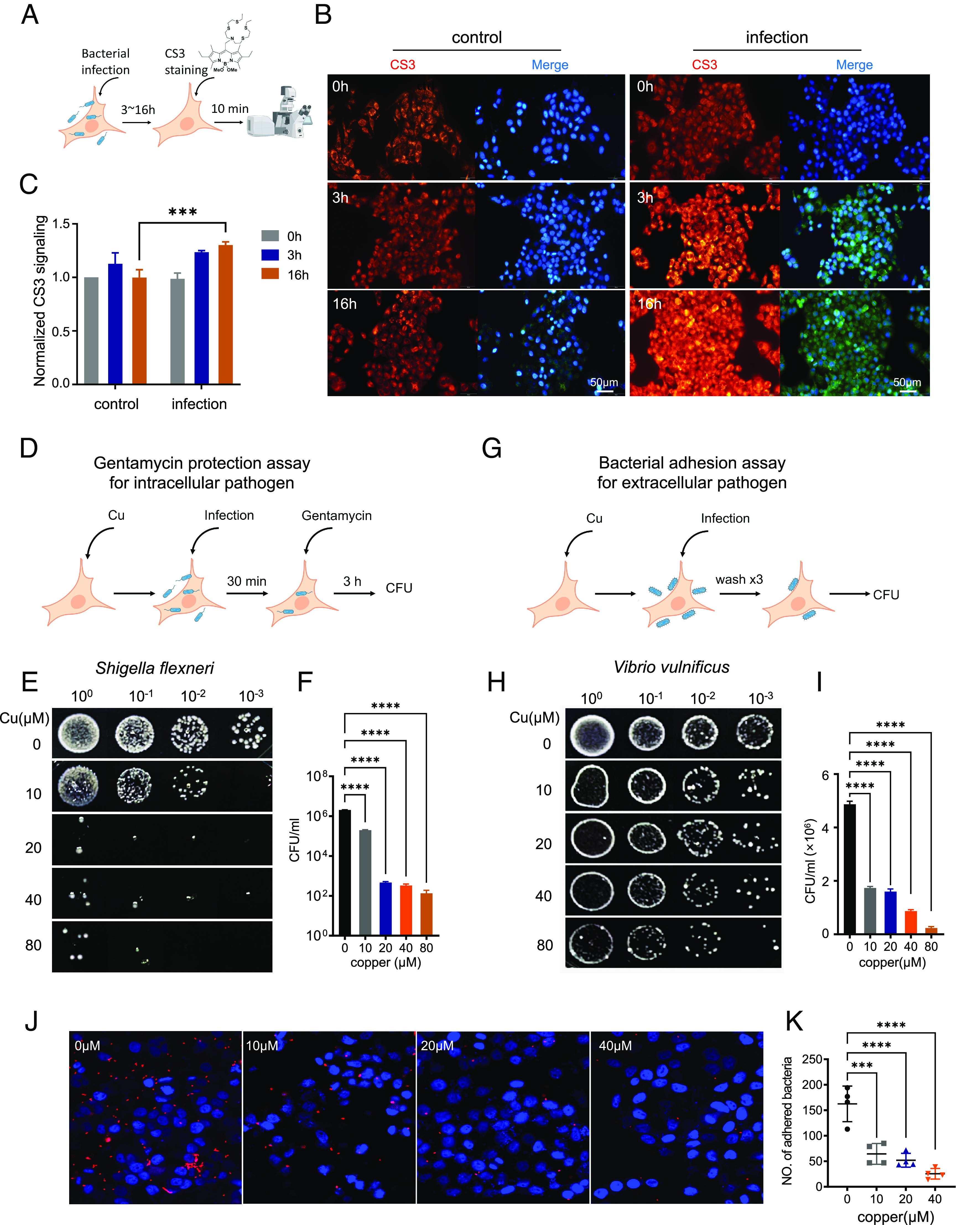
Accumulated copper in the host cell cytosol during bacterial infection enhances host cell defense against evading bacteria. (A) Schematic overview for (B and C). (B) Fluorescence imaging of cytosol Cu in HeLa cell with CS3 staining. HeLa cells were stained with 2 μM CS3 for 10 min at 37 °C in DMEM without (ctrl) or with Salmonella infection (MOI of 100) after 3 or 16 h. (Scale bar, 50 μm.) (C) The quantification of mean fluorescence intensity of each condition in (B) (n = 3 fields of cells per condition). Infection increased Cu levels to a similar extent as additional copper supplementation as shown in SI Appendix, Fig. S1. (D) Schematic overview for (E and F). (E and F) Assessing survival of intracellular pathogen S. flexneri inside HeLa cell by gentamycin protection assay. HeLa cells were pretreated with Cu at indicated concentrations; excess copper was removed by replacing culture with fresh medium before infection with S. flexneri for 30 min. Infected cells were incubated with 100 μg/mL gentamycin for 1 h to kill extracellular bacteria and then in 10 μg/mL gentamycin for the remainder of the experiment. Intracellular bacteria were released from HeLa cells and assessed by spot assay. (G) Schematic overview for (H and I). (H and I) The effect of Cu on host cell defense against extracellular pathogen V. vulnificus assessed by bacterial adhesion assay. HeLa cells were pretreated with Cu; excess copper was removed by replacing culture with fresh medium before infection with V. vulnificus. The unadhered bacteria were washed away with phosphate-buffered saline (PBS), and the number of bacteria adhered on the cell membrane was assessed by spot assay. (J) Representative imaging of mCherry-labeled bacterial pathogen in adherence assay. E. coli expressing mCherry infected HeLa cell with or without Cu pretreatment. The unadhered bacteria were removed by PBS. mCherry signal was observed with fluorescence microscopy analysis. (K) The quantification of mean bacterial numbers of each condition in (J) (n = 3 fields of cells per condition). Data are shown as mean ± SEM (n ≥ 3). ns, not significant, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Given that copper uptake into host cells depends heavily on copper transporter CTR1 (1), we therefore knocked out CTR1 in mammalian cell HeLa using the CRISPR-Cas9 system (23) and found that CTR1 deficiency nearly eliminates cytosolic Cu accumulation via whole-cell ICP-MS and CS3 assay (SI Appendix, Figs. S2 and S3). Furthermore, we determined the CTR1 messenger RNA (mRNA) level by RT-qPCR and found about threefold increases in its level in host cells at 3 h after bacterial infection (SI Appendix, Fig. S1C). Moreover, several other Cu homeostasis-related proteins (1) including copper chaperone ATOX1, copper transporter ATP7b and metallothionein proteins MT1H, MT2, were also up-regulated at mRNA levels in response to bacterial challenge (SI Appendix, Fig. S1C), confirming that host cells actively regulate Cu homeostasis responding to bacterial infection.
We next studied whether copper redistribution inside cells also contributes to cytosolic copper accumulation during infection. Considering mitochondria as a main metal ion pool, which releases metal ions from the mitochondrial matrix into the cytosol when membrane potential falls (24, 25), we determined the mitochondrial membrane potential during bacterial infection by JC-1 assay and found that the bacterial infection triggered a significant reduction of mitochondrial membrane potential at a postinfection time as early as 2 h (SI Appendix, Fig. S1 D and E). We next measured the copper content in mitochondria by ICP-MS, which reduced rapidly from around 1.6 to 0.7 pg/1 × 106 cells after 3 hours postinfection (hpi) and further to around 0.3 pg/1 × 106 cells after 24 hpi (SI Appendix, Fig. S1F), which is consistent with the JC-1 assay results. Remarkably, we showed that copper releasing from mitochondria could be induced by different bacterial pathogens (SI Appendix, Figs. S4 and S5). Furthermore, we observed that the knockout (KO) of SLC25A3, a mitochondrial copper transporter (26), resulted in a reduction of the basic cytosolic copper level in the absence of infection (SI Appendix, Fig. S2). Although bacterial challenge still induced the cytosolic copper accumulation in SLC25A3-deficient host cells, wild-type (WT) cells accumulated significantly higher copper content than SLC25A3 mutants (SI Appendix, Figs. S2 and S3). These findings suggest that the mitochondrial copper pool contributes, at least partially, to the bacteria-induced elevation of cytosolic copper. Collectively, we demonstrate that the host cell actively promotes copper accumulation in the cytoplasm in response to bacterial infection.
To evaluate the effects of increased cytosolic copper on host defense against bacterial infection, host cells were pretreated with the exogenous copper ions in cell culture medium to increase the cytosolic copper level and the excess copper was then removed from medium. Subsequently, host cells were infected with intracellular pathogen S. flexneri (Fig. 1D). Neither the viability of host cells (SI Appendix, Fig. S6A) nor all tested bacterial strains (SI Appendix, Fig. S6 C–I) were affected by the treatment of copper. As shown in Fig. 1 E and F, gentamycin protection assay showed a copper dose-dependent reduction of colony-forming units (CFUs) of S. flexneri in the host cytoplasm, and almost 4 log unit drops in the CFUs was observed in the presence of 20 μM of copper, indicating that copper-treated cells were more resistant to S. flexneri infection.
To exclude bacterial type specificity, copper-treated HeLa cells were infected with several other bacterial strains. We found that copper pretreatment enhanced host cell resistance to all tested intracellular bacterial strains (SI Appendix, Fig. S7A), including S. enterica and Salmonella Typhimurium, in consistence with the previous reports that copper might work as an antibacterial agent in the lysosomes of phagosomes (11, 27). Unexpectedly, copper-treated host cells also showed higher resistance to extracellular pathogens, including Vibrio vulnificus (Fig. 1 H and I) and pathogenic adhesive Escherichia coli (SI Appendix, Fig. S7B), as revealed by bacterial adhesion assay (Fig. 1G), suggesting an alternative role of copper beyond the direct bactericidal action inside host cells. To further confirm this, host cells were infected with engineered E. coli expressing mCherry. Consistently, fewer bacteria with mCherry signals were detected on the surface of host cells pretreated with copper (Fig. 1 J and K). Moreover, we treated the host cell with a specific copper chelator BCS (bathocuproine disulfonate) and observed that BCS treatment abrogated host defense against both extracellular and intracellular bacterial infections (SI Appendix, Fig. S8). These results collectively suggest that the elevated cytosolic copper acts to inhibit the infections from either intracellular or extracellular bacterial pathogens.
Copper Induces Host Cell NF-κB Response via the ALPK1–TIFA Pathway.
As copper promotes host resistance to both intracellular and extracellular pathogens, the underlying mechanism on how copper is involved in the host–pathogen interfaces remains incompletely understood. In view of the central role of NF-κB in mediating host cell immune response against invading microbial pathogens (16, 17, 19), we studied the potential effect of copper on NF-κB activation. HEK293t cells harboring NF-κB reporter were incubated with gradient concentrations of copper for 24 h. Interestingly, NF-κB in HEK293t was markedly activated by copper in a dose-dependent manner when copper concentration is lower than 100 μM (Fig. 2B). However, the relatively high concentration of copper (>200 μM) resulted in suppressed NF-κB signaling, which, we reasoned, likely resulted from the toxicity of copper overdosage to mammalian cells (Fig. 2B). In view of a long incubation time needed for the accumulation of secreted reporter in NF-κB reporter assay, we further used Western blot analysis to determine the activation of NF-κB pathway in HeLa cell, in which treatment of copper at concentrations ranging from 0 to 100 μM did not induce detectable cell apoptosis for 8 h (SI Appendix, Fig. S6B). As shown in Fig. 2C, Western blot analysis showed that copper induced NF-κB activation as indicated by the phosphorylation of p65, even at a concentration as low as 10 μM. Interestingly, we found that copper could also induce an elevated level of p-TIFA (Fig. 2C). We further detected the expression of NF-κB-dependent genes by RT-qPCR upon copper stimulation in cell, including IL8, IL1B, TNFα, RelB, IL6, and antimicrobial peptide hBD2. The upregulation of these NF-κB-dependent genes upon copper treatment was detectable for as early as 3 h (Fig. 2 D–H and SI Appendix, Fig. S9A). Taken together, we demonstrate that copper can stimulate NF-κB inflammation signaling.
Fig. 2.

Copper induces host cell NF-κB response via ALPK1. (A) Schematic of the ALPK1–NF-κB signaling pathway in host cells responding to bacterial infection. (B) NF-κB signaling of HEK293t cell assessed by secreted alkaline phosphatase (SEAP) assay. HEK293t cells harboring NF-κB SEAP reporter were treated with Cu at indicated concentrations. NF-κB activation was accessed at 24 h after Cu treatment. Cell viability was determined via 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) assay. (C) Western blot analysis of p-p65 and p-TIFA levels in HeLa cell. HeLa cells were treated with indicated concentrations of Cu for 8 h. p-p65 and p-TIFA were revealed by immunoblotting with anti-p-p65 and anti-p-TIFA antibody, respectively. (D–H) Quantitative real-time PCR analysis of IL8 (D), IL1B (E), TNFα (F), RELB (G), and IL6 (H) expression in AGS cells with mock treatment or Cu treatment for 3 h. (I) Quantitative real-time PCR analysis of IL8 expression in AGS cells (WT or ALPK KO) with/without treatment of Cu. (J) Quantitative real-time PCR analysis of IL1B expression in AGS cells (WT or ALPK KO) with/without treatment of Cu. (K) Quantitative real-time PCR analysis of IL8 expression in AGS cells (WT or TIFA KO) with/without treatment of Cu. Data are shown as mean ± SEM (n ≥ 3). ns, not significant, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
It has been demonstrated previously that ALPK1 serves as a master regulator in epithelial cells to mediate NF-κB signaling against various bacterial pathogens (Fig. 2A) (16, 17, 19–21). We generated an ALPK1-deficient cell line based on mammalian cell AGS using CRISPR-Cas9 system (23) (SI Appendix, Fig. S10). Surprisingly, knocking out ALPK1 abrogated copper-induced expression of IL8 (Fig. 2I) and IL1B (Fig. 2J). As ALPK1 depends on TIFA to activate downstream NF-κB signaling (16, 28), we thus targeted TIFA via the CRISPR-Cas9 method in AGS cells and examined whether copper-induced NF-κB signaling relies on TIFA. RT-qPCR analysis revealed that knocking out TIFA also dampened the expression of IL8 upon copper treatment (Fig. 2K), suggesting that copper-induced NF-κB activation is critically dependent on the integrity of the ALPK1–TIFA pathway.
TLR4 is also an important PRR, working in innate immune response of host cells to bacterial infection. Interestingly, a previous study showed that TLR4 is responsible for allergies to nickel-releasing metal alloys (29). We thus examined whether copper activates NF-κB via TLR4. However, RT-qPCR analysis revealed that knocking out TLR4 caused no changes in copper-mediated IL8 production (SI Appendix, Fig. S9B), implying copper-mediated NF-κB signaling is independent of TLR4, in consistence with a previous report (30).
Collectively, we demonstrate that copper is involved in the regulation of the ALPK1–TIFA–NF-κB axis.
Copper Binds to ALPK1 Directly and Is Required for ALPK1 Kinase Activity.
We have demonstrated a regulator role of copper in ALPK1-mediated NF-κB signaling, but a direct mechanistic link is still lacking. We next investigated the underlying mechanism of copper-induced NF-κB activation via ALPK1. We first examined whether copper binds to ALPK1 in cell by Cellular Thermal Shift Assay (CETSA) (31) to study thermal stabilization of ALPK1 in cell treated with copper. The treatment of cells with copper resulted in the melting point of ALPK1 shifts from ca. 49 °C to 58 °C with ΔTm = 8.6 °C, indicating that copper bound to and stabilized ALPK1 (Fig. 3 A and B). Similarly, bacterial infection induced an increased thermal stability of ALPK1 (SI Appendix, Fig. S11). However, copper chelator BCS and NCP (neocuproine) led to a reduction in ALPK1 thermal stability (SI Appendix, Fig. S11), suggesting the copper-ALPK1 binding in response to bacterial exposure.
Fig. 3.
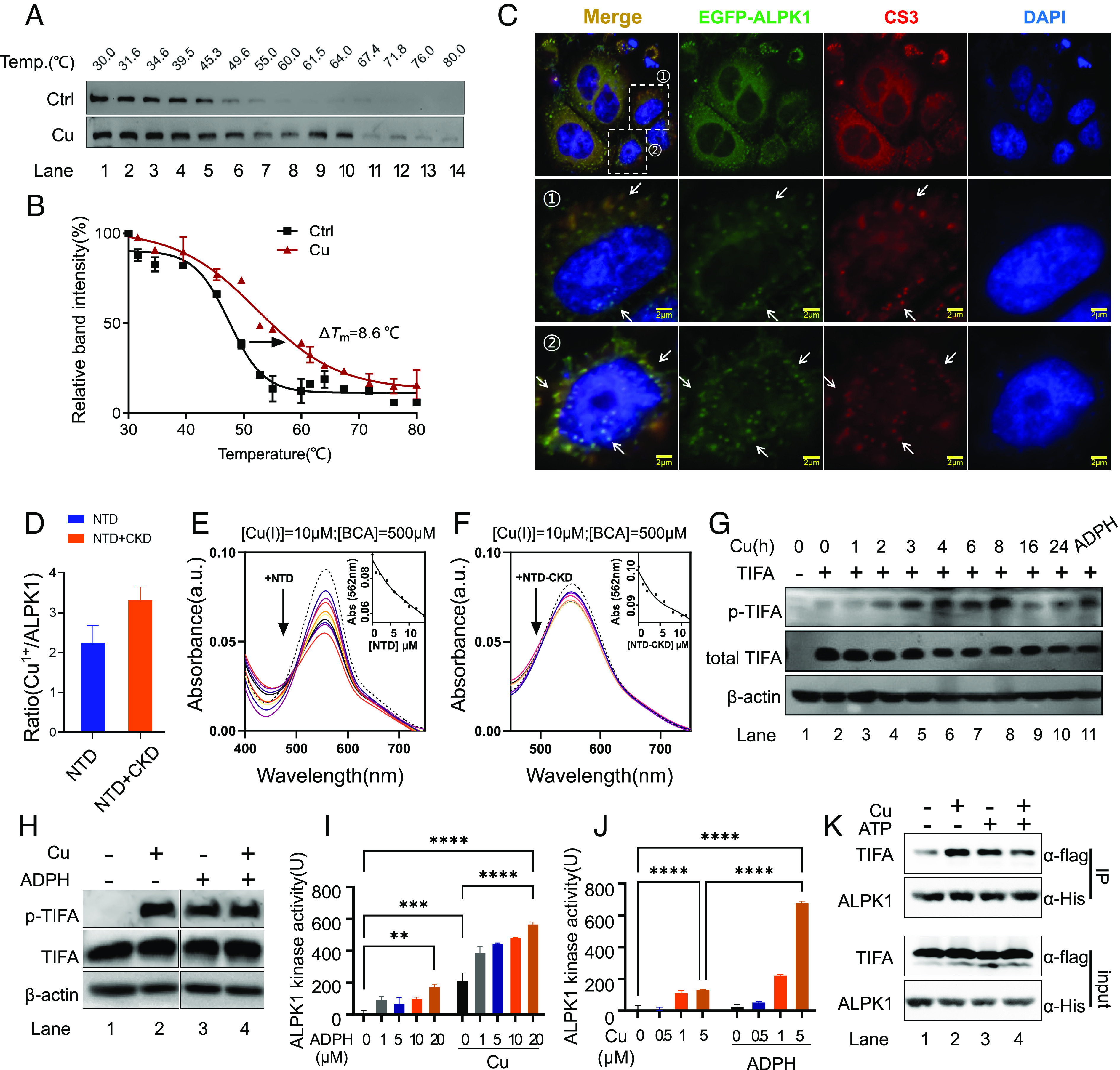
Copper binds to ALPK1 and regulates its kinase activity. (A) Representative CETSA blot for Cu-ALPK1 binding in cell. HEK293t cells expressing ALPK1-flag were pretreated with Cu following indicated heat shocks. Soluble ALPK1-flag in sample supernatant was revealed by immunoblotting with anti-Flag antibody. (B) CETSA melt curves for Cu-ALPK1 binding in cell. The melting temperatures shift (ΔTm) between treated and control samples was measured. (C) Enhanced green fluorescent protein (EGFP)-labeled ALPK1 localization compared with Cu sensor CS3 in HeLa cell. (Scale bar, 2 μm.) Arrows indicate overlap. (D) Cu content in NTD and NTD-CKD recombination proteins determined with ICP-MS. (E) Ultraviolet–visible (UV–vis) absorption spectra of Cu-BCA complex with the addition of gradient molar equivalents of NTD protein. (F) UV–vis absorption spectra of Cu-BCA complex with the addition of gradient molar equivalents of NTD-CKD complex protein. (G) Representative Western blot for p-TIFA in cell. Cells were treated with copper (20 μM) or ADP-heptose (1 μM, 2 h). At the indicated time points, samples were harvested for Western blot. (H) Representative western blot for ALPK1–TIFA assay in vitro using purified recombination ALPK1 and TIFA proteins. Recombination ALPK1 and TIFA proteins were mixed with Cu1+ (0 or 1 μM) and ADP-heptose (0 or 20 μM) in the presence of 5 mM GSH. Kinase reactions were initiated by the addition of adenosine triphosphate (ATP) and stopped by adding 3× SDS loading buffer. Phosphorylated TIFA was revealed by immunoblotting with anti-p-TIFA antibody. (I) Kinase assay for ALPK1 kinase activity in vitro using purified recombination ALPK1 and TIFA proteins in the presence of gradient concentrations of ADP-heptose. Recombination ALPK1 and TIFA proteins were mixed with indicated concentrations of ADP-heptose in the presence of 5 mM GSH, with/without Cu1+ (5 μM). Kinase reactions were initiated by the addition of ATP. Kinase activity was assessed as the manufacturer’s protocol indicated. (J) Kinase assay for ALPK1 kinase activity in vitro using purified recombination ALPK1 and TIFA proteins in the presence of gradient concentrations of Cu1+. Recombination ALPK1 and TIFA proteins were mixed with indicated concentrations of Cu1+ in the presence of 5 mM GSH, with/without ADP-heptose (5 μM). Kinase reactions were initiated by addition of ATP. Kinase activity was assessed as the manufacturer’s protocol indicated. The unit of ALPK1 kinase activity was defined as the amounts of ATP (ng) consumed by ALPK1 (1 μg) protein per hour (ng/[μg × h]). (K) Coimmunoprecipitation of ALPK1 and TIFA treated with or without Cu1+ (1 μM). Data are shown as mean ± SEM (n ≥ 3). ns, not significant, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To further investigate the binding of ALPK1 to copper in the cell, we transiently overexpressed ALPK1 fused to EGFP in host cells, which were sequentially treated with 20 μM copper and labeled with CS3 probe. We observed the colocalization of copper sensor CS3 with EGFP-ALPK1 in a relatively specific manner (Fig. 3C). Moreover, we overexpressed ALPK1 fused to Flag tag (ALPK1-Flag) in HEK293t cells treated with or without copper, ALPK1-flag was extracted and subsequently subjected to ICP-MS analysis for copper content. We found that a significantly elevated level of copper with more than four folds was detected in the ALPK1 sample extracted from mammalian cells treated with copper, compared to the one without copper treatment (SI Appendix, Fig. S12), demonstrating that copper binds to ALPK1 directly in the cell.
We next prepared recombinant ALPK1 proteins (NTD and NTD-CKD complex) (SI Appendix, Fig. S13) to study the potential copper-ALPK1 interaction in vitro. Considering it is cuprous (Cu1+), rather than cupric (Cu2+) ions, that are transported into cytoplasm by hCTR1 (1, 32), and there are high levels of GSH maintaining the reduced intracellular environment (33), we incubated ALPK1 proteins with 10 molar equivalents of Cu1+ in the presence of 5 mM GSH to mimic the cellular physiological condition, and the samples were subjected to ICP-MS analysis after removal of excess copper. Interestingly, ca. 2.2 ± 0.4 and 3.3 ± 0.3 molar equivalences of Cu1+ were found to bind to NTD and NTD-CKD respectively (Fig. 3D), indicating ALPK1 might contain multimetal binding sites for Cu1+.
We further performed copper-competition assay using bicinchoninic acid (BCA) to study Cu1+-ALPK1 binding. Specific Cu1+ chelator BCA formed a color complex with Cu1+, with a main absorption peak at ca. 562 nm. Titration of recombination NTD or NTD-CKD protein into Cu1+-(BCA)2 solution resulted in a dose-dependent decrease in the absorption peak at 562 nm, indicative of the reduction in Cu1+-(BCA)2 complex owing to the competitive binding of ALPK1 to Cu1+ from Cu1+-(BCA)2 complex (Fig. 3 E and F). The average dissociation constants between Cu1+ and NTD or NTD-CKD were calculated to be ca. 1.82 × 10−17 μM or 0.88 × 10−17 μM respectively, based on the reported Cu1+-BCA formation constant [log β2(BCA) = 17.7] (34) and UV titration curve fitting as described previously (35). Collectively, these cellular and in vitro data indicate that ALPK1 is a copper-binding protein.
Copper is involved in various physiological processes via its regulation role for many enzymes (1). Given that ALPK1 is a kinase, the CKD of which phosphorylates and activates TIFA for downstream NF-κB activation upon recognition of ADP-heptose by NTD of ALPK1 (17). We therefore hypothesized that copper may regulate ALPK1 kinase activity. To validate this, we first monitored phosphorylation of TIFA (p-TIFA), an indicator of ALPK1 activity, by using Western blot. We observed that copper resulted in the phosphorylation of TIFA in a time-dependent manner in host cells, and high levels of p-TIFA could be detected upon the supplementation of copper into culture medium at around 3 to 8 h (Fig. 3G).
We further incubated recombinant ALPK1 and TIFA proteins with copper (Cu1+) and ADP-heptose for reaction. In line with a previous report (17), incubation of apo-ALPK1 and TIFA did not result in phosphorylation of TIFA in vitro (Fig. 3H, Lane 1), while ALPK1 phosphatized TIFA in the presence of a relatively high concentration of ADP-heptose (20 μM) in vitro (Fig. 3H, Lane 3). Noticeably, a low concentration of ADP-heptose (1 μM), a concentration that is present in bacteria as indicated in previous reports (16, 17, 36), was not able to induce p-TIFA in this reaction (SI Appendix, Fig. S14), implying ALPK1 might need additional cofactor(s) for its sensitivity to ADP-heptose. We further observed that copper alone is sufficient in inducing ALPK1-mediated p-TIFA in the absence of ADP-heptose in vitro (Fig. 3H, Lane 2). Surprisingly, the introduction of a phosphor-mimetic of TIFA (T9D or T9E) without ALPK1 activation is insufficient to elicit NF-κB signaling in host cells (SI Appendix, Fig. S15), in consistence with a previous report (37). Copper-induced ALPK1 activation was also confirmed in native gel, which showed that copper alone led to an increased level of TIFA oligomer in vitro (SI Appendix, Fig. S16 A and B, Lanes 1 to 3). Interestingly, the combination of copper (5 μM) and ADP-heptose exhibited a synergistic effect on the oligomerization of TIFA (SI Appendix, Fig. S16 A and B).
To further study the effect of copper on ALPK1 function, we used an in vitro kinase assay to quantify ALPK1 kinase activity. As shown in Fig. 3I, ADP-heptose activated ALPK1 kinase in a dose-dependent manner, and 20 μM of ADP-heptose stimulated the ALPK1 kinase activity to ca. 200 units (Fig. 3I). In line with Western blot results, copper alone (5 μM) could elicit ALPK1 kinase activity to the extent that 20 μM of ADP-heptose did (Fig. 3I), whereas the combination of ADP-heptose (1 to 5 μM) and copper (5 μM) promoted the ALPK1 kinase activity by almost two folds (Fig. 3 I and J), implicating the synergistic effect of copper and ADP-heptose on ALPK1 kinase. Interestingly, the enhanced effect of copper on ALPK1 could only be observed in the presence of 5 mM GSH, which mimics the cellular physiological condition. In contrast, additional CuSO4 (Cu2+) suppressed ALPK1 kinase activity (SI Appendix, Fig. S16C) in the absence of GSH.
We next investigated the potential effect of copper on the substrate-binding capacity of ALPK1 using recombinant ALPK1 and TIFA proteins via coimmunoprecipitation. We observed a relatively weak interaction between ALPK1 and TIFA in the absence of copper and ATP (Fig. 3K, Lane 1). The ALPK1–TIFA interaction was enhanced by the addition of copper (Fig. 3K, Lane 2). However, the presence of ATP resulted in slightly reduced copper-induced ALPK1–TIFA interaction (Fig. 3K, Lanes 3 and 4). In view of that copper could mediate the ALPK1-dependent phosphorylation of TIFA in the presence of ATP (Fig. 3H), we speculate that copper might play a subtle regulation role in TIFA/p-TIFA subtract binding for ALPK1 kinase activity. We further investigated ALPK1–TIFA interaction in an infection scenario. As depicted in SI Appendix, Fig. S17, similar to copper treatment, bacterial infection enhanced ALPK1–TIFA interaction in host cells, while BCS treatment attenuated the association between ALPK1 and TIFA, suggesting that the ALPK1–TIFA complexation during infection is dependent on copper availability (SI Appendix, Fig. S17). Taken together, we demonstrate that copper functions as an essential cofactor for the regulation of kinase activity of ALPK1.
Copper Sensitizes the ADP-Heptose–ALPK1–NF-κB Pathway of Host Cells.
Upon infection, ALPK1 in the host cytoplasm recognizes the metabolite molecule ADP-heptose from bacteria and mediates downstream NF-κB signaling (17). We therefore investigated the effect of copper on ALPK1-mediated host cell response to ADP-heptose. We stimulated host cells with 1 μM ADP-heptose, which mimics the bacterial infection (16, 17, 36), with or without copper at various concentrations. By using NF-κB reporter assay, we showed that ADP-heptose alone induced a twofold change of NF-κB activation (Fig. 4A). However, in the presence of copper (20, 40, or 100 μM), ADP-heptose stimulated stronger NF-κB activation with a ca. fourfold change (Fig. 4A). We also evaluated the potential synergistic effect of ADP-heptose and copper on NF-κB signaling in host cells by Western blot analysis and found that ADP-heptose together with copper induced a higher level of p-p65 (Fig. 4B), as well as p-TIFA (Fig. 4C), than ADP-heptose alone. Similarly, RT-qPCR analysis revealed that more than 10 folds of IL8 production of host cell was stimulated by ADP-heptose for 3 h in the presence of copper, compared to that by ADP-heptose alone (Fig. 4D), confirming that copper sensitizes ADP-heptose–ALPK1–NF-κB pathway in host cells. Furthermore, we treated host cells with BCS and found that BCS attenuated the IL8 expression upon ADP-heptose stimulation (SI Appendix, Fig. S18), Confirming that copper, under normal conditions, is indeed essential for the ADP-heptose–ALPK1–NF-κB pathway.
Fig. 4.
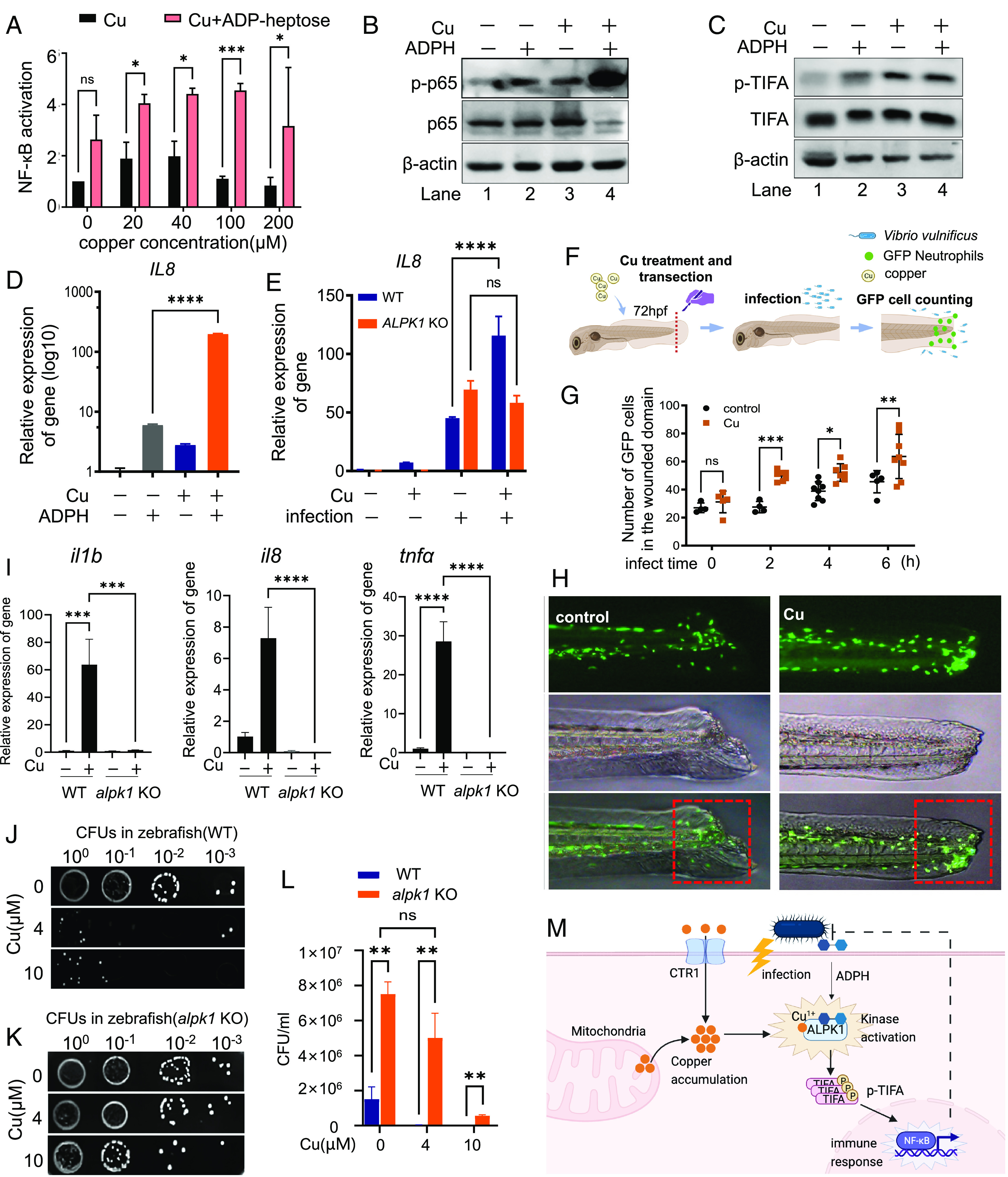
Copper sensitizes the ADP-heptose–ALPK1–NF-κB pathway of host cells and is required for host defense against bacterial infection in vivo. (A) NF-κB signaling of HEK293t cell upon ADP-heptose in the presence of various concentrations of copper assessed by SEAP assay. HEK293t cells were transfected with plasmids encoding a NF-κB SEAP reporter gene and were treated with Cu and ADP-heptose as indicated. NF-κB activity was measured by SEAP assay. (B) Western blot analysis of p-p65 level in HeLa cell upon ADP-heptose with/without copper treatment. HeLa cells were treated with/without Cu following 1 μM ADP-heptose stimulation for 3 h. p-p65 were revealed by immunoblotting with anti-p-p65 antibody. (C) Western blot analysis of the p-TIFA level in the HeLa cell upon ADP-heptose with/without copper treatment. p-TIFA were revealed by immunoblotting with anti-p-TIFA antibody, respectively. (D) Quantitative real-time PCR analysis of IL8 expression in HeLa cell upon ADP-heptose with/without copper treatment. HeLa cells were treated with/without Cu following 1 μM ADP-heptose stimulation for 3 h. Total RNA in samples was extracted, and the mRNA level of IL8 was revealed by RT-qPCR. (E) Quantitative real-time PCR analysis of IL8 expression in AGS cells upon bacterial infection (MOI of 100) with/without copper pretreatment. AGS cells (WT or ALPK1 KO) were treated with/without Cu following bacterial infection for 3 h. Total RNA in samples was extracted, mRNA level of IL8 was revealed by RT-qPCR. (F) Schematic overview for neutrophils recruitment assay in the zebrafish model. (G) The quantification of neutrophil numbers around wound upon V. vulnificus infection (MOI of 100). (H) Fluorescence imaging of GFP-positive neutrophils in zebrafish showing the recruitment of neutrophils around wound upon V. vulnificus infection. (I) Quantitative real-time PCR analysis of NF-κB-dependent gene expression in zebrafish (WT or alpk1 KO) upon copper. (J) Assessing colonization of pathogen S. enterica inside WT zebrafish with/without copper treatment. (K) Assessing colonization of pathogen S. enterica inside alpk1 KO zebrafish with/without copper treatment. (L) The quantification of CFUs in (J and K). (M) The working model of the mechanism by which copper regulates ALPK1 kinase for host defense against bacterial infection. Data are shown as mean ± SEM (n ≥ 3). ns, not significant, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To further evaluate the effect of copper on ALPK1–NF-κB response of host cells in an infection scenario, we infected host cells by S. enterica and examined the IL8 expression in the presence or absence of copper. Bacterial infection alone triggered IL8 production of host cells by about 50 folds, whereas a dramatically enhanced IL8 expression by more than 120 folds was detected by RT-qPCR in the presence of copper (Fig. 4E). Out of our expectation, deficiency of ALPK1 gene did not abolish NF-κB response of host cells to infection, as indicated by the elevated IL8 expression in ALPK1 KO cells comparable to that in the WT cells (Fig. 4E), which may be due to the expression of other PRRs mediating antibacterial immune response in the host cells (38, 39). Nevertheless, the additional copper did not further enhance IL8 expression in ALPK1 KO cells during infection (Fig. 4E), confirming copper regulates the NF-κB pathway via ALPK1. We further challenged CTR1- or SLC25A3-deficient host cells with ADP-heptose or S. enterica and found that KOs of CTR1 and SLC25A3 attenuated the NF-κB response of host cells to ADP-heptose stimulation and infection (SI Appendix, Fig. S19), confirming the critical role of copper in the ALPK1–NF-κB pathway. These data collectively suggest that copper ions sensitize ALPK1-mediated NF-κB response of host cells to ADP-heptose and bacterial infection.
Copper Is Required for Host Defense against Bacterial Infection In Vivo.
ALPK1 is relatively conserved across various vertebrates, including zebrafish (Danio rerio) (17). Taking the advantage of the optical transparency of zebrafish in the study of infectious diseases (40), we investigated the effect of copper on innate immune response using a transgenic zebrafish line that expresses GFP under the neutrophil-specific myeloperoxidase promoter, in view of that NF-kB signaling plays a critical role in the recruitment of neutrophils to infection sites (41). The zebrafish larvae were pretreated with copper (4 μM) and transected at the tailfin. Pathogen V. vulnificus (MOI of 100) was used to challenge the zebrafish with injured tails (Fig. 4F). We observed that treatment of copper induced massive neutrophil recruitment to the wound area in response to bacterial infection (Fig. 4 G and H). To further investigate the role of ALPK1 in this process, we next generated alpk1 KO fish by utilizing a CRISPR-Cas9 method (SI Appendix, Fig. S20) (42). Copper induced a significant NF-κB activation in zebrafish, as indicated by the elevated IL1B, IL8, and TNFα expression (Fig. 4I). Noticeably, alpk1 KO abrogated copper-induced NF-κB signaling, as evidenced by the abolished IL1B, IL8, and TNFα expression upon copper treatment in alpk1 KOs (Fig. 4I). Furthermore, we observed that 4 μM of copper has provided an almost complete protection to the host from the infection, as evidenced by the sharply decreased bacterial loads in WT zebrafish larvae in the presence of copper (Fig. 4J) compared with those without copper treatment (Fig. 4K). However, alpk1 KO resulted in much higher bacterial loads (around five folds) in the host, compared with WT fish. The pretreatment of copper (4 μM) did not reduce the bacterial loads obviously in alpk1 KOs, which are far more vulnerable to bacterial infection compared to the WT host (Fig. 4L). Although 10 μM copper showed some protection against bacteria, this ALPK1-independent antimicrobial effect, which, we reasoned, is likely attributable to the direct bactericidal effect of copper at relatively high concentration (Fig. 4L). Nevertheless, taken together, we demonstrate clearly that the copper–ALPK1 axis plays a critical role in the host defense against bacterial pathogens in vivo.
Discussion
PRRs play a critical role in the recognition of pathogens and in mediating the downstream immune signaling (15). ALPK1 is a cytosolic PRR, which plays a central role in mediating the activation of the NF-κB signaling pathway upon its recognition of the bacterial metabolite ADP-heptose during infection (16, 17, 36). In this study, we demonstrate that in response to bacterial infection, host cells actively increase cytosolic copper to enhance ALPK1 kinase activity, as well as the downstream ALPK1-mediated NF-κB signaling responsive to ADP-heptose or infection (Fig. 4M).
It has long been known that copper is important for the maintenance of the normal functions of the host immune system against microbial infection (6, 12). Copper deficiency often results in the susceptibility of hosts to bacterial infection (6). However, the molecular basis for copper-facilitating immune response remains elusive. Owing to its potential toxic properties, copper has been considered as a direct bactericidal agent in the phagolysosome of macrophage, neutrophils and other immune cell types (7, 43). This hypothesis well explains how phagosomes combat with bacterial pathogens, especially for intracellular bacteria. However, it is not in line with our observation that copper also bolstered host epithelial cells to fight against extracellular microbial bacteria, which suggests that copper’s immune effect extends farther than previously thought; this metal has an important extracellular inhibitive role to play beyond its role in the cytosolic phagolysosome. In other words, copper can serve as an indirect/noncontact bactericide and not just a direct one. In this study, we uncover the mechanism of such an event, that the increased cytosolic copper activates ALPK1-mediated immune signaling for host defense (Fig. 4).
Copper acts as a cofactor in the active sites of various enzymes, such as cytochrome c oxidase, peptidyl-α-monooxygenase, and SOD1 (1). Alternatively, copper is also involved in many human physiological processes via binding to the nonactive site of a protein (1). For example, copper was found to regulate ULK1/2 activity to drive lung adenocarcinoma (44). Moreover, the binding of copper to lipoylated components in the tricarboxylic acid cycle has been shown to be a trigger for Cuproptosis, a form of regulated programmed cell death (45). In this study, we found that cytosolic copper, likely as its cuprous (Cu1+) form, binds to ALPK1 directly to enhance kinase activity. The molecular basis for how copper regulates ALPK1 may warrant further study in the future. The crystal structure of ALPK1 with copper bound will provide an insight into the detailed mechanism. Nevertheless, we found that copper enhances the thermal stabilization of ALPK1 (Fig. 3 A and B), as well as the interaction of ALPK1 with its substrate TIFA (Fig. 3K). Moreover, we observed that the increased cytosolic copper induces the unexpected formation of ALPK1 foci in the cell (Fig. 3C). All of these observations suggested that copper may induce the conformational change of ALPK1 protein, which might be critical for activation of ALPK1 kinase.
Maintenance of copper homeostasis in host cells depends on a battery of proteins, including transmembrane copper transporters (CTR1), copper chaperones (ATOX1), copper storage molecules (metallothioneins), etc. (46, 47). We observed a remarkably up-regulated expression of genes encoding copper transporter and metalloproteins/metallomolecules upon bacterial infection (SI Appendix, Fig. S1C), which may promote cytosolic copper uptake and accumulation (Fig. 1 A–C). Consistently, a previous report showed that levels of the Ctr1 importer are elevated in macrophages upon interferon-gamma (IFN-γ) and LPS stimulation, which mimics infection (43). The increased copper in the cytoplasm may also be released from mitochondria and/or other membrane-enclosed organelles, which are the intracellular reservoirs for copper, as evidenced by the decreased mitochondria membrane potential and reduced copper content in mitochondria upon infection (SI Appendix, Fig. S1 D–F). Eventually, the accumulated copper in the cytoplasm from the extracellular space and intracellular pools together primes the ALPK1 signaling pathway.
Although a copper homeostasis is indispensable for normal physiological activity of host cells, excess copper is harmful to the host (6). Disordered copper metabolism has been reported to be associated with inflammation, which contributes to the development of diseases, such as Wilson disease (13), Alzheimer’s disease and Parkinson’s disease (14). However, the underlying molecular mechanism involved for copper-mediated inflammation in these diseases remains inconclusive. In this study, we showed that increased cytosolic copper induced the production of inflammation cytokines via stimulating the innate immune signaling pathway (Figs. 2 and 4). Interestingly, a recent study reported that ALPK1 knockdown showed a neuroprotective effect in mice (48). Whether the detrimental immune activation induced by the pathological copper accumulation is responsible for the cellular and tissue damage in Wilson disease and neurodegenerative disorders needs further investigation. Alleviating such copper-induced immune activation might serve as a potential therapeutic strategy.
In summary, we have shown that copper serves as a signal molecule to regulate the innate immune pathway in host cells during infection. We validated that copper binds to cytosolic PRR ALPK1 directly and regulates its kinase activity for the downstream signal conduction, which is distinct from the direct bactericidal action of copper in the host–pathogen interface. These findings shed light on the cross talk between cytosolic copper homeostasis and the innate immune pathway.
Materials and Methods
Bacterial Strains and Culture Conditions.
Salmonella, S. flexneri, V. vulnificus, and E. coli strains were obtained from the Department of Infectious Diseases (The Fifth Affiliated Hospital, Sun Yat-sen University). Bacteria were grown in proper medium at 37 °C with 200 rpm shaking unless otherwise stated. Log-phase bacterial pathogens were resuspended in PBS for infection. Multiplicity of infection (MOIs) are indicated on each figure legend.
Cell Culture.
All cells were cultured at 37 °C in an atmosphere of 5% (v/v) CO2. HEK293t and HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS), penicillin (100 μg/mL), and streptomycin (100 μg/mL). AGS cells were cultured in RPMI 1640 supplemented with 10% FBS and antibiotics. All cells were tested for mycoplasma with the standard PCR method at regular intervals.
CS3 Staining and Cell Imaging.
Where indicated, cells were challenged with bacterial pathogen. At indicated time points (0, 3, and 16 hpi), cells were washed with PBS and loaded with CS3 (purchased from Toronto Research Chemicals, B874750-1) (2 μM, 10 min) and DAPI (5 μM, 10 min) in DMEM at 37 °C in dark. After an additional wash step, cells were imaged in fresh DMEM (λEx = 538 nm, λEm = 548 nm for CS3; λEx = 359 nm, λEm = 457 nm for DAPI).
Zebrafish Lines and Maintenance.
All of the adult zebrafish including WT AB and Tg(coro1a:eGFP) zebrafish lines were obtained from Zebrafish International Resource Center (ZIRC) and were raised under standard conditions (28.5 °C, 14 h light/10 h dark). All fish were fed with hatched fairy shrimps three times each day. Male and female zebrafish were kept separately until mating and spawning. Eggs were kept in E3 medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, and 0.33 mM MgSO4) with 0.003% 1-phenyl 2-thiourea (PTU) to reduce melanin deposition and staged as hour postfertilization and day postfertilization.
Gentamycin Protection Assay.
HeLa cells were pretreated with CuSO4 for 2 h and starved in antibiotic-free medium before challenge with logarithmic phase (OD600 = 0.8 to 1.0) bacteria (Salmonella, Shigella strains) with an MOI of 100 for 30 min at 37 °C. Cells were then washed three times in PBS and placed in medium containing 100 μg/mL of gentamycin and incubated for another 2.5 h (3 hpi) at 37 °C to kill the extracellular bacteria. At the indicated time points, cells were washed and harvested. Cell lysates were serially diluted and plated onto agar plates. After overnight incubation at 37 °C, the number of internalized bacteria was determined by counting the CFUs.
Bacterial Adhesion Assay.
HeLa cells were pretreated with CuSO4 for 2 h and starved in antibiotic-free medium before challenge with logarithmic phase (OD600 = 0.8 to 1.0) bacteria (V. vulnificus and pathogenic E. coli strains) with an MOI of 100 at 37 °C. At indicated time points, cells were then washed three times in PBS and harvested. Cell lysates were serially diluted and plated onto agar plates. After overnight incubation at 37 °C, the number of adhesion bacteria on the host cell surface was determined by counting CFUs.
p-TIFA In Vitro Assay.
ALPK1 kinase activity was determined by p-TIFA in vitro assay. Briefly, recombinant NTD-CKD complex or full-length ALPK1 (800 ng/reaction) and subtract TIFA-His (2 μg/reaction) proteins were incubated with copper or ADP-heptose as indicated in a 50 μL reaction assay buffer (45 mM HEPES, pH 7.4, and 4 mM MgCl2) supplemented with 5 mM glutathione (GSH). ATP (100 μM final) was added to initial the reaction. After incubation at 30 °C for 60 min, the reaction was terminated by the addition of 3× sodium dodecyl sulfate (SDS) loading buffer. The samples were subjected to western blot assay. Phosphorylated TIFA was revealed by immunoblotting with anti-p-TIFA antibody (Abcam).
ALPK1 Kinase Assay.
ALPK1 kinase activity was quantified by the Kinase-Lumi™ Plus Luminescent Kinase Assay Kit (Beyotime) according to the manufacturer’s protocol by determination of the amounts of consumed ATP. Briefly, recombination full-length ALPK1 (800 ng/reaction) and TIFA-His (2 μg/reaction) proteins were incubated with copper or ADP-heptose as indicated in the presence of 5 mM GSH. ATP (100 μM final concentration) was added to initial the reaction. After incubation at 30 °C for 60 min, the reaction was terminated by the addition of assay buffer from the kit. Luminescent signaling was recorded after another 10-min incubation with background correction. The unit of ALPK1 kinase activity was defined as the amounts of ATP (ng) consumed by ALPK1 (1 μg) protein per hour (ng/[μg × h]).
CETSA.
HEK293t cells expressing ALPK1-Flag were treated with or without CuSO4 in the culture medium for 24 h and harvested. Equal volumes of cell suspensions from control and copper-treated groups were aliquoted into PCR strip tubes. The heating procedure was carried out in a PCR Cycler (BioRad) with a gradient-temperature program (30 to 80 °C) for 3 min followed by immediate cooling on ice. After three freeze–thaw cycles using liquid nitrogen, samples were centrifuged at 15,000 × g for 30 min at 4 °C to pellet the denatured protein precipitation. The soluble ALPK1 proteins in the supernatants were analyzed by following Western blot analysis using anti-Flag antibody (Abmart).
Measurement of Cell Viability.
Mammalian cell viability was determined by the MTT Cell Proliferation and Cytotoxicity Assay Kit (Beyotime), and bacterial viability was determined by the formation of CFUs on agar plates.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
This study was financially supported by the Guangdong-Hong Kong-Macao University Joint Laboratory of Interventional Medicine Foundation of Guangdong Province (2023LSYS001) and the Research Grants Council of Hong Kong (2122-7S04 and 17306323). We thank Prof. Feng Shao (National Institute of Biological Sciences, Beijing, China) for alpha-kinase 1 expression vectors and Prof. Wei Xia (Sun Yat-sen University, Guangzhou, China) and Dr. Lennart Pfannkuch (Charité Universitätsmedizin, Berlin, DE) for helpful comments and discussion. We also thank the staff at Molecular Imaging Center for helpful support. We are grateful to Ms. Chung Yan Sherry Chan for critical reading of the manuscript.
Author contributions
J.L., X. Liu, and X.Y. designed research; J.L., X. Liu, Xinghua Li, L.S., and X.X. performed research; B.-l.H., T.F.M., and Xiaofeng Li contributed new reagents/analytic tools; J.L., X. Liu, and X.Y. analyzed data; B.-l.H., T.F.M., and Xiaofeng Li provide consult and helpful discussion; and H.L., H.S., and X.Y. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Xiaofeng Li, Email: lixiaofeng@mail.sysu.edu.cn.
Hongzhe Sun, Email: hsun@hku.hk.
Xinming Yang, Email: yangxm57@mail.sysu.edu.cn.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Kim B. E., Nevitt T., Thiele D. J., Mechanisms for copper acquisition, distribution and regulation. Nat. Chem. Biol. 4, 176–185 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Ge E. J., et al. , Connecting copper and cancer: From transition metal signalling to metalloplasia. Nat. Rev. Cancer 22, 102–113 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brady D. C., et al. , Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature 509, 492–496 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krishnamoorthy L., et al. , Copper regulates cyclic-AMP-dependent lipolysis. Nat. Chem. Biol. 12, 586–592 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiao T., et al. , Copper regulates rest-activity cycles through the locus coeruleus-norepinephrine system. Nat. Chem. Biol. 14, 655–663 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Festa R. A., Thiele D. J., Copper at the front line of the host-pathogen battle. PLoS Pathog. 8, e1002887 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hodgkinson V., Petris M. J., Copper homeostasis at the host-pathogen interface. J. Biol. Chem. 287, 13549–13555 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Subashchandrabose S., Mobley H. L., Back to the metal age: Battle for metals at the host-pathogen interface during urinary tract infection. Metallomics 7, 935–942 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Subashchandrabose S., et al. , Host-specific induction of Escherichia coli fitness genes during human urinary tract infection. Proc. Natl. Acad. Sci. U.S.A. 111, 18327–18332 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Santamarina S., et al. , A lytic polysaccharide monooxygenase-like protein functions in fungal copper import and meningitis. Nat. Chem. Biol. 16, 337–344 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murdoch C. C., Skaar E. P., Nutritional immunity: The battle for nutrient metals at the host–pathogen interface. Nat. Rev. Microbiol. 20, 657–670 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Djoko K. Y., Ong C. L., Walker M. J., McEwan A. G., The role of copper and zinc toxicity in innate immune defense against bacterial pathogens. J. Biol. Chem. 290, 18954–18961 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee S., et al. , Copper capture in a thioether-functionalized porous polymer applied to the detection of Wilson’s disease. J. Am. Chem. Soc. 138, 7603–7609 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bandmann O., Weiss K. H., Kaler S. G., Wilson’s disease and other neurological copper disorders. Lancet Neurol. 14, 103–113 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawai T., Akira S., The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 11, 373–384 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Zimmermann S., et al. , ALPK1- and TIFA-dependent innate immune response triggered by the Helicobacter pylori type IV secretion system. Cell Rep. 20, 2384–2395 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Zhou P., et al. , Alpha-kinase 1 is a cytosolic innate immune receptor for bacterial ADP-heptose. Nature 561, 122–126 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Maubach G., et al. , TIFA has dual functions in Helicobacter pylori-induced classical and alternative NF-κB pathways. EMBO Rep. 22, e52878 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Milivojevic M., et al. , ALPK1 controls TIFA/TRAF6-dependent innate immunity against heptose-1,7-bisphosphate of gram-negative bacteria. PLoS Pathog. 13, e1006224 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.García-Weber D., et al. , ADP-heptose is a newly identified pathogen-associated molecular pattern of Shigella flexneri. EMBO Rep. 19, e46943 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cui J., et al. , The ALPK1 pathway drives the inflammatory response to Campylobacter jejuni in human intestinal epithelial cells. PLoS Pathog. 17, e1009787 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dodani S. C., et al. , Calcium-dependent copper redistributions in neuronal cells revealed by a fluorescent copper sensor and X-ray fluorescence microscopy. Proc. Natl. Acad. Sci. U.S.A. 108, 5980–5985 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ran F. A., et al. , Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vest K. E., et al. , Overlap of copper and iron uptake systems in mitochondria in Saccharomyces cerevisiae. Open Biol. 6, 150223 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dodani S. C., Leary S. C., Cobine P. A., Winge D. R., Chang C. J., A targetable fluorescent sensor reveals that copper-deficient SCO1 and SCO2 patient cells prioritize mitochondrial copper homeostasis. J. Am. Chem. Soc. 133, 8606–8616 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boulet A., et al. , The mammalian phosphate carrier SLC25A3 is a mitochondrial copper transporter required for cytochrome c oxidase biogenesis. J. Biol. Chem. 293, 1887–1896 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Focarelli F., Giachino A., Waldron K. J., Copper microenvironments in the human body define patterns of copper adaptation in pathogenic bacteria. PLoS Pathog. 18, e1010617 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bauer M., et al. , The ALPK1/TIFA/NF-κB axis links a bacterial carcinogen to R-loop-induced replication stress. Nat. Commun. 11, 5117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmidt M., et al. , Crucial role for human Toll-like receptor 4 in the development of contact allergy to nickel. Nat. Immunol. 11, 814–819 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Oblak A., Pohar J., Jerala R., MD-2 determinants of nickel and cobalt-mediated activation of human TLR4. PLoS One 10, e0120583 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jafari R., et al. , The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 9, 2100–2122 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Ren F., et al. , X-ray structures of the high-affinity copper transporter Ctr1. Nat. Commun. 10, 1386 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong Y., Lai Y. T., Chan G. C., Sun H., Glutathione and multidrug resistance protein transporter mediate a self-propelled disposal of bismuth in human cells. Proc. Natl. Acad. Sci. U.S.A. 112, 3211–3216 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bagchi P., Morgan M. T., Bacsa J., Fahrni C. J., Robust affinity standards for Cu(I) biochemistry. J. Am. Chem. Soc. 135, 18549–18559 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang X., Li H., Lai T. P., Sun H., UreE-UreG complex facilitates nickel transfer and preactivates GTPase of UreG in Helicobacter pylori. J. Biol. Chem. 290, 12474–12485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pfannkuch L., et al. , ADP heptose, a novel pathogen-associated molecular pattern identified in Helicobacter pylori. FASEB J. 33, 9087–9099 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Snelling T., Shpiro N., Gourlay R., Lamoliatte F., Cohen P., Co-ordinated control of the ADP-heptose/ALPK1 signalling network by the E3 ligases TRAF6, TRAF2/c-IAP1 and LUBAC. Biochem. J. 479, 2195–2216 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Broz P., Monack D. M., Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol. 13, 551–565 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Cao X., Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat. Rev. Immunol. 16, 35–50 (2016). [DOI] [PubMed] [Google Scholar]
- 40.van der Sar A. M., et al. , Zebrafish embryos as a model host for the real time analysis of Salmonella typhimurium infections. Cell Microbiol. 5, 601–611 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Tak P. P., Firestein G. S., NF-kappaB: A key role in inflammatory diseases. J. Clin. Invest. 107, 7–11 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kroll F., et al. , A simple and effective F0 knockout method for rapid screening of behaviour and other complex phenotypes. eLife 10, e59683 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.White C., Lee J., Kambe T., Fritsche K., Petris M. J., A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J. Biol. Chem. 284, 33949–33956 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsang T., et al. , Copper is an essential regulator of the autophagic kinases ULK1/2 to drive lung adenocarcinoma. Nat. Cell Biol. 22, 412–424 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsvetkov P., et al. , Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 375, 1254–1261 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Banci L., et al. , Affinity gradients drive copper to cellular destinations. Nature 465, 645–648 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Wang J., et al. , Inhibition of human copper trafficking by a small molecule significantly attenuates cancer cell proliferation. Nat. Chem. 7, 968–979 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li J. Y., et al. , Neuroprotective effect of alpha-kinase 1 knockdown against cerebral ischemia through inhibition of the NF-κB pathway and neuroinflammation. Int. Immunopharmacol. 113, 109330 (2022). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.