

Significance
Elucidating the cell death mechanisms involved in bone marrow failure (BMF) syndromes is crucial to develop prevention and treatment strategies. Here, we identify the mechanisms underlying the cell death pathways in hematopoietic stem and progenitor cells (HSPCs) that lead to BMF syndromes. We demonstrate that in mice lacking RIPK1, we detect increased Zbp1 expression and show that expression of a mutant form of Zbp1 significantly delays BMF, suggesting that ZBP1-mediated HSPC death contributes to BMF. Our results identify a key mechanism underlying the pathogenesis of BMF in the mouse that may be operative in human HSPCs.
Keywords: bone marrow failure, hematopoiesis, necroptosis, apoptosis, ZBP1
Abstract
Human bone marrow failure (BMF) syndromes result from the loss of hematopoietic stem and progenitor cells (HSPC), and this loss has been attributed to cell death; however, the cell death triggers, and mechanisms remain unknown. During BMF, tumor necrosis factor-α (TNFα) and interferon-γ (IFNγ) increase. These ligands are known to induce necroptosis, an inflammatory form of cell death mediated by RIPK1, RIPK3, and MLKL. We previously discovered that mice with a hematopoietic RIPK1 deficiency (Ripk1HEM KO) exhibit inflammation, HSPC loss, and BMF, which is partially ameliorated by a RIPK3 deficiency; however, whether RIPK3 exerts its effects through its function in mediating necroptosis or other forms of cell death remains unclear. Here, we demonstrate that similar to a RIPK3 deficiency, an MLKL deficiency significantly extends survival and like Ripk3 deficiency partially restores hematopoiesis in Ripk1HEM KO mice revealing that both necroptosis and apoptosis contribute to BMF in these mice. Using mouse models, we show that the nucleic acid sensor Z-DNA binding protein 1 (ZBP1) is up-regulated in mouse RIPK1-deficient bone marrow cells and that ZBP1’s function in endogenous nucleic acid sensing is necessary for HSPC death and contributes to BMF. We also provide evidence that IFNγ mediates HSPC death in Ripk1HEM KO mice, as ablation of IFNγ but not TNFα receptor signaling significantly extends survival of these mice. Together, these data suggest that RIPK1 maintains hematopoietic homeostasis by preventing ZBP1 activation and induction of HSPC death.
Our previous study demonstrated a role for receptor-interacting protein kinase 1 (RIPK1) during bone marrow failure (BMF) and demonstrated that a deficiency in the related receptor-interacting protein kinase 3 (RIPK3) partially rescued BMF, suggesting that necroptosis may underlie the pathogenesis of BMF syndromes (1). As RIPK1 is a critical regulator of multiple forms of cell death, including pyroptosis, apoptosis, and necroptosis (PAN-optosis), we sought to determine the cell death mechanisms involved. Cell death pathways have unique features: Pyroptosis and necroptosis are considered inflammatory forms of cell death because the process involves cell lysis and the release of cytokines and other cellular contents that drive inflammation, whereas apoptosis has been considered immunologically silent, although plasma membrane rupture may also occur in apoptotic cells if they are not efficiently cleared in vivo (2).
Necroptosis is mediated by RIPK1 and/or RIPK3 and mixed lineage kinase-like pseudokinase (MLKL). RIPK3 phosphorylates MLKL on serine 345, leading to its oligomerization and translocation to the plasma membrane. At the plasma membrane, MLKL leads to the release of cellular contents that trigger innate immune activation. Necroptosis is induced by TNFα, Toll-like receptor (TLR)3/4 ligands, and nucleic acids in the Z conformation (Z-NA) sensed by the antiviral sensor Z-DNA-binding protein 1 (ZBP1). ZBP1 contains multiple RIP Homotypic Interaction Motifs (RHIMs) that interact with RIPK1 and RIPK3. Studies have shown that ZBP1 functions in innate antiviral immunity by recognizing viral RNA/DNA and complexing with RIPK3 and MLKL to mediate mutant murine cytomegalovirus-induced necroptosis and influenza A virus-induced PANoptosis (3–6). In addition to alerting the host to viral infection, ZBP1 has been suggested to recognize endogenous retroelements to induce necroptosis and skin inflammation (7, 8).
Studies in mouse models have uncovered specific roles for RIPK1 in different cell death pathways. Ripk1 knock-out mice undergo multiorgan inflammation and die during the perinatal period due to unrestrained necroptosis and apoptosis (9–11). Studies in conditional Ripk1 mice revealed tissue- and stage-specific roles for RIPK1 as a negative regulator of necroptosis and apoptosis (12). For example, RIPK1 prevents TNFα-mediated apoptosis in the intestinal epithelium, whereas in keratinocytes, it primarily restrains ZBP1-mediated necroptosis (7, 13–15). RIPK1 deletion in keratinocytes or a mutation in the RIPK1 RHIM (Ripk1mRhim) activates ZBP1 to induce necroptosis and skin inflammation (7, 8, 13, 15). ZBP1-mediated necroptosis also contributes to the inflammation and perinatal lethality associated with a complete RIPK1 deficiency or with a RIPK1 expressing a mutant RHIM (8, 13, 15). These studies suggest that in the absence of RIPK1 or a functional RHIM, ZBP1 engages RIPK3 to activate MLKL to induce necroptosis.
Recent studies suggest that ZBP1 may interact with endogenous RNAs (16) and that expression of a mutant form of ZBP1 (N46A, Y50A, N122A, and Y126A) that cannot bind/sense Z-nucleic acids prevents necroptosis and inflammation in mice with keratinocyte-specific RIPK1 deficiency (7, 8). RNA sequencing of RIPK1-deficient keratinocytes implicates dsRNAs from endogenous retroelements as potential ZBP1 ligands (8), suggesting that in keratinocytes, RIPK1 prevents ZBP1-mediated necroptosis triggered by endogenous Z-RNAs. This alternate role for ZBP1 as an endogenous nucleic acid sensor has yet to be demonstrated in other cell types besides keratinocytes.
We showed that mice with a hematopoietic RIPK1 deficiency (designated Vav-iCre Ripk1fl/fl or Ripk1HEM KO) exhibit HSPC loss and inflammation that results in BMF (1). In fetal liver and bone marrow transplant studies, we and others showed that RIPK1 is essential for HSPC survival/function (1, 11, 17). Like human BMF syndromes, disease in Vav-iCre Ripk1fl/fl mice was associated with increases in serum TNFα and IFNγ, and RIPK1-deficient HSPCs were sensitive to TNFα and type I and II IFN-induced cell death in vitro (1). Survival of Vav-iCre Ripk1fl/fl mice was significantly increased and cytokine/chemokine levels were significantly reduced when placed on a RIPK3-deficient background (1); however, HSPC numbers remained reduced, suggesting that a RIPK1 deficiency in mouse HSPCs triggers additional forms of cell death. Here, we sought to determine the cell death pathways behind the RIPK1-mediated loss of HSPCs.
We provide evidence that although the survival of mice with a hematopoietic RIPK1 deficiency is significantly extended by a RIPK3 or MLKL deficiency, HSPC numbers remain suppressed, indicating that other forms of cell death contribute to BMF in this model. Surprisingly, we find that TNF receptor 1 and 2 deficiencies had no effect on survival, whereas an IFNGR1 deficiency significantly extended survival. We demonstrate that the expression of a ZBP1 mutant that cannot sense Z-NA increases mouse HSPC numbers and significantly extends the survival of Vav-iCre Ripk1fl/fl mice. Thus, RIPK1 exerts its anti-inflammatory function(s) in mouse skin and blood systems by preventing ZBP1 activation. However, unlike in skin, where a RIPK1 deficiency results primarily in ZBP1-mediated necroptosis, a hematopoietic RIPK1 deficiency triggers multiple forms of IFNγ and/or ZBP1-mediated cell death.
Results
Mice with a Hematopoietic RIPK1 Deficiency undergo BMF that Is Partially Rescued by a RIPK3 or MLKL Deficiency.
We previously published that conditional deletion of Ripk1 in the hematopoietic system leads to HSPC loss and BMF, revealing that RIPK1 prevents HSPC death by unclear mechanisms (1). Deleting Ripk3 in these hematopoietic RIPK1-deficient mice rescued the inflammation, pancytopenia, anemia, and bone marrow hypocellularity; however, HSPC numbers remained reduced at 5 wk, suggesting that the overall survival may be compromised. To test whether survival is compromised, we examined survival in Vav-iCre Ripk1fl/fl and Vav-iCre Ripk1fl/fl Ripk3−/− (as well as littermate controls), we found that an absence of RIPK3 extended overall survival with the Vav-iCre Ripk1fl/fl Ripk3−/− mice surviving significantly longer than Vav-iCre Ripk1fl/fl mice with a median latency of 127 d (Fig. 1A; P < 0.001). We next sought to determine whether the Ripk3−/− rescue of survival was mediated through its necroptosis function by examining the downstream effector MLKL.
Fig. 1.

A Ripk3 or Mlkl deficiency stimulates hematopoiesis and significantly extends the survival of Vav-iCre Ripk1fl/fl mice. (A) Kaplan–Meier survival curves reveal median latencies of 127 d for Vav-iCre Ripk1fl/fl Ripk3−/− mice (n = 16) and 144 d for Vav-iCre Ripk1fl/fl Mlkl−/− mice (n = 10) compared to the average latency of 35 d for Vav-iCre Ripk1fl/fl mice P < 0.0001 [Vav-iCre Ripk1fl/fl data presented herein have been published previously (1)]. (B) Total weight, (C) bone marrow cellularity, and (D) white and red blood cell counts and hematocrits of Vav-iCre Ripk1fl/fl Mlkl−/− (n = 4) are compared to control Vav-iCre Ripk1fl/+Mlkl−/− mice (n = 2 to 3) at day 35. [Vav-iCre Ripk1fl/fl (n = 11) and Vav-iCre Ripk1fl/+ (n = 14) mice are from ref. 1.] (E) Representative LSK and L86K flow cytometry plots of a WT and Vav-iCre Ripk1fl/fl Mlkl−/− mouse. (F) Absolute numbers of HSPCs in WT (n = 5) and Vav-iCre Ripk1fl/fl Mlkl−/− mice (n = 4) determined by flow cytometry using the LSK or L86K method. (G) Hematopoietic colony number from the bone marrow of day 35 Vav-Cre Ripk1fl/fl Mlkl−/− (n = 3) compared to control Vav-iCre Ripk1fl/+ Mlkl−/− (n = 3). Values compared to colony number from day 35 Vav-iCre Ripk1fl/+ (n = 4) and Vav-iCre Ripk1fl/fl (n = 5) bone marrow from ref. 1. (H) Representative images of BM at day 35 stained with H&E. (I) Graphs showing serum cytokine and chemokine levels for Vav-iCre Ripk1fl/fl (n = 9) and Vav-iCre Ripk1fl/fl Mlkl−/− (n = 3) mice. Error bars represent SEM, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Although the protection provided by RIPK3 deficiency suggested that RIPK1-deficient HSPCs undergo necroptosis (1), RIPK3 also has necroptosis-independent functions in cytokine regulation and inflammasome activation (18, 19). To provide genetic evidence that RIPK1-deficient HSPCs undergo necroptosis, we generated and analyzed Vav-iCre Ripk1fl/fl Mlkl−/− mice. Once phosphorylated by RIPK3, MLKL undergoes a conformational change and translocates to the plasma membrane where it forms pores and releases intracellular contents. Like the Vav-iCre Ripk1fl/fl Ripk3−/− mice, the Vav-iCre Ripk1fl/fl Mlkl−/− mice developed normally and reached adulthood without showing any macroscopic or histologic abnormalities. Vav-iCre Ripk1fl/fl Mlkl−/− mice survived significantly longer than Vav-iCre Ripk1fl/fl mice with a median survival of 144 d (Fig. 1A; P < 0.001); however, no significant difference in overall survival was observed between Vav-iCre Ripk1fl/fl Ripk3−/− and Vav-iCre Ripk1fl/fl Mlkl−/− mice (Fig. 1A), suggesting that blocking necroptosis (as opposed to other RIPK3-dependent forms of cell death) is sufficient to extend survival in these mice. Body weights of Vav-iCre Ripk1fl/fl Mlkl−/− mice were comparable to littermate controls; however, bone marrow cellularity and total white blood cell (WBC) count remained suppressed in 5-wk-old Vav-iCre Ripk1fl/fl Mlkl−/− mice (Fig. 1 B–D). Interestingly, a RIPK3- or MLKL deficiency both significantly increased red blood cell (RBC) numbers in the peripheral blood and improved the hematocrit, suggesting that erythroid necroptosis may underlie the anemia associated with a hematopoietic RIPK1 deficiency (ref. 1 and Fig. 1D).
Because MLKL extended the survival time but did not rescue the loss of HSPCs, we sought to characterize the effects of the MLKL deficiency on specific HSPC lineages. We analyzed the HSPC compartments in the bone marrow of 5-week-old wild-type (WT) and Vav-iCre Ripk1fl/fl Mlkl−/− mice. Flow analysis was performed to determine the percentages and absolute cell numbers of lineage-negative SCA-1 and c-KIT-positive (LIN−SCA-1+c-KIT+, LSK) cells, long-term and short-term hematopoietic stem cells (LT-HSC, ST-HSC), and multipotential progenitors (MPP) in these mice. We observed significant increases in the percentages of LSK HSPCs, LT-HSCs, and ST-HSCs in Vav-iCre Ripk1fl/fl Mlkl−/− mice compared to age-matched WT controls (Fig. 1 E and F). The apparent increase in these populations was inconsistent with reduced in vitro hematopoietic colony formation (Fig. 1G) and the bone marrow histopathology observed in Vav-iCre Ripk1fl/fl Mlkl−/− mice (Fig. 1H). We examined cytokine and chemokine levels in Vav-iCre Ripk1fl/fl Mlkl−/− mice and as we observed with Ripk3 deficiency (1) found cytokine/chemokine levels reduced in the absence of Mlkl (Fig. 1I). These data suggest that necroptosis is responsible for the cytokine/chemokine dysregulation in the Vav-iCre Ripk1 fl/fl mice.
Although cytokines were reduced on Mlkl−/− background, we detected evidence of SCA-1 deregulation in the LSK flow plots of Vav-iCre Ripk1fl/fl Mlkl−/− mice (Fig. 1 E, Upper Right) but not in age-matched WT or littermate controls (Fig. 1 E, Left and data not shown). Thus, to quantify HSPC numbers, we used the CD86 marker known to be constitutively expressed on HSPCs and unaffected by inflammation or stress hematopoiesis (20). The LSK and LIN− CD86+ c-KIT+ (L86K) populations were similar in WT mice and, unlike the LSK analysis, which showed increases in LSK, LT-HSC, and ST-HSC populations but decreases in MPPs, L86K analysis revealed decreases in the L86K population and MPPs, with no significant changes in ST-HSC populations (Fig. 1F). We did observe significant increases in Vav-iCre Ripk1fl/fl Mlkl−/− LT-HSC compared to age-matched WT controls, leading us to speculate that inflammation may be responsible for the apparent increase in LT-HSCs (21). Together, these data indicate that the specific HSPC types that are lost due to the RIPK1 defect are the L86K population and MPPs. Thus, our data suggest that HSPCs and MPPs depend on RIPK1 for survival.
To identify additional changes that would account for the extended survival of the Vav-iCre Ripk1fl/fl Mlkl−/− mice, we examined other cell types in the thymus, spleen, bone marrow, and blood during different stages of development. An absence of MLKL significantly increased thymic and splenic cellularity compared to Vav-iCre Ripk1fl/fl mice (SI Appendix, Fig. S1). Thymocyte numbers were significantly increased at all developmental stages (SI Appendix, Fig. S1B). A lineage analysis of 5-wk-old Vav-iCre Ripk1fl/fl Mlkl−/− mice found lymphoid and myeloid lineage development suppressed in peripheral blood (Fig. 1D) and bone marrow (SI Appendix, Fig. S1C), whereas numbers of splenic B220+ B cells, Mac1+ myeloid cells, and neutrophils were similar to littermate controls (SI Appendix, Fig. S1D). However, CD8 T cells, monocytes, and inflammatory monocytes remained significantly decreased in spleens (SI Appendix, Fig. S1D). Importantly, erythroid lineage development was rescued in the bone marrow and spleen of Vav-iCre Ripk1fl/fl Mlkl−/− mice (SI Appendix, Fig. S1 C and D), potentially explaining the extended survival of Vav-iCre Ripk1fl/fl Mlkl−/− mice (Fig. 1A) and implicating necroptosis in erythropoiesis.
The partial rescue of hematopoiesis observed in Vav-iCre Ripk1fl/fl Ripk3−/− (1) and Mlkl−/− mice (Fig. 1) suggests that when necroptosis is blocked by an absence of RIPK3 or MLKL, a hematopoietic RIPK1 deficiency triggers other forms of cell death. To examine this possibility, we prepared bone marrow lysates from 5-wk-old Vav-iCre Ripk1fl/fl on a WT or Ripk3−/− or Mlkl−/− background and probed with antibodies to phospho-MLKL or cleaved Caspase-3 (CC3) to detect necroptosis or apoptosis, respectively. We detected phospho-MLKL in one of three Vav-iCre Ripk1fl/fl samples examined but were unable to detect total MLKL in the other 2 samples due to low cell numbers (Fig. 2A). We detected CC3 reactivity in murine embryonic fibroblasts (MEF) treated with TNFα (10 ng/mL) and cycloheximide (1 mg/mL) and in three of five Vav-iCre Ripk1fl/fl bone marrow samples examined (Fig. 2B). We also detected CC3 in the four Vav-iCre Ripk1fl/fl Ripk3−/− and five Vav-iCre Ripk1fl/fl Mlkl−/− bone marrow samples examined (Fig. 2 C and D). These data suggest that RIPK1 is essential for HSPC survival by preventing apoptosis and necroptosis, thereby explaining why a RIPK3 or MLKL deficiency was insufficient to completely rescue HSPC numbers and prevent BMF in these mice.
Fig. 2.

Necroptosis and apoptosis are detected in the bone marrow of Vav-iCre Ripk1fl/fl mice, whereas apoptosis is detected in Vav-iCre Ripk1fl/fl Ripk3−/− and Vav-iCre Ripk1fl/fl Mlkl−/− mice. (A) To detect necroptosis, L929 cells were left untreated or treated with TNFa and Z-VAD-fmk. Lysates were prepared from L929 cells and from bone marrow samples from Vav-iCre Ripk1fl/fl or Ripk1fl/fl littermate controls and probed with phospho-Mlkl, total Mlkl, or Erk1/2. (B−D) To detect apoptosis, lysates were prepared from wild-type murine embryonic fibroblasts (MEFs) left untreated or treated with TNFa (10 ng/mL) and CHX (1 µg/mL) and from bone marrow cells from Vav-iCre Ripk1fl/fl (n = 5), Vav-iCre Ripk1fl/fl Ripk3−/− (n = 4), or Vav-iCre Ripk1fl/fl Mlkl−/− mice (n = 5). Lysates were probed for cleaved Caspase-3 (c. caspase 3; p19 and p17) or Erk1/2 by immunoblotting.
IFNγ, not TNFα, Triggers HSPC Death and BMF in Ripk1HEM KO Mice.
To identify the receptors/ligands that trigger apoptosis and necroptosis in RIPK1-deficient HSPCs, we focused on TNFα and IFNγ, which we found increased in the serum of Vav-iCre Ripk1fl/fl mice prior to signs of pancytopenia, anemia, or reductions in HSPC numbers (1). TNFα and IFNγ affect HSPC quiescence, and overproduction of these two cytokines is associated with human BMF syndromes, such as aplastic anemia and myelodysplastic syndrome (21–23).
To examine the contribution of IFNγ in this BMF model, we generated Vav-iCre Ripk1fl/fl Ifngr1−/− mice and littermate controls. We show that ablation of IFNγ signaling rescued the lethal BMF of Vav-iCre Ripk1fl/fl mice, increasing median survival to 133 d (Fig. 3A; P < 0.001). Vav-iCre Ripk1fl/fl Ifngr1−/− mice were of typical weight and exhibited near typical bone marrow cellularity and total WBC counts (Fig. 3 B−D and G). Remarkably, red blood cell numbers and the hematocrit were at normal levels in the 5-wk-old Vav-iCre Ripk1fl/fl Ifngr1−/− mice examined (Fig. 3D). In the absence of IFNGR1, we did not observe SCA-1 deregulation and, therefore, used LSK analysis to determine HSPC numbers. Absolute numbers of LSK, ST-HSC, and MPPs were also significantly increased when compared to Vav-iCre Ripk1fl/fl mice at day 35 (Fig. 3E). A Ripk1 and Ifngr1 deficiency reduced LT-HSC numbers compared to littermate controls; LT-HSC numbers were not significantly different when compared to Vav-iCre Ripk1fl/fl mice (Fig. 3E). An absence of IFNγ signaling also rescued the colony-forming activity of RIPK1-deficient HSPCs (Fig. 3F). Interestingly, an absence of IFNγ signaling appeared to protect the ST-HSC and MPP populations and overall led to a better rescue of hematopoietic colony growth than either a RIPK3- (1) or MLKL deficiency (Figs. 1 F and G and 3 E and F), indicating that apoptosis induced by IFNγ signaling plays a role in RIPK1-deficient HSPC loss.
Fig. 3.
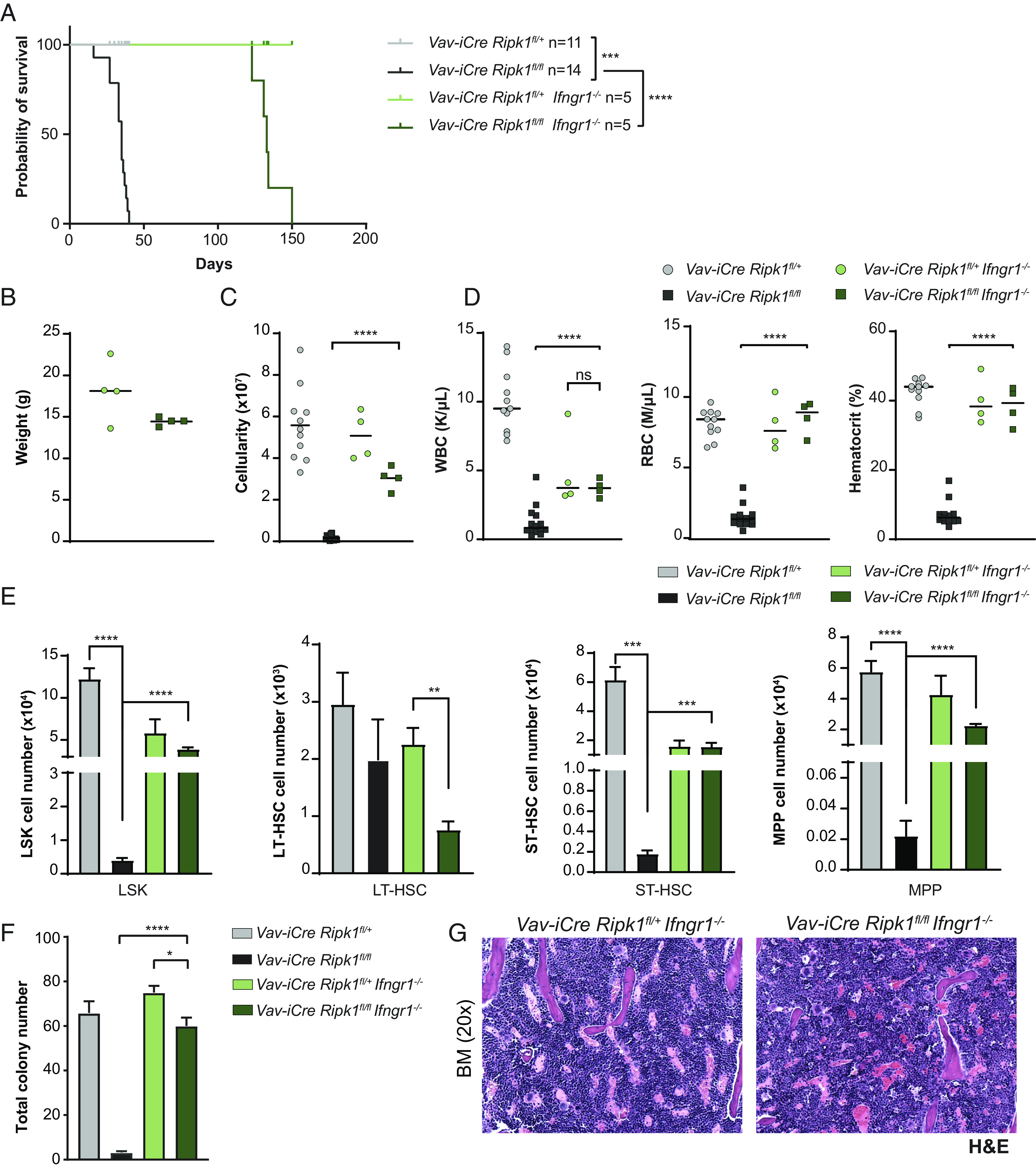
IFNGR1 deficiency rescues hematopoiesis and significantly extends the survival of Vav-iCre Ripk1fl/fl mice. (A) The Kaplan–Meier survival curve reveals an increased median latency of 133 d for Vav-iCre Ripk1fl/fl Ifngr1−/− mice (n = 5) compared to the average latency of 35 d for Vav-iCre Ripk1fl/fl mice (1). (B) Weight, (C) bone marrow cellularity, and (D) blood cell counts and hematocrits of Vav-iCre Ripk1fl/fl Ifngr1−/− mice compared to littermate controls Vav-iCre Ripk1fl/+ Ifngr1−/− mice at day 35 [#Bone marrow cellularity of Vav-iCre Ripk1fl/fl (n = 11) and Vav-iCre Ripk1fl/+ (n = 14) mice is from ref. 1.] (E) Absolute numbers of HSPCs determined by flow cytometry of Vav-iCre Ripk1fl/fl Ifngr1−/− (n = 4) and control Vavi-Cre Ripk1fl/+ Ifngr1−/− (n = 3 to 4) mice are compared to Vav-iCre Ripk1fl/fl, # (n = 3) and control Vav-Cre Ripk1fl/+ # (n = 3) mice at day 35# (#LSK, LT-HSC, ST-HSC, and MPP values are from ref. 1) (F) Hematopoietic colony numbers from the bone marrow of day 35 control Vav-iCre Ripk1fl/+ Ifngr1−/− (n = 4) and Vav-iCre Ripk1fl/fl Ifngr1−/− (n = 4) mice, values compared to the bone marrow from day 35 Vav-iCre Ripk1fl/+ (n = 3) and Vav-iCre Ripk1fl/fl (n = 3) mice (1). (G) Representative images of the day 35 bone marrow stained with H&E from control Vavi-Cre Ripk1fl/+ Ifngr1−/− and Vav-iCre Ripk1fl/fl Ifngr1−/− mice. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
In addition to BM cellularity (Fig. 3C), cellularity in the thymus and spleen was increased in Vav-iCre Ripk1fl/fl Ifngr1−/− mice (SI Appendix, Fig. S2A). Thymocyte cellularity was significantly rescued at each developmental stage (SI Appendix, Fig. S2B). Absolute numbers of B lymphoid and myeloid cells were all increased in the Vav-iCre Ripk1fl/fl Ifngr1−/− bone marrow and, in some cases, were similar to littermate controls (SI Appendix, Fig. S2C). Similarly, in the spleen, B cells, macrophages, monocytes, neutrophils, and inflammatory monocytes were all significantly increased in the Vav-iCre Ripk1fl/fl Ifngr1−/− mice examined at day 35, whereas splenic CD4 and CD8 T cell numbers remained reduced (SI Appendix, Fig. S2D). And as observed in Vav-iCre Ripk1fl/fl Mlkl−/− mice, erythropoiesis was improved in the bone marrow and restored in the spleen of Vav-iCre Ripk1fl/fl Ifngr1−/− mice (SI Appendix, Fig. S2 C and D). Overall, blocking IFNγ signaling appeared to rescue hematopoiesis more effectively than either a RIPK3 or MLKL deficiency (1) and Figs. 1 and 3 and SI Appendix, Figs. S1 and S2), suggesting that IFNγ drives HSPC death in Vav-iCre Ripk1fl/fl mice.
The detection of cleaved Caspase-3 in the Vav-iCre Ripk1fl/fl, Vav-iCre Ripk1fl/fl Ripk3−/−, and Vav-iCre Ripk1fl/fl Mlkl−/− bone marrow (Fig. 2) led us to hypothesize that the hematopoietic defects observed in these mice reflected TNFα-mediated apoptosis. To examine the contribution of TNFα in vivo, we generated Vav-iCre Ripk1fl/fl mice on the Tnfr1−/− and Tnfr1−/−Tnfr2−/− genetic backgrounds, as TNFR2 is expressed on mouse HSPCs (24) and recent evidence suggests that ligation of both TNFR1 and TNFR2 are needed to induce necroptosis (25). Surprisingly, an absence of TNF signaling failed to prevent HSPC death in vivo, and consequently, both Vav-iCre Ripk1fl/fl Tnfr1−/− and Vav-iCre Ripk1fl/fl Tnfr1−/− Tnfr2−/− mice rapidly succumbed to BMF with median latencies of 35 and 29 d, respectively (Fig. 4A). Vav-iCre Ripk1fl/+ Tnfr1−/− and Vav-iCre Ripk1fl/fl Tnfr1−/− mice exhibited no differences in weight (Fig. 4B). However, Vav-iCre Ripk1fl/fl Tnfr1−/− Tnfr2−/− mice were lymphopenic and exhibited hypocellularity in the BM due to significant decreases in the numbers of HSPCs (Fig. 4 C–F). Although an absence of TNF signaling appeared to rescue erythropoiesis in 2 of 3 Vav-iCre Ripk1fl/fl Tnfr1−/− Tnfr2−/− mice examined at 5 wk (Fig. 4D), this had no effect on overall survival (Fig. 4A). We reasoned that BMF in Vav-iCre Ripk1fl/fl mice may reflect apoptosis and necroptosis and generated Vav-iCre Ripk1fl/fl Ripk3−/−Tnfr1−/−Tnfr2−/− mice to test this possibility. Although these mice exhibited normal bone marrow cellularity at day 35, they developed hematological disease and died significantly earlier than Vav-iCre Ripk1fl/fl Ripk3−/− mice (Karna and Kelliher, unpublished data). Hence, blocking TNF and RIPK3 signaling prevents BMF but alters disease pathogenesis. Taken together, these mouse genetic data provide evidence that IFNγ drives HSPC death in this BMF model, and this cell death is only partially dependent on RIPK3 and MLKL.
Fig. 4.
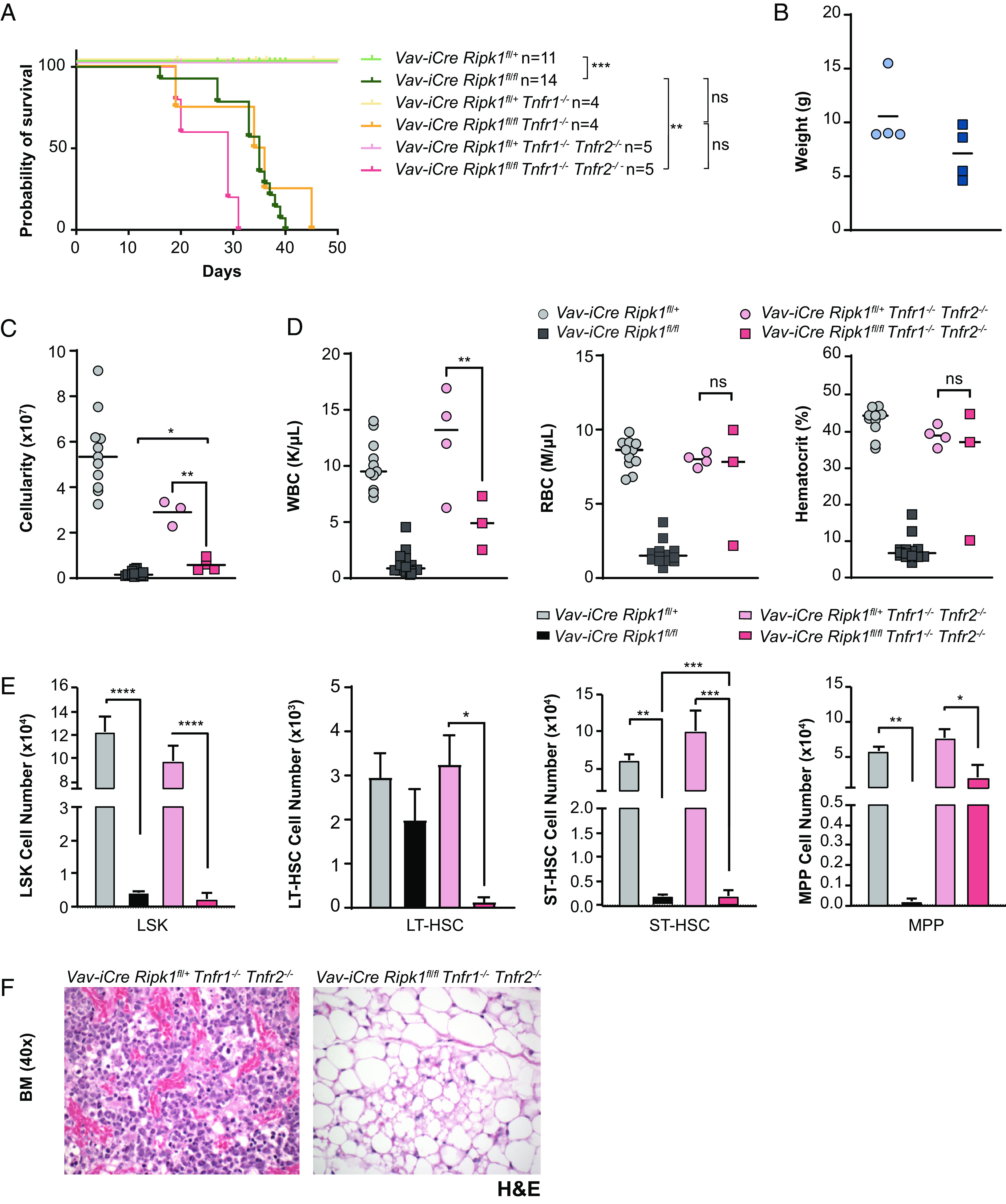
TNFR1/2 deficiencies fail to rescue HSPC death in vivo or prevent bone marrow failure in Vav-iCre Ripk1fl/fl mice. (A) The Kaplan–Meier survival curve reveals a median latency of 35 d for Vav-iCre Ripk1fl/fl Tnfr1−/− and a median latency of 29 d for Vav-iCre Ripk1fl/fl Tnfr1−/− Tnfr2−/− mice compared to the average latency of Vav-iCre Ripk1fl/fl at 35 d (1). (B) Weight, (C) bone marrow cellularity, and (D) blood cell counts and hematocrits of Vav-iCre Ripk1fl/fl Tnfr1−/− Tnfr2−/− mice (n = 3) compared to littermate controls Vav-iCre Ripk1fl/+ Tnfr1−/− Tnfr2−/− (n = 3 to 4) at the time of disease. [#Bone marrow cellularity of Vav-iCre Ripk1fl/fl (n = 11) and Vav-iCre Ripk1fl/+ (n = 14) mice is from ref. 1]. (E) Flow cytometry showing the number of HSPCs in control Vav-iCre Ripk1fl/+ Tnfr1−/− Tnfr2−/− (n = 3) and Vav-iCre Ripk1fl/fl Tnfr1−/− Tnfr2−/− (n = 3) mice at the time of disease. (F) Representative image of BM stained with H&E from Vav-iCre Ripk1fl/+ Tnfr1−/− Tnfr2−/− and Vav-iCre Ripk1fl/fl Tnfr1−/− Tnfr2−/− mice at the time of disease. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
A Role for the Nucleic Acid Sensor ZBP1 in HSPC Death and BMF in RIPK1HEM KO Mice.
During development, RIPK1 functions via its RIP Homotypic Interacting Motif (RHIM) to prevent necroptosis mediated by the nucleic-acid sensor ZBP1. In keratinocytes that lack RIPK1, ZBP1 binds endogenous Z-RNAs and activates RIPK3 and MLKL to induce necroptosis and skin inflammation (7, 8). We found Zbp1 and the IFNγ target Stat1 up-regulated in the absence of Ripk1 (Fig. 5A). Treatment with IFNb significantly increased Zbp1 and Stat1 expression as expected but increases were also observed with IFNγ treatment (Fig. 5A). This result could reflect a recently described feed-forward loop by which ZBP1 activation can promote type I IFN signaling (26).
Fig. 5.

Expression of Zbp1 DNA binding mutant rescues hematopoiesis and extends survival of Vav-iCre Ripk1fl/fl mice. (A) qPCR of Zbp1 and Stat1 in lineage-negative HSPCs isolated from Ripk1HEM KO mutant mice (n = 5) or littermate controls (n = 4) treated with PBS, IFNγ, or IFNβ (20 ng/mL). (B) The Kaplan–Meier survival curve reveals an increased median latency of 106 d for Vav-iCre Ripk1fl/fl Zbp1 Za1a2/Za1a2 mice (n = 7) compared to the average latency of 35 d for Vav-iCre Ripk1fl/fl mice (1) (***P < 0.001). (C) Weight, (D) bone marrow cellularity, and (E) blood cell counts and hematocrits of Vav-iCre Ripk1fl/fl Zbp1Za1a2/Za1a2 mice (n = 4) compared to littermate control Vav-iCre Ripk1fl/+ Zbp1 Za1a2/Za1a2 mice (n = 4) at day 35 [#Bone marrow cellularity of Vav-iCre Ripk1fl/fl (n = 11) and Vav-iCre Ripk1fl/+ (n = 14) mice is from ref. 1.] (F) Absolute numbers of HSPCs determined by flow cytometry of Vav-iCre Ripk1fl/fl Zbp1Za1a2/Za1a2 (n = 4) and littermate control Vav-iCre Ripk1fl/+ Zbp1 Za1a2/Za1a2 (n = 4) mice are compared to Vav-iCre Ripk1fl/fl (n = 3) and control Vav-iCre Ripk1fl/+ (n = 3) mice at day 35 (LSK, LT-HSC, ST-HSC, and MPP values are for Vav-iCre Ripk1fl/fl mice from ref. 1. (G) Hematopoietic colony numbers from the bone marrow of Vav-iCre Ripk1fl/+ Zbp1Za1a2/Za1a2 (n = 4) and Vav-iCre Ripk1fl/fl Zbp1Za1a2/Za1a2 (n = 4) mice, values compared to the bone marrow from day 35 Vav-iCre Ripk1fl/+ (n = 3) and Vav-iCre Ripk1fl/fl (n = 3) mice (1). (H) Representative images of the bone marrow stained with H&E from control Vav-iCre Ripk1fl/+ Zbp1Za1a2/Za1a2 and Vav-iCre Ripk1fl/fl Zbp1Za1a2/Za1a2 mice. (I) Graphs showing serum cytokine and chemokine levels for Vav-iCre Ripk1fl/fl (n = 9) and Vav-iCre Ripk1fl/fl Zbp1Za1a2/Za1a2(n = 4) mice. Error bars represent SEM, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. (J) qPCR of ZBP1 and MLKL gene expression in human CD34+ progenitors isolated from cord blood donors and treated with vehicle or IFNγ (1000 IU) for 18 h (n = 4 independent cord blood donors). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To determine whether ZBP1 sensing contributes to BMF, we generated Vav-iCre Ripk1fl/fl and littermate controls on a ZBP1 DNA-binding domain mutant background where the two Za-domain-coding regions were mutated (Zbp1Za1a2/Za1a2), resulting in the expression of a mutant ZBP1 that is unable to interact with nucleic acids in the Z conformation (Z-NA) (7, 16). Expression of a Z-nucleic acid–binding domain mutant of ZBP1 significantly extended the survival of Vav-iCre Ripk1fl/fl mice from 35 d to 106 d (Fig. 5B; P < 0.001). Compared to littermate controls, body weight was significantly reduced in Vav-iCre Ripk1fl/fl Zbp1Za1a2/Za1a2 mice (Fig. 5C). Although mouse weight decreased, bone marrow cellularity, white blood cell, red blood cell, and hematocrit were all significantly increased compared to Vav-iCre Ripk1fl/fl mice (Fig. 5 D and E). Absolute numbers of LSK, LT-HSC, and ST-HSC in Vav-iCre Ripk1fl/fl Zbp1Za1a2/Za1a2 mice were all significantly increased and approximated numbers observed in littermate controls (Fig. 5F), indicating that, in the absence of RIPK1, ZBP1-mediated HSPC death contributes to BMF. Consistent with these HSPC analyses, in vitro colony-forming assays revealed that expression of a ZBP1 nucleic acid–binding mutant completely rescued the hematopoietic colony defect and stimulated multilineage differentiation in the Vav-iCre Ripk1fl/fl Zbp1Za1a2/Za1a2 bone marrow (Fig. 5 G and H). We also analyzed inflammatory cytokine/chemokine levels in these mice and as observed with an Mlkl deficiency (Fig. 1I) found IFNγ and IL-6 reduced with significant reductions in IP-10 and G-CSF (Fig. 5I).
We analyzed erythroid, myeloid, and lymphoid lineages in the thymus, bone marrow, and spleen of 5-week-old Vav-iCre Ripk1fl/fl Zbp1Za1a2/Za1a2 mice. Thymic and splenic cellularity was significantly increased (SI Appendix, Fig. S3A) with increased numbers of double-negative (DN), CD4 and CD8 double-positive (DP), and CD4 or CD8 single-positive (SP) thymocytes (SI Appendix, Fig. S3B). As observed in Mlkl-deficient mice, we found that B lymphoid and myeloid lineage development remained suppressed in the bone marrow, with significant decreases in B cells, Mac1+ myeloid cells, neutrophils, and inflammatory monocytes (SI Appendix, Fig. S3C). However, significant increases in bone marrow erythroblasts were observed (SI Appendix, Fig. S3C). Like an Mlkl or Ifngr1 deficiency, hematopoiesis in the spleen showed the most significant rescue with mutant ZBP1 expression resulting in normal numbers of B lymphoid, myeloid, and erythroid cells (SI Appendix, Fig. S3D). Mature single-positive CD4 and CD8 T cell numbers remained low (SI Appendix, Fig. S3D).
These data implicate nucleic acid sensing in mouse HSPC death and BMF leading us to query whether ZBP1 may also regulate human hematopoiesis. To test this, we treated human CD34+ cells isolated from cord blood from four different donors with vehicle or IFNγ and observed a trend of increased ZBP1 expression in IFNγ-treated human CD34+ cells, whereas MLKL expression was not affected (Fig. 5J). Although ZBP1 upregulation may not be sufficient to induce cell death, its expression is up-regulated in mammary tumor models (27) and correlates with COVID-19 patient outcome (28), suggesting a link between ZBP1 levels and pathogenesis.
While an Mlkl or Ifngr1 deficiency or expression of a Zbp1 Za mutant led to increases in HSPC numbers and significantly extended the survival of Vav-iCre Ripk1fl/fl mice (Figs. 1A, 3A, and 5B), the partial rescue observed in MPPs and in the myeloid and B lymphoid lineages in the BM suggests that hematopoietic stem and progenitor activity remained compromised. Consistent with these data, Vav-iCre Ripk1fl/fl mice on the Ripk3−/−, Mlkl−/−, Ifngr1−/− or mutant Zbp1Za1a2/Za1a2 backgrounds all succumbed to BMF (SI Appendix, Fig. S4). At killing, these mice exhibited significant decreases in thymic and splenic cellularity and in the number of myeloid, B, and T lymphoid cells in the bone marrow, spleen, and thymus (SI Appendix, Figs. S5–S8). These data reveal that other TLRs or nucleic acid sensors also contribute to hematopoietic failure in these mice.
Discussion
A hematopoietic RIPK1 deficiency resulted in lethal BMF with Vav-iCre Ripk1fl/fl mice exhibiting a median survival of 35 d (1). Combining a RIPK3 or MLKL deficiency with the hematopoietic RIPK1 deficiency significantly extended survival; however, hematopoiesis remained suppressed in these mice. An IFNGR1 deficiency or expression of mutant ZBP1 that cannot sense Z-NA also significantly extended survival in these mice, whereas an absence of TNFR1 and TNFR2 had no significant effects on the survival of Vav-iCre Ripk1fl/fl mice. These genetic data implicate IFNγ and/or ZBP1-mediated cell death in the Ripk1-deficient bone marrow.
It is possible that BMF reflects non-cell-autonomous effects of the RIPK3 or MLKL deficiencies in the bone marrow microenvironment. We think the latter scenario is unlikely as transplant of Vav-iCre Ripk1fl/fl Ripk3−/− bone marrow cells into lethally irradiated WT recipients results in hematopoietic failure, whereas mice transplanted with bone marrow cells from age-matched littermate controls (Vav-iCre Ripk1fl/+ Ripk3−/− or Mlkl−/− or Ripk1fl/fl Ripk3−/− or Mlkl−/−) exhibit normal bone marrow cellularity/histology and successfully reconstitute irradiated WT hosts (1).
An IFNGR1 deficiency or mutant ZBP1 expression restored hematopoiesis and rescued intrinsic survival defects of RIPK1-deficient HSPCs to a greater degree than deficiencies in RIPK3 (1) or MLKL (Figs. 1, 3, and 5). These data suggest that the effects of RIPK1 deficiency are cell-autonomous and that RIPK1-deficient HSPCs undergo additional forms of cell death. We detected cleaved Caspase-3 reactivity in the Vav-iCre Ripk1fl/fl, Vav-iCre Ripk1fl/fl Ripk3−/−, or Mlkl−/− bone marrow (Fig. 2), indicating that apoptosis is also induced. The fact that TNFR1/2 deficiencies had no effect on hematopoiesis or survival (Fig. 4) suggests that the apoptosis is TNFα-independent.
Hematopoiesis is best restored by IFNGR1 deficiency or expression of mutant ZBP1, indicating that in RIPK1-deficient HSPCs, IFNγ and/or ZBP1 induces necroptosis and apoptosis. An IFNGR1 deficiency or mutant ZBP1 expression rescued the Vav-iCre Ripk1fl/fl ST-HSC and MPPs, whereas an MLKL deficiency rescued the ST-HSC but not the MPPs (Figs. 1F, 3E, and 5F). Taken together, these data suggest that RIPK1-deficient ST-HSCs undergo IFNγ/ZBP1-mediated necroptosis, whereas IFNγ/ZBP1-dependent apoptosis depletes the MPPs. ZBP1-mediated activation of Caspase-8 has been observed in RIPK1-deficient murine embryonic fibroblasts (29). In response to influenza A virus (IAV) infection, ZBP1 recruits RIPK3 to activate MLKL, NLRP3, and Caspase-3/-6/-7 to induce necroptosis, pyroptosis, and apoptosis, respectively (4, 30). Precisely how ZBP1 engages Caspases to induce apoptosis in the absence of RIPK1 is unclear, although RIPK3 has been shown to engage Caspase-6 during IAV infection (31).
Hematopoiesis in the spleen was rescued to a greater degree than in the bone marrow with significant increases in splenic myeloid, B lymphoid, and erythroid cells in Mlkl−/−, Ifngr1−/−, or mutant ZBP1 backgrounds (SI Appendix, Figs. S1–S3 C and D). This likely reflects, in part, the splenic extramedullary hematopoiesis induced by BMF. We also observed significant increases in the number of RIPK1-deficient erythroblasts and/or erythrocytes in the absence of MLKL or IFNGR1 or in the presence of mutant ZBP1 (SI Appendix, Figs. S1D, S2D, and S3D). These data suggest that RIPK1-deficient mouse erythroid progenitors may be prone to IFNγ/ZBP1-mediated necroptosis. Interestingly, a deficiency in the related Adenosine deaminase acting on RNA 1 (ADAR1), which also contains a Za domain that recognizes endogenous Z-RNAs, and a C-terminal deaminase domain that converts adenosine (A) to inosine (I), results in embryonic lethality due to failed erythropoiesis (32). The erythroid cell death is thought not to involve apoptosis (33), raising the possibility that unedited RNAs trigger erythroid necroptosis.
These genetic studies demonstrate that mutations in the Za nucleic acid–sensing domains of ZBP1 rescue HSPC death and significantly delay BMF in Vav-iCre Ripk1fl/fl mice, indicating that endogenous Z-NAs have the potential to trigger cell death and inflammation in RIPK1-deficient hematopoietic cells as has been observed in keratinocytes (7, 13, 15). Unlike a keratinocyte RIPK1 deficiency, which results primarily in necroptosis rescued by a RIPK3 or MLKL deficiency (8, 15), a hematopoietic RIPK1 deficiency triggers necroptosis and apoptosis in the bone marrow that is not completely rescued by a RIPK3-, MLKL-, or TNFR1/2 deficiency. These death outcomes may reflect differential expression of FADD, Caspase-8, c-FLIP, or cIAP1/2 levels in mouse keratinocytes vs. HSPC. However, keratinocyte-specific deletion of the Linear Ubiquitin chain Assembly Complex (responsible for preventing cell death) triggers TNFα-mediated apoptosis (34, 35) rather than ZBP1-mediated necroptosis, indicating that other factors determine the predominant cell death mode, not the lineage, per se. We show that IFNγ levels are elevated in the serum of Vav-iCre Ripk1fl/fl mice (1), and we detect increased Zbp1 expression in the RIPK1-deficient bone marrow (Fig. 5A). These findings led us to speculate that IFNγ activates endogenous retroelements that are sensed by ZBP1. Mice with a keratinocyte RIPK1 deficiency are also rescued by mutant ZBP1 expression, and treatment with reverse transcriptase inhibitors partially ameliorates the skin inflammation (8, 13). Some retroelements may have highly conserved sequences that favor Z-RNA formation (36), structures sensed and edited by the ADAR1 deaminase to prevent innate immune activation (37). Although these studies do not identify the nature of the Z-NAs sensed by ZBP1 due to the limited number of viable HSPCs, they suggest that endogenous retroelement-derived dsRNAs may trigger ZBP1 activation.
These mouse genetic data demonstrate that RIPK1 prevents ZBP1 activation to ensure hematopoietic cell survival, but whether human RIPK1 functions similarly is unclear. Biallelic loss-of-function variants in RIPK1 have been detected in unrelated families, and these individuals exhibit lymphopenia and immune deficiency that in one patient was corrected with a hematopoietic stem cell transplant (38). These data suggest that RIPK1 is also required for human HSPC survival/differentiation and may have implications for hematopoietic stem cell therapy, where virus reactivation may trigger ZBP1-mediated cell death and contribute to hematopoietic failure. Additional data are also emerging to link necroptosis to human BMF syndromes, such as myelodysplastic syndrome (MDS). MLKL is up-regulated in MDS patients, and MLKL levels correlate with response to hypomethylating agent therapy (39, 40). MDS is also associated with mutations in splicing factors (U2AF1, SF3B1, and SRSF2), formation of R-loops, and an IFN signature (41, 42), raising the possibility that aberrant nucleic acid sensing triggers human HSPC death in MDS.
Materials and Methods
Mice.
Ripk1 conditional mice (Ripk1fl/fl) described previously (14) or Ripk1fl/fl Ripk3−/− mice were crossed with Vav-iCre mice (Jackson Laboratory 035670), Mlkl−/− mice (43) (gift of Warren Alexander), Zbp1Za1a2/Za1a2 mice (16) (gift of Jan Rehwinkel), Infgr1−/− (Jackson Laboratory 003288), Tnfr1−/− (Jackson Laboratory 003242) and Tnfr1−/−Tnfr2−/− mice (Jackson Laboratory 003243) were purchased from Jackson Labs. All mouse experiments and procedures were approved by the UMass Chan Medical School’s Institutional Care and Animal Use Committee.
Histology.
Tissues were fixed in 10% formalin (Fisher Scientific), and the bone marrow was decalcified in Cal-Rite (Richard Allen Scientific) for 48 h. Samples were stained with H&E. Images were taken at 20× magnification on an Olympus BX41 microscope using an Evolution MP 5.0 Mega-Pixel Camera (MediaCybernetics) and QCapture Pro software (QImaging).
Blood Analysis and Flow Cytometry.
Complete blood counts and hematocrits were performed on a Hemavet 950FS analyzer (Drew Scientific). Single-cell suspensions of peripheral blood, bone marrow, spleen, and thymus were stained with cell surface antibodies for myeloid (Gr-1, Ly6-c, and CD11b) and lymphoid (CD4, CD8, and B220) markers. For LSK or L86K analyses, bone marrow cells were stained with a biotin lineage mixture, SCA-1, CD86, c-KIT, CD34, FLK2, and CD150. All samples were run on a BD LSRII or a Cytek Aurora flow cytometer (BD Bioscience) and analyzed using FlowJo software (Tree Star). A complete list of antibodies, including clone numbers, is given in SI Appendix, Table S1.
Colony-Forming Assay.
Bone marrow cells were seeded in MethoCult medium M3434 (STEMCELL Technologies), and the total colony number was determined following the manufacturer’s protocol. Ligands were added at the time of plating at the following concentrations: mTNF-α, 10 ng/mL (R&D), and mIFN-γ, 10 ng/mL (Pepro Tech). zVAD-FMK (Enzo) was added at a concentration of 20 μM.
Mouse HSPC and Human CD34 Isolation and Treatment.
Lineage-negative HSPCs were isolated from the mouse bone marrow using the Stem Cell Lineage-Depletion Kit. CD34 cells were isolated from cord blood obtained from the UMass Memorial Cord Blood Bank. Cells were selected for using the human CD34 Microbead Kit (Miltenyi Biotec). Cells were expanded in Stemspan SFEM II (STEMCELL Technologies) supplemented with StemSpan CD34+ Expansion Supplement (10×) (STEMCELL Technologies) for 1 to 2 d. Cells were treated with 1,000 U of human interferon-gamma (STEMCELL Technologies) for 18 h and then harvested in TRIzol for gene expression analysis.
Gene Expression Analysis.
RNA was prepared using TRIzol (Invitrogen). cDNA was prepared using the SuperScript First-Strand Synthesis System (Invitrogen). Quantitative real-time PCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems) and run on the AB7300 Detection System (Applied Biosystems). Relative gene expression was normalized to β-actin and determined using the ΔΔCT method. The primers used to amplify genes are listed in SI Appendix, Table S2.
Western Blotting.
Bone marrow cells were lysed in RIPA buffer (Thermo Scientific™, 89900) supplemented with 1× halt protease and phosphatase inhibitor cocktail (Thermo Scientific™, 78440) for 30 min on ice, followed by mixing every 10 min. Cell lysates were centrifuged at 14,000 rpm for 20 min at 4 °C. Protein concentration was determined by Bradford protein assay, and 20 μg of total protein was separated by electrophoresis through 4 to 20% gradient polyacrylamide gels (Bio-Rad, #4568094) and transferred to PVDF membranes (Millipore, 78440). The membrane was blocked with 5% nonfat milk for an hour at 4 °C and incubated overnight at 4 °C with antibodies to cleaved Caspase-3 (CST, #9664, 1:1,000), phospho-MLKL (Abcam #196436), total MLKL (Abcam #184718), or ERK1/2 (CST, #9102, 1:1,000). The membrane was washed with Tris-buffered saline containing 0.01% Tween 20 (TBST, pH 7.2) three times and then incubated with a secondary anti-rabbit antibody (Amersham, NA934V 1:5,000) at room temperature for 1 h. Blot images were developed using Femto chemiluminescent substrates (Thermo Fisher, 34095) and acquired with a ChemiDocTM XRS system (Bio-Rad).
Statistics.
Statistical analyses were performed using GraphPad Prism software, version 6.0. Kaplan–Meier survival curves were analyzed using a log-rank test with a 95% CI. A two-sided P < 0.05 was considered statistically significant for Student’s t tests and nonparametric Mann–Whitney U tests. For comparisons of 3 or more groups, a one-way ANOVA test was performed.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We would like to thank Kelliher lab members for critical reading of the manuscript. We thank the UMass Chan Flow Cytometry Core for flow cytometry analysis. This work was supported by the NIH/National Institute of Allergy and Infectious Diseases Grant AI075118 and an Edward P. Evans MDS Discovery Research Grant to M.A.K. S.S. was supported by a Damon Runyon Physician Scientist Award.
Author contributions
J.E.R-R., S.-E.L., L.H.C., and M.A.K. designed research; J.E.R-R., S.-E.L., S.S., M.H.A., R.S., K.K., and J.O. performed research; J.S., J.L., J.M., and J.R. contributed new reagents/analytic tools; J.E.R-R., S.-E.L., and M.A.K. analyzed data; J.S. and J.L. figure preparation; J.M. and J.R. provided advice; M.A.K. obtained funding for the research; and M.A.K. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix. Previously published data were used for this work (1).
Supporting Information
References
- 1.Roderick J. E., et al. , Hematopoietic RIPK1 deficiency results in bone marrow failure caused by apoptosis and RIPK3-mediated necroptosis. Proc. Natl. Acad. Sci. U.S.A. 111, 14436–14441 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kayagaki N., et al. , NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 591, 131–136 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Upton J. W., Kaiser W. J., Mocarski E. S., DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang T., et al. , Influenza virus Z-RNAs induce ZBP1-mediated necroptosis. Cell 180, 1115–1129.e3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thapa R. J., et al. , DAI senses Influenza A virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe 20, 674–681 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuriakose T., et al. , ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 1, aag2045 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Devos M., et al. , Sensing of endogenous nucleic acids by ZBP1 induces keratinocyte necroptosis and skin inflammation. J. Exp. Med. 217, e20191913 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiao H., et al. , Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature 580, 391–395 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelliher M. A., et al. , The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 8, 297–303 (1998). [DOI] [PubMed] [Google Scholar]
- 10.Dillon C. P., et al. , RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157, 1189–1202 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rickard J. A., et al. , RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 157, 1175–1188 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Pasparakis M., Vandenabeele P., Necroptosis and its role in inflammation. Nature 517, 311–320 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Lin J., et al. , RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 540, 124–128 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dannappel M., et al. , RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 513, 90–94 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newton K., et al. , RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 540, 129–133 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Maelfait J., et al. , Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 36, 2529–2543 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cusson N., Oikemus S., Kilpatrick E. D., Cunningham L., Kelliher M., The death domain kinase RIP protects thymocytes from tumor necrosis factor receptor type 2-induced cell death. J. Exp. Med. 196, 15–26 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawlor K. E., et al. , RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 6, 6282 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Najjar M., et al. , RIPK1 and RIPK3 kinases promote cell-death-independent inflammation by Toll-like Receptor 4. Immunity 45, 46–59 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanayama M., et al. , CD86-based analysis enables observation of bona fide hematopoietic responses. Blood 136, 1144–1154 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Baldridge M. T., King K. Y., Goodell M. A., Inflammatory signals regulate hematopoietic stem cells. Trends Immunol. 32, 57–65 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baldridge M. T., King K. Y., Boles N. C., Weksberg D. C., Goodell M. A., Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 465, 793–797 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitagawa M., et al. , Overexpression of tumor necrosis factor (TNF)-alpha and interferon (IFN)-gamma by bone marrow cells from patients with myelodysplastic syndromes. Leukemia 11, 2049–2054 (1997). [DOI] [PubMed] [Google Scholar]
- 24.Wajant H., Siegmund D., TNFR1 and TNFR2 in the control of the life and death balance of macrophages. Front. Cell Dev. Biol. 7, 91 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Legarda D., et al. , CYLD proteolysis protects macrophages from TNF-mediated auto-necroptosis induced by LPS and licensed by type I IFN. Cell Rep. 15, 2449–2461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez D. A., et al. , Caspase-8 and FADD prevent spontaneous ZBP1 expression and necroptosis. Proc. Natl. Acad. Sci. U.S.A. 119, e2207240119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baik J. Y., et al. , ZBP1 not RIPK1 mediates tumor necroptosis in breast cancer. Nat. Commun. 12, 2666 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karki R., et al. , ZBP1-dependent inflammatory cell death, PANoptosis, and cytokine storm disrupt IFN therapeutic efficacy during coronavirus infection. Sci. Immunol. 7, eabo6294 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ingram J. P., et al. , ZBP1/DAI drives RIPK3-mediated cell death induced by IFNs in the absence of RIPK1. J. Immunol. 203, 1348–1355 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kesavardhana S., et al. , The Zalpha2 domain of ZBP1 is a molecular switch regulating influenza-induced PANoptosis and perinatal lethality during development. J. Biol. Chem. 295, 8325–8330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng M., Kanneganti T. D., Newly identified function of Caspase-6 in ZBP1-mediated innate immune responses, NLRP3 inflammasome activation, PANoptosis, and host defense. J. Cell Immunol. 2, 341–347 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liddicoat B. J., et al. , Adenosine-to-inosine RNA editing by ADAR1 is essential for normal murine erythropoiesis. Exp. Hematol. 44, 947–963 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walkley C. R., Kile B. T., Cell death following the loss of ADAR1 mediated A-to-I RNA editing is not effected by the intrinsic apoptosis pathway. Cell Death Dis. 10, 913 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumari S., et al. , Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. Elife 3, e03422 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taraborrelli L., et al. , LUBAC prevents lethal dermatitis by inhibiting cell death induced by TNF, TRAIL and CD95L. Nat. Commun. 9, 3910 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herbert A., To “Z” or not to “Z”: Z-RNA, self-recognition, and the MDA5 helicase. PLoS Genet. 17, e1009513 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liddicoat B. J., et al. , RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349, 1115–1120 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cuchet-Lourenco D., et al. , Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science 361, 810–813 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wagner P. N., et al. , Increased Ripk1-mediated bone marrow necroptosis leads to myelodysplasia and bone marrow failure in mice. Blood 133, 107–120 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Montalban-Bravo G., et al. , Transcriptomic analysis implicates necroptosis in disease progression and prognosis in myelodysplastic syndromes. Leukemia 34, 872–881 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sperling A. S., Gibson C. J., Ebert B. L., The genetics of myelodysplastic syndrome: From clonal haematopoiesis to secondary leukaemia. Nat. Rev. Cancer 17, 5–19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ganan-Gomez I., et al. , Deregulation of innate immune and inflammatory signaling in myelodysplastic syndromes. Leukemia 29, 1458–1469 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murphy J. M., et al. , The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix. Previously published data were used for this work (1).