

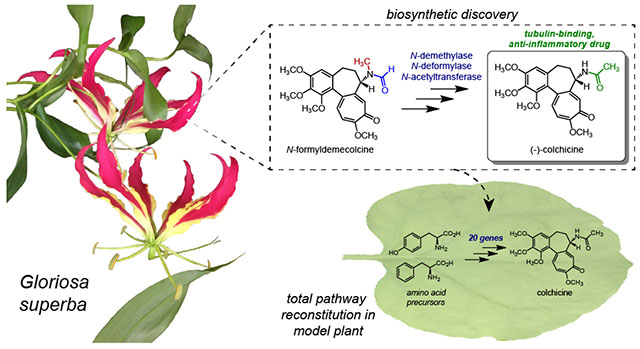
Abstract
Colchicine (1) is a bioactive plant alkaloid from Colchicum and Gloriosa species that is used as a pharmaceutical treatment for inflammatory diseases, including gouty arthritis and familial Mediterranean fever. The activity of this alkaloid is attributed to its ability to bind tubulin dimers and inhibit microtubule assembly, which not only promotes anti-inflammatory effects, but also makes colchicine a potent mitotic poison. The biochemical origins of colchicine biosynthesis have been investigated for over fifty years, but only recently has the underlying enzymatic machinery become clear. Here, we report the discovery of multiple pathway enzymes from Gloriosa superba that allow for the reconstitution of a complete metabolic route to 1. This includes three enzymes that process a previously established tropolone-containing intermediate into 1 via tailoring of the nitrogen atom. We further demonstrate the total biosynthesis of enantiopure (−)-1 from primary metabolites via heterologous production in a model plant, thus enabling future efforts for the metabolic engineering of this medicinal alkaloid. Additionally, our results provide insight into the timing and tissue specificity for the late stage modifications required in colchicine biosynthesis, which are likely connected to the biological functions for this class of medicinal alkaloids in native producing plants.
Graphical Abstract
INTRODUCTION
One of the oldest known treatments for inflammation is colchicine (1), a plant alkaloid that is currently used as a pharmaceutical for the treatment of gouty arthritis and familial Mediterranean fever.1 Although FDA-approval for colchicine was only achieved in 2009,1 plant extracts from Colchicum and Gloriosa species have served as treatments for inflammation for hundreds, if not thousands, of years.2 Colchicine is best known for its ability to bind to tubulin dimers, thereby leading to microtubule destabilization.3,4 Within the context of inflammation, it is thought that this microtubule destabilization leads to a disruption in neutrophil cell mobility and functionality,5 as well an inhibition in the formation and function of the NLRP3 inflammasome complex.6 However, mechanisms of colchicine bioactivity within the context of inflammation seem to be multifaceted, and are not yet fully understood.5 Nevertheless, through its ability to promote microtubule destabilization, colchicine is effectively able to inhibit chromosomal segregation, which has made colchicine an effective biochemical tool for studying the dynamics of microtubules, chromosomes, and cell division,7 as well as to promote the formation of polyploidy in plants, which is widely utilized within agriculture to generate beneficial crop traits.8
Since the 1960’s, the chemical origins of 1 have been thoroughly studied through structural characterization of isolated alkaloids, as well as through meticulous radio-isotope labeling studies that helped to define a well-established biosynthetic hypothesis.9 This foundational work facilitated our previous study of the colchicine biosynthetic pathway, in which we were able to determine a set of metabolic enzymes that convert the amino acid precursors L-tyrosine (L-Tyr) and L-phenylalanine (L-Phe) first into a phenethylisoquinoline scaffold (2), and ultimately to N-formyldemecolcine (3), a precursor to 1 that has the same tropolone ring structure (Figure 1a).10 The bioactivity of 1 is largely attributed to this unique tropolone ring system, as multiple precursors to colchicine with this scaffold exhibit tubulin-binding activity and cytotoxicity,11 while precursors lacking the tropolone moiety (e.g. O-methylandrocymbine) seem to have limited cytotoxic effects.12,13 Though tropolone formation is critical to the pharmaceutical properties of these alkaloids, specific modification of the lone nitrogen atom is also important, as 1, which possesses an N-acetyl group, has been shown to have the highest tubulin affinity and corresponding bioactivity of the colchicine-related alkaloids.11 Additionally, 1 is the most abundant alkaloid present in the native Colchicum and Gloriosa species,14 which implies its importance within the chemical ecology of these plants. Therefore, processing of this nitrogen atom to form the N-acetyl moiety, and thus 1, is paramount not only for establishing the full biosynthesis of this natural product, but also for better understanding the role that these late stage nitrogen-tailoring reactions have on the biological effects of this group of alkaloids. In this work, we leverage biochemical assays in native plant extracts and transcriptomic analysis to identify and characterize four enzymes within colchicine biosynthesis, including three late stage tailoring enzymes that complete a biosynthetic route to the pharmaceutical alkaloid (−)-1, as well as an oxidase that is critical for producing an early colchicine precursor.
Figure 1. Proposed biosynthetic pathway for colchicine.
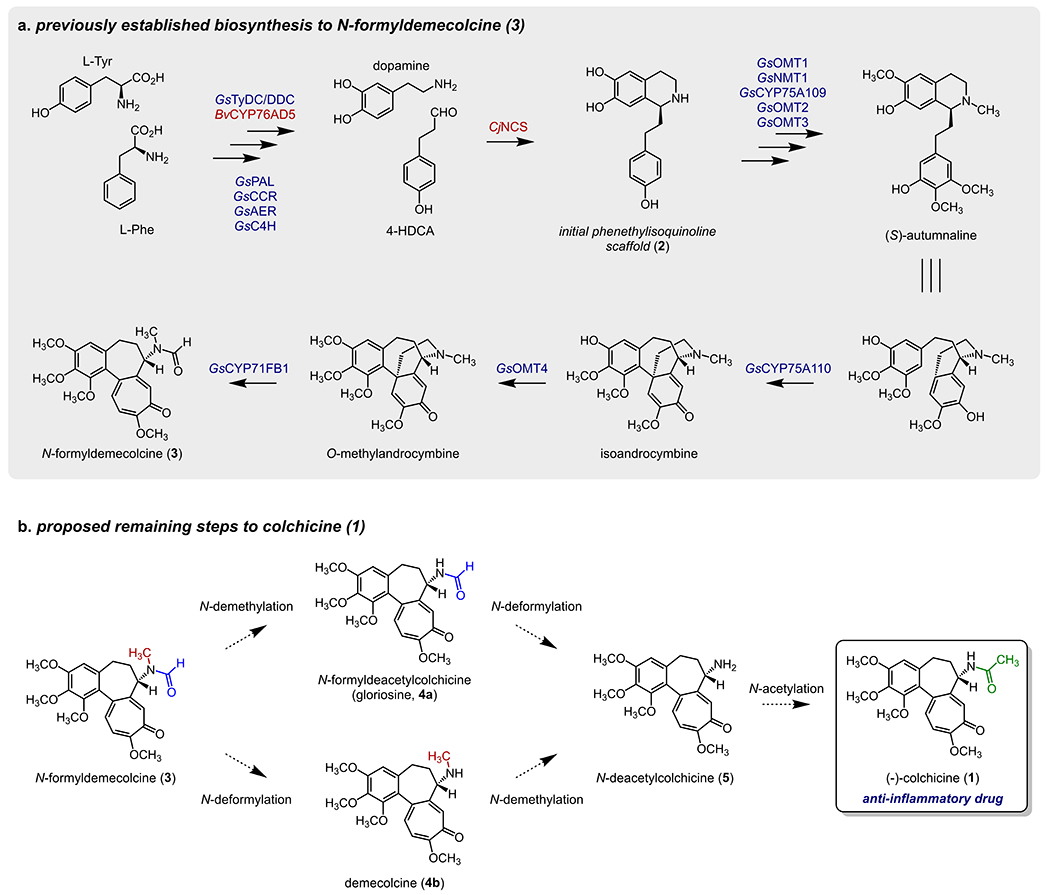
a) Previously established biosynthetic pathway for the engineered production of 3.10 Native Gloriosa superba genes are shown in blue, while those from other plant sources are in red. b) Proposed remaining biosynthetic transformations to produce 1 from 3.
RESULTS
We had previously established a biosynthetic route (Figure 1a) using enzymes from G. superba that convert a phenethylisoquinoline precursor (2) into N-formyldemecolcine (3)10. 3 possesses the same tropolone ring-containing scaffold as colchicine (1), and has been confirmed as a metabolic precursor to 1 through isotope labeling studies.15 The eight biosynthetic enzymes necessary for this process were identified via correlation analysis of transcript expression across G. superba tissue types. Notably, while this method revealed a set of highly co-expressed genes for colchicine biosynthesis, we were not able to initially identify the final enzymes necessary to convert 3 into 1 within this group.
The proposed steps for the final processing of 3 presumably require N-demethylation, N-deformylation, and N-acetylation (Figure 1b). In theory, this could occur either via initial N-demethylation to form N-formyldeacetylcolchicine (a.k.a gloriosine, 4a), or through initial N-deformylation to produce demecolcine (4b). It has previously been determined in Colchicum autumnale plants that radio-labeled 3 is incorporated into 4b, which lacks the N-formyl group, and that 4b is subsequently incorporated into 1.15,16 This suggested that N-deformylation precedes N-demethylation, at least within C. autumnale. However, our prior work had focused almost exclusively on 1 biosynthesis in G. superba, and it was not known whether this same order of events was predominant in this species. Therefore, we first opted to investigate these downstream biosynthetic routes within the native G. superba plant.
The rhizome of G. superba, an underground tuber-like structure, has been determined to be enriched for 1, as well as the requisite biosynthetic enzymes.10 We reasoned that controlled biochemical reactions with rhizome tissue lysates could provide insight into the order of biosynthetic events, as well as the class of enzymes responsible for a given chemical transformation. Oxidative demethylations of heteroatoms are known to be catalyzed by cytochrome P450 (CYP) enzymes, which in eukaryotes are usually tethered to the endoplasmic reticulum (ER) through a transmembrane domain,17 and rely upon a redox partner enzyme (cytochrome P450 reductase) that is reduced via NADPH. Previous work has shown that the three final chemical transformations in colchicine biosynthesis (N-deformylation, N-demethylation, and N-acetylation) could be reconstituted in ER-derived microsomal protein fractions purified from C. autumnale seeds when NADPH and acetyl-CoA are supplied as cofactors.18 This suggested the likely involvement of a CYP for the requisite N-demethylation reaction, which we further wanted to probe in G. superba. To investigate endogenous biosynthetic reactions in G. superba, we prepared cell-free lysates and microsomal protein fractions from leaf and rhizome tissue, and incubated these fractions with potential N-demethylation substrates in the presence or absence of cofactors. Incubating 3 in these fractions without cofactors did not lead to an obvious increase in any downstream products. However, incubating 3 and NADPH in the rhizome microsomal fraction led to an increase in both 4a and N-deacetylcolchicine (5), but no increase in 4b (Figure S1). This not only suggested that biosynthesis proceeds through initial N-demethylation of 3, but also that this reaction is likely CYP-catalyzed. The observed increase in 5 further suggested that the subsequent N-deformylation could either be NADPH-dependent or does not require another supplemented cofactor. Accordingly, incubation of 4a in the microsomal fraction led to a modest increase in 5 that was independent of NADPH (Figure S1). We were not able to observe the production of 5 when 4b was incubated in this microsomal fraction, regardless of the inclusion of NADPH. This further supports N-demethylation preceding N-deformylation in G. superba, and demonstrates that the order of these chemical transformations in G. superba is likely crucial. We also observed acetyl-CoA-dependent consumption of 5 and a corresponding increase in 1 within the cell-free lysates, consistent with the activity of a soluble, acetyl-CoA transferase enzyme (Figure S1). Collectively, these results suggested that the major order of events in G. superba is CYP-catalyzed N-demethylation of 3, thereby producing 4a, followed by NADPH-independent N-deformylation of 4a to yield 5, and finally, N-acetylation to produce 1 (Figure 1b).
Guided by this information, we focused on identifying a CYP enzyme capable of catalyzing the N-demethylation of 3. Because our initial RNA-seq correlation analysis did not identify any putative N-demethylase enzymes, we shifted our strategy for locating the final biosynthetic genes. In particular, we had previously observed that the tropolone-forming enzyme, GsCYP71FB1, does not correlate as strongly with the rest of the pathway genes,10 which hinted at potentially different regulation of downstream reactions relative to the upstream acting genes of the pathway. As such, we utilized GsCYP71FB1 as a query gene for correlation analysis within our transcriptome expression data to find other highly co-expressing CYPs. This identified 11 CYPs with a Pearson correlation coefficient greater than 0.75 (Figure S2), including the previously characterized pathway genes GsCYP75A109 and GsCYP75A110, as well as several previously untested CYPs. Each of the newly identified CYPs were cloned from G. superba cDNA and tested for activity via Agrobacterium-mediated transient expression in N. benthamiana with substrate co-infiltration. Transiently expressing one of these gene candidates (GsCYP71DA12) in N. benthamiana with co-infiltration of 3 as substrate led to consumption of 3 and concurrent production of 4a, the N-demethylated product (Figure S3). We also observed the accumulation of a compound with a mass increase of 16 Da (m/z 416.1704), consistent with the addition of a hydroxyl group (Figure S3). Based upon MS/MS analysis, we predict this molecule to be the hemiaminal intermediate (3’) that precedes carbon loss (Figure S3). This is consistent with archetypical CYP-catalyzed N-demethylations, which are proposed to precede via initial hydroxylation of the N-methyl to form a hemiaminal intermediate that can non-enzymatically decompose via loss of formaldehyde.19 Activity of GsCYP71DA12 was confirmed through in vitro assays with microsomal protein purified from GsCYP71DA12-expressing yeast cells, which exhibited NADPH-dependent consumption of 3, and a corresponding production of the hemiaminal intermediate (3’) (Figure S4). This intermediate was only modestly stable, as incubating quenched reactions containing 3’ at room temperature for several hours led to loss of 3’ and an accumulation of 4a. Thus, we propose a canonical N-demethylation in which GsCYP71DA12 catalyzes a hydroxylation of the methyl group of 3 to generate the hemiaminal (3’), which can non-enzymatically decompose to release formaldehyde, resulting in the formation of 4a (Figure S4). In agreement with our in vitro reactions with G. superba protein, no N-demethylation activity from GsCYP71DA12 was observed with 4b as substrate.
The next logical step in the production of colchicine would be N-deformylation to form 5. Multiple enzyme families have been implicated to catalyze N-deformylation reactions, including those with redox-neutral mechanisms (e.g. hydrolases), as well as those with oxidative mechanisms (e.g. peptide deformylases or aldehyde dehydrogenases). Our biochemical assays with G. superba cell-free lysates indicated that the N-deformylation of 4a occurs equivalently regardless of NADPH (Figure S1), suggesting that this enzyme is likely a cofactor-independent hydrolase. We first considered the alpha/beta hydrolase (ABH) protein family, as a representative member of this family has been found to catalyze the N-deformylation of N-formyl-kynurenine within mammalian tyrosine catabolism.20 We selected five ABH genes to test based upon their co-expression with known biosynthetic genes (Figure S5; Pearson’s r > 0.8 when compared to GsTyDC/DDC, the first gene in the pathway10). We first tested these genes by generating the proposed substrate, 4a, in situ by transiently co-expressing GsCYP71DA12 with each candidate gene and co-infiltrating 3 as substrate. The transient co-expression of only one of the tested ABH genes (GsABH1) resulted in a reduction in 4a and a concomitant accumulation of 5 (Figure S6). While N-deformylase activity of GsABH1 was also detected when 4a was co-infiltrated directly as a substrate, the accumulation of 5 was much lower relative to assays where substrate was generated in situ via co-expression of GsCYP71DA12 with infiltration of 3 as substrate (Figure S6). Deformylation of 3 by GsABH1 to produce 4b could also be detected in transient expression experiments, but this activity was low as well (Figure S6).
In a parallel set of experiments, we were able to purify a small amount of soluble GsABH1 from heterologous production in E. coli, but we did not observe activity on 4a or 3 with this purified protein (Figure S7). Similarly, 4a and 3 were not deformylated in cell-free lysates prepared from N. benthamiana leaves transiently expressing GsABH1. One possible explanation for the apparent low activity observed when GsABH1 was tested on 4a directly is that the co-occurrence of GsCYP71DA12 may be crucial for activity (e.g. via protein-protein interaction). However, conversion of co-infiltrated 4a was not improved when GsCYP71DA12 was included in the transient expression system (Figure S7). Collectively, these results suggested that 4a may not be the preferred substrate for GsABH1. Because the direct product of GsCYP71DA12 activity on 3 appears to be the hemiaminal (3’), we hypothesized that this compound may instead be the substrate for N-deformylation. The potential of 3’ as the direct substrate for this transformation is supported by two criteria: a) the intramolecular hydrogen bond between the formamide oxygen and the hemiaminal proton of 3’ would make the formamide carbon a better electrophile, and thus more susceptible to hydrolysis, and b) the subsequent deformylated hemiaminal (3’’) is a stronger base than 3’ and could therefore more readily promote decomposition into 5 (Figure S7).21 To test this hypothesis, we prepared in vitro reactions with both GsCYP71DA12-enriched microsomes and the previously purified GsABH1 enzyme. When 3 and NADPH were included in the reaction, we observed not only the accumulation of 3’ and 4a, but also GsABH1-dependent accumulation of 5 (Figure S7). Conversely, incubation of 4a and NADPH in this two-enzyme system did not lead to an accumulation of 5. Although it is difficult to show direct activity of GsABH1 on 3’ (as this intermediate is unstable and thus difficult to isolate for use as a direct substrate), the clear production of this molecule by GsCYP71DA12 and the lack of GsABH1 activity on 4a in vitro would suggest that 3’ is likely the major substrate for GsABH1. Though activity of GsABH1 on 4a was not observed in vitro, we speculate that this may be due to a suboptimal reaction environment for the purified protein, as low levels of 4a turnover to 5 were evident in both G. superba rhizomes and N. benthamiana transient expression experiments (Figures S1 & S6). Taken together, our data suggests that GsABH1 may preferably act on the transient hemiaminal (3’) that is primed/activated for N-deformylation through oxidation of 3 by GsCYP71DA12.
Having uncovered the N-demethylase and N-deformylase enzymes to produce 5, the only remaining step for the generation of 1 was acetylation of the generated primary amine. Enzymatically, this reaction could feasibly be catalyzed by either a BAHD acyltransferase or an N-acetyltransferase (NAT) superfamily protein, as each of these families have representative enzymes that catalyze N-acylations.22,23 Within plant secondary metabolism, BAHD acyltransferases seem to be the more prevalent of these two protein families, so we initially considered 10 BAHD acyltransferase candidates based upon co-expression with the rest of the colchicine pathway genes, but none of these candidates were able to acetylate 5 within N. benthamiana transient expression experiments. This led us to more strongly consider the NAT family of enzymes. Overall, the NAT superfamily has not been widely assessed for roles within plant secondary metabolism. However, NAT enzymes have been shown to carry out N-acetylations within the biosynthesis of melatonin in rice,24 and thus were considered for the N-acetylation in colchicine biosynthesis. We identified only one candidate gene (GsNAT1) that shared reasonably high expression correlation with downstream biosynthetic genes (Figure S8; Pearson’s r > 0.6 when compared to GsCYP71FB1) in both available G. superba transcriptomes (public and in-house). Accordingly, we found that transiently-expressed GsNAT1 efficiently consumed 5 to produce 1 (Figure S9), which was also confirmed via in vitro reactions with purified enzyme (Figure S10). We further observed that this enzyme is likely specific to 5, and therefore colchicine biosynthesis, as activity was not detected with either 4b or tyramine, a common biogenic amine.
Altogether, we demonstrated that the three-enzyme system of GsCYP71DA12, GsABH1, and GsNAT1 could act consecutively to convert 3 into 1, thereby uncovering the final required steps for reconstituting a full biosynthetic pathway to 1 (Figure 2). Our results further defined the probable order of events in G. superba, wherein N-demethylation precedes N-deformylation. As a final proof of principle, we were able to add these three genes into our previously established in planta metabolic engineering system,10 which allowed for the total biosynthesis of 1 in N. benthamiana (Figure 3 & Figure S11, final yield 268 ± 72 ng/g plant dry weight). We confirmed the biosynthetic 1 produced in this system to be (−)-colchicine (Figure S12), which is the native enantiomer of 1 that is produced by Gloriosa and Colchicum plants and utilized as a pharmaceutical. Ultimately, this heterologous system includes a combination of 20 genes from G. superba (17) and other plants (3), and allows for the conversion of primary plant metabolites from a heterologous host into the bioactive structure of 1, a pharmaceutical alkaloid.
Figure 2. Discovery of three enzymes that transform N-formyldemecolcine (3) into colchicine (1).
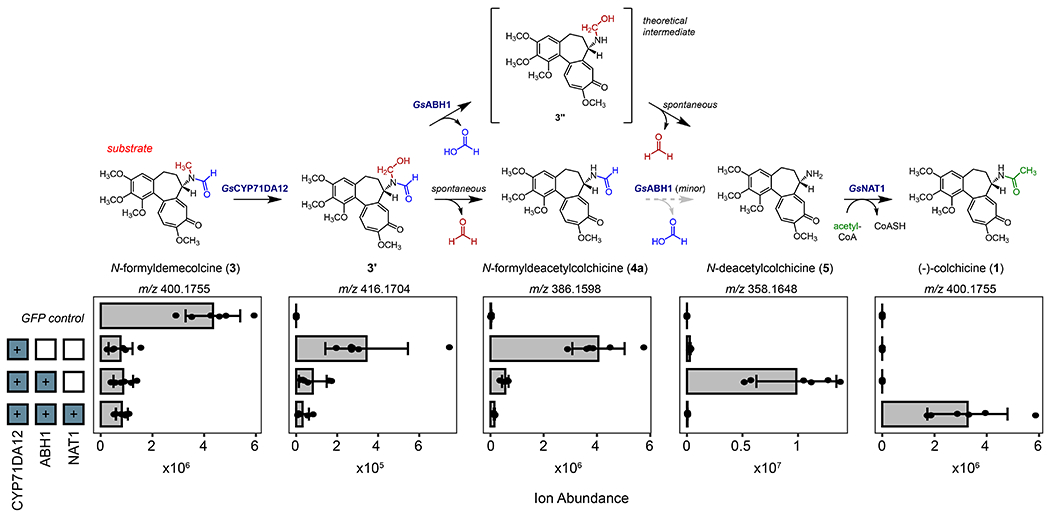
Transiently co-expressed combinations of G. superba genes in N. benthamiana with co-infiltration of 3 as substrate leads to stepwise transformation into 1. Shown here are the quantified ion abundances (m/z, [M+H]+) of the verified pathway intermediates/products observed upon the addition of each biosynthetic gene, as measured by LC-MS analysis. Shown above the quantified ion abundances is the corresponding biosynthetic proposal for the stepwise formation of 1 from 3.
Figure 3. Complete engineered biosynthesis of colchicine in N. benthamiana.
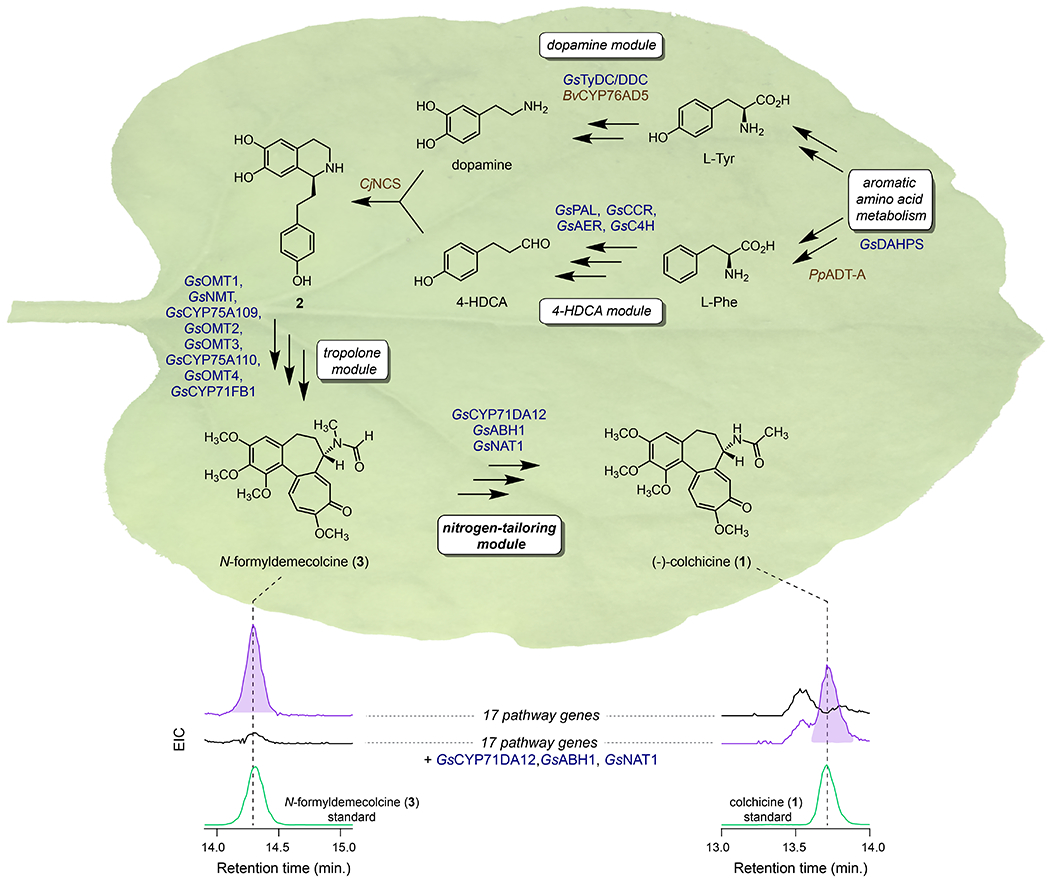
Addition of the three identified enzymes to the previously engineered biosynthetic pathway to 3 allows for total biosynthesis of 1 in N. benthamiana, as shown here via LC-MS chromatograms. EIC = extracted ion chromatogram (EIC = m/z 400.1755 [M+H]+ and m/z 422.1574 [M+Na]+ for both compounds). Native Gloriosa superba genes are shown in blue, while those from other plant sources are brown.
Our fully engineered pathway makes use of three enzymes that did not originate from G. superba. One of these enzymes, arogenate dehydratase A from Pinus pinaster (PpADT-A)25 was included to increase the level of Phe precursor supply, and thus downstream alkaloid yield (Figure S13). The other two enzymes, however, were necessary for catalyzing chemical transformations within the colchicine biosynthetic pathway. Specifically, we utilized BvCYP76AD5 to catalyze 3’-hydroxylation of L-Tyr within the engineered production of dopamine, and (S)-norcoclaurine synthase from Coptis japonica (CjNCS) to catalyze the Pictet-Spengler condensation of dopamine and 4-hydroxydihydrocinnamaldehyde (4-HDCA). 10,26,27 To better understand the initial stages of native colchicine biosynthesis, we first considered enzymes for the 3’-hydroxylation of L-Tyr. To our knowledge, the BvCYP76AD isoforms from the common beet (Beta vulgaris) are the only plant-derived enzymes that have been biochemically confirmed as L-Tyr 3’hydroxylases,26,28,29 and thus we initially tested several CYP enzymes that co-express with the rest of the colchicine pathway. However, none of these enzymes were able to catalyze this reaction. It has also been suggested that polyphenol oxidase (PPO) family enzymes in plants may play a prominent role in this hydroxylation, though this does not seem to have been functionally verified.30–32 Regardless, the implication for PPO enzymes in catalyzing this chemistry led us to consider this protein class within colchicine biosynthesis. By using both our transcriptome with the publicly-available G. superba transcriptome, we identified a single PPO gene (GsPPO1) that correlated with the L-Tyr/L-DOPA decarboxylase from G. superba (GsTyDC/DDC, Pearson’s r > 0.7), which would generate the necessary substrate for 3’-hydroxylase activity (Figure S14). Accordingly, we found that GsPPO1 could hydroxylate tyramine to produce dopamine in our transient expression system. This is distinct from BvCYP76AD5, which seems to preferentially acts on L-Tyr to produce L-DOPA (Figure S15).26 However, because GsTyDC/DDC is capable of acting on act on either L-Tyr or L-DOPA (Figure S15), the tyramine hydroxylase activity of GsPPO1 is plausible within the context of colchicine biosynthesis.
DISCUSSION
When studying secondary metabolism, a common assumption is that genes from the same biosynthetic pathway will exhibit similar spatial and temporal expression patterns.33 While whole-pathway coordination if often observed, there are instances in which distinct portions of a given secondary metabolic pathway experience differential regulation.34,35 In hindsight, this appears to be the case for the colchicine biosynthetic pathway. In particular, while the early and middle portions of the pathway share extremely high co-expression,10 the steps downstream of tropolone formation exhibit differing patterns of expression across G. superba (Figure 4). In particular, the final four genes in the pathway (GsCYP71FB1, GsCYP71DA12, GsABH1, and GsNAT1) show much higher relative expression within the leaves than the rest of the genes in this pathway within our G. superba transcriptome, which collectively have low expression in this tissue (Figure 4). This change in expression patterning made it difficult to identify pathway genes using transcriptional correlation analysis alone. Though we do not know the biological relevance for this differential expression, it does suggest a possibility for transport of a late stage precursor to all parts of the plant, at which point the final genes of the pathway can convert this intermediate into 1. In support of this possibility, we previously observed that most upstream colchicine intermediates only accumulated to high levels in the rhizome, while 1 can be found at relatively high levels in all of the tissues examined (rhizome, root, stem, and leaf).10 Compartmentalization of specific biosynthetic modules and transport of intermediates are both crucial aspects within the production of many plant molecules,36 and future evaluation of colchicine alkaloid transport in G. superba (e.g. through identification of potential transporters) may yield insight into the basis of the observed differential spatial patterns of colchicine biosynthetic gene expression.
Figure 4. Expression variation of colchicine biosynthetic pathway genes.
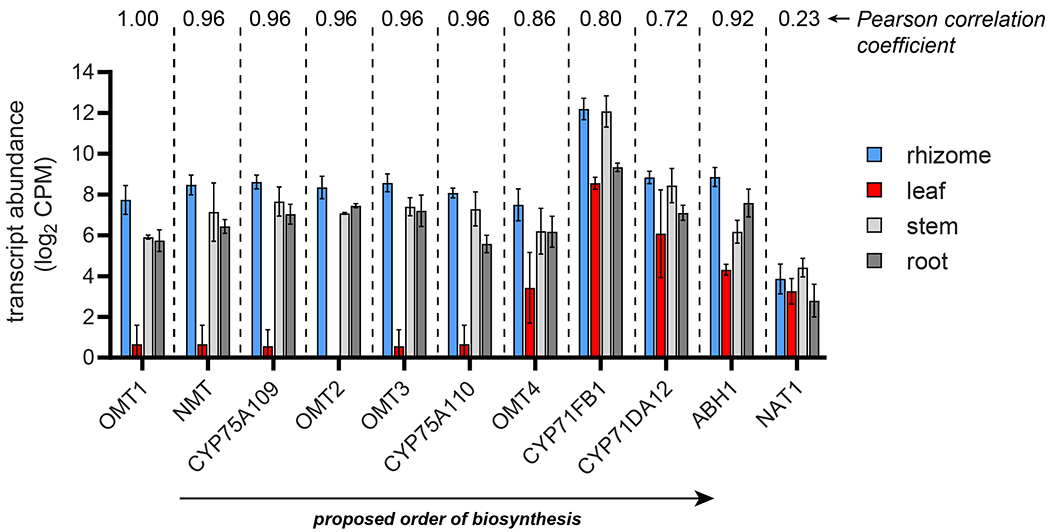
Transcript abundances in four sequenced tissues are shown for each biosynthetic pathway gene that acts downstream of 1-phenethylisoquinoline scaffold (2) formation. Shown for each tissue is the trimmed mean of M (TMM)-normalized, log2-transformed transcript counts per million (CPM) with ± standard deviation. n = 5 transcript libraries for rhizome, n = 2 for the three other tissue types. Shown above the bar graphs for each gene is the Pearson correlation coefficient (Pearson’s r) when comparing its transcript expression profile to that of GsOMT1.
By combining in vitro biochemistry from the native plant with transcriptomic analysis, we have identified three enzymes that modify the nitrogen atom of 3 to ultimately produce 1. Although removal of the N-methyl and N-formyl groups is necessary for the final N-acetylation to occur, it is not immediately evident why Gloriosa and Colchicum plants would have evolved to carry out these biosynthetic transformations, as formation of the tropolone scaffold seems to be sufficient for tubulin-binding activity.11 Some of the colchicine-related alkaloids, particularly 4a and 4b, can accumulate to relatively high levels in Gloriosa and Colchicum species,14,37 respectively, suggesting that these molecules may have a specific role in the fitness of these plants beyond acting as precursors to 1. Though the potent cytotoxicity of 1 via microtubule destabilization supports its potential role as a natural anti-herbivory molecule, it is possible that 1 has a separate, distinct mechanism of action within the natural biological interactions experienced by these plants. Alternatively, given the well-documented chemical “arms race” that occurs between plants and their pests/pathogens,38 it may be that precursors to 1 can be metabolized or resisted by certain organisms to overcome the Gloriosa/Colchicum defense system. Thus, 1 may be the latest variant in a series of tubulin-binding alkaloids within these plants that have evolved to ward off predation and/or infection.
Our results suggest that the order of events for the downstream nitrogen tailoring reactions in G. superba is different from what has previously been proposed in C. autumnale. The majority of isotope tracer studies in C. autumnale have indicated that N-deformylation likely occurs prior to N-demethylation in this species,15,16 and the high levels of 4b found in C. autumnale seem to support this.14 Conversely, 4a has been noted to accumulate to high levels in G. superba, which is indicative of initial N-demethylation of 3.37 Our ability to detect N-demethylation, but not N-deformylation, of 3 within G. superba rhizome extracts, as well as our subsequent enzymatic characterizations, support a major metabolic route within G. superba in which N-demethylation precedes N-deformylation. Although potential orthologs (≥84% amino acid identity) of GsCYP71DA12 and GsABH1 appear to be present within the C. autumnale transcriptome (https://medplantrnaseq.org/), further biochemical characterization will be necessary to determine whether these enzymes function similarly within colchicine biosynthesis in C. autumnale. Additionally, we cannot rule out the possibility that both biosynthetic routes can occur in either plant, and that we have only captured one of these pathways under the conditions in which we have analyzed G. superba.
The addition of alkyl or acyl groups to heteroatoms is a commonplace within the biosynthesis of natural products, and many of these chemical transformations seem to serve as biosynthetic protecting groups.39–41 Within the biosynthesis of 1, two particular reactions stand out as potential protecting group strategies: the early O-methylation of the catechol moiety of 2, thereby protecting against formation of the reactive ortho-quinone,42 and the subsequent N-methylation of this phenethylisoquinoline intermediate. Both of these transformations occur relatively early after formation of the phenethylisoquinoline scaffold,10 and thus may have evolved to limit the reactivity of these chemical moieties. Interestingly, analogous heteroatom methylations occur within the initial biosynthetic stages of structurally-related benzylisoquinoline alkaloids,43 suggesting that these early heteroatom alkylations are important for the chemical integrity of hydroxylated isoquinoline intermediates in general. While the aforementioned O-methyl group is retained throughout the full biosynthetic pathway of 1, the N-methyl is removed after formation of the tropolone ring, suggesting that it may be important to limit amine reactivity and ensure chemical selectivity during the downstream phenol coupling and tropolone-forming ring expansion reactions. The presence of the N-methyl group is required for biosynthesis of the tropolone scaffold to proceed,10 and thus it is likely that N-methylation evolved prior to the downstream biosynthetic pathway(s) to produce tropolone alkaloids. Therefore, the N-methyl may have served as a general protecting group of this class of phenethylisoquinoline-derived alkaloids, rather than as a dedicated means to ensure biosynthetic integrity for the specific production of 1. Accordingly, the acquisition of GsCYP71DA12 as the N-demethylating enzymes would have evolved later, thereby allowing for subsequent functionalization of the nitrogen (e.g. acetylation) to occur, which could feasibly act as a strategy by the plant to increase or change the bioactivity of the tropolone scaffold.
Beyond the identification of a biosynthetic pathway for 1, our results further expand upon the repetoire of enzyme families known to be involved within plant secondary metabolism. While the ability of CYPs to catalyze heteroatom demethylations and ABH family enzymes to hydrolyze acyl groups are well documented,19,44 the functions of PPO and NAT family enzymes within natural product biosynthesis in plants have been less elaborated. For example, our identification of GsPPO1 as a tyramine hydroxylase suggests that PPO enzymes may have a more widespread function within dopamine-related metabolism in plants, which has previously been speculated.30–32 Similarly, although NAT family enzymes had been identified within the biosynthesis of melatonin in rice,24 it was not clear whether this family would have an expanded role within plant secondary metabolism, especially since most heteroatom acylating enzymes have been found within the BAHD acyltransferase enzyme family.22 The study and elucidation of biosynthetic pathways relies upon our ability to identify the requisite biosynthetic genes, and our identification of PPO and NAT family enzymes within colchicine biosynthesis will help to guide future investigations into specialized metabolic pathways in plants.
CONCLUSION
Through the discovery of three nitrogen-tailoring enzymes, we have completed a fully engineered biosynthetic system to produce 1. The pathway we have established is almost completely composed of genes derived from Gloriosa superba, a native producer of 1, and represents a plausible route by which 1 is synthesized from Tyr and Phe in the native plant. Based upon the current biosynthetic hypothesis, the only specialized metabolic enzyme not accounted for is the presumed Pictet-Spenglerase that produces the initial phenethylisoquinoline scaffold (2). Enzymatic Pictet-Spengler activity seems to have evolved independently for the biosynthesis of multiple different natural products,45 and thus we speculate that the relevant Pictet-Spenglerase in G. superba will belong to a novel enzyme family. Our inability to find this missing piece highlights a major remaining challenge in plant specialized metabolism - identification of enzymes that have novel activity or belong to previously undefined enzymatic families. Though correlation analyses have proven to be extremely powerful for the elucidation of biosynthetic pathways, they still rely upon our ability to narrow down candidate genes via chemical/enzymatic logic, and this strategy breaks down if the class of enzyme is not yet known. This challenge, which will become increasingly relevant as more diverse and less-studied classes of plant natural products are investigated, will require complementary approaches such as genomic analyses of potential gene clusters,46 traditional biochemical assays (e.g. activity-guided fractionation),36 or newer, untargeted techniques to identify protein-small molecule interactions.47
MATERIALS AND METHODS
Chemicals and reagents
All standard molecular biology reagents and chemicals were purchased from commercial vendors, unless noted otherwise. Other than N-formyldemecolcine (3), which was previously synthesized from 4b,10 all colchicine-related alkaloids were purchased from chemical vendors, as follows: N-formyldeacetylcolchicine (4a, Toronto Research Chemicals), demecolcine (4b, MilliporeSigma), N-deacetylcolchicine (5, Santa Cruz Biotechnology), (−)-colchicine (1, MilliporeSigma), (+/−)-colchicine (Toronto Research Chemicals).
Plant growth
Gloriosa superba plants were propagated from rhizomes generated previously.10 Specifically, new rhizomes produced from seed-germinated G. superba plants were sown into PRO MIX HP Mycorrhizae soil (Premier Tech Horticulture) in pots and grown under ambient lab temperature and lighting. The soil was watered lightly for several months until new vegetative growth emerged, at which point individual plants (including rhizome and roots) were removed for subsequent protein lysate preparation. The Nicotiana benthamiana plants utilized for transient gene expression were grown exactly as previously described,10 with ambient lab temperature and a 16:8 light:dark cycle.
In vitro feeding experiments with Gloriosa superba tissue lysates
G. superba plants with several feet of green, vegetative growth were uprooted and dissected into major tissue types (rhizome, roots, leaves, and stem), which were then snap frozen in liquid nitrogen and stored at −80 °C for future use. To isolate crude protein lysates, frozen tissue samples of rhizome (13 g) and green leaf tissue (3 g) were each separately ground in 28 mL of ice-cold tricine buffer (100 mM tricine, pH 7.5, 20 mM 2-mercaptoethanol) with polyvinylpolypyrrolidone (PVPP) and acid-washed sand (1.3 g of each for rhizome, 0.3 g of each for leaves) using a mortar and pestle until the tissue was fully homogenized. The homogenate was transferred to a pre-chilled test tube and the mortar and pestle were each rinsed with an additional 10 mL of ice-cold tricine buffer, which was added to the bulk homogenate for each tissue. Samples were centrifuged at 3000 x g and 4 °C for 5 min to pellet plant debris. The supernatant was decanted into new, pre-chilled tubes, and centrifuged again at the same parameters. A portion of the supernatant, which represents the “soluble” protein fraction, was aliquoted into individual, pre-chilled microfuge tubes, snap frozen in liquid N2, and saved at −80 °C.
To prepare a microsomal protein fraction, the remaining protein lysate (26 mL) was ultra-centrifuged at 50,000 x g and 4 °C for 1 hour using a using a Type 70 Ti rotor (Beckman, part # 337922) on an Optima™ XE-90 - IVD ultracentrifuge (Beckman). The supernatants from these samples were discarded, and the pellets were resuspended in ice-cold tricine buffer (1.6 mL for rhizome microsomes, 0.374 mL for leaf microsomes). Protein abundance was measured using the BioRad Protein Assay kit, and the microsomal fractions, which represented the “insoluble” protein fraction, were aliquoted into pre-chilled microfuge tubes, snap frozen in liquid N2, and stored at −80 °C. We note that neither protein fraction (soluble or insoluble) was further purified to remove small molecules, and thus protein samples contain natively-produced colchicine alkaloids from G. superba.
Enzyme assays with the soluble and insoluble protein fractions contained variable colchicine-related substrates [N-formyldemecolcine (3), N-formyldeacetylcolchicine (4a), demecolcine (4b), and N-deacetylcolchicine (5)] and variable cofactors (NADPH and acetyl-CoA). For the insoluble protein assays, 100 μL reactions were set up using phosphate buffer (50 mM potassium phosphate, 100 mM sodium chloride, pH 7.8) or glycine buffer (100 mM glycine, pH 8.5), and contained 40 μg microsomal protein, 25 μM substrate, and cofactor (500 μM for NADPH, 200 μM for acetyl-CoA; control reactions with no cofactor were included). Soluble protein assays were performed similarly, except that the undiluted soluble protein lysate made up the bulk volume of the reaction (i.e. substrate and cofactors were added directly to the soluble protein lysate). Reactions were run overnight at 30 °C, after which they were quenched by the addition of 50 μL acetonitrile (ACN) with 0.1% formic acid. Samples were then filtered and prepared for LC-MS analysis.
Analysis of G. superba transcriptomic data
The analysis and selection of candidate genes was performed using our previously generated G. superba transcriptome, as well as the publicly-available G. superba transcriptome generated by the Medicinal Plants Consortium (https://www.medplantrnaseq.org), much as previously described.10 Transcript abundances within our in-house generated G. superba transcriptome were calculated as trimmed mean of M (TMM)-normalized, log2-transformed counts per million (CPM), while transcript abundances from the public G. superba transcriptome were calculated as the fragments per kilobase of transcript per million mapped reads (FPKM). Pearson correlation analyses of transcript expression was performed using the standard stats package in R (v4.1.0).
Cloning of biosynthetic genes
All candidate biosynthetic genes described in this study were cloned via PCR amplification of the coding sequence (CDS) from cDNA generated from G. superba rhizome mRNA. For transient expression in N. benthamiana, gene constructs were cloned into the pEAQ-HT plasmid via isothermal DNA assembly and subsequently transformed into Agrobacterium tumefaciens GV3101 vis the freeze-thaw method, as previously described in detail.10
To express GsCYP71DA12 in yeast, the CDS was PCR amplified from its pEAQ-HT construct using primers with 5’ and 3’ overhangs for subsequent assembly into the pYeDP60 plasmid (CarbR for E. coli selection and ADE2 for yeast selection), which was previously digested with BamHI and EcoRI restriction enzymes. The amplified product was analyzed via agarose gel electrophoresis, purified using the Zymo Gel Purification Kit (Zymo), and inserted into digested pYeDP60 using isothermal assembly with NEBuilder HiFi DNA Assembly 2x Mix (New England Biolab, NEB). Assembled constructs were transformed into E. coli NEB 10β cells, which were plated on LB agar with 100 μg/mL carbenicillin (carb) and grown overnight at 37 °C. The presence of assembled plasmid in transformants was confirmed via colony PCR, after which a positive clone was used to inoculate a 4 mL liquid LB culture (100 μg/mL carb), which was grown on a rotary culture drum overnight at 37 °C. Plasmid DNA was isolated using the Zymo Mini prep kit and the sequence was verified using Sanger Sequencing (ELIM BioPharm). Verified plasmid was then used to transform the chemically-competent yeast strain Saccharomyces cerevisiae WAT11 (ade2; contains the Arabidopsis thaliana cytochrome P450 reductase I gene, ATR1)48 using the Frozen-EZ Yeast Transformation II Kit (Zymo). Transformed yeast were plated on synthetic drop-out media plates without adenine (6.7 g/L yeast nitrogen base without amino acids, 20 g/L glucose, 2 g/L drop-out mix minus adenine [-Ade], 20 g/L agar) and grown at 30 °C for two days to select for positive transformations, which were confirmed via colony PCR. A positive yeast transformant was then used to inoculate a 4 mL liquid yeast dropout (-Ade) culture, which was grown with shaking at 250 RPM and 30 °C for two days, after which a 25% glycerol stock was made and stored at −80 °C.
For E. coli expression, the CDS for GsABH1 and GsNAT1 were PCR amplified from their respective pEAQ-HT constructs using primers with 5’ and 3’ overhangs for subsequent assembly into pET-28a(+) (KanR), which was digested with NdeI and XhoI such that the resulting inserts would have an N-terminal 6x His tag. Amplified products were gel purified, assembled into pET-28a, transformed into E. coli NEB 10β, and verified by Sanger sequencing as described above. Positive transformants were grown in liquid LB media with 50 μg/mL kanamycin (kan) overnight on a culture rotary drum at 4 °C, after which plasmid DNA was isolated, as described above. Plasmid constructs were then transformed into E. coli BL21 (DE3) cells (NEB) as the host strain for subsequent heterologous protein production, and were plated on selective LB agar (50 μg/mL kan). Positive transformants were confirmed via colony PCR and were then used to inoculate 4 mL liquid LB cultures (50 μg/mL kan), which were grown overnight on a culture rotary drum at 37 °C. These cultures were then used to make 25% glycerol stocks that were stored at −80 °C.
Transient gene expression in Nicotiana benthamiana
Candidate biosynthetic genes were expressed in N. benthamiana via Agrobacterium-mediated transient expression, as previously described.10 Briefly, Agrobacterium strains harboring gene constructs of interest were thickly plated on LB agar (50 μg/mL kan, 30 μg/mL gentamycin [gent]) and grown at 30 °C for two days, after which a lawn of bacteria had developed. Each bacterial lawn was removed from the plate with a sterile pipette tip, resuspended in 500 μL LB media, and centrifuged at 8,000 x g for 5 min to pellet cells. Cell pellets were then resuspended in 500 μL of Agrobacterium induction buffer (10 mM MES, pH 5.6, 10 mM MgCl2, 150 μM acetosyringone) and incubated at room temperature for 1-2 hours. Individual stains were diluted to an OD600 of 0.3; when gene constructs were tested in combination, strains were mixed together in equal concentration such that each strain had an OD600 of 0.3. Agrobacterium strain mixtures were then infiltrated into the abaxial side of N. benthamiana leaves, with approximately 300 μL per leaf. In a typical experiment, three individual leaves were used as replicates for each strain combination being tested, with each leaf belonging to a different plant in order to randomize any batch effects. In general, the infiltrated portions of leaves (approximately 1/3 of each leaf) were excised 4-5 days post-infiltration, snap frozen in liquid N2, and lyophilized to dryness overnight. To test for enzymatic activity on colchicine-related substrates, at three days post-Agrobacterium infiltration, N. benthamiana leaves transformed with the gene constructs of interest were infiltrated into their abaxial side with approximately 100 μL of a 25 μM solution of substrate in water, and this infiltrated area was marked for future harvest. After one day, the marked area was excised, snap frozen, and lyophilized. The dried tissue was then weighed and prepared for metabolite extraction, as described below.
Preparation of crude protein extracts from N. benthamiana
To assess the functionality of soluble enzymes produced in N. benthamiana in a more controlled environment, cell-free protein lysates were prepared from N. benthamiana leaves transiently expressing genes of interest, as follows. Leaves were harvested 4 days after infiltration with the Agrobacterium strain of interest and snap frozen in liquid nitrogen. The tissue was homogenized using a mortar and pestle with liquid N2 and a small scoop of polyvinylpolypyrrolidone (PVPP). The powdered tissue (~0.9 g) was resuspended in 5 mL of ice-cold Tris buffer (100 mM Tris-HCl pH 7.4, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride and 10 mM β-mercaptoethanol), and this mixture was incubated on ice for 20 min with periodic gentle inversions of the sample. Following incubation, samples were centrifuged at 12,000 x g and 4 °C for 10 min to pellet plant cell debris. The supernatant was removed and aliquoted into pre-chilled microfuge tubes, which were then snap frozen in liquid N2 and stored at −80 °C. In general, this crude protein lysate was used as the bulk buffer for in vitro reactions, to which other reagents (enzymes, cofactors, and substrate) were added. See In vitro reactions with purified enzymes for additional details.
Heterologous expression in yeast
Expression of GsCYP71DA12 in yeast was performed much as described previously.10,48 The S. cerevisiae WAT11 strain harboring the GsCYP71DA12 plasmid construct was streaked from its glycerol stock onto synthetic drop-out media (-Ade) plates and grown for two days at 30 °C. After this, a single colony was used to inoculate a 4 mL starter culture of liquid drop-out media (-Ade), which was grown on an orbital shaker at 28 °C with 250 RPM for 2 days. Following this growth, 2 mL of the starter culture was used to inoculate 500 mL YPGE media (10 g/L Bacto yeast extract, 10 g/L Bacto peptone, 5 g/L glucose and 3% [v/v] ethanol), which was shaken at 28 °C and 250 RPM until reaching a cell density of approximately 5×107 cells/mL, which was estimated via OD600 measurements. After reaching this density, gene expression was induced by the addition of a sterile galactose solution (200 g/L) to a final concentration of 10% (v/v), after which the culture was grown at 28 °C and 250 RPM for another 20 hours, at which point the cell density was approximately 5×108 cell/mL. The culture was then immediately prepared for microsomal protein isolation.
Isolation of yeast microsomal protein
Isolation of yeast microsomal protein followed a previously described protocol.48 After reaching the desired cell density, yeast cultures expressing GsCYP71DA12 were centrifuged at 5000 x g for 10 min to pellet cells. The spent media was discarded, and cells were resuspended in 1 mL TEK buffer (50 mM Tris-HCl, 1 mM EDTA, 1 mM potassium chloride, pH 7.4) per 0.5 g of wet cell pellet mass, and were then incubated at room temperature for 5 min. Cells were then centrifuged again as above, after which the supernatant was discarded, and the cell pellet was resuspended in 5 mL of ice-cold TES B buffer (50 mM Tris-HCl, 1 mM EDTA, 600 mM sorbitol, pH 7.4). From this point on, every step was performed on ice and/or in a 4 °C room. Approximately 5 mL of glass beads (0.5 mm) were added to the cell resuspension in a 500 mL centrifugation tube, after which the tube was shaken vigorously at a rate of two up-and-down motions per second for 10 min (30 sec shaking followed by 30 sec on ice) in order to lyse yeast cells. The lysis was verified by the change in color and consistency of the yeast cell resuspension. Following lysis, 10 mL of TES B buffer was added to the lysate and glass bead suspension, the tube was mixed, and the lysate (without glass beads) was pipetted into a new chilled tube. This process was repeated twice more, with the resulting lysates combined into a single tube. This combined lysate was then centrifuged at 23,000 x g for 10 min at 4 °C to pellet cell debris. The supernatant was removed and saved, while the pellet was discarded. The remaining clarified lysate was diluted two-fold in TES B, and microsomes were precipitated via the addition of sodium chloride to a concentration of 150 mM, and polyethylene glycol (PEG)-4000 to a concentration of 0.1 g/mL. This solution was incubated on ice for approximately 30 min with periodic swirling to dissolve the solutes, after which it was centrifuged at 10,000 x g for 10 min to pellet the microsomal fraction. The supernatant was removed and discarded, and the resulting pellet was resuspended in 2 mL of ice-cold TEG buffer (50 mM Tris-HCl, 1 mM EDTA, 20% [v/v] glycerol, pH 7.4). The protein abundance in the microsomal fraction was measured using the Bio-Rad Protein Assay Kit, and 100 μL aliquots were snap frozen in liquid N2 and stored at −80 °C.
Production and purification of protein in E. coli
E. coli BL21 strains harboring GsNAT1 and GsABH1 N-terminal 6xHis tag constructs were streaked on LB agar (50 μg/mL kan) and were grown overnight at 37 °C. A single colony for each strain was then used to inoculate a 2 mL starter culture of LB (50 μg/mL kan), which was grown overnight at 37 °C on a culture rotary drum. A portion of this starter culture (0.5 mL) was used to inoculate 200 mL LB media (50 μg/mL kan) in a 500 mL Erlenmeyer flask. Cultures were grown on an orbital shaker at 37 °C with 250 RPM until reaching an OD600 of approximately 0.6, at which point the temperature of the shaker was lowered to 18 °C. After shaking at this temperature for 1 hour, IPTG was added to the culture at a final concentration of 0.5 mM to induce heterologous gene expression, and cultures were continued to shake at this temperature overnight. After approximately 15 hours, cultures were centrifuged (5000 x g at 4 °C for 10 min) to pellet cells and the supernatant was discarded. All remaining steps were performed on ice or within a 4 °C room. Cell pellets (approximately 2 g) were resuspended in 8 mL binding buffer (50 mM potassium phosphate pH 7.8, 100 mM NaCl, 10 mM imidazole), and cells were lysed using an EmulsiFlex®-B15 High-Pressure Homogenizer with pressure set at 15,000 psi, which was performed three times for complete lysis. The resulting cell lysates were then centrifuged at 10,000 x g and 4 °C for 30 min to pellet cell debris. Each clarified lysate was added to 2 mL of Ni-NTA agarose resin (Invitrogen; R901-15), and this mixture was incubated with gentle, continuous mixing for 1 hour. After this incubation, the bound protein was eluted using increasing concentrations of imidazole (20-500 mM) in potassium phosphate buffer (50 mM potassium phosphate, 100 mM NaCl, pH 7.8). Collected fractions were each analyzed for purified protein via sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) with Coomassie Blue staining. Fractions containing purified protein (typically the 100 mM and 200 mM imidazole fractions) were combined and concentrated using Amicon® Ultra-15 Centrifugal Filter Units (10 kDa molecular weight cut-off) via centrifugation at 4000 x g for 20 min at 4 °C. After being concentrated to less than 0.5 mL, the samples were diluted with 10 mL of ice-cold potassium phosphate buffer, and centrifuged again as above, with the eluent being discarded after each centrifugation. This was repeated at least 6 times to dilute imidazole out from the purified protein. The final lysate was diluted in approximately 2 mL ice-cold potassium phosphate buffer, and glycerol was added to a final concentration of 10% (v/v). Protein concentrations were estimated via A280 measurements on a NanoDrop and with the BioRad Protein Assay Kit. The prepared protein solutions were then aliquoted into pre-chilled microfuge tubes, snap frozen in liquid N2, and stored at −80 °C.
In vitro reactions with purified enzymes
All in vitro reactions were carried out in potassium phosphate buffer (50 mM potassium phosphate, 100 mM sodium chloride, pH 7.8). To test activity of GsCYP71DA12-enriched microsomes, approximately 40 μg of microsomal protein was added to a 100 μL reaction volume (0.4 μg/μL microsomal protein) containing 25 μM of N-formyldemecolcine and 1 mM NADPH (which was omitted for control reactions). Initial reactions were allowed to proceed overnight, after which they were quenched by the addition of 100 μL ACN +0.1% formic acid, and filtered for subsequent LC-MS analysis. To perform kinetic analysis, it was first determined that 50 ng/μL of microsomal protein demonstrated a linear rate of product formation (observed as the hemiaminal, 3’) within 20 min, with less than 10% of substrate consumed, and thus this amount of enzyme was used for all subsequent reactions. To estimate Michaelis-Menten kinetic parameters, N-formyldemecolcine (3) substrate was added at varying concentrations (1-200 μM) along with NADPH (1 mM) into 3 × 100 μL reaction volumes, and the reactions were initiated via the addition of microsomal protein. After 20 min, the reactions were quenched as above and filtered for LC-MS analysis. It was observed that the hemiaminal intermediate (3’) would decay at room temperature over the course of several hours to produce 4a. In order to accurately estimate kinetic parameters, the quenched, filtered reactions were incubated at room temperature for several hours in order to allow for full conversion of 3’ into 4. The abundance of 4a, which is stable and can be quantified with a standard curve generated using the authentic standard, was then used as a proxy to infer the reaction rate of 3’ generation. Michaelis-Menten constants were calculated using GraphPad Prism (v9.1).
To assess activity of purified GsABH1, 100 μL reactions were run in potassium phosphate buffer with 0.1 μg/μL of enzyme and 25 μM of substrate (3 or 4a). In order to determine whether GsABH1 acted on 3’ as a substrate, 0.4 μg/μL GsCYP71DA12 microsomal protein and 1 mM NADPH were included in the reaction, and either 3 or 4a were added as a substrate at 25 μM. Reactions were incubated at 30 °C for a 24-hour period, during which individual aliquots were quenched with an equal volume of ACN +0.1% (v/v) formic acid at specific time points (0.5, 2, 4, and 24 hrs), and subsequently prepared for LC-MS analysis. Similar reactions were run in which whole cell protein lysate from N. benthamiana leaves expressing GsABH1 (see Preparation of crude protein extracts from N. benthamiana for details) was used in place of purified GsABH1. In these reactions, the protein lysate constituted the bulk buffer for the reaction, to which GsCYP71DA12 microsomal protein (0.4 μg/μL), NADPH (1 mM), and 3 or 4a (25 μM) were added. These reactions were run and quenched as described above.
Initial in vitro assays with purified GsNAT1 were run in potassium phosphate buffer with 50 ng/μl enzyme, 100 μM acetyl-CoA (omitted in control reactions), and 25 μM 5 as substrate in a 100 μL reaction volume. Reactions were incubated at 30 °C for 1 hr, after which they were quenched via the addition of 100 μL MeOH and prepared for LC-MS analysis. It was subsequently determined that 25 pg/μL of purified GsNAT1 demonstrated a linear rate of product formation and less than 10% substrate consumption after 20 min, and thus this enzyme concentration was utilized for subsequent kinetic analysis. To assess the Michaelis-Menten kinetics of this enzyme, 3 × 100 μL reactions in potassium phosphate buffer containing variable concentrations of 5 (1-200 μM) and a fixed concentration of acetyl-CoA (100 μM) were incubated at 30 °C for 20 min, after which they were quenched as described above, and prepared for LC-MS analysis. The reaction rate was assessed via the accumulation of 1 as a product, which was quantified by generating a standard curve with an authentic standard of 1. Michaelis-Menten kinetics were measured in GraphPad Prism (v9.1) using the substrate inhibition model.
Metabolite extraction and LC-MS sample preparation
For N. benthamiana transient expression experiments, harvested tissue was placed in a 2 mL SafeLock tube, snap frozen in liquid N2, lyophilized to dryness, and weighed to calculate dry mass. Tissue was then homogenized to a fine powder on a ball mill homogenizer (Retsch) using 5-mm steel beads, with shaking at 25 Hz for 2 min. To extract metabolites, 80% methanol (MeOH) in water was added to dried tissue at a ratio of 20 μL solvent per milligram of tissue mass. After mixing, samples were incubated at room temperature for at least 20 min, after which samples were centrifuged at 10,000 x g to pellet solid plant debris. The supernatant was filtered through MultiScreen Solvinert 96 Well Filter Plates (part no. MSRLN0450, MilliporeSigma) and pipetted into LC-MS vials for subsequent analysis. Quenched in vitro reactions were filtered in the same manner. For the analysis of polar metabolites (e.g. substrates and products of GsPPO1), methanolic extracts were diluted 10-fold in ACN prior to filtration in order to better match the starting mobile phase conditions for LC-MS analysis.
LC-MS data analysis
Metabolites were analyzed using an Agilent 1260 high-performance liquid chromatography (HPLC) instrument paired with a coupled Agilent 6520 Accurate Mass quadrupole time-of-flight (Q-TOF) electrospray ionization (ESI) mass spectrometer in positive ion mode. For standard separation of nonpolar metabolites, reversed-phase chromatography with a Gemini NX-C18 column (Phenomenex, 5 μm, 2 × 100 mm) was utilized with water +0.1% formic acid (buffer A) and ACN +0.1% formic acid (buffer B) as mobile phases at a flow rate of 0.4 mL/min. In general, a 2 μL injection volume was used for each sample with a 33-min gradient method, much as previously described.10 For targeted analysis of metabolites (e.g. in vitro enzymatic reactions) a shorter 23-min or 18-min gradient was utilized. See Supporting Information for details of HPLC method parameters. For the separation of polar metabolites associated with GsPPO1 enzymatic activity, hydrophilic liquid chromatography (HILIC) analysis was utilized with a Poroshell 120 HILIC-Z column (Agilent, 2.7 μm, 2.1 × 150 mm) using water +0.1% formic acid, 10 mM ammonium formate (buffer A) and ACN +0.1% formic acid, 10 mM ammonium formate (buffer B) as the mobile phase components at a flow rate of 0.25 mL/min. Standard HILIC analysis consisted of a 2 μL sample injection that was separated using a 22-min gradient method, as described in the Supporting Information.
Mass spectrometry (MS) data for both C18 and HILIC analyses was collected in positive ion mode with the following parameters: mass range, 100-1700 m/z; drying gas, 300 °C, 11 l/min; nebulizer, 25 psig; capillary, 3,500 V; fragmentor, 150 V; skimmer, 65 V; octupole 1 RF Vpp, 750 V; 1,000 ms per spectrum. To avoid salt contamination of the MS, the initial 0.5 min of each run was discarded to waste. For tandem mass spectrometry (MS/MS) analysis, defined m/z features, within an m/z tolerance of 1.3 centered around the ion being analyzed, were selected and fragmented using 10-, 20- and 40-V collision energies. In general, MS/MS spectra were directly compared to those of authentic standards for compound verification.
The relative abundances for compounds described in this manuscript were quantified using the MassHunter Qualitative Analysis software via automated integration (‘Agile2’ method with default settings) of extracted ion chromatograms (EICs) with a 20-50 ppm mass range tolerance. Unless noted otherwise, all compounds described herein were verified via retention time and MS/MS comparison to an authentic standard.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Prof. G. Lomonossoff (John Innes Centre) for providing the pEAQ-HT vector and Prof. D. Nelson (University of Tennessee) for his assistance in cytochrome P450 nomenclature. We further thank Dr. K. Smith, N. Mehta, J. Liu, and Y. Dho (Stanford University) for useful discussion and feedback on this work. We acknowledge the Stanford Genetics Bioinformatics Service Center for the use of computational resources supported by NIH S10 Instrumentation Grant S10OD023452. The research in this manuscript was supported by an R01 GM121527 (to E.S.S.). R.S.N. is a Howard Hughes Medical Institute Fellow of the Life Sciences Research Foundation.
Footnotes
Supporting Information. Supplementary figures describing the identification and characterization of each enzyme in this study and detailed LC-MS method parameters.
DATA AVAILABILITY
Coding sequences for the genes functionally characterized in this study have been deposited in GenBank under the accession numbers OK571350-OK571353. All other data is available upon request.
REFERENCES
- (1).Yang LPH Oral colchicine (Colcrys®). Drugs 2010, 70 (12), 1603–1613. [DOI] [PubMed] [Google Scholar]
- (2).Hartung EF History of the use of colchicum and related medicaments in gout; with suggestions for further research. Ann. Rheum. Dis. 1954, 13 (3), 190–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ravelli RBG; Gigant B; Curmi PA; Jourdain I; Lachkar S; Sobel A; Knossow M Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428 (6979), 198–202. [DOI] [PubMed] [Google Scholar]
- (4).Margolis RL; Wilson L Addition of colchicine tubulin complex to microtubule ends: The mechanism of substoichiometric colchicine poisoning. Proc. Natl. Acad. Sci. U. S. A. 1977, 74 (8), 3466–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Leung YY; Yao Hui LL; Kraus VB Colchicine - Update on mechanisms of action and therapeutic uses. Semin. Arthritis Rheum. 2015, 45 (3), 341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Misawa T; Takahama M; Kozaki T; Lee H; Zou J; Saitoh T; Akira S Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14 (5), 454–460. [DOI] [PubMed] [Google Scholar]
- (7).Peterson JR; Mitchison TJ Small molecules, big impact: A history of chemical inhibitors and the cytoskeleton. Chem. Biol. 2002, 9 (12), 1275–1285. [DOI] [PubMed] [Google Scholar]
- (8).Sattler MC; Carvalho CR; Clarindo WR The polyploidy and its key role in plant breeding. Planta 2016, 243 (2), 281–296. [DOI] [PubMed] [Google Scholar]
- (9).Herbert RB The biosynthesis of plant alkaloids and nitrogenous microbial metabolites. Nat. Prod. Rep. 2001, 20 (5), 494–508. [DOI] [PubMed] [Google Scholar]
- (10).Nett RS; Lau W; Sattely ES Discovery and engineering of colchicine alkaloid biosynthesis. Nature 2020, 584, 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Rósner M; Capraro HG; Jacobson AE; Atwell L; Brossi A; Lorio MA; Williams TH; Sik RH; Chignell CF Biological effects of modified colchicines. Improved preparation of 2-demethylcolchicine, 3-demethylcolchicine, and (+)-colchicine and reassignment of the position of the double bond in dehydro-7-deacetamidocolchicines. J. Med. Chem. 1981, 24 (3), 257–261. [DOI] [PubMed] [Google Scholar]
- (12).Nicolaou KC; Valiulin RA; Pokorski JK; Chang V; Chen JS Bio-inspired synthesis and biological evaluation of a colchicine-related compound library. Bioorganic Med. Chem. Lett. 2012, 22 (11), 3776–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Al-Mahmoud MS; Alali FQ; Tawaha K; Qasaymeh RM Phytochemical study and cytotoxicity evaluation of Colchicum stevenii Kunth (Colchicaceae): A Jordanian meadow saffron. Nat. Prod. Res. 2006, 20 (2), 153–160. [DOI] [PubMed] [Google Scholar]
- (14).Wildman WC Chapter 8 - Colchicine and Related Compounds. In Alkaloids: Chemistry and Physiology; Manske, R. H. F, Ed.; Academic Press, Inc.: New York, NY, 1960; Vol. 6, pp 247–288. [Google Scholar]
- (15).Barker AC; Julian DR; Ramage R; Woodhouse RN; Hardy G; McDonald E; Battersby AR Biosynthesis. Part 28. Colchicine: definition of intermediates between O-methylandrocymbine and colchicine and studies on speciosine. J. Chem. Soc. Perkin Trans. 1 1998, No. 18, 2989–2994. [Google Scholar]
- (16).Barker AC; Battersby AR; McDonald E; Ramage R; Clements JH Biosynthesis of colchicine: ring expansion and later stages. Structure of speciosine. Chem. Commun. 1967, No. 8, 390b–392. [Google Scholar]
- (17).Ortiz de Montellano PR Substrate Oxidation by Cytochrome P450 Enzymes. In Cytochrome P450: Structure, Mechanism, and Biochemistry; Ortiz De Montellano PR, Ed.; Springer International Publishing: Switzerland, 2015; pp 111–176. [Google Scholar]
- (18).Rueffer M; Zenk MH Microsome-mediated transformation of O-methylandrocymbine to demecolcine and colchicine. FEBS Lett. 1998, 438 (1–2), 111–113. [DOI] [PubMed] [Google Scholar]
- (19).Hu D; Gao YH; Yao XS; Gao H Recent advances in dissecting the demethylation reactions in natural product biosynthesis. Curr. Opin. Chem. Biol. 2020, 59, 47–53. [DOI] [PubMed] [Google Scholar]
- (20).Wogulis M; Chew ER; Donohoue PD; Wilson DK Identification of formyl kynurenine formamidase and kynurenine aminotransferase from Saccharomyces cerevisiae using crystallographic, bioinformatic and biochemical evidence. Biochemistry 2008, 47 (6), 1608–1621. [DOI] [PubMed] [Google Scholar]
- (21).Tao J; Kang Y; Xue Z; Wang Y; Zhang Y; Chen Q; Chen Z; Xue Y Theoretical study on the N-demethylation mechanism of theobromine catalyzed by P450 isoenzyme 1A2. J. Mol. Graph. Model. 2015, 61, 123–132. [DOI] [PubMed] [Google Scholar]
- (22).D’Auria JC Acyltransferases in plants: a good time to be BAHD. Curr. Opin. Plant Biol. 2006, 9 (3), 331–340. [DOI] [PubMed] [Google Scholar]
- (23).Vetting MW; Luiz LP; Yu M; Hegde SS; Magnet S; Roderick SL; Blanchard JS Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys. 2005, 433 (1), 212–226. [DOI] [PubMed] [Google Scholar]
- (24).Kang K; Lee K; Park S; Byeon Y; Back K Molecular cloning of rice serotonin N-acetyltransferase, the penultimate gene in plant melatonin biosynthesis. J. Pineal Res. 2013, 55 (1), 7–13. [DOI] [PubMed] [Google Scholar]
- (25).El-Azaz J; Cánovas FM; Barcelona B; Ávila C; de la Torre F Deregulation of phenylalanine biosynthesis evolved with the emergence of vascular plants. Plant Physiol. 2021, kiab454, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Sunnadeniya R; Bean A; Brown M; Akhavan N; Hatlestad G; Gonzalez A; Symonds VV; Lloyd A Tyrosine hydroxylation in betalain pigment biosynthesis is performed by cytochrome P450 enzymes in beets (Beta vulgaris). PLoS One 2016, 11 (2), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Luk LYP; Bunn S; Liscombe DK; Facchini PJ; Tanner ME Mechanistic studies on norcoclaurine synthase of benzylisoquinoline alkaloid biosynthesis: An enzymatic Pictet-Spengler reaction. Biochemistry 2007, 46 (35), 10153–10161. [DOI] [PubMed] [Google Scholar]
- (28).Hatlestad GJ; Sunnadeniya RM; Akhavan NA; Gonzalez A; Goldman IL; McGrath JM; Lloyd AM The beet R locus encodes a new cytochrome P450 required for red betalain production. Nat. Genet. 2012, 44 (7), 816–820. [DOI] [PubMed] [Google Scholar]
- (29).Polturak G; Breitel D; Grossman N; Sarrion-Perdigones A; Weithorn E; Pliner M; Orzaez D; Granell A; Rogachev I; Aharoni A Elucidation of the first committed step in betalain biosynthesis enables the heterologous engineering of betalain pigments in plants. New Phytol. 2016, 210 (1), 269–283. [DOI] [PubMed] [Google Scholar]
- (30).Araji S; Grammer TA; Gertzen R; Anderson SD; Mikulic-Petkovsek M; Veberic R; Phu ML; Solar A; Leslie CA; Dandekar AM; Escobar MA Novel roles for the polyphenol oxidase enzyme in secondary metabolism and the regulation of cell death in walnut. Plant Physiol. 2014, 164 (3), 1191–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Schenck CA; Maeda HA Tyrosine biosynthesis, metabolism, and catabolism in plants. Phytochemistry 2018, 149, 82–102. [DOI] [PubMed] [Google Scholar]
- (32).Sullivan ML Beyond brown: Polyphenol oxidases as enzymes of plant specialized metabolism. Front. Plant Sci. 2015, 5, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Nett RS; Dho Y; Low Y-Y; Sattely ES A metabolic regulon reveals early and late acting enzymes in neuroactive Lycopodium alkaloid biosynthesis. Proc. Natl. Acad. Sci. 2021, 118 (24), e2102949118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kellner F; Kim J; Clavijo BJ; Hamilton JP; Childs KL; Vaillancourt B; Cepela J; Habermann M; Steuernagel B; Clissold L; Mclay K; Buell CR; O’Connor SE Genome-guided investigation of plant natural product biosynthesis. Plant J. 2015, 82 (4), 680–692. [DOI] [PubMed] [Google Scholar]
- (35).Li Q; Ramasamy S; Singh P; Hagel JM; Dunemann SM; Chen X; Chen R; Yu L; Tucker JE; Facchini PJ; Yeaman S Gene clustering and copy number variation in alkaloid metabolic pathways of opium poppy. Nat. Commun. 2020, 11 (1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Jacobowitz JR; Weng JK Exploring uncharted territories of plant specialized metabolism in the postgenomic era. Annu. Rev. Plant Biol. 2020, 71 (1), 631–658. [DOI] [PubMed] [Google Scholar]
- (37).Jana S; Shekhawat GS Critical review on medicinally potent plant species: Gloriosa superba. Fitoterapia 2011, 82 (3), 293–301. [DOI] [PubMed] [Google Scholar]
- (38).Mithöfer A; Boland W Plant defense against herbivores: Chemical aspects. Annu. Rev. Plant Biol. 2012, 63, 431–450. [DOI] [PubMed] [Google Scholar]
- (39).Li Y; Llewellyn NM; Giri R; Huang F; Spencer JB Biosynthesis of the unique amino acid side chain of butirosin: Possible protective-group chemistry in an acyl carrier protein-mediated pathway. Chem. Biol. 2005, 12 (6), 665–675. [DOI] [PubMed] [Google Scholar]
- (40).Shinohara Y; Kudo F; Eguchi T A natural protecting group strategy to carry an amino acid starter unit in the biosynthesis of macrolactam polyketide antibiotics. J. Am. Chem. Soc. 2011, 133 (45), 18134–18137. [DOI] [PubMed] [Google Scholar]
- (41).Dang TTT; Chen X; Facchini PJ Acetylation serves as a protective group in noscapine biosynthesis in opium poppy. Nat. Chem. Biol. 2015, 11 (2), 104–106. [DOI] [PubMed] [Google Scholar]
- (42).Bruins JJ; Albada B; van Delft F ortho-quinones and analogues thereof: highly reactive intermediates for fast and selective biofunctionalization. Chem. - A Eur. J. 2018, 24 (19), 4749–4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Singh A; Menéndez-Perdomo IM; Facchini PJ Benzylisoquinoline alkaloid biosynthesis in opium poppy: an update. Phytochem. Rev. 2019, 18 (6), 1457–1482. [Google Scholar]
- (44).Mindrebo JT; Nartey CM; Seto Y; Burkart MD; Noel JP Unveiling the functional diversity of the alpha/beta hydrolase superfamily in the plant kingdom. Curr. Opin. Struct. Biol. 2016, 41, 256–257. [DOI] [PubMed] [Google Scholar]
- (45).Roddan R; Ward JM; Keep NH; Hailes HC Pictet–Spenglerases in alkaloid biosynthesis: Future applications in biocatalysis. Curr. Opin. Chem. Biol. 2020, 55 (2), 69–76. [DOI] [PubMed] [Google Scholar]
- (46).Guo L; Winzer T; Yang X; Li Y; Ning Z; He Z; Teodor R; Lu Y; Bowser TA; Graham IA; Ye K The opium poppy genome and morphinan production. Science 2018, 362 (6412), 343–347. [DOI] [PubMed] [Google Scholar]
- (47).Venegas-Molina J; Molina-Hidalgo FJ; Clicque E; Goossens A Why and how to dig into plant metabolite–protein interactions. Trends Plant Sci. 2021, 26 (5), 472–483. [DOI] [PubMed] [Google Scholar]
- (48).Pompon D; Louerat B; Bronine A; Urban P Yeast expression of animal and plant P450s in optimized redox environments. Methods Enzymol. 1996, 272, 51–64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coding sequences for the genes functionally characterized in this study have been deposited in GenBank under the accession numbers OK571350-OK571353. All other data is available upon request.