

Summary
Background:
Thromboelastography (TEG) is used for real-time determination of hemostatic status in patients with acute risk of bleeding. Thrombin is thought to drive clotting in TEG through generation of polymerized fibrin and activation of platelets through PARs. However, the specific role of platelet agonist receptors and signaling in TEG has not been reported.
Objectives:
Here we investigated the specific receptors and signaling pathways required for platelet function in TEG using genetic and pharmacological inhibition of platelet proteins in mouse and human blood samples.
Methods:
Clotting parameters (R, α, MA) were determined in recalcified, kaolin-triggered citrated blood samples using a TEG 5000 analyzer.
Results:
We confirmed the requirement of platelets, platelet contraction, and αIIbβ3 integrin function for normal α-angle (α) and maximal amplitude (MA). Loss of the integrin adaptor Talin1 in megakaryocytes/platelets (Talin1mKO) also reduced α and MA, but only minimal defects were observed in samples from mice lacking Rap1 GTPase signaling. PAR4mKO samples showed impaired α but normal MA. However, impaired TEG traces similar to platelet-depleted samples were observed with samples from PAR4mKO mice depleted of GPVI on platelets or with addition of a Syk inhibitor. We reproduced these results in human blood with combined inhibition of PAR1, PAR4, and Syk.
Conclusions:
Our results demonstrate that standard TEG is not sensitive to platelet signaling pathways critical for integrin inside-out activation and platelet hemostatic function. Furthermore, we provide first evidence that PARs and GPVI play redundant roles in platelet-mediated clot contraction in TEG.
Keywords: Thromboelastography, blood platelets, protease-activated receptor, fibrin, bleeding
Introduction
Thromboelastography (TEG) is used clinically at point-of-care to determine the risk of bleeding and/or thrombosis [1,2]. The assay accounts for the contribution of coagulation factors, fibrinogen, and platelets to clot formation, and generates parameters representing the speed and strength of clot formation. The primary clinical application is to rapidly determine a patient’s hemostatic status and guide administration of blood products [3]. Clotting parameters include R time (time to first inflection of the trace), α-angle (angle between the midline and the developing trace), and MA (maximum amplitude-maximum force transmitted to the pin). The R parameter is dependent on soluble coagulation factors, while α and MA are dependent on fibrin(ogen) and platelets, and these associations are used to guide transfusion of blood products [3]. TEG-guided transfusions can reduce the use of blood products by preventing unnecessary transfusions in the setting of surgery (cardiac, liver transplant) and trauma [4–7]; however, the impact on clinical outcomes (re-exploration, morbidity/mortality) is often limited [5,8,9] and unfortunately, low quality evidence (low numbers, poor methodology) often hampers analysis of the real impact of viscoelastic tests on patient outcomes [4,10].
Platelets mediate both hemostasis and thrombosis, and express agonist receptors and signaling molecules to rapidly respond to damaged blood vessels. Von Willebrand factor (VWF) recruits platelets to the site of injury via glycoprotein (GP)Ibα, and platelets are subsequently activated by collagen via GPVI and by thrombin via protease activated receptors (PARs). Receptor stimulation induces Ca2+ influx and activation of CalDAG-GEFI, a guanine nucleotide exchange factor (GEF) for the small GTPase Rap1. Ultimately, Rap1 and Talin1 drive integrin αIIbβ3 into an active conformation, leading to fibrinogen binding and platelet aggregation [11]. Thrombin also cleaves fibrinogen which assembles into polymerized fibrin, binds αIIbβ3, and induces platelet outside-in signaling to mediate clot contraction [12]. Recently, GPVI has also been shown to bind fibrinogen and fibrin, although the consequences of this interaction on hemostasis in vivo are not entirely understood [13,14]. Importantly, loss of PAR, CalDAG-GEFI, Rap1, Talin1, or αIIbβ3 lead to severe bleeding in mice [15–18], and humans [19,20].
In TEG, MA is sensitive to thrombocytopenia and inhibition of αIIbβ3 integrin and platelet contraction [21,22]. It is assumed that platelet activation is mediated by activation of PARs by thrombin [23]. However, there is little information on the receptors and signaling proteins that mediate platelet function in TEG. Prostacyclin, which prevents platelet activation by inducing inhibitory signaling, has no impact on MA [24]. Inhibition of PAR1 with vorapaxar does not modify TEG parameters, even in the presence of dual antiplatelet therapy [25,26]. Viscoelastic data on patients lacking CalDAG-GEFI/Rap1 signaling has not been reported. Additionally, the importance of the novel interaction between GPVI and fibrin(ogen) to platelet-dependent viscoelastic parameters has not yet been assessed. Here, we investigated the specific receptors and signaling pathways required for platelet function in TEG.
Methods
Mice:
The following strains were included in this study: C57BL/6J, Caldaggef1−/− [16], P2ry12−/− [27], Rap1bfl/fl × Pf4-Cre+ (Rap1bmKO) [17], Talin1fl/fl × Pf4-Cre+ (Talin1mKO) [18], Par4fl/fl × Pf4-Cre+ (Par4mKO) [15], Talin1fl/R35E,R118E × Pf4-Cre+(Tln1mR35E,R118E) [11], and Fibrinogen γ chainΔ5/Δ5 (Fgnγ Δ5/γΔ5) [28]. All mice were bred in-house and are on a C57BL/6/J background. Male and female mice were used as blood donors between 8–20 weeks of age, but males were used whenever possible due to greater blood volume. All procedures were approved by the Animal Care and Use Committee of the University of North Carolina at Chapel Hill.
Mouse blood collection:
Blood was collected from anesthetized mice via retroorbital plexus (RO) or inferior vena cava (IVC). RO collection was performed by puncturing the retroorbital plexus with a non-heparinized capillary tube, wasting the first two drops of blood to limit tissue factor contamination and then dripping blood into a microcentrifuge tube containing 3.2% citrate (9:1 volume ratio). IVC collection was performed by exposing the IVC and gently drawing blood through a 25-G needle into a 10 mL syringe containing 3.2% citrate (9:1 volume ratio). For platelet-depleted samples, donor mice were injected intravenously with 1 mg/kg anti-GPIbα antibody (Emfret Analytics, clone R300) 3 hours prior to blood collection. For GPVI-deficient samples, donor mice were injected intravenously with 50 μg of the anti-GPVI antibody JAQ1 (Emfret Analytics) 5 days before blood collection [29]. Cytochalasin D (5 μg/ml, Sigma) [21], anti-αIIbβ3 antibody (75 μg/ml, Emfret Analytics, clone Leo.H4), and PRT-062607 (20 μM, Selleck Chem) were added directly to whole blood samples prior to starting TEG. PRT-2607 was used at a supratherapeutic concentration of 20 μM to achieve complete inhibition of whole blood platelet aggregation induced by a high concentration of the GPVI-specific agonist convulxin; 20 μM PRT-2607 did not affect PAR4-mediated platelet aggregation (not shown).
Human blood collection:
Whole blood was collected from healthy subjects (male and female subjects between the ages of 20–50 who had not taken aspirin/NSAIDs within two weeks) using a 21-G needle vacutainer butterfly into 3.2% citrate tubes (BD). For inhibition of αIIbβ3, PAR1, PAR4, or Syk, samples were treated with abciximab (20 μg/ml, provided by Dr. Rick Stouffer), vorapaxar (5 μM, Med Chem Express), BMS-986120 (10 μM, Cayman Chemical), or PRT-062607 (20 μM), respectively. Vorapaxar and BMS-986120 were used at concentrations which completely prevented thrombin-induced human platelet aggregation when combined (not shown). Blood collection from healthy donors was performed with informed consent in accordance with a protocol approved by the Institutional Review Board at the University of North Carolina at Chapel Hill.
Flow cytometry analysis:
Platelet counts were determined in 2 μL whole blood stained with anti-GPIX antibody (Emfret Analysis), and diluted samples in PBS were analyzed on an Accuri C6 Plus flow cytometer (BD). To confirm GPVI deficiency by JAQ1 administration, platelets in whole blood were diluted in modified Tyrode’s buffer containing 1 mM Ca2+ and activated with the GPVI agonist convulxin (Cvx) for 10 minutes in the presence of JON/A-PE (Emfret Analytics) and anti-P-selectin-AlexaFluor647 (BD) antibodies to measure activated αIIbβ3 integrin and α-granule secretion, respectively.
TEG analysis:
All samples underwent TEG analysis (TEG 5000 Thrombelastograph Hemostasis Analyzer System, Haemonetics) using a citrated kaolin assay following manufacturer instructions. Blood samples were allowed to rest at room temperature for 15–20 mins prior to TEG analysis. 16 μL of kaolin reagent was pipetted from the vial and added to 384 μL of citrated mouse or human blood (after addition of inhibitors when applicable) in a microcentrifuge tube and mixed by inversion. Then, 340 μL blood was pipetted into a plain cup containing 20 μL of CaCl2 (0.2 M) and inserted into the TEG machine. Samples were run for approximately one hour for calculation of R time (mins), α-angle (degrees), and MA (mm). Representative traces for each condition are shown overlaid in figures; for groups of data in which R times were not significantly different, traces were aligned so that the first inflection point of all traces are superimposed for a more direct comparison of α-angle and MA.
Statistical analysis:
Data are presented as mean ± standard deviation (SD). Normality of data was determined by Shapiro-Wilk test. Parametric data were analyzed using a 2-tailed student’s t-test for two groups or a 1-way ANOVA with Tukey’s post-hoc test for three or more groups. Non-parametric data were analyzed by Mann-Whitney test for two groups or Kruskal-Wallis test with Dunn’s post-hoc test for three or more groups. Analyses were performed using GraphPad Prism 9 software. A P-value of 0.05 or less was considered significant.
Results
Thromboelastography (TEG) was performed with mouse citrated whole blood samples collected via retroorbital (RO) blood collection, and clotting was initiated with kaolin using a TEG 5000 analyzer. For this study we analyzed 3 parameters: R time (R), α-angle (α), and maximum amplitude (MA). K time (a secondary measure of clot kinetics) was not included because it is not calculated for traces with MA <20, and LY30 (extent of fibrinolysis at 30 mins) was not included because mouse blood samples did not undergo spontaneous fibrinolysis. Using wild-type (WT) C57BL/6J mice, we did not observe any differences in parameters between males and females (Figure S1). Although inferior vena cava (IVC) blood collection was demonstrated to be ideal for TEG when studying coagulation factor deficiencies in mice [30] and may be more comparable to human venipuncture, direct comparison of IVC vs RO blood collection methods with WT mice showed significant but only small differences in R and α, and no difference in MA (Figure S2). We therefore used RO blood draws for subsequent studies as this method is non-terminal but should be comparable to clinical sample collection for TEG.
Platelets are required for clot formation [31], and thrombocytopenia impairs viscoelastic clotting parameters [22]. To determine the contribution of platelets to TEG parameters under our experimental conditions, we analyzed blood from WT mice depleted of virtually all circulating platelets by administration of an anti-GPIbα antibody which reduced platelets counts by >99%. Platelet depletion did not alter the R value but significantly reduced α and MA (Figure 1). Pre-treatment of whole blood with cytochalasin D, an inhibitor of actin-mediated platelet contraction [32], reduced α and MA to a similar degree as in platelet-depleted samples without affecting R (Figure 1), suggesting the platelet contribution to α and MA is predominantly dependent on platelet contraction. This was also supported by normal TEG parameters in blood samples from mice expressing a mutant fibrinogen (FgnγΔ5/γΔ5) which impairs platelet aggregation but leaves platelet contraction intact [28] (Figure S3). The ability of platelets to interact with fibrin and contract the fibrin clot is dependent on integrin αIIbβ3 [33]. R was not affected by integrin inhibition, but consistently, significant reductions in α and MA were observed in WT blood treated with a blocking antibody to αIIbβ3 integrin and in blood from mice with megakaryocyte(MK)/platelet-specific deficiency in the integrin adaptor protein Talin1 (Tln1mKO) (Figure 2). Although the requirement for αIIbβ3 integrin in TEG was known [21], the requirement for inside-out signaling has not been directly demonstrated. Therefore, we next determined if platelet intracellular signaling upstream of Talin is critical in TEG. The small GTPase Rap1 is a key regulator of αIIbβ3 activation [17,34]. Mice lacking the major isoform of Rap1 (Rap1bmKO) [34] or the two major pathways for Rap1 activation (Cdg1−/− × P2ry12−/−) [35] had normal TEG parameters (Figure 3). Mice lacking both isoforms of Rap1 (Rap1a/bmKO) [17] showed significant reductions in α and MA (Figure 3), however these mice also have significant thrombocytopenia (<50% of WT; Figure S4) [17]. It was recently demonstrated that a direct interaction between Rap1 and Talin1 is critical for αIIbβ3 activation in platelets [11]. TEG parameters were also normal in Tln1mR35E,R118E mice lacking this interaction (Figure S5), demonstrating a primarily Rap1-independent but talin/integrin-dependent function of platelets in TEG.
Figure 1:
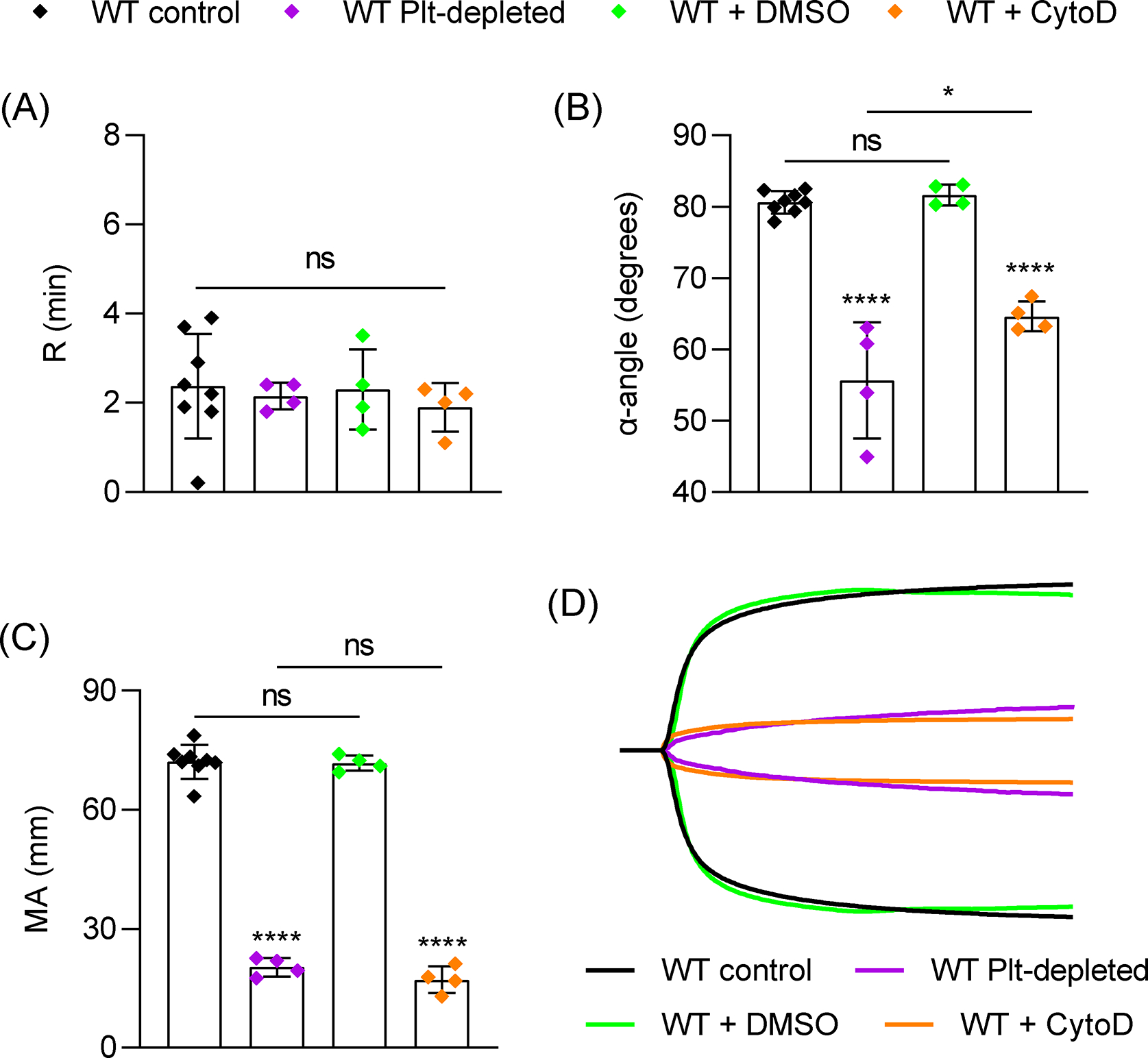
Role of platelets and platelet contraction in TEG. Citrated blood samples were collected by retroorbital bleed (RO) from wild-type (WT) mice (n=8) or WT mice depleted of circulating platelets (WT Plt-depleted) by injection of an anti-GPIbα antibody (R300, 1 mg/kg, n=4). For inhibition of platelet-mediated contraction, WT samples were treated with DMSO (n=4) or cytochalasin D (5 μg/ml, n=4) for 10 mins prior to TEG assay. Samples were mixed with CaCl2 and kaolin in plastic TEG cup and run immediately in a TEG 5000 analyzer and recorded for 1 hour. (A-C) TEG parameters: R time (A), α-angle (B) and MA (C). (D) Representative TEG traces. Data shown as mean ± SD. Statistical significance was determined by unpaired Student’s t-test (A) or one-way ANOVA with Tukey’s multiple comparison test. Symbols directly over bars represent significance compared to WT control. *P < .05, ****P < .0001.
Figure 2:
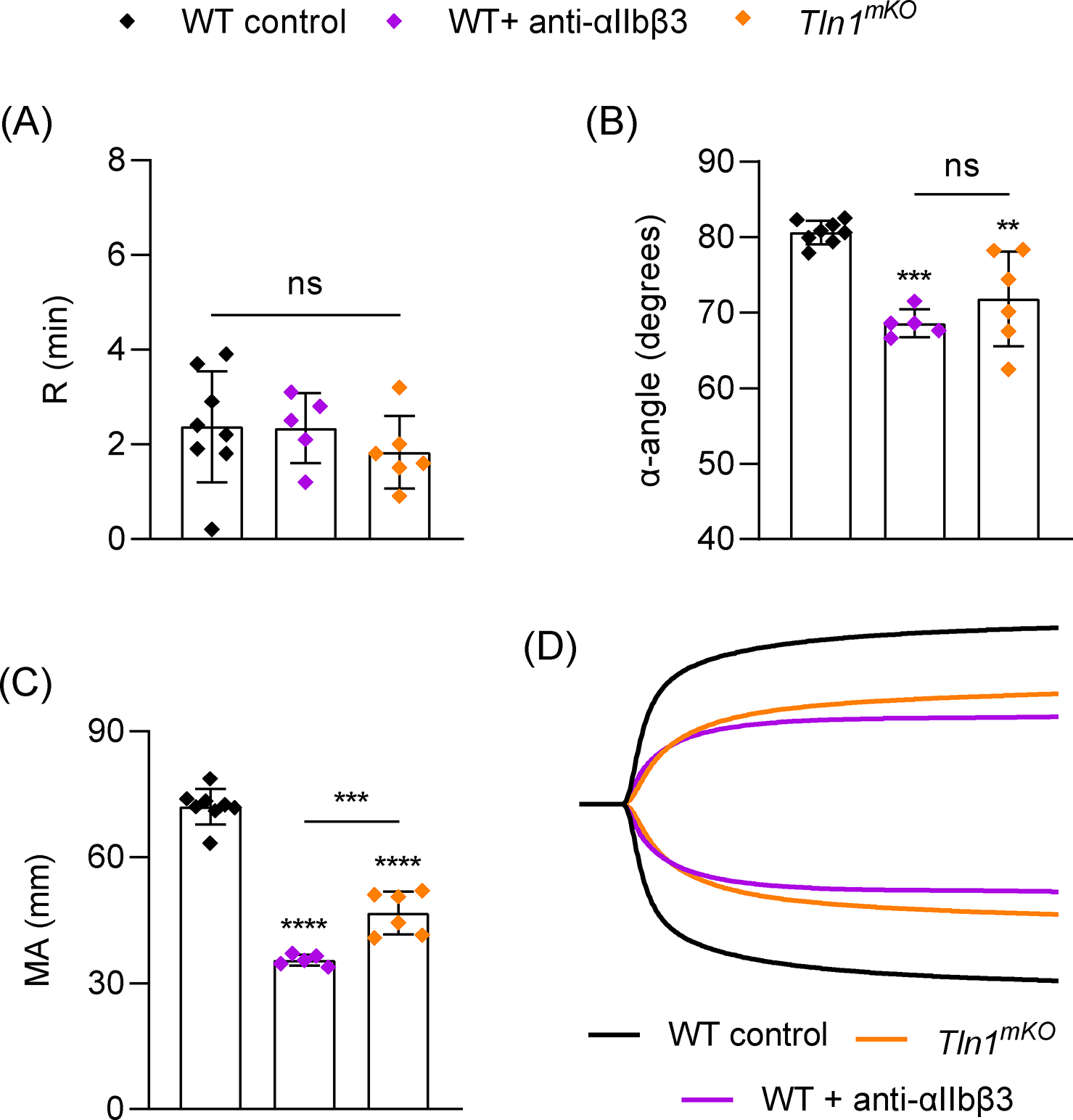
Role of αIIbβ3 integrin activation and ligand binding in TEG. Blood samples were analyzed from WT mice (n=8) or mice with megakaryocyte/platelet-specific deletion of Talin1 (Tln1mKO, n=5). αIIbβ3 ligand binding was inhibited by treating WT samples with anti-αIIbβ3 antibody (Leo.H4, 75 μg/ml, n=6) for 10 mins prior to TEG assay. (A-C) TEG parameters: R time (A), α-angle (B) and MA (C). (D) Representative TEG traces. Data shown as mean ± SD. Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparison test. Symbols directly over bars represent significance compared to control. **P< .01, ***P<0.001, ****P < .0001.
Figure 3:
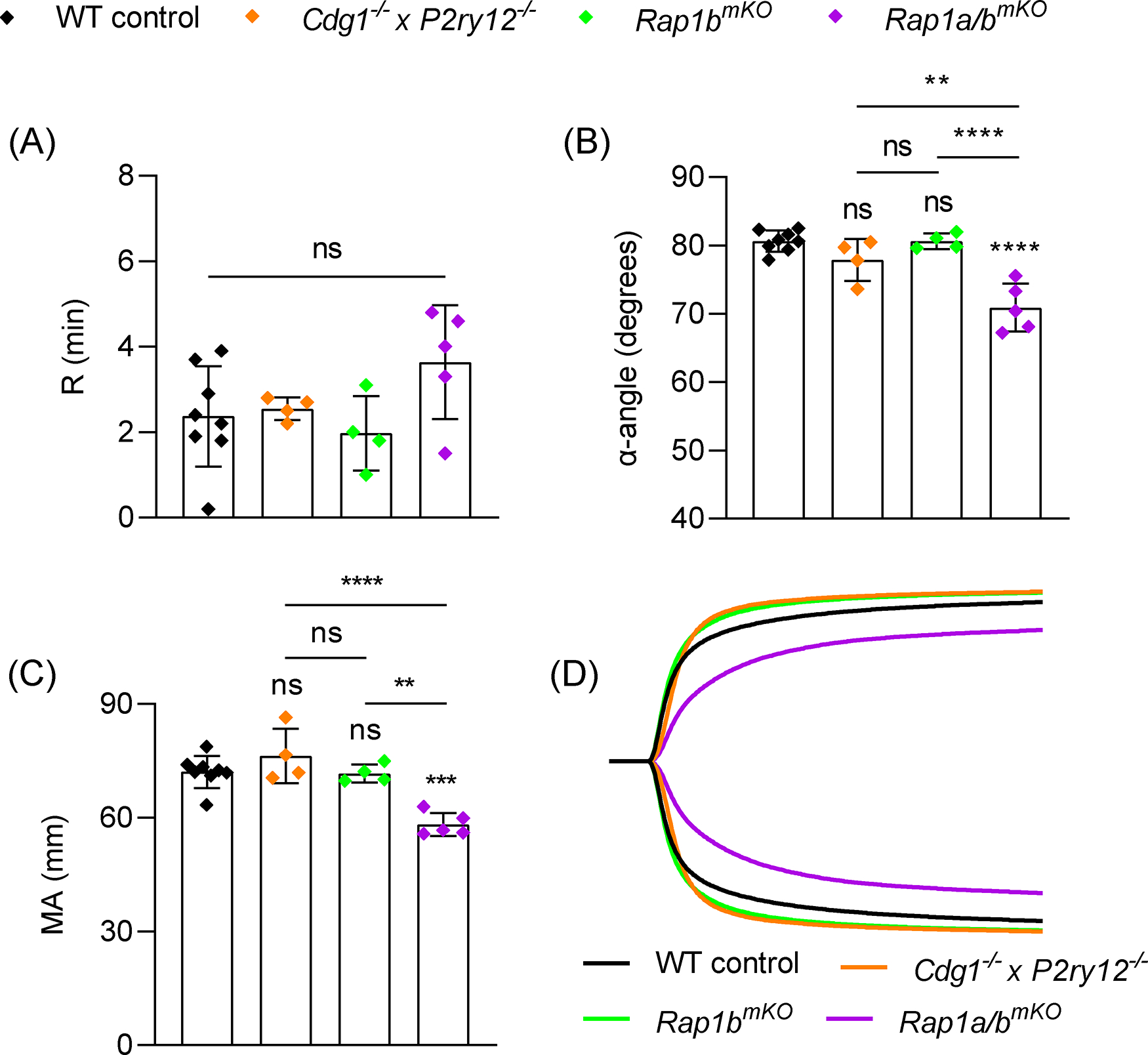
Role of Rap1 GTPase signaling in TEG. Blood samples were analyzed from WT mice (n=8), mice with global deficiency in CalDAG-GEFI and P2Y12 (Cdg1−/− × P2ry12−/−, n=4) or mice with megakaryocyte/platelet-specific deletion of Rap1b alone (Rap1bmKO, n=4) or both Rap1a and Rap1b (Rap1a/bmKO, n=5). (A-C) TEG parameters: R time (A), α-angle (B) and MA (C). (D) Representative TEG traces. Data shown as mean ± SD. Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparison test. Symbols directly over bars represent significance compared to control. **P< .01, ***P<0.001, ****P < .0001.
Thrombin is assumed to activate platelets in TEG [23], but the requirement for thrombin-mediated platelet activation has not been directly demonstrated. Interestingly, PAR1 inhibition on human platelets with vorapaxar had no effect on TEG parameters [25,26]. Mouse platelets express 2 thrombin receptors, protease activated receptor (PAR) 4 and 3. PAR4 is the main activating receptor and deletion of PAR4 completely eliminates the platelet response to thrombin [15,36]. However, when we tested blood from mice lacking PAR4 in MKs/platelets (PAR4mKO), we only observed a significant reduction in α, while R and MA were comparable to controls (Figure 4). We confirmed the absolute requirement for thrombin activity by treating WT samples with hirudin and observed no initiation of clotting up to 60 minutes (R >60 min; not shown). Based on these findings, we considered whether a different ligand-receptor interaction can drive platelet contraction in the absence of PAR4. One candidate we considered was GPVI, which has recently been identified as an important receptor for fibrin on platelets [13,14], and this interaction can initiate platelet intracellular signaling [14]. We tested blood from mice treated with an anti-GPVI antibody (JAQ1) which depletes GPVI from the surface of all circulating platelets [29]. GPVI deficiency alone had no impact on R, α, or MA (Figure 4). However, when we tested blood from JAQ1-treated PAR4mKO mice, which have platelets that are unresponsive to both PAR4 and GPVI agonists (Figure S6), α was reduced to a similar extent as in PAR4mKO samples while MA was dramatically reduced (Figure 4). In fact, MA was reduced to a similar extent observed with platelet depletion or inhibition of platelet contraction (Figure 1). The tyrosine kinase Syk is critical for GPVI-mediated platelet activation [37] and also plays a role in αIIbβ3 outside-in signaling [38]. Inhibiting Syk with PRT-2607 (PRT) slightly reduced MA compared to controls but had no effect on R or α (Figure 5). However, PAR4mKO samples treated with PRT displayed the near-maximal reduction in MA observed in PAR4mKO + JAQ1 samples, as well as a significant reduction in α (Figure 5).
Figure 4:
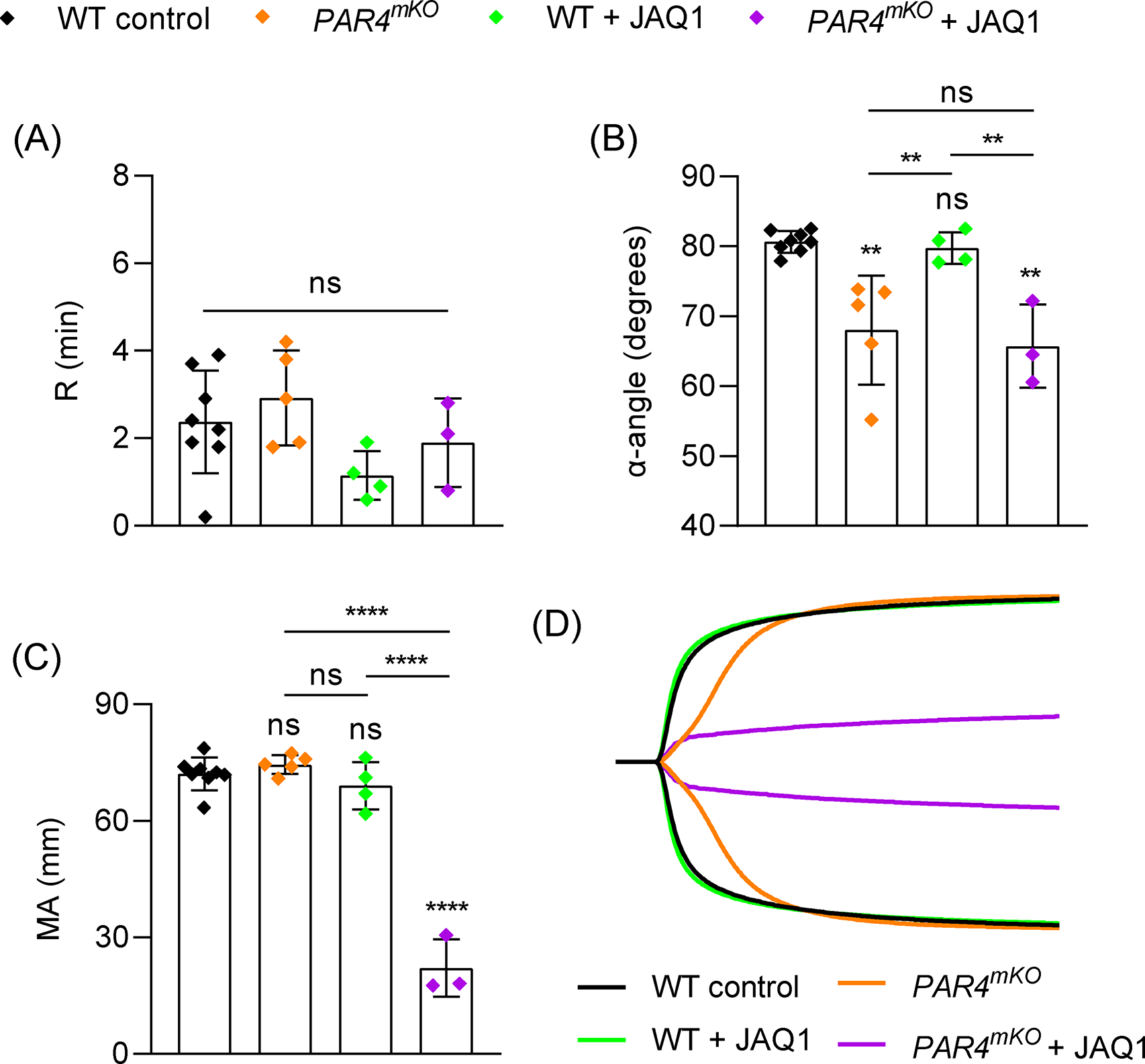
Role of platelet PAR4 and GPVI in TEG. Blood samples were analyzed from WT mice (n=8), mice with megakaryocyte/platelet-specific deletion of PAR4 (PAR4mKO, n=5), WT mice treated with anti-GPVI antibody to deplete GPVI on circulating platelets (JAQ1, 50 μg/mouse, n=4), or JAQ1-treated PAR4mKO mice (n=3). (A-C) TEG parameters: R time (A), α-angle (B) and MA (C). (D) Representative TEG traces. Data shown as mean ± SD. Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparison test. Symbols directly over bars represent significance compared to control. **P< .01, ****P < .0001.
Figure 5:
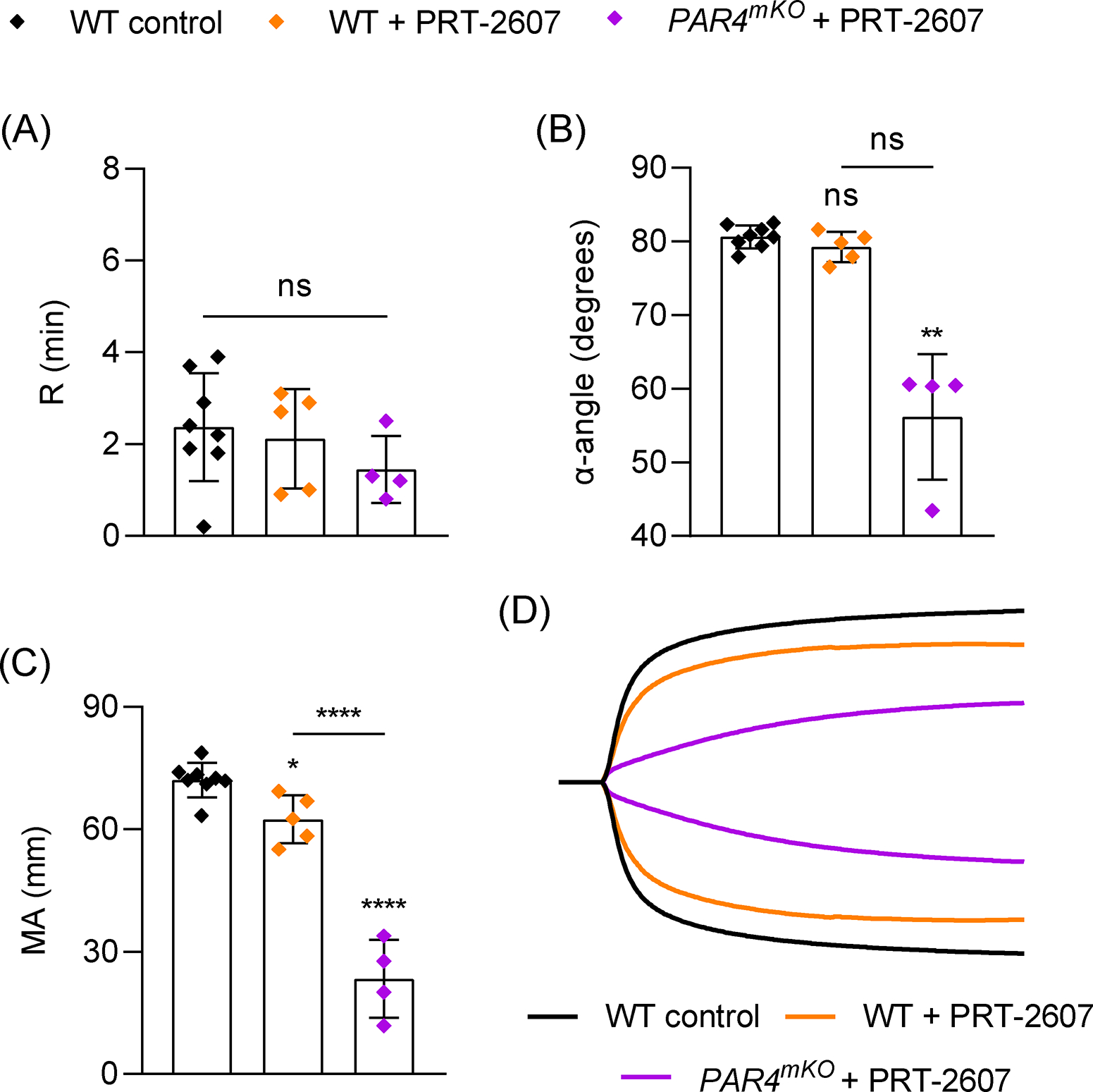
Role of Syk tyrosine kinase signaling in TEG. Blood samples were analyzed from WT mice (n=8), or WT (n=5) or PAR4mKO (n=4) mice with addition of the Syk inhibitor PRT-2607 (20 μM) 10 mins prior to TEG assay. (A-C) TEG parameters: R time (A), α-angle (B) and MA (C). (D) Representative TEG traces. Data shown as mean ± SD. Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparison test (A,C) or Kruskal-Wallis test with Dunn’s multiple comparison (B). Symbols directly over bars represent significance compared to control. **P < .01, ****P < .0001.
Human platelets express PAR4 but also express PAR1, both of which induce platelet signaling independently but can also heterodimerize [39]. To characterize the contribution of thrombin signaling to TEG in human blood, we simultaneously inhibited both PAR1 and PAR4 with the specific inhibitors vorapaxar (Vora) and BMS-986120 (BMS), respectively. Vora/BMS-treated human blood samples were comparable to DMSO vehicle samples in all parameters (Figure 6), demonstrating that thrombin activation through PAR1/4 is not the only mechanism that drives the platelet component in TEG. Similar results were observed in samples treated with PRT to inhibit Syk (Figure 6). As a positive control, αIIbβ3 ligand binding was blocked with an anti-αIIbβ3 antibody (abciximab), which significantly reduced MA (Figure S7). However, when human samples were simultaneously inhibited with Vora/BMS/PRT, we observed a marked reduction in α and MA (Figure 6). Collectively, these data reveal functional redundancy in mouse and human PARs and GPVI that drives platelet activation and contraction in TEG (Figure 7).
Figure 6:
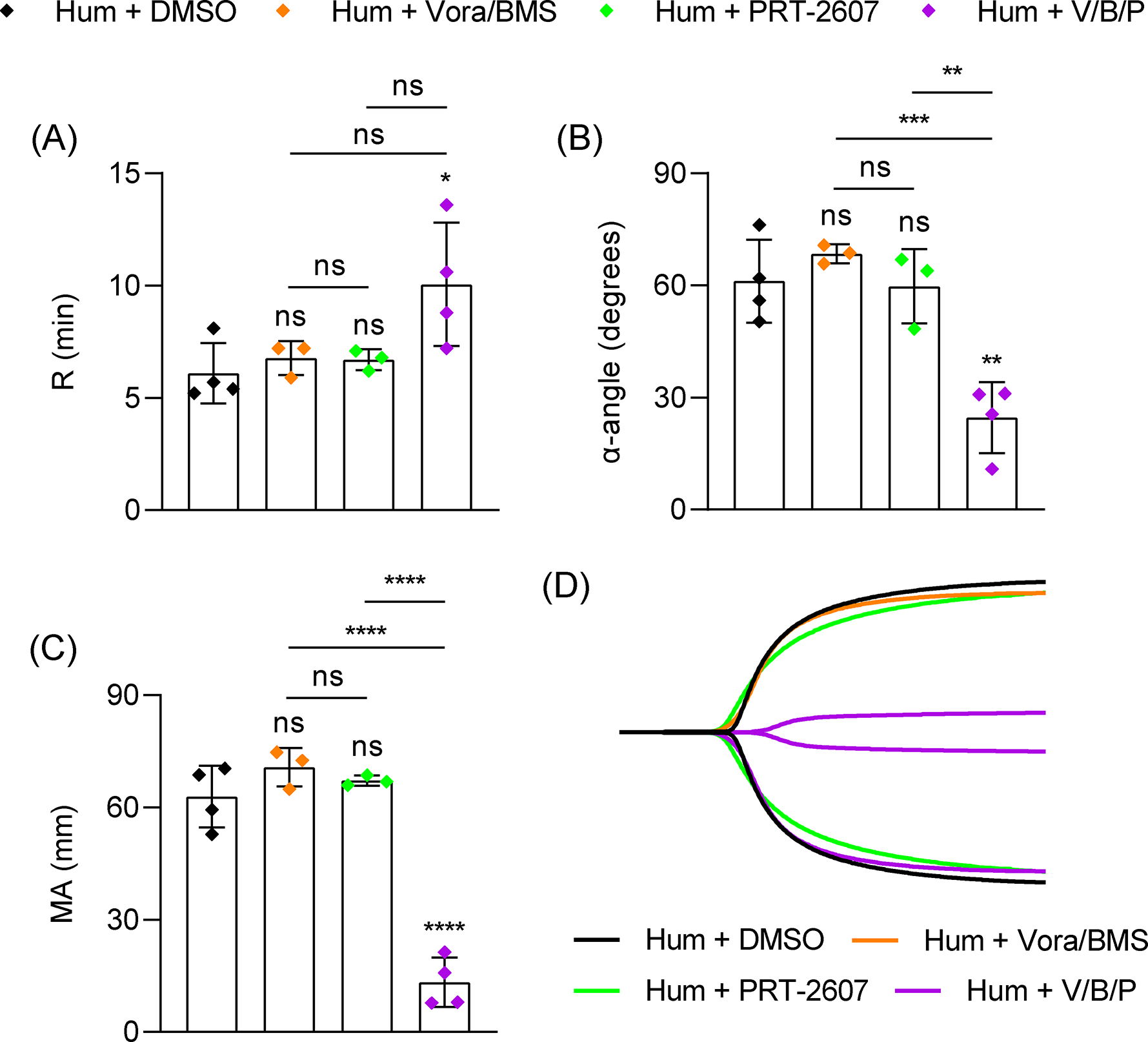
Role of PAR1/PAR4 and Syk in human blood TEG. Healthy volunteer blood samples were analyzed with addition of DMSO (n=4), vorapaxar (Vora, 5 μM, n=3), BMS-986120 (BMS, 10 μM, n=3) and PRT-2607 (PRT, 20 μM, n=4) to inhibit PAR1, PAR4 and Syk, respectively, 10 mins prior to TEG assay. (A-C) TEG parameters: R time (A), α-angle (B) and MA (C). (D) Representative TEG traces. Data shown as mean ± SD. Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparison test. Symbols directly over bars represent significance compared to control. *P < .05, **P< .01, *** P<0.001, ****P < .0001.
Figure 7:
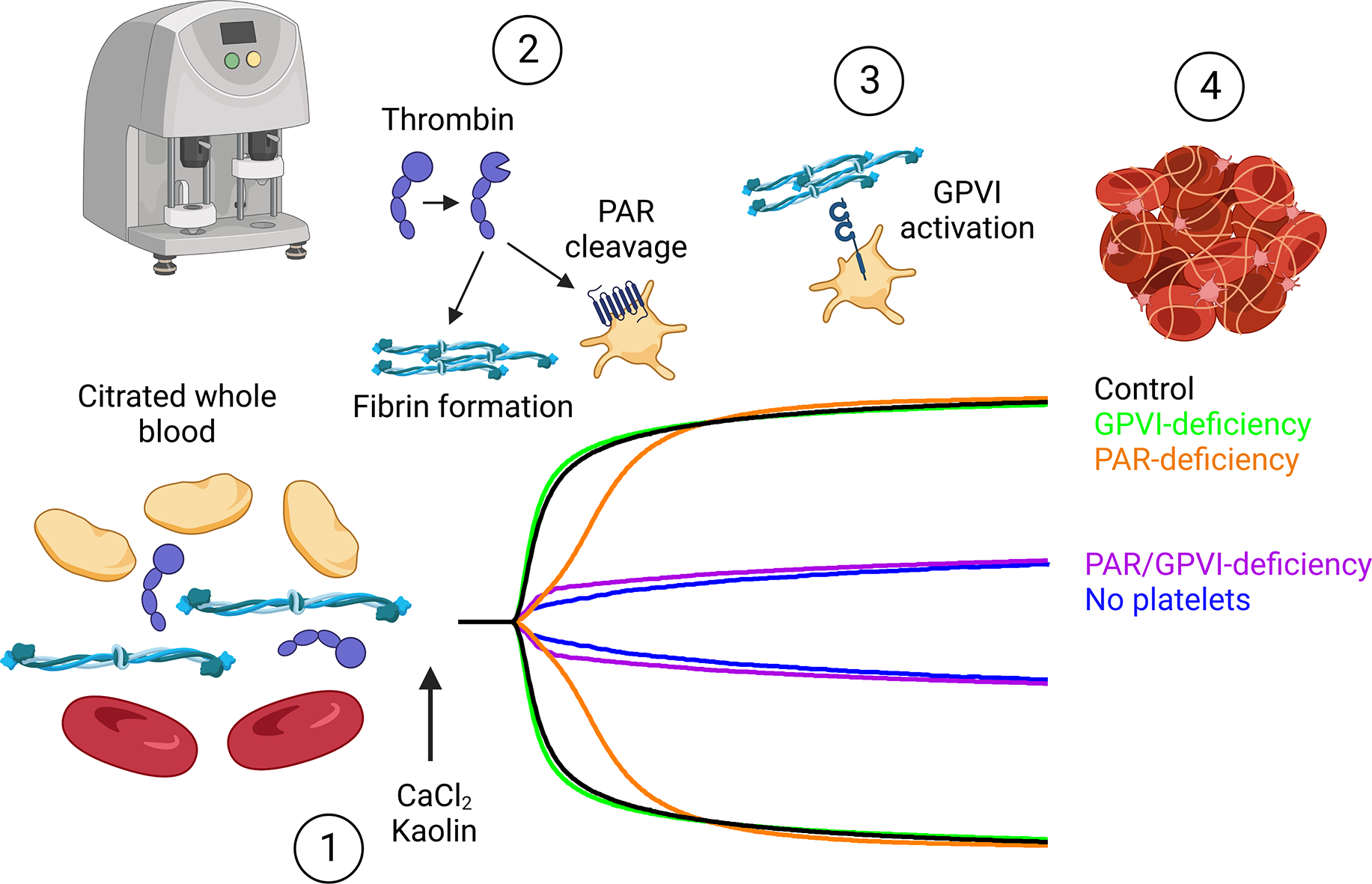
PAR1/4 and GPVI play redundant roles in platelet activation and contraction in TEG. (1) Analysis of clot formation speed and clot strength in TEG is performed by recalcifying citrated whole blood and activating with kaolin to initiate coagulation. (2) Within several minutes, thrombin generation initiates fibrinogen cleavage to fibrin, which polymerizes and crosslinks. Additionally, thrombin activates platelets through protease activated receptors (PARs) (PAR4 on mouse platelets, PAR1 and PAR4 on human platelets). Both fibrin polymerization and platelet activation contribute to the speed of clot formation (α-angle). (3) As more fibrin is generated, platelets can also bind fibrin via GPVI for additional activation signaling, resulting in robust αIIbβ3 integrin activation and platelet-mediated contraction of the fibrin clot. Clot strength (MA) is entirely dependent on platelet contraction. (4) The end result is a tightly contracted whole blood clot with contracting platelets bound to fibrin. While loss of either platelet PARs or GPVI alone has limited impact on TEG parameters, loss of both leads to TEG traces similar to platelet-depleted blood samples. Created with BioRender.com.
Discussion
The primary clinical utility of TEG is to guide transfusion of blood products in patients with active hemorrhage. Platelet Mapping TEG, which generates fibrin independently of thrombin and directly activates platelets [40], can identify defects in COX-1 and P2Y12 [41], but identifying other forms of platelet dysfunction relies on standard TEG. While TEG utilization may reduce unnecessary transfusions and costs, the impact on patient outcomes has not been universally demonstrated [3]. Regarding platelet transfusions, the lack of universal improvements in outcomes may be due to a disconnect between platelet function in TEG versus platelet function during hemostasis in vivo, and an incomplete understanding of the platelet contribution to TEG parameters. Our study uncovers several novel aspects of platelet activation and function in TEG: 1) platelet activation largely bypasses Rap1 GTPase signaling, which is critical for platelet integrin activation and aggregation under flow conditions, 2) PAR4 and GPVI play redundant roles in driving the platelet-dependent parameters α-angle and MA, and 3) standard TEG can effectively identify platelet contraction defects. We also observed redundant roles for PAR1/PAR4 and GPVI in human blood samples, demonstrating that this is not a species-specific phenomenon. While this study exclusively utilized TEG we anticipate that similar mechanisms would mediate the ROTEM equivalent of MA when using INTEM (maximum clot firmness, MCF), although this requires future investigation.
Integrin inside-out signaling through the Rap1/Talin1/αIIbβ3 axis is critical for platelet aggregation and hemostasis at sites of injury and deficiencies in this pathway are associated with moderate to severe bleeding in mice and humans [17,18,42]. For example, humans with mutations in CalDAG-GEFI can suffer from severe epistaxis and bleeding after dental extractions despite intact P2Y12 signaling [42]. However, we found that MA is driven by Talin1 and αIIbβ3 but not Rap1 signaling. TEG parameters were normal in mice with various Rap1 signaling defects, although Rap1a/bmKO mice had reduced α-angle and MA. However, Rap1a/bmKO mice also suffer from substantial thrombocytopenia [17], which may contribute to reduced parameters either independently or in conjunction with the platelet function defect in these mice. When activated with high concentrations of thrombin or PAR4p, platelets with defective Rap1 signaling demonstrate residual aggregation and clot contraction [11,17], and this may be sufficient to generate normal TEG values. In standard TEG, clotting is initiated with kaolin to drive rapid and robust activation of the contact pathway and virtually all prothrombin is converted to thrombin, so all platelets will be exposed to high concentrations of thrombin. In contrast to TEG, platelets at sites of injury in vivo during hemostasis and thrombosis undergo heterologous spatiotemporal activation by various agonists such as collagen, thrombin, and ADP [43]. Brass and Stalker described thrombi as containing a “core” of highly activated, degranulated platelets activated by thrombin and collagen surrounded by a “shell” of less strongly activated platelets with some platelets remaining discoid, heterogenous degranulation and activation by feedback mediators rather than primary agonists [44]. In our saphenous vein laser injury model, “core” platelets can be found around the edge of the injury whereas “shell” platelets bridge the center of the plug (unpublished observation), which is sensitive to antiplatelet therapy [45,46]. A similar arrangement of platelet phenotypes was reported in a larger venous puncture model [47]. In this context, platelet function in TEG is representative of a subset of highly activated core platelets, but it does not give a picture of overall platelet function.
Knowing that TEG is driven entirely by thrombin and that PAR4mKO platelets are unresponsive to this agonist [15], we expected to see markedly reduced MA in PAR4mKO samples, yet only observed a mild reduction in α. This was also unexpected as we and others have shown a critical role for PAR4 in hemostasis in mice [15,48]. The recent identification of a GPVI-fibrin(ogen) interaction [13,14] led us to investigate a potential role for GPVI in platelet activation and contraction during TEG. We found that GPVI/Syk can compensate for the lack of PAR4 in mouse platelets or inhibition of both PAR1 and PAR4 in human platelets. The literature describing GPVI-fibrin(ogen) interaction and function continues to evolve and is not lacking in controversy [49]. GPVI has been shown to bind fibrinogen [50], fibrin [13,14], and fibrin degradation products [51] with some species differences [50]. Candidate binding sites for GPVI on fibrin(ogen) include the αC and D regions [51–53]. Interestingly, one study suggested that GPVI can bind fibrin formed from recombinant fibrinogen but not from blood-sourced fibrinogen [54], and another demonstrated a lack of GPVI dimer binding to fibrin [55]. Studies to address whether the requirement for GPVI in TEG is indeed due to binding on fibrin will require specific tools to disrupt this interaction without affecting fibrin(ogen) structure or expression levels [56].
The finding that GPVI has a redundant role with PARs was surprising to us, as the activation of GPVI by fibrin(ogen) binding has not been described as a primary pathway for platelet integrin activation, but rather for supporting thrombus stability and thrombin generation [13,51,57]. We do not suspect that platelet-mediated thrombin generation is critical in kaolin TEG as we observed no differences in TEG parameters in samples from mice lacking cyclophilin D (unpublished observation), which mediates platelet procoagulant activity [58]. However, we expect the majority of fibrinogen to be converted to fibrin, and therefore the platelets are exposed to an extremely dense and abundant fibrin network which may drive GPVI clustering and greater activation [52]. However, the relevance of this interaction during hemostatic plug formation is unknown. Fibrin is mainly generated at the periphery or extravascular face of platelet plugs [45,47] and may only contact a subset of platelets. GPVI does play a minor supporting role during hemostasis in PAR4mKO mice [15], but whether this is due to GPVI interaction with collagen or fibrin(ogen) has not been determined. There may also be a role for active regulation of the platelet cytoskeleton by PARs and GPVI. PAR4 activates the heterotrimeric Gα13 protein which manipulates the cytoskeleton through regulation of Rho GTPases [59]. Engagement of PAR4 and GPVI triggers release of thromboxane A2, and the TxA2 receptor also connects to G13/Rho signaling [60].
Our results demonstrate that the platelet contribution to MA is almost entirely mediated through platelet contraction, as demonstrated by maximal reduction in MA with cytochalasin D. Interestingly, MA was not reduced to the maximal extent observed with platelet depletion or cytochalasin D when we inhibited αIIbβ3 function, suggesting receptors other than αIIbβ3 may contribute to clot contraction. Platelet contractile forces are critical for hemostasis and reduced platelet forces strongly associate with bleeding risk [61], but our data suggest that these defects may be masked in standard TEG due to robust thrombin activation and multiple redundant pathways for activation. Importantly, TEG is a static assay and does not account for defects in platelet function under flow conditions. Platelet interaction with VWF via GPIbα is a critical first step in the sequence of platelet adhesion and activation under flow, and platelet surface expression of GPIbα as well as GPVI may be altered in trauma [62].
We expect that our findings are relevant to clinical scenarios where kaolin (standard) TEG is used to determine the necessity for platelet transfusion. First, in trauma-associated hemorrhage, transfusion algorithms include MA thresholds which would trigger platelet transfusion [63]. Taken in the context of our findings, significant reductions in standard TEG MA would require substantial platelet dysfunction, affecting either multiple inside-out signaling pathways, integrin outside-in signaling, or a direct effect on αIIbβ3 itself. However, we and others have shown that platelet transfusion is ineffective in reversing platelet integrin dysfunction when platelet counts are within the normal range unless enough units are given to achieve specific transfused to endogenous platelet ratios [64]. Second, a patient with a normal MA by standard TEG may have platelet defects that contribute to ongoing hemorrhage but are masked in the TEG assay. The risk of missing these patients can be mitigated, at least in part, by the use of platelet mapping TEG; however, that assay is still specific to platelet dysfunction affecting ADP and thromboxane activation pathways. Inclusion of a GPVI function-blocking antibody as a TEG reagent to remove the GPVI/fibrin interaction may help unmask underlying platelet dysfunction and increase the sensitivity of TEG to additional pathological mechanisms. Interestingly, the GPVI blocking antibody fragment glenzocimab was recently shown to inhibit both collagen and fibrin-dependent GPVI activation [65].
Overall, our findings are in agreement with the literature that standard TEG is sensitive to thrombocytopenia and direct inhibition of αIIbβ3, but we demonstrate that multiple redundant pathways mediate platelet integrin activation and contraction in TEG. Moreover, these data suggest that TEG outcomes have little correlation with platelet function in vivo. We also demonstrate a novel role for GPVI in TEG, suggesting that further research is needed on the impact of GPVI-fibrin(ogen) interaction in various clotting assays.
Supplementary Material
Essentials.
Thromboelastography (TEG) helps evaluate bleeding risk at point-of-care and guide transfusion.
The platelet receptors and signaling pathways mediating TEG have not been fully investigated.
TEG is largely insensitive to inside-out signaling defects associated with major bleeding.
The receptors PAR1/4 and GPVI play redundant roles in the platelet contribution to TEG parameters.
Acknowledgements
We would like to acknowledge Dr. Valerie Tutwiler for providing helpful comments and Summer Jones for assistance with mouse husbandry.
Funding
This work was supported by the National Institutes of Health [R01 HL142799 (SA), R01 HL160046 (MJF), R01 HL126974 (ASW), P01 HL151433 (MG, WB), R35 HL144976 (WB)]; the American Society of Hematology [Scholar Award (RHL)]; and the National Blood Foundation [Early Career Grant (RHL)].
Footnotes
Conflict of Interest
The authors report no conflicts of interest.
References
- 1.Erdoes G, Koster A, Levy JH. Viscoelastic Coagulation Testing: Use and Current Limitations in Perioperative Decision-making. Anesthesiology; 2021; 135: 342–9. [DOI] [PubMed] [Google Scholar]
- 2.Raval JS, Burnett AE, Rollins-Raval MA, Griggs JR, Rosenbaum L, Nielsen ND, Harkins MS. Viscoelastic testing in COVID-19: a possible screening tool for severe disease? Transfusion (Paris); 2020; 60: 1131–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmidt AE, Israel AK, Refaai MA. The Utility of Thromboelastography to Guide Blood Product Transfusion. Am J Clin Pathol; 2019; 152: 407–22. [DOI] [PubMed] [Google Scholar]
- 4.Wikkelsø A, Wetterslev J, Møller AM, Afshari A. Thromboelastography (TEG) or thromboelastometry (ROTEM) to monitor haemostatic treatment versus usual care in adults or children with bleeding. Cochrane Database of Systematic Reviews; 2016; 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serraino GF, Murphy GJ. Routine use of viscoelastic blood tests for diagnosis and treatment of coagulopathic bleeding in cardiac surgery: Updated systematic review and meta-analysis. Br J Anaesth; 2017; 118: 823–33. [DOI] [PubMed] [Google Scholar]
- 6.Fleming K, Redfern RE, March RL, Bobulski N, Kuehne M, Chen JT, Moront M. TEG-directed transfusion in complex cardiac surgery: Impact on blood product usage. Journal of Extra-Corporeal Technology; 2017; 49: 283–90. [PMC free article] [PubMed] [Google Scholar]
- 7.Meco M, Montisci A, Giustiniano E, Greco M, Pappalardo F, Mammana L, Panisi P, Roscitano C, Cirri S, Donatelli F, Albano G . Viscoelastic Blood Tests Use in Adult Cardiac Surgery: Meta-Analysis, Meta-Regression, and Trial Sequential Analysis. J Cardiothorac Vasc Anesth; 2020; 34: 119–27. [DOI] [PubMed] [Google Scholar]
- 8.Franchini M, Mengoli C, Cruciani M, Marietta M, Marano G, Vaglio S, Pupella S, Veropalumbo E, Masiello F, Liumbruno GM. The use of viscoelastic haemostatic assays in non-cardiac surgical settings: A systematic review and meta-analysis. Blood Transfusion; 2018; 16: 235–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baksaas-Aasen K, Gall LS, Stensballe J, Juffermans NP, Curry N, Maegele M, Brooks A, Rourke C, Gillespie S, Murphy J, Maroni R, Vulliamy P, Henriksen HH, Pedersen KH, Kolstadbraaten KM, Wirtz MR, Kleinveld DJB, Schäfer N, Chinna S, Davenport RA, et al. Viscoelastic haemostatic assay augmented protocols for major trauma haemorrhage (ITACTIC): a randomized, controlled trial. Intensive Care Med; 2021; 47: 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drumheller BC, Stein DM, Moore LJ, Rizoli SB, Cohen MJ. Thromboelastography and rotational thromboelastometry for the surgical intensivist: A narrative review. Journal of Trauma and Acute Care Surgery; 2019; 86: 710–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lagarrigue F, Paul DS, Gingras AR, Valadez AJ, Sun H, Lin J, Cuevas MN, Ablack JN, Lopez-Ramirez MA, Bergmeier W, Ginsberg MH. Talin-1 is the principal platelet Rap1 effector of integrin activation. Blood 2020; 136: 1180–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Osdoit S, Rosa JP. Fibrin Clot Retraction by Human Platelets Correlates with αIIbβ3Integrin-dependent Protein Tyrosine Dephosphorylation *. Journal of Biological Chemistry; 2001; 276: 6703–10. [DOI] [PubMed] [Google Scholar]
- 13.Mammadova-Bach E, Ollivier V, Loyau S, Schaff M, Dumont B, Favier R, Freyburger G, Latger-Cannard V, Nieswandt B, Gachet C, Mangin PH, Jandrot-Perrus M. Platelet glycoprotein VI binds to polymerized fibrin and promotes thrombin generation. Blood 2015; 126: 683–91. [DOI] [PubMed] [Google Scholar]
- 14.Alshehri OM, Hughes CE, Montague S, Watson SK, Frampton J, Bender M, Watson SP. Fibrin activates GPVI in human and mouse platelets. Blood 2015; 126: 1601–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee RH, Kawano T, Grover SP, Bharathi V, Martinez D, Cowley DO, Mackman N, Bergmeier W, Antoniak S. Genetic deletion of platelet PAR4 results in reduced thrombosis and impaired hemostatic plug stability. Journal of Thrombosis and Haemostasis; 2022; 20: 422–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crittenden JR, Bergmeier W, Zhang Y, Piffath CL, Liang Y, Wagner DD, Housman DE, Graybiel AM. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat Med 2004; 10: 982–6. [DOI] [PubMed] [Google Scholar]
- 17.Stefanini L, Lee RH, Paul DS, O’Shaughnessy EC, Ghalloussi D, Jones CI, Boulaftali Y, Poe KO, Piatt R, Kechele DO, Caron KM, Hahn KM, Gibbins JM, Bergmeier W. Functional redundancy between RAP1 isoforms in murine platelet production and function. Blood 2018; 132: 1951–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petrich BG, Marchese P, Ruggeri ZM, Spiess S, Weichert RAM, Ye F, Tiedt R, Skoda RC, Monkley SJ, Critchley DR, Ginsberg MH. Talin is required for integrin-mediated platelet function in hemostasis and thrombosis. Journal of Experimental Medicine 2007; 204: 3103–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morrow DA, Braunwald E, Bonaca MP, Ameriso SF, Dalby AJ, Fish MP, Fox KAA, Lipka LJ, Liu X, Nicolau JC, Ophuis AJO, Paolasso E, Scirica BM, Spinar J, Theroux P, Wiviott SD, Strony J, Murphy SA. Vorapaxar in the Secondary Prevention of Atherothrombotic Events. New England Journal of Medicine; 2012; 366: 1404–13. [DOI] [PubMed] [Google Scholar]
- 20.Canault M, Ghalloussi D, Grosdidier C, Guinier M, Perret C, Chelghoum N, Germain M, Raslova H, Peiretti F, Morange PE, Saut N, Pillois X, Nurden AT, Cambien F, Pierres A, van den Berg TK, Kuijpers TW, Alessi MC, Tregouet DA. Human CalDAG-GEFI gene (RASGRP2) mutation affects platelet function and causes severe bleeding. Journal of Experimental Medicine 2014; 211: 1349–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khurana S, Mattson JC, Westley S, O’Neill WW, Timmis GC, Safian RD. Monitoring platelet glycoprotein IIb/IIIa-fibrin interaction with tissue factor-activated thromboelastography. Journal of Laboratory and Clinical Medicine; 1997; 130: 401–11. [DOI] [PubMed] [Google Scholar]
- 22.Dias JD, Lopez-Espina CG, Bliden K, Gurbel P, Hartmann J, Achneck HE. TEG®6s system measures the contributions of both platelet count and platelet function to clot formation at the site-of-care. Platelets; 2020; 31: 932–8. [DOI] [PubMed] [Google Scholar]
- 23.Racine-Brzostek SE, Asmis LM. Assessment of platelet function utilizing viscoelastic testing. Transfusion (Paris); 2020; 60: S10–20. [DOI] [PubMed] [Google Scholar]
- 24.Van Heerden P V, Gibbs NM, Michalopoulos N. Effect of low concentrations of prostacyclin on platelet function in vitro. Anaesth Intensive Care; 1997; 25: 343–6. [DOI] [PubMed] [Google Scholar]
- 25.Franchi F, Rollini F, Faz G, Rivas JR, Rivas A, Agarwal M, Briceno M, Wali M, Nawaz A, Silva G, Shaikh Z, Maaliki N, Fahmi K, Been L, Pineda AM, Suryadevara S, Soffer D, Zenni MM, Baber U, Mehran R, et al. Pharmacodynamic effects of vorapaxar in prior myocardial infarction patients treated with potent oral p2y12 receptor inhibitors with and without aspirin: Results of the vora-pratic study. J Am Heart Assoc; 2020; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bliden K, Chaudhary R, Kuliopulos A, Tran H, Taheri H, Tehrani B, Rosenblatt A, Navarese E, Tantry US, Gurbel PA. Effects of vorapaxar on clot characteristics, coagulation, inflammation, and platelet and endothelial function in patients treated with mono- and dual-antiplatelet therapy. Journal of Thrombosis and Haemostasis; 2020; 18: 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.André P, Delaney SM, LaRocca T, Vincent D, DeGuzman F, Jurek M, Koller B, Phillips DR, Conley PB. P2Y12 regulates platelet adhesion/activation, thrombus growth, and thrombus stability in injured arteries. Journal of Clinical Investigation 2003; 112: 398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holmbäck K, Danton MJS, Suh TT, Daugherty CC, Degen JL. Impaired platelet aggregation and sustained bleeding in mice lacking the fibrinogen motif bound by integrin α(IIb)β3. EMBO Journal 1996; 15: 5760–71. [PMC free article] [PubMed] [Google Scholar]
- 29.Nieswandt B, Schulte V, Bergmeier W, Mokhtari-Nejad R, Rackebrandt K, Cazenave JP, Ohlmann P, Gachet C, Zirngibl H. Long-term antithrombotic protection by in vivo depletion of platelet glycoprotein VI in mice. Journal of Experimental Medicine 2001; 193: 459–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schroeder JA, Kuether EA, Fang J, Jing W, Weiler H, Wilcox DA, Montgomery RR, Shi Q. Thromboelastometry assessment of hemostatic properties in various murine models with coagulopathy and the effect of factor VIII therapeutics. Journal of Thrombosis and Haemostasis; 2021; 19: 2417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen I, Gerrard JM, White JG. Ultrastructure of clots during isometric contraction. Journal of Cell Biology; 1982; 93: 775–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Osdoit S, Rosa JP. Fibrin Clot Retraction by Human Platelets Correlates with α IIbβ3 Integrin-dependent Protein Tyrosine Dephosphorylation. Journal of Biological Chemistry; 2001; 276: 6703–10. [DOI] [PubMed] [Google Scholar]
- 33.Höök P, Litvinov RI, Kim OV., Xu S, Xu Z, Bennett JS, Alber MS, Weisel JW. Strong Binding of Platelet Integrin αiIbβ3 to Fibrin Clots: Potential Target to Destabilize Thrombi. Sci Rep; 2017; 7: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chrzanowska-Wodnicka M, Smyth SS, Schoenwaelder SM, Fischer TH, White GC. Rap1b is required for normal platelet function and hemostasis in mice. Journal of Clinical Investigation 2005; 115: 680–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cifuni SM, Wagner DD, Bergmeier W. CalDAG-GEFI and protein kinase C represent alternative pathways leading to activation of integrin αIIbβ3 in platelets. Blood 2008; 112: 1696–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sambrano GR, Weiss EJ, Zheng YW, Huang W, Coughlin SR. Role of thrombin signalling in platelets in haemostasis and thrombosis. Nature 2001; 413: 74–8. [DOI] [PubMed] [Google Scholar]
- 37.Zheng TJ, Lofurno ER, Melrose AR, Lakshmanan HHS, Pang J, Phillips KG, Fallon ME, Kohs TCL, Ngo ATP, Shatzel JJ, Hinds MT, McCarty OJT, Aslan JE. Assessment of the effects of Syk and BTK inhibitors on GPVI-mediated platelet signaling and function. Am J Physiol Cell Physiol; 2021; 320: C902–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woodside DG, Obergfell A, Leng L, Wilsbacher JL, Miranti CK, Brugge JS, Shattil SJ, Ginsberg MH. Activation of Syk protein tyrosine kinase through interaction with integrin β cytoplasmic domains. Current Biology; 2001; 11: 1799–804. [DOI] [PubMed] [Google Scholar]
- 39.Han X, Nieman MT, Kerlin BA. Protease-activated receptors: An illustrated review. Res Pract Thromb Haemost; 2021; 5: 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Craft RM, Chavez JJ, Bresee SJ, Wortham DC, Cohen E, Carroll RC. A novel modification of the Thrombelastograph assay, isolating platelet function, correlates with optical platelet aggregation. Journal of Laboratory and Clinical Medicine; 2004; 143: 301–9. [DOI] [PubMed] [Google Scholar]
- 41.Nekludov M, Bellander BM, Blombäck M, Wallen HN. Platelet dysfunction in patients with severe traumatic brain injury. J Neurotrauma; 2007; 24: 1699–706. [DOI] [PubMed] [Google Scholar]
- 42.Palma-Barqueros V, Ruiz-Pividal J, Bohdan N, Vicente V, Bastida JM, Lozano M, Rivera J. RASGRP2 gene variations associated with platelet dysfunction and bleeding. Platelets; 2019; 30: 535–9. [DOI] [PubMed] [Google Scholar]
- 43.Welsh JD, Poventud-Fuentes I, Sampietro S, Diamond SL, Stalker TJ, Brass LF. Hierarchical organization of the hemostatic response to penetrating injuries in the mouse macrovasculature. Journal of Thrombosis and Haemostasis; 2017; 15: 526–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stalker TJ, Traxler EA, Wu J, Wannemacher KM, Cermignano SL, Voronov R, Diamond SL, Brass LF. Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood 2013; 121: 1875–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Getz TM, Piatt R, Petrich BG, Monroe D, Mackman N, Bergmeier W. Novel mouse hemostasis model for real-time determination of bleeding time and hemostatic plug composition. Journal of Thrombosis and Haemostasis 2015; 13: 417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paul DS, Bergmeier W. Novel mouse model for studying hemostatic function of human platelets. Arterioscler Thromb Vasc Biol 2020; 40: 1891–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tomaiuolo M, Matzko CN, Poventud-Fuentes I, Weisel JW, Brass LF, Stalker TJ. Interrelationships between structure and function during the hemostatic response to injury. Proc Natl Acad Sci; 2019; 116: 2243–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hamilton JR, Cornelissen I, Coughlin SR. Impaired hemostasis and protection against thrombosis in protease-activated receptor 4-deficient mice is due to lack of thrombin signaling in platelets. Journal of Thrombosis and Haemostasis 2004; 2: 1429–35. [DOI] [PubMed] [Google Scholar]
- 49.Slater A, Perrella G, Onselaer MB, Martin EM, Gauer JS, Xu RG, Heemskerk JWM, Ariëns RAS, Watson SP. Does fibrin(ogen) bind to monomeric or dimeric GPVI, or not at all? Platelets 2019; 30: 281–9. [DOI] [PubMed] [Google Scholar]
- 50.Mangin PH, Onselaer MB, Receveur N, Le Lay N, Hardy AT, Wilson C, Sanchez X, Loyau S, Dupuis A, Babar AK, Miller JLC, Philippou H, Hughes CE, Herr AB, Ariëns RAS, Mezzano D, Jandrot-Perrus M, Gachet C, Watson SP. Immobilized fibrinogen activates human platelets through glycoprotein VI. Haematologica; 2018; 103: 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Induruwa I, Moroi M, Bonna A, Malcor JD, Howes JM, Warburton EA, Farndale RW, Jung SM. Platelet collagen receptor Glycoprotein VI-dimer recognizes fibrinogen and fibrin through their D-domains, contributing to platelet adhesion and activation during thrombus formation. Journal of Thrombosis and Haemostasis; 2018; 16: 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu RG, Gauer JS, Baker SR, Slater A, Martin EM, McPherson HR, Duval C, Manfield IW, Bonna AM, Watson SP, Ariëns RAS. GPVI (Glycoprotein VI) Interaction With Fibrinogen Is Mediated by Avidity and the Fibrinogen αC-Region. Arterioscler Thromb Vasc Biol; 2021; 41: 1092–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Onselaer MB, Hardy AT, Wilson C, Sanchez X, Babar AK, Miller JLC, Watson CN, Watson SK, Bonna A, Philippou H, Herr AB, Mezzano D, Ariëns RAS, Watson SP. Fibrin and D-dimer bind to monomeric GPVI. Blood Adv; 2017; 1: 1495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang D, Ebrahim M, Adler K, Blanchet X, Jamasbi J, Megens RTA, Uhland K, Ungerer M, Münch G, Deckmyn H, Weber C, Elia N, Lorenz R, Siess W. Glycoprotein VI is not a Functional Platelet Receptor for Fibrin Formed in Plasma or Blood. Thromb Haemost; 2020; 120: 977–93. [DOI] [PubMed] [Google Scholar]
- 55.Ebrahim M, Jamasbi J, Adler K, Megens RTA, M’Bengue Y, Blanchet X, Uhland K, Ungerer M, Brandl R, Weber C, Elia N, Lorenz R, Münch G, Siess W. Dimeric Glycoprotein VI Binds to Collagen but Not to Fibrin. Thromb Haemost; 2018; 118: 351–61. [DOI] [PubMed] [Google Scholar]
- 56.Hur WS, Paul DS, Bouck EG, Negrón OA, Mwiza JM, Poole LG, Cline-Fedewa HM, Clark EG, Juang LJ, Leung J, Kastrup CJ, Ugarova TP, Wolberg AS, Luyendyk JP, Bergmeier W, Flick MJ. Hypofibrinogenemia with preserved hemostasis and protection from thrombosis in mice with an Fga truncation mutation. Blood; 2022; 139: 1374–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ahmed MU, Kaneva V, Loyau S, Nechipurenko D, Receveur N, Le Bris M, Janus-Bell E, Didelot M, Rauch A, Susen S, Chakfé N, Lanza F, Gardiner EE, Andrews RK, Panteleev M, Gachet C, Jandrot-Perrus M, Mangin PH. Pharmacological Blockade of Glycoprotein VI Promotes Thrombus Disaggregation in the Absence of Thrombin. Arterioscler Thromb Vasc Biol; 2020; 40: 2127–42. [DOI] [PubMed] [Google Scholar]
- 58.Jobe SM, Wilson KM, Leo L, Raimondi A, Molkentin JD, Lentz SR, Di Paola J. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood; 2008; 111: 1257–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aslan JE, Mccarty OJT. Rho GTPases in platelet function. J Thromb Haemost; 2013; 11: 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Djellas Y, Manganello JM, Antonakis K, Le Breton GC. Identification of Gα13 as one of the G-proteins that couple to human platelet thromboxane A2 receptors. Journal of Biological Chemistry; 1999; 274: 14325–30. [DOI] [PubMed] [Google Scholar]
- 61.Ting LH, Feghhi S, Taparia N, Smith AO, Karchin A, Lim E, John AS, Wang X, Rue T, White NJ, Sniadecki NJ. Contractile forces in platelet aggregates under microfluidic shear gradients reflect platelet inhibition and bleeding risk. Nat Commun; 2019; 10: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vulliamy P, Montague SJ, Gillespie S, Chan MV., Coupland LA, Andrews RK, Warner TD, Gardiner EE, Brohi K, Armstrong PC. Loss of GPVI and GPIbα contributes to trauma-induced platelet dysfunction in severely injured patients. Blood Adv; 2020; 4: 2623–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stettler GR, Sumislawski JJ, Moore EE, Nunns GR, Kornblith LZ, Conroy AS, Callcut RA, Silliman CC, Banerjee A, Cohen MJ, Sauaia A. Citrated kaolin thrombelastography (TEG) thresholds for goal-directed therapy in injured patients receiving massive transfusion. J Trauma Acute Care Surg; 2018; 85: 734–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee RH, Piatt R, Dhenge A, Lozano ML, Palma-Barqueros V, Rivera J, Bergmeier W. Impaired hemostatic activity of healthy transfused platelets in inherited and acquired platelet disorders: Mechanisms and implications. Sci Transl Med 2019; 11: eaay0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Billiald P, Jiacomini I, Rose SARomics Biostructures N, Kristell Lebozec Acticor Biotech SS, F Elie Toledano Acticor-Biotech, Deborah François Acticor Biotech FS, Steve Watson F, Slater A, Welin M, Clark JC, Loyau S, Pugnière M, Jiacomini IG, Rose N, Lebozec K, Toledano E, François D, Watson SP, Jandrot-Perrus M. Targeting platelet GPVI with glenzocimab: a novel mechanism for inhibition. Blood Adv; 2022; Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.