

Abstract
Platelet transfusions can fail to prevent bleeding in patients with inherited platelet function disorders (IPDs), such as Glanzmann’s Thrombasthenia (GT; integrin αIIbβ3 dysfunction), Bernard-Soulier Syndrome (BSS; GPIb/IX/V dysfunction), and the more recently identified non-syndromic RASGRP2 variants. Here we used IPD mouse models and real-time imaging of hemostatic plug formation to investigate whether dysfunctional platelets impair the hemostatic function of healthy donor (wild-type, WT) platelets. In Rasgrp2−/− mice or mice deficient in the integrin adaptor protein TALIN1 (Talin1f/f x Pf4-Cre+; “GT-like”), WT platelet transfusion was ineffective unless the ratio between mutant and WT platelets was ~ 2:1. In contrast, thrombocytopenic mice or mice lacking the extracellular domain of GPIbα (“BSS-like”) required very few transfused WT platelets to normalize hemostasis. Both Rasgrp2−/− and “GT-like”, but not “BSS-like” platelets effectively localized to the injury site. Mechanistic studies identified at least two mechanisms of interference by dysfunctional platelets in IPDs: (1) delayed adhesion of WT donor platelets due to reduced access to GPIbα ligands exposed at sites of vascular injury, and (2) impaired consolidation of the hemostatic plug. We also investigated the hemostatic activity of transfused platelets in the setting of dual anti-platelet therapy (DAPT), an acquired platelet function disorder (APD). “DAPT” platelets did not prolong the time to initial hemostasis, but plugs were unstable and frequent rebleeding was observed. Thus, we propose that the endogenous platelet count and the ratio of transfused versus endogenous platelets should be considered when treating select IPD and APD patients with platelet transfusions.
One Sentence Summary:
In select platelet disorders, dysfunctional endogenous platelets affect the hemostatic activity of transfused platelets by multiple mechanisms.
Introduction
Platelets are the essential cellular mediators of primary hemostasis. Following blood vessel damage, platelets make transient contacts with von Willebrand Factor (vWF) exposed in the extracellular matrix(1), mediated by the glycoprotein (GP) Ib/V/IX complex. Reduced platelet velocity then allows for platelet activation via engagement of immune-type and G protein-coupled receptors(2, 3). Activation of these receptors triggers a rapid increase in cytosolic Ca2+ concentrations, leading to the activation of calcium and diacylglycerol-regulated guanine nucleotide exchange factor (CalDAG-GEFI; gene name RASGRP2) and in turn, the small GTPase RAP1. Active RAP1 engages the cytoskeletal adapter protein, TALIN, to induce a conformational change in αIIbβ3 integrin, resulting in the high affinity state required for fibrinogen binding and platelet aggregation(4, 5).
Both clinical and experimental observations have shown that loss of expression or function of any of these molecular components leads to impaired platelet-mediated hemostasis and bleeding. Bernard-Soulier Syndrome (BSS), a rare but severe bleeding diathesis, results from molecular pathology in genes encoding the subunits of GPIb/V/IX, leading to loss of expression or function of the major platelet receptor for vWF, and is often associated with macrothrombocytopenia(6). Glanzmann’s Thrombasthenia (GT) is caused by mutations to either subunit of the αIIbβ3 integrin, leading to loss of function/expression of the complex, abolished platelet aggregation, and severe bleeding(7, 8). More recently, patients have been identified with mutations in RASGRP2(9–17). In agreement with the mouse model(18), human platelets lacking CalDAG-GEFI function exhibit a markedly impaired integrin activation response to various agonists. As a result, patients with RASGRP2 mutations present with a moderate to severe bleeding diathesis, including bleeding following tooth extraction, excessive menstrual bleeding and bleeding complications following pregnancy.
To prevent or reverse bleeding in RASGRP2 patients, a number of treatments have been used, with varying success. The two major treatment options are administration of recombinant active FVII (rFVIIa; NovoSeven®)(19) and platelet transfusions, or both. Unlike BSS or GT patients, RASGRP2 patients have normal surface glycoprotein expression(15) and are unlikely to generate anti-platelet antibodies against the major platelet glycoproteins following transfusions; because of this, and the high cost of recombinant coagulation factors, platelet transfusion is a desired treatment option. To date, however, transfusion is reported to be ineffective in some patients. For example, a recent study of a family with a truncation deletion in the C-terminal regulatory domain of RASGRP2 (c.1490delT)(15) demonstrated that perioperative platelet transfusion was unsuccessful in controlling bleeding in two homozygous family members. Cessation of bleeding occurred only after administration of rFVIIa.
Besides inherited disorders, patients on anti-platelet therapy may also require platelet transfusion. Platelet inhibitors are widely used in the prevention of thrombotic events but carry an inherent risk of bleeding. The current gold standard in the prevention of secondary cardiovascular events is dual anti-platelet therapy (DAPT) with low-dose aspirin and a P2Y12 inhibitor (typically clopidogrel, prasugrel or ticagrelor)(20, 21). Patients on DAPT who require emergency surgical intervention often receive platelet transfusion ahead of the procedure to prevent bleeding(22), but the effectiveness of transfusion in these patients is not entirely understood(23). The inability to control peri- and postoperative bleeding is a major concern in patients with platelet function disorders(24). Therefore, it is of great clinical relevance to understand why certain patients with platelet dysfunction do not benefit from platelet transfusion.
In this work, we used the mouse model system to investigate the interaction of dysfunctional platelets with healthy (wild-type, WT) platelets at sites of vascular injury. To analyze transfusion effectiveness in vivo, we used two experimental approaches: (1) transfusion of WT platelets into murine models of inherited platelet disorders and (2) recipient mice were rendered thrombocytopenic (TP) and transfused with specific ratios of WT and dysfunctional platelets. Our studies suggest that platelets with impaired integrin activation compete with WT platelets for GPIbα binding sites on vWF, exposed at sites of vascular injury, or other GPIb ligands, and that this competition negatively affects the hemostatic activity of transfused WT platelets. Spinning disk confocal analysis of hemostatic plugs further identified negative effects of dysfunctional platelets on the composition of the plugs. Additionally, in the presence of “DAPT” platelets, hemostatic plugs formed rapidly but demonstrated instability and reopening. We conclude that the endogenous platelet count and the ratio of transfused versus endogenous platelets would be useful in predicting the success of platelet transfusion therapy in patients with platelet integrin signaling defects.
Results
Healthy transfused platelets have reduced hemostatic efficacy in the presence of Rasgrp2−/− platelets
Deficiency in CalDAG-GEFI causes severe bleeding in mice(18), and moderate to severe bleeding in humans often requiring platelet transfusions(17). In RASGRP2 patients, transfused healthy platelets need to function in the presence of a normal number of dysfunctional platelets. We sought to examine the potential competition between transfused and endogenous platelets during hemostasis. To investigate aspects of hemostatic plug formation in vivo, we performed top-down penetrating laser injury of the murine saphenous vein coupled with intravital microscopy(25). This model allows for the quantification of bleeding time (time-to-hemostasis) with simultaneous visualization of fluorescently labeled platelets and other blood components. We first determined the hemostatic impact of wild-type (WT) platelets transfused into Rasgrp2−/− mice (Fig. 1A). WT mice formed a hemostatic plug in 20–30 seconds (Fig. 1B,C, purple circles/line)(25). In contrast, Rasgrp2−/− mice typically bled for the entire observation period (270 sec)(26), with only few injuries reaching hemostasis (Fig. 1B,C, red circles/line). As expected, platelet-depleted mice (thrombocytopenic IL-4R/GPIb-Tg mice; TP Tg) also bled throughout the entire observation period (Fig. 1B,C, black circles/line). Consistent with recent studies demonstrating that a low peripheral platelet count (~5–10% of normal) is sufficient for hemostasis in mice(27), we were able to prevent bleeding in TP mice by transfusion of WT platelets to achieve a peripheral count of ~5×107/mL (Fig. 1B,C, open black circles/dashed line). Interestingly, transfusing twice that number of WT platelets into non-depleted Rasgrp2−/− mice (resulting in a ~10:1 ratio of mutant (endogenous) to WT (transfused) platelets) had only a minimal effect on the bleeding time following laser injury (Fig. 1B,C, blue circles/line). To secure hemostasis, Rasgrp2−/− mice had to be transfused with considerably more WT platelets than were required in TP mice (Fig. 1B,C, orange circles/line). A ratio of ~ 3:1 Rasgrp2−/−:WT platelets was established as the threshold required to normalize hemostasis when transfusing WT platelets into Rasgrp2−/− mice.
Fig. 1: Rasgrp2−/− mice require a large number of transfused WT platelets for hemostasis.
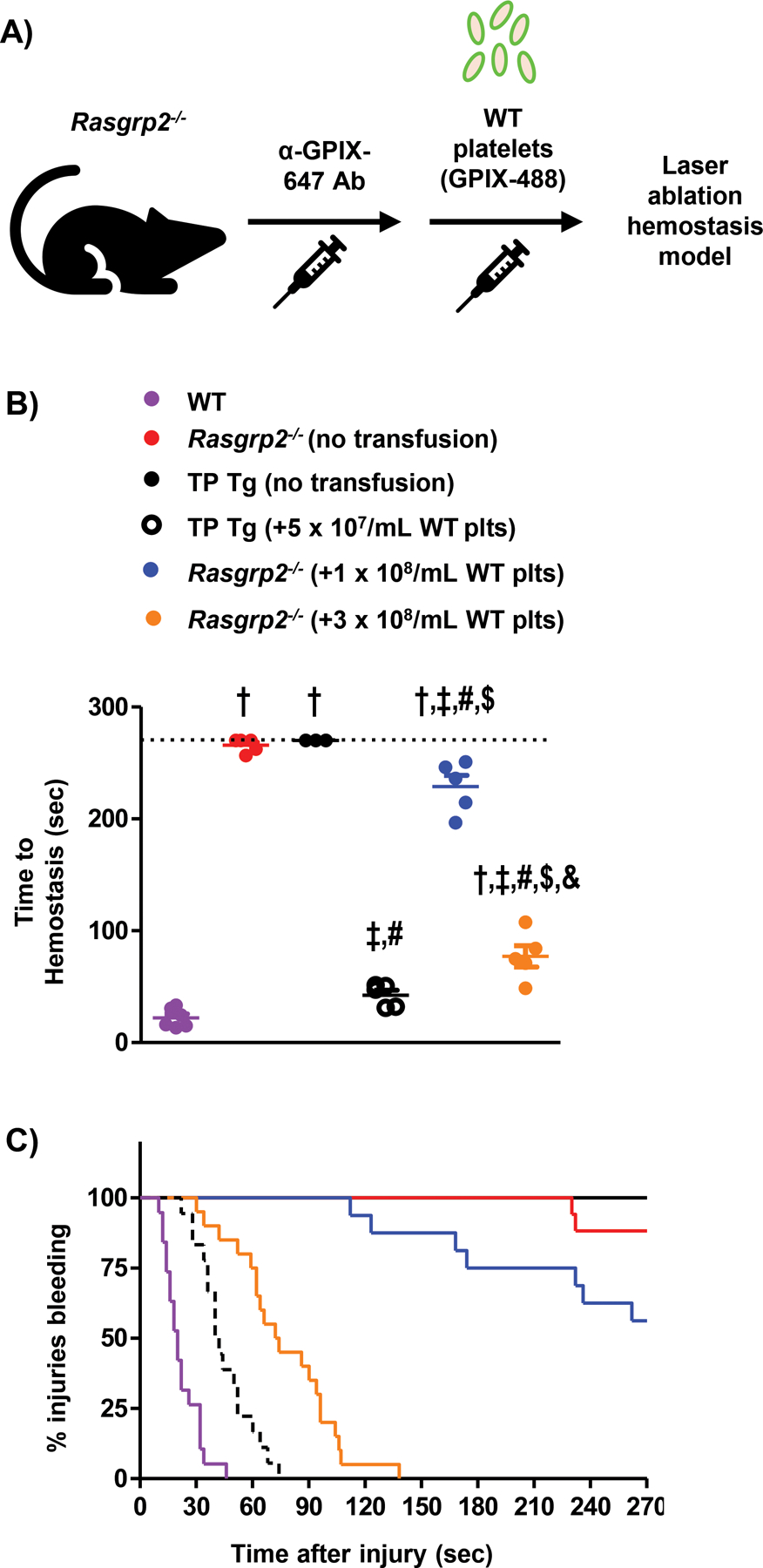
(A) Model depicting platelet (plt) transfusion scheme in Rasgrp2−/− mice. Endogenous plts were labeled by injection of anti-GPIX-647 antibody prior to transfusion of GPIX-488 labeled WT plts. (B) Comparison of bleeding times in the saphenous vein laser ablation model. Rasgrp2−/− mice were transfused, or not, with a low (1 × 108/mL) or high (3 × 108/mL) number of WT plts, and compared with WT mice or plt-depleted IL4R-GPIb-Tg mice (thrombocytopenic (TP) Tg) transfused, or not, with a very low (5 × 107/mL) number of WT plts. Each dot represents the average time-to-hemostasis for 4–6 individual injuries in one mouse; n=3–6 mice per group, data shown as mean ± SEM. P<0.05: † vs. WT, ‡ vs. Rasgrp2−/− (no transfusion), # vs. TP Tg (no transfusion), $ vs. TP Tg (+5 × 107/mL WT plts), & vs. Rasgrp2−/− (+1 × 108/mL WT plts). (C) Kaplan-Meier curve representation of bleeding time data, including all individual injury sites. Note that >50% of injuries are still bleeding at the end of the experiment in Rasgrp2−/− mice receiving 1 × 108/mL WT plts.
We next performed adoptive transfer studies with differentially labeled WT and Rasgrp2−/− platelets in TP IL-4R/GPIb-Tg recipient mice (TP Tg)(Fig. 2A)(28). Flow cytometry dot plots show depletion and reconstitution of Tg recipient mice, and both the WT and Rasgrp2−/− (KO) platelet populations were clearly distinguishable by their fluorescence signals with no double-staining of platelets (Fig. 2B). Furthermore, WT and Rasgrp2−/− platelets in whole blood from transfused mice demonstrated similar integrin activation profiles to endogenous platelets from WT and Rasgrp2−/− mice, suggesting that platelet function was minimally affected by the labeling and/or transfusion process (fig. S1). Thrombocytopenic mice receiving WT platelets alone rapidly achieved hemostasis following laser injury (Fig. 2C,D, purple circles/line; movie S1), while mice receiving Rasgrp2−/− platelets alone bled for almost the entirety of the observation period (Fig. 2C,D red circles/line; movie S2). Guided by our findings from WT platelet transfusions into Rasgrp2−/− mice (Fig. 1), we transfused mutant and WT platelets at a ratio of either 5:1 or 2:1 into TP Tg mice (with the absolute number of WT platelets kept constant). In agreement with the results obtained in Rasgrp2−/− mice, a ratio of 2:1 Rasgrp2−/−:WT platelets was required to normalize time-to-hemostasis (Fig. 2C,D, blue and orange circles/lines; movies S3 & S4, respectively). Importantly, these results demonstrated that increasing numbers of Rasgrp2−/− platelets led to prolonged bleeding times in mice transfused with the same absolute number of WT platelets, strongly suggesting that Rasgrp2−/− platelets impair the hemostatic efficacy of WT platelets.
Fig. 2: Co-transfusion of 2:1 Rasgrp2−/−:WT platelets is required for normal hemostasis.
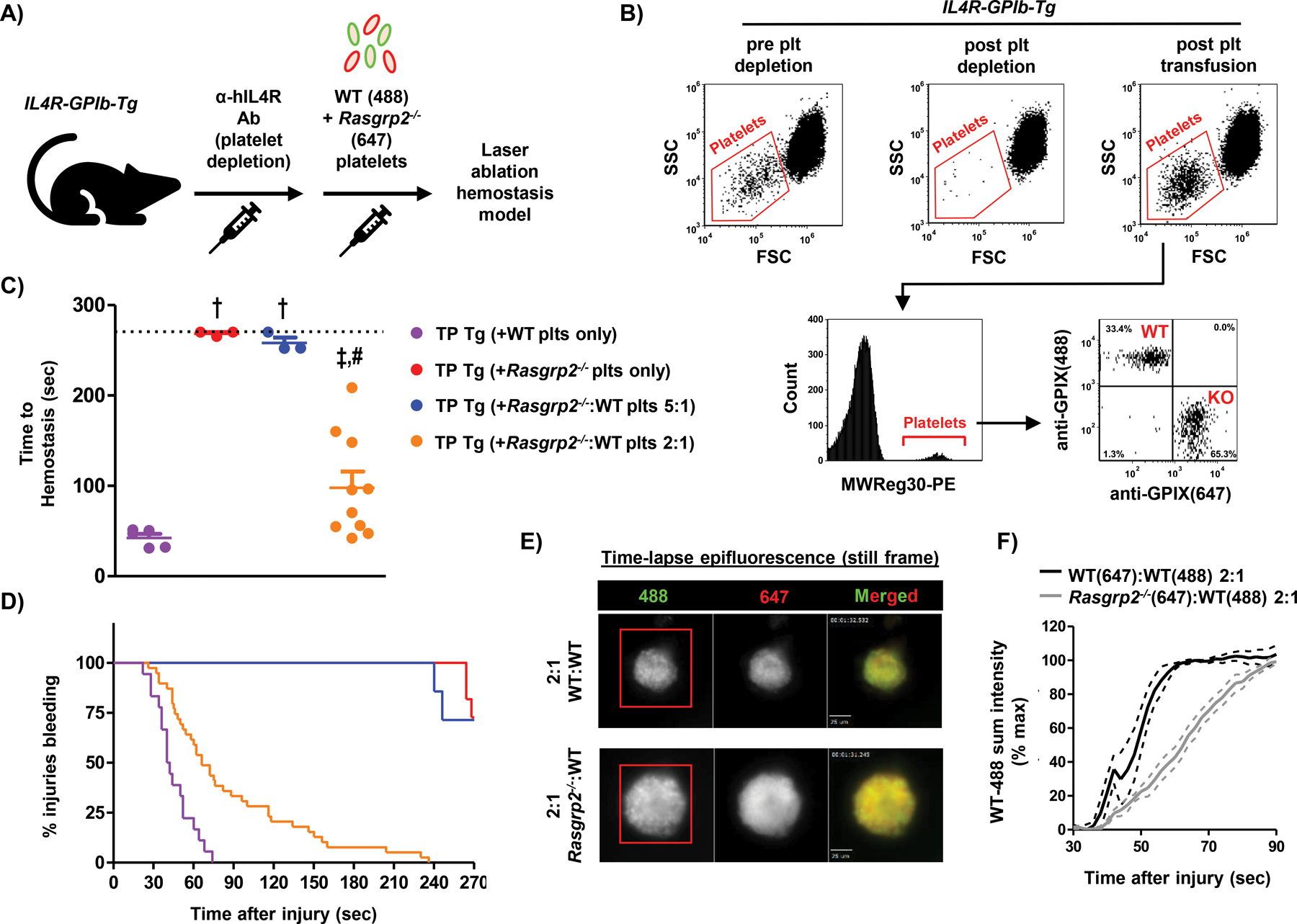
(A) Model depicting platelet (plt) transfusion scheme. IL4R-GPIb-Tg mice were depleted of endogenous plts (thrombocytopenic (TP) Tg) and transfused with labeled WT and/or Rasgrp2−/− plts at the desired ratios, before undergoing the laser ablation hemostasis model. (B) Flow cytometry dot plots of whole blood from IL4R-GPIb-Tg mice before and after endogenous plt depletion, and after WT/Rasgrp2−/− plt transfusion. In blood collected post-transfusion, the anti-CD41 antibody MWReg30 was used to label all transfused platelets and distinct populations of differentially labeled WT(488) and Rasgrp2−/−(647) plts were observed. (C) TP Tg mice were transfused with WT or Rasgrp2−/− plts only, or ratios of Rasgrp2−/−:WT plts, and time-to-hemostasis was determined in the saphenous vein laser ablation model (movies S1–S4). n=3–10 per group, data shown as mean ± SEM. P<0.05: † vs. TP Tg (+WT plts only), ‡ vs. TP Tg (+Rasgrp2−/− plts only), # vs. TP Tg (+Rasgrp2−/−:WT plts 5:1). (D) Kaplan-Meier curve representation of bleeding time data, including all individual injury sites. (E) Still frame images from epifluorescence videos at ~60 seconds post-laser injury in TP Tg mice transfused with WT(647):WT(488) (top panel) or Rasgrp2−/−(647):WT(488) plts (bottom panel) at a ratio of 2:1. Scale bar represents 25 μm. (F) Quantification of WT(488) plt adhesion (sum fluorescence intensity) in WT:WT or Rasgrp2−/−:WT plt transfused TP Tg mice following laser injury. Red boxes in (E) define area where 488 intensity was measured. Sum intensity was normalized to the maximum intensity at time-to-hemostasis for each injury. n=3, data graphed as mean ± SEM.
To substantiate this conclusion, we quantified the adhesion of anti-GPIX-AlexaFluor488-labeled WT platelets in TP Tg mice co-transfused with Rasgrp2−/−(AF647):WT(AF488) platelets at a ratio of 2:1, and, as a control, TP Tg mice co-transfused with WT(647):WT(488) platelets at the same ratio. Representative still-frame images from epifluorescence videos at ~60 seconds post-laser injury demonstrate successful hemostatic plug formation in both groups of recipient mice (Fig. 2E). However, WT(488) platelets showed markedly delayed adhesion to the site of laser injury in the presence of Rasgrp2−/−(647) platelets compared to co-transfusion with WT(647) platelets (Fig. 2F). Additionally, Rasgrp2−/− platelets adhered to the site of injury at a similar rate to WT platelets when co-transfused (fig. S2), suggesting that WT platelets can also enhance the recruitment of dysfunctional platelets to the site of injury.
Dysfunctional platelets alter hemostatic plug structure and integrity
At the conclusion of time-lapse epifluorescence imaging, spinning disk confocal (SDC) microscopy was used to acquire Z-stacks of hemostatic plugs. Epifluorescence imaging established that both WT and Rasgrp2−/− platelets rapidly accumulated at the site of vascular injury when co-transfused into TP mice, and both platelet populations seemed randomly distributed throughout the plug immediately after hemostasis was achieved (Fig. 2E). However, SDC Z-stacks revealed a specific pattern whereby WT platelets consolidated into distinct areas within the plug, and Rasgrp2−/− platelets filled areas devoid of WT platelets (Fig. 3A, top panel; movie S5). Segregation between differentially labeled populations of platelets was not observed in TP mice injected with WT platelets only (Fig. 3A, middle panel; movie S6). Additionally, when WT platelets were in excess (TP Tg mice transfused with 1:3 Rasgrp2−/−:WT platelets), the WT platelets did not segregate and formed a normal hemostatic plug (Fig. 3A, bottom panel).
Fig. 3: Rasgrp2−/− platelets disrupt hemostatic plug architecture.
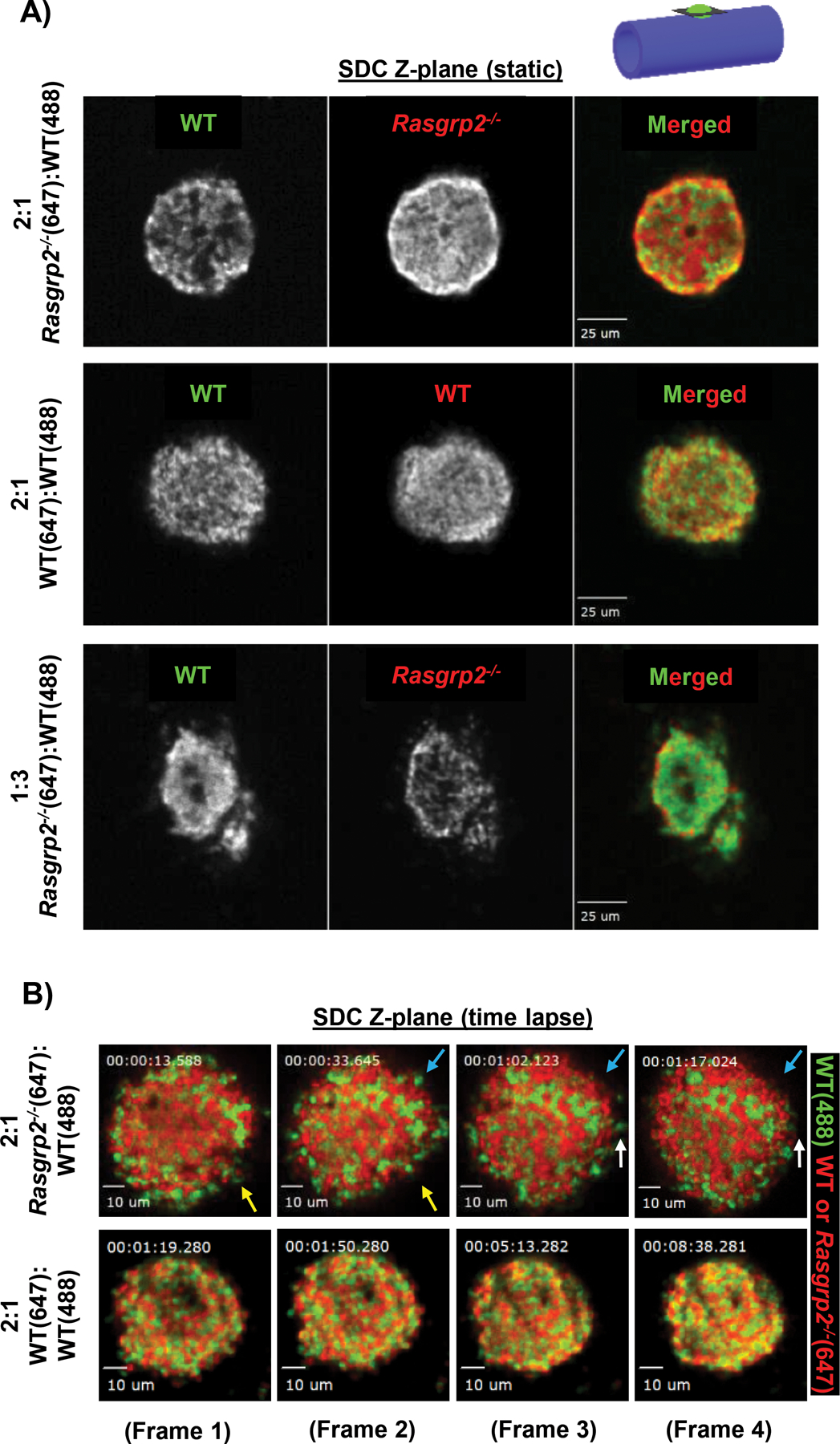
(A) TP Tg recipient mice were transfused with differentially labeled WT and Rasgrp2−/− platelets, in the combinations and ratios noted to the left of the image panels. Laser injury was performed and hemostatic plugs allowed to form. Spinning disk confocal (SDC) Z-stacks were then acquired at the conclusion of time-lapse imaging. Shown here are representative single Z-planes from the center of the hemostatic plug (see cartoon in upper right), with individual channels shown in greyscale along with a colored merged image (see movies S5 and S6 for Z-stack series). Scale bars represent 25 μm. (B) Still frames from time-lapse SDC imaging in TP Tg mice transfused with either WT:WT or Rasgrp2−/−:WT plts at a 2:1 ratio. Colored arrows indicate several instances of loosely adhered platelets being shed from the edges of the Rasgrp2−/−:WT hemostatic plug (movie S7), whereas the WT:WT plug retains its structure with little to no platelet shedding (movie S8). Scale bars represent 10 μm.
To monitor platelet dynamics during plug formation at single platelet resolution, we performed real-time SDC imaging in a single plane immediately after laser ablation. Despite achieving hemostasis within 60–90 seconds, plugs containing 2:1 Rasgrp2−/−:WT platelets were loosely packed, with single platelets shedding off from the edges and constant plug remodeling occurring (Fig. 3B, top panels; movie S7). Enrichment of WT and mutant platelets in distinct areas of the plug was observed. In contrast, differentially labeled WT platelets integrated randomly and firmly throughout the plug; no shedding of platelets or plug remodeling was observed (Fig 3B, bottom panels; movie S8).
Platelets with impaired integrin activation compete with healthy platelets for GPIbα ligand binding sites
CalDAG-GEFI and RAP1 are critically important for αIIbβ3 activation and firm platelet adhesion, but not for GPIbα-mediated transient adhesion to vWF(29). Thus, we hypothesized that Rasgrp2−/− platelets interfere with the function of healthy transfused platelets by competing for GPIbα binding sites in the damaged vascular wall. To test this hypothesis, we evaluated the hemostatic efficacy of WT platelets in two other mouse models of inherited platelet disorders: a model of Bernard-Soulier Syndrome (BSS) with mice lacking the extracellular domain of GPIbα (IL-4R/GPIb-Tg, “BSS-like”)(30), and a model of Glanzmann’s Thrombasthenia (GT) with mice lacking the integrin adapter protein, TALIN1, specifically in platelets (Talin1f/f x Pf4-Cre+, “GT-like”)(31) (Fig. 4A). Both “BSS-like” and “GT-like” mice were previously shown to exhibit a severe hemostatic defect in the saphenous vein laser injury model(25). Transfusion of a low number of WT platelets (1 × 108/mL) normalized time-to-hemostasis in “BSS-like”, but not “GT-like” mice (Fig. 4B). Consistent with our hypothesis, “BSS-like” platelets showed very poor incorporation into the hemostatic plug (Fig. 4C, top panel; movie S9). Similar results were obtained when we co-transfused normal WT platelets with WT platelets treated ex vivo with O-sialoglycoprotein endopeptidase (OSGE) to cleave the 45 kDa N-terminus of GPIbα (movie S10)(32). In contrast, “GT-like” platelets incorporated into the growing plug together with WT platelets (Fig. 4C, bottom panel; movie S11). Similar to our findings with Rasgrp2−/− platelets, “GT-like” platelets also affected the composition of the hemostatic plug, leading to shedding of platelets and segregation of WT and “GT-like” platelets (movie S12).
Fig. 4: Interference by dysfunctional platelets is dependent on GPIbα.
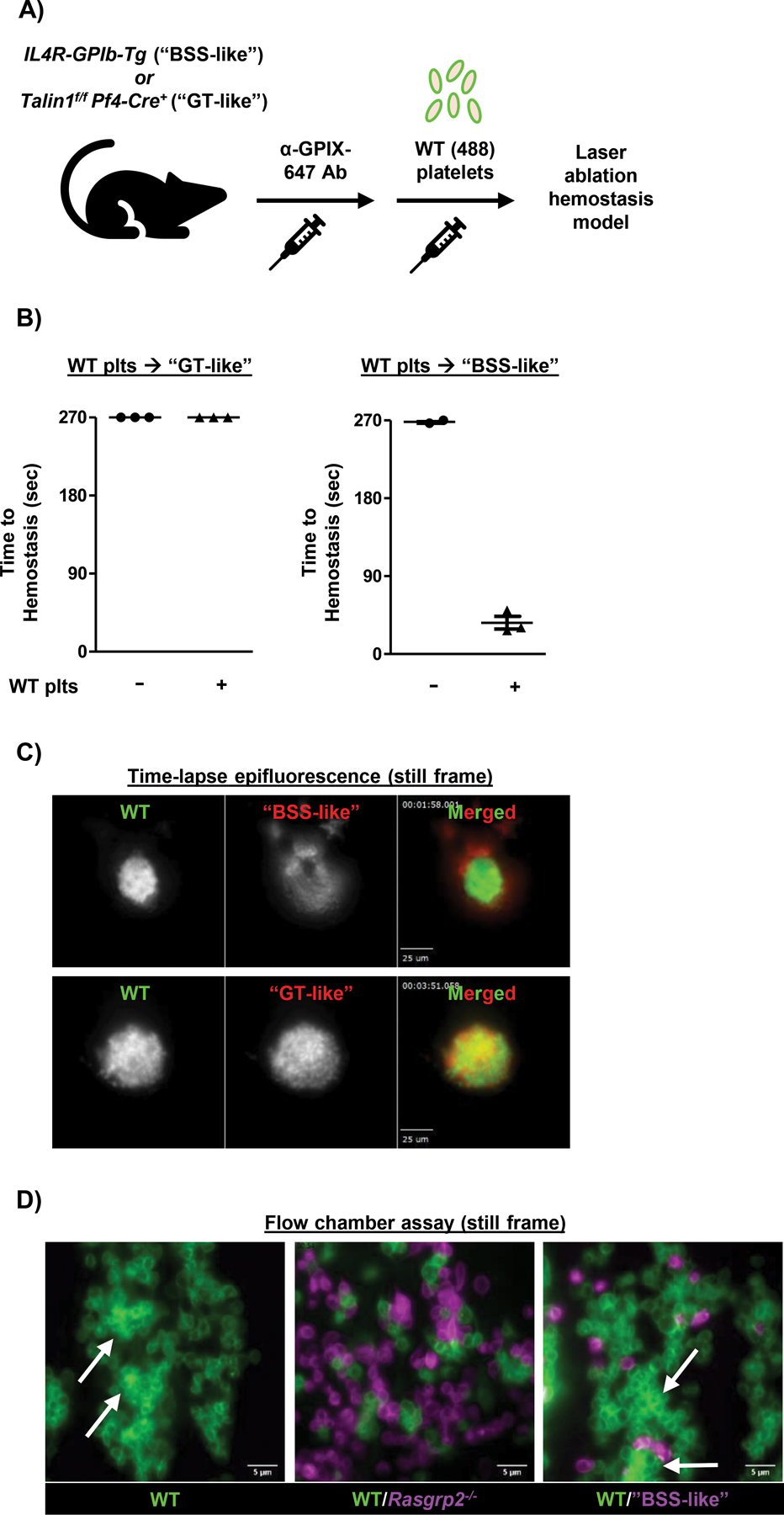
A) Model depicting platelet (plt) transfusion scheme. Endogenous plts in “GT-like” or “BSS-like” mice were labeled by injection of anti-GPIX-647 antibody prior to transfusion of GPIX-488 labeled WT plts. B) “GT-like” or “BSS-like” mice were transfused with WT plts to reach a circulating count of 1 × 108/mL, for a ratio of ~ 7:1 endogenous:transfused plts in both mouse models. Time-to-hemostasis was then assessed in the laser injury model. n=3 per group. C) Mice were transfused with WT plts to reach a ratio ~ 2:1 endogenous:transfused plts, to allow for hemostatic plug formation in “GT-like” mice. Still frame images from epifluorescence videos (movies S9 and S11) demonstrate incorporation of “GT-like” (bottom panel) but not “BSS-like” (top panel) plts within the hemostatic plug. Some “BSS-like” plts can be seen adhered to the luminal side of the WT plug. Scale bars represent 25 μm. D) Flow chamber assay was performed on collagen-coated (200 μg/mL) coverslips at a shear rate of 1600 s-1. WT blood was flowed alone or after mixing at a 3:1 mutant:WT ratio with Rasgrp2−/− or “BSS-like” blood for 5 minutes over the collagen surface and visualized with a 100X oil objective. Plts were labeled with anti-GPIX antibody prior to mixing. Representative still frame images are shown. White arrows denote areas of WT thrombus formation. Scale bars represent 5 μm.
To validate our findings ex vivo, we studied platelet adhesion to a collagen surface under arterial flow conditions in a microfluidics chamber system. Although our bleeding model uses the venous system, the shear rate substantially increases where blood flows from a penetrating injury(33) which increases dependence on GPIb/vWF. Heparinized whole blood was obtained from WT, Rasgrp2−/−, and “BSS-like” mice, differentially labeled with Alexa488- and Alexa647-conjugated anti-platelet antibodies and mixed at defined ratios before perfusion at a shear rate of 1600 s-1. Thrombus formation observed in WT blood (Fig. 4D, left panel) was markedly impaired in the presence of an excess in Rasgrp2−/− (Fig. 4D, middle panel), but not “BSS-like” platelets (Fig. 4D, right panel). Consistent with our in vivo studies, Rasgrp2−/− platelets were also successfully recruited to adherent WT platelets. These results demonstrate a mechanism by which platelets with dysfunctional integrin activation compete for GPIbα-mediated binding to reduce the adhesion and hemostatic efficacy of healthy platelets.
Human GT platelets interfere with healthy platelets
To expand our findings to the human clinical situation, we performed ex vivo competition studies with human platelets from healthy volunteers and GT patients. Platelet function was assessed using the Impact-R method, where shear stress is applied to blood in a polystyrene well and platelet surface coverage is measured. Importantly, platelet adhesion in this assay is dependent on both GPIbα/vWF and αIIbβ3/fibrinogen interactions(34). Healthy whole blood was supplemented with 33 or 66% (by volume) platelet concentrate from another healthy patient, or from a GT patient. In accordance with our findings in the “GT-like” mouse model, increasing numbers of GT platelets impaired the function of normal platelets in whole blood in a ratio-dependent manner (Fig. 5). These results demonstrate that competition between healthy and dysfunctional platelets also occurs in humans.
Fig. 5: GT patient platelets interfere with the function of healthy donor platelets.
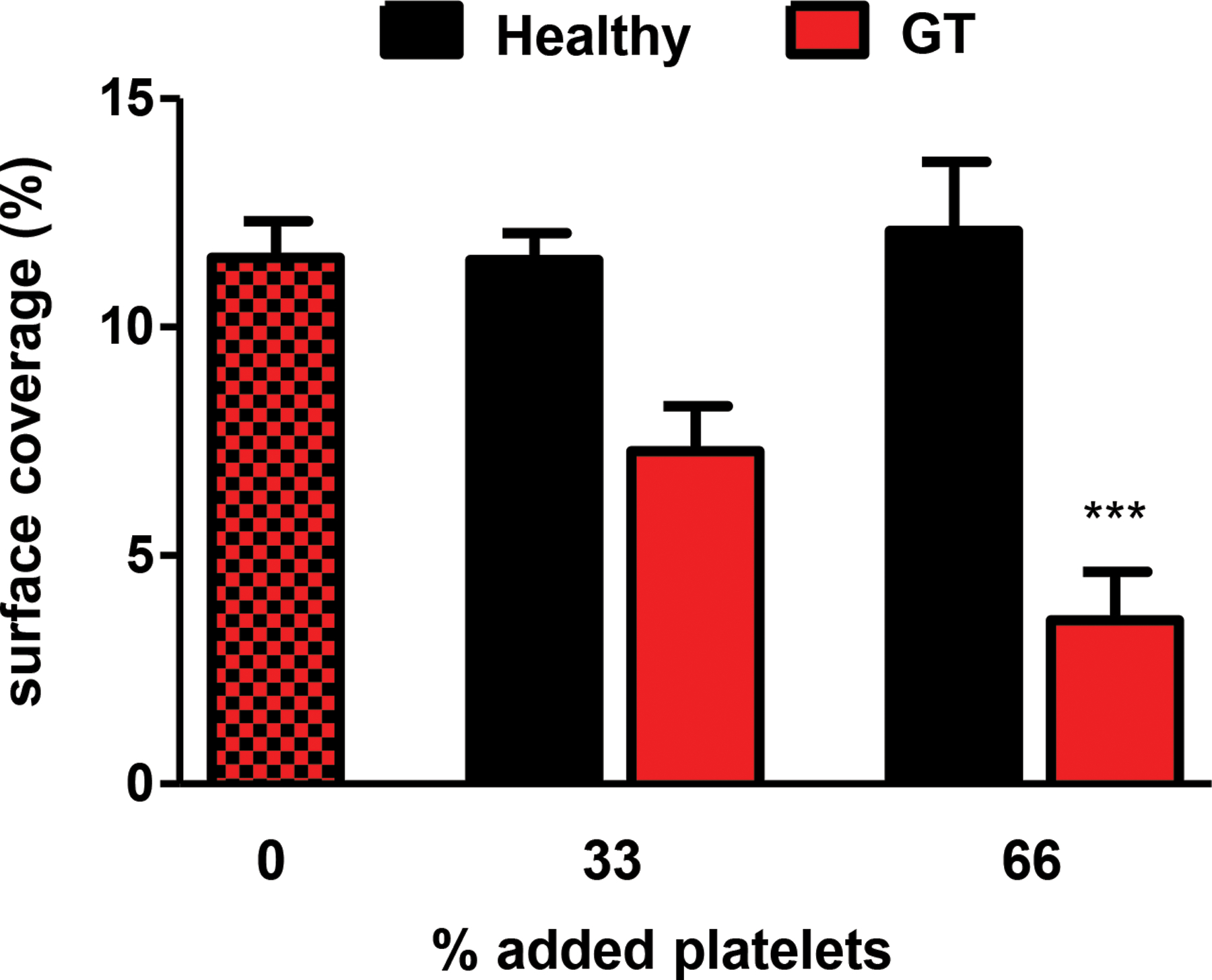
Blood collected from healthy donors was supplemented with platelet (plt) concentrates from other healthy donors, or from GT patients. Plt function was then tested using the Impact-R cone and plate analyzer. Plt surface coverage was determined by light microscopy following May-Grünwald staining of the well. n=6–7 per condition, data shown as mean ± SEM. ***P<0.001 compared to 0% added platelets. Red/black checkered bar represents control for both healthy and GT plts added, where healthy blood was run with no plt concentrations added.
“Dual anti-platelet therapy” platelets impair the hemostatic activity of WT platelets
The use of platelet transfusions in patients on anti-platelet drugs is not well standardized, and the efficacy is not completely understood(23). To investigate whether platelets with acquired dysfunction also impair the hemostatic activity of fully functional platelets, we co-transfused TP Tg mice with WT and aspirin-treated P2ry12−/− (“DAPT”) platelets (Fig. 6A), and then monitored hemostatic plug formation following laser injury. At a ratio of 2:1 “DAPT”:WT platelets, the adhesion kinetics of WT platelets and the initial time-to-hemostasis was comparable to mice receiving 2:1 WT:WT platelets (Fig. 6B,C). However, hemostatic plugs containing “DAPT” platelets showed instability at the center of the plug, leading to visible remodeling and rebleeding (Fig. 6D,E; movie S13). Thus, platelets with an acquired function defect, such as seen with anti-platelet therapy, also interfere with the hemostatic efficacy of transfused WT platelets, at least in the mouse system.
Fig. 6: “Dual anti-platelet therapy” platelets impair plug stability in the presence of WT platelets.
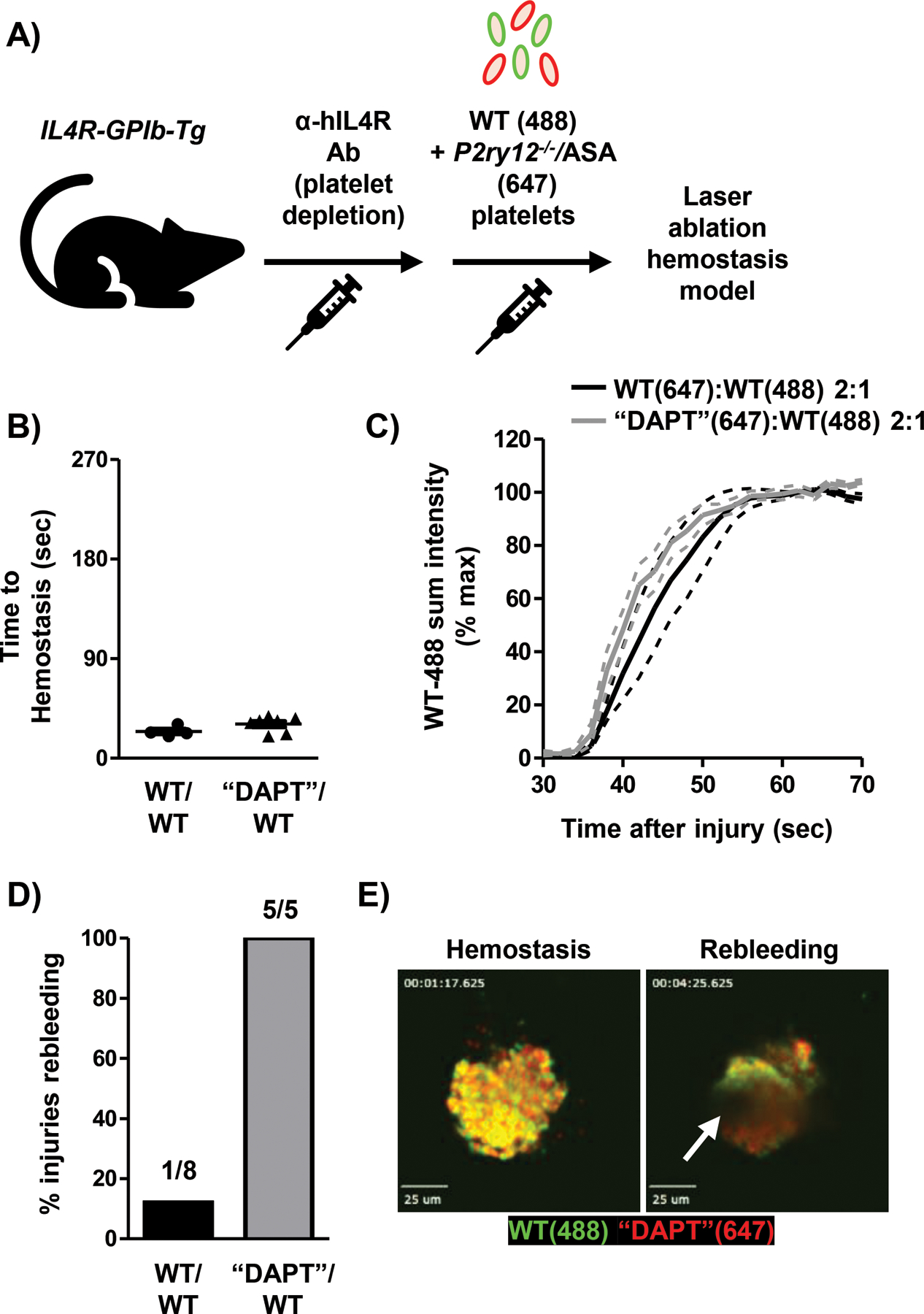
A) Model depicting platelet (plt) transfusion scheme. P2ry12−/− plts were treated with 2 mM acetylsalicylic acid (“DAPT”) ex vivo prior to transfusion. B) Time-to-hemostasis in TP Tg mice transfused with 2:1 “DAPT”:WT plts or 2:1 WT:WT plts was determined following laser injury. C) Adhesion of WT-488 plts was quantified in WT:WT or “DAPT”:WT plt transfused TP Tg mice. Data shown as mean ± SEM. D) Re-bleeding events (%) at individual injury sites during real-time SDC imaging. 5–8 injuries from 2–3 mice per group. E) Still frame images from real-time SDC video showing hemostasis and rebleeding at the same injury site (movie S13). White arrow indicates site of blood flow from hemostatic plug. Scale bars represent 25 μm.
Discussion
Inherited and acquired platelet function disorders (IPDs and APDs, respectively) are often associated with bleeding which necessitates transfusion of blood products and administration of procoagulant and/or antifibrinolytic agents(35). Interestingly, platelet transfusions can fail to correct hemostasis in these patients. In this study, we used various murine models of both IPDs and APDs in combination with in vivo, real-time imaging of hemostatic plug formation to establish the underlying mechanism(s) for the limited hemostatic activity of fully functional (normal) transfused platelets in these situations. The main conclusions from our work are: (1) normal platelets are less hemostatically active in the presence of dysfunctional platelets, (2) dysfunctional platelets compete with normal platelets for adhesion to GPIbα ligands in the vessel wall and the growing thrombus, (3) dysfunctional platelets disrupt the architecture and integrity of the plug, and (4) the post-transfusion ratio between normal and dysfunctional platelets determines whether platelet transfusion will correct hemostasis. Our findings have important clinical implications as they provide a basis for guidelines to optimize platelet transfusions for patients with platelet dysfunction but normal platelet counts.
The clinical usefulness of platelet transfusions for the treatment or prevention of bleeding in thrombocytopenic patients is well established(36). Largely based on clinical experience, it is believed that platelet counts greater than 10–20 × 109/L are sufficient to prevent spontaneous bleeding, while counts greater than 50 × 109/L are required to prevent bleeding following major surgery(37–39). Prophylactic platelet transfusions are recommended when platelet counts drop below 10 × 109/L. The corrected count increment (CCI), which compares the post-transfusion to the pre-transfusion platelet counts, provides a fairly reliable method to estimate post-transfusion platelet survival in patients(40). With the help of the CCI and other methods it was established that the transfusion of 3 × 1011 platelets, the equivalent of one unit of either apheresis platelets or pooled whole blood-derived platelets, increases the peripheral platelet count in a patient by ~ 30 × 109/L. Thus, transfusion of one apheresis or whole blood-derived pooled platelet unit is recommended to prevent bleeding in patients suffering from severe thrombocytopenia(36). However, the efficacy of platelet transfusion in patients with IPDs/APDs is less clear. Compared to thrombocytopenic patients, peripheral platelet counts in IPD and APD patients are generally in the normal range. Whether or not these patients require the same number of transfused platelets as thrombocytopenic patients to restore hemostasis is not known. A case report by Jennings et al.(41) describes findings in a GT patient that suggest that platelet transfusion protocols may need to be adjusted in patients with a platelet function disorder. In a first, unsuccessful attempt to control bleeding associated with tonsillectomy and adenoidectomy, the GT patient received apheresis platelets from a single donor, yielding a post-transfusion GT:normal (endogenous:transfused) platelet ratio of ~5:1. Hemostasis was restored after an additional transfusion with apheresis platelets from four donors, leading to a post-transfusion GT:normal platelet ratio of ~1:1(41). Another indication that transfusion requirements are different in the presence of dysfunctional platelets comes from recent reports of patients with mutations in RASGRP2, which show a poor response to platelet transfusion therapy(15, 16, 42). Similar to GT, RASGRP2 patients exhibit marked defects in integrin-mediated platelet adhesion and hemostatic plug formation. To date, 19 unrelated pedigrees with mutations in RASGRP2 have been reported(17). All of these patients present with moderate to severe bleeding symptoms, which often require combinatorial treatments including platelets, desmopressin and rFVIIa(15, 16). Of note, the pro-hemostatic mechanism of rFVIIa in platelet function disorders (most commonly GT and RASGRP2 patients) is not entirely understood. While platelets from these patients have impaired aggregation, they are still able to adhere and likely provide some surface for coagulation factor complex formation. Therefore, rFVIIa may function by enhancing platelet surface thrombin generation in vivo(43). Moreover, RASGRP2 mutant platelets typically have normal or only minimally reduced response to thrombin(11, 13), and therefore may benefit from increased thrombin generation not only due to fibrin formation(44) but also via thrombin receptor signaling. Future studies should address the mechanism by which procoagulants improve hemostatic plug formation in vivo in the setting of platelet dysfunction.
Our study investigated in vivo if and how endogenous (dysfunctional) platelets affect the hemostatic activity of transfused platelets. Given our expertise in CalDAG-GEFI signaling, we first investigated the platelet transfusion requirements for correcting hemostasis in Rasgrp2−/− mice. Like RASGRP2 patients, knockout mice are characterized by a markedly impaired platelet aggregation response, caused by a defect in the near-immediate activation of integrin receptors(45). Our studies demonstrate that, compared to thrombocytopenic mice, hemostasis in Rasgrp2−/− mice requires considerably higher numbers of transfused WT (healthy) platelets. Consistent with findings in the GT patient reported by Jennings et al.(41), the time to hemostasis following vascular injury was markedly improved at post-transfusion Rasgrp2−/−:WT ratios of 2:1, but not 5:1. We showed that both WT and mutant platelets localized to the injury site with similar kinetics. However, high resolution SDC imaging revealed that their distribution within the plug was not random, as we observed areas rich in either WT platelets or Rasgrp2−/− platelets. At this point, it is not clear why WT and Rasgrp2−/− platelets segregate into different areas within the hemostatic plug. Impaired platelet-platelet cohesion and contraction, mediated by activated integrin receptors, seems the most likely explanation. Segregation of procoagulant platelet subpopulations within clots has recently been demonstrated(46), and thus could be an alternative explanation for our findings. However, we have shown that CalDAG-GEFI/RAP1 signaling contributes to the platelet procoagulant response(47), and procoagulant platelets enrich on the surface of the plug while Rasgrp2−/− platelets were found surrounding clusters of WT platelets within the plug.
Consistent with an integrin-centered explanation, we also found platelet segregation within hemostatic plugs consisting of WT and TALIN1-deficient platelets. TALIN1 is crucial for integrin signaling, and thus Talin1f/f x Pf4-Cre+ mice show a platelet phenotype similar to that described for GT patients (“GT-like”)(31). Talin1f/f x Pf4-Cre+ platelets were recruited into the plug and drastically impaired the hemostatic activity of WT platelets. GT is the most severe form of IPD, causing major bleeds in 50% of deliveries(48). In several patients, platelet transfusion failed to prevent these pregnancy-related bleeds. Importantly, we could confirm the relevance of our in vivo findings for human patients, as we observed that platelets from GT patients impaired the adhesion of healthy donor platelets in whole blood when assessed by the Impact-R method(34). Similar findings were recently reported with the PFA-100 method(49). We acknowledge that our studies in mice are limited in that we exclusively focused on the saphenous vein hemostasis model, with the translational aspect coming from a single ex vivo assay using human platelets. However, the general principles of hemostatic plug formation are conserved between various vessel types and models(50, 51), suggesting that our mechanistic conclusions are transferrable to other vascular beds while the required transfusion ratio between dysfunctional and functional platelets may slightly vary dependent on the vessel and type of injury. Validation of these concepts in patients will require additional empirical data from the clinic, and may also be studied in mouse models of human platelet transfusion(52).
Using a mouse model for Bernard-Soulier syndrome (IL4R/GPIb-Tg, “BSS-like”)(30), we further demonstrated that the recruitment of dysfunctional platelets (Rasgrp2−/− or “GT-like”) to the forming hemostatic plug is likely mediated by GPIbα binding to its main ligands (most likely vWF), in the ECM and potentially on the surface of WT platelets within the growing plug(53). Platelets lacking GPIbα did not interfere with transfused WT platelets during hemostatic plug formation, even when the ratio of mutant:WT platelets exceeded 5:1. A similar mechanism was previously described in arterial thrombosis in mice; platelets lacking the GPIbα extracellular domain or the 45 kDa N-terminal domain were entirely unable to incorporate into WT arterial thrombi(54). In BSS and GT, platelet transfusions are reserved for patients with serious bleeding due to the potential risk of alloimmunization and platelet refractoriness after repeated exposure to transfused blood products(55). Unlike for GT, however, there are no reports in the literature that platelet transfusion was ineffective in BSS patients. Given that BSS patients also exhibit a marked macrothrombocytopenia, a phenotype that is not seen in RASGRP2 and GT patients, it is possible that the better hemostatic efficacy of transfused healthy platelets in BSS is, at least in part, a reflection of a lower ratio between dysfunctional and healthy platelets. However, our studies in BSS-like and GT-like mice, which have similar peripheral platelet counts, argue in favor of a mechanistic explanation.
While patients with IPDs are comparatively rare, the use of anti-platelet drugs and the associated bleeding complications are common in the clinic setting. The efficacy and use of platelet transfusion for the acute reversal of DAPT is not well understood(23, 56), and the AABB (formerly called American Association of Blood Banks) has not found convincing evidence supporting the usefulness of platelet transfusion for patients receiving anti-platelet therapy who suffer from intracranial hemorrhage(36). The consensus thinking is that in patients on aspirin and clopidogrel/prasugrel, irreversible inhibitors of cyclooxygenase-1 and the P2Y12 receptor, platelet transfusion might be beneficial once the drugs are eliminated from circulation. However, by co-transfusing healthy and dual-inhibited platelets and excluding circulating inhibitors, we here provide evidence that “DAPT” platelets themselves also impair the hemostatic activity of fully functional platelets by markedly destabilizing the hemostatic plug. Unlike Rasgrp2−/− or “GT-like” platelets, which delayed WT platelet adhesion and hemostasis, “DAPT” platelets did not affect the initial adhesion of WT platelets and the time-to-hemostasis. Consistent with the key role of TxA2 and ADP in sustained platelet activation(57, 58), however, hemostatic plugs consisting of WT and “DAPT” platelets were unstable and frequently reopened. Importantly, patients on anti-platelet therapy typically have platelet counts within a normal range, similar to RASGRP2 patients, increasing the dysfunctional:healthy platelet ratio.
An important conclusion of our study is that hemostasis can be achieved in IPDs associated with integrin dysfunction if the posttransfusion ratio of dysfunctional/normal platelets is ≤ 2:1, at least in saphenous vein laser injury hemostasis model. Obviously, studies in patients will be required to validate this number but given the case report by Jennings et al.(41) we believe that our number is a good starting point to guide future studies. An important variable to be considered, however, is the age of the platelet product. Our experiments used only freshly washed WT platelets, which were transfused within several hours of blood collection and platelet washing. An extended platelet storage time, however, reduces the hemostatic capacity of donor platelets in humans(59). Therefore, the ratio of dysfunctional:normal platelets required for hemostasis could be altered when transfusing platelets stored for several days. Another important variable is the nature of the platelet function defect. Our studies utilized mice with severe defects in platelet integrin signaling. Additional studies will be required to determine whether healthy platelets are equally impaired in their hemostatic function in other IPDs and APDs. Studies in mouse models will provide a great tool for highly controlled studies. In humans, an improved understanding of the transfusion efficacy will require more detailed reporting practices, such as the inclusion of the CCI and its correlation with the success of platelet transfusion in individual patients. In disorders like GT and BSS, where the expression of surface glycoproteins is altered, flow cytometry has been used to more accurately determine the ratio between endogenous and transfused platelets(60, 61). For other patients, such as those with mutations in RASGRP2, whose platelets express normal numbers of surface glycoproteins(11–13), distinguishing endogenous from transfused platelets would require either platelet pre-labeling(62) or an intracellular staining protocol for flow cytometry, the latter of which could be standardized for use in the clinical laboratory. Additionally, future studies will be necessary to determine whether the required ratio would be affected by transfusing platelets prophylactically vs. therapeutically. Our experimental design modeled prophylactic transfusion, but we speculate that therapeutic transfusion may be less effective as the dysfunctional platelets would have the first opportunity to absorb vWF binding sites.
In summary, we here present systematic in vivo analysis of the interaction between endogenous dysfunctional and healthy transfused platelets in murine models of inherited and acquired platelet function disorders. Our studies provide clear evidence that dysfunctional platelets can interfere with the hemostatic activity of transfused platelets by at least two mechanisms: 1) early interference by competition for GPIbα ligands at the site of injury, and 2) late interference by impaired consolidation of the hemostatic plug. Our finding that a critical ratio of ~ 2:1 endogenous (dysfunctional) to transfused (healthy) platelets is required to prevent prolonged injury-induced bleeding suggests that patients with select qualitative platelet function disorders will require greater numbers of transfused platelets than thrombocytopenic patients to achieve successful hemostasis. However, current guidelines, which are mostly based on clinical experience in thrombocytopenic patients, suggest the transfusion of up to one apheresis unit (or equivalent) into patients with bleeding complications(36). We propose that multiple apheresis units (or equivalents) are required to control bleeding in patients with IPD or APD, and that the pre-transfusion platelet count and the post-transfusion platelet ratio are important variables for determining the required number of platelet concentrates and the predicted effect on hemostasis, respectively. Future studies should be directed towards the development of simple clinical tests that help establish the post-transfusion platelet ratio.
Materials and methods
Study Design
Our experimental design aimed to determine the effectiveness of healthy (wild-type, WT) platelet transfusion in the setting of inherited and acquired platelet disorders. Bleeding time was assessed using the saphenous vein laser injury model. The translational relevance of this model is that it is a penetrating injury, and is sensitive to both platelet and coagulation defects. The hemostatic efficacy of WT platelets was tested in two ways: transfusion of WT platelets into Rasgrp2−/− mice (representative of the clinical situation) or co-transfusion of specific amounts/ratios of WT/Rasgrp2−/− platelets into thrombocytopenic recipient mice. For WT and Rasgrp2−/− platelet mixing, n ≥ 3 mice per group were included for statistical analysis. Each data point represents the average time-to-hemostasis for at least 3 individual injuries along the saphenous vein for a single mouse. The cut-off for bleeding time in this study was set at 270 seconds, similar to the length of the first of 3 consecutive injuries at the same injury site described in the first publication of this model(25). Only injuries ~50–70 μm in diameter were included. In the laser injury model, the researcher typically performed the platelet transfusion and laser injury model in one mouse per day and was not blinded to the condition. Post-experiment determination of bleeding time, assessed by visual cessation of blood flow from the site of laser ablation, was confirmed by two independent assessors. For proof-of-principle experiments where WT platelets were transfused into “GT-like” or “BSS-like” mice, n = 2–3 per condition. For co-transfusion of WT/WT or WT/“DAPT” platelets, n ≥ 4. Images of flow chamber assay were representative of 3 independent experiments. For the Impact-R assay using human platelets, n = 6–7 samples were tested at least in duplicate.
Reagents
Low molecular weight heparin (enoxaparin sodium, Fresenius Kabi) and isofluorane (Piramal Critical Care) were purchased from UNC Hospitals pharmacy. Antibodies recognizing GPIX (clone Xia.B4), GPIbα (clone Xia.G5, PE-conjugated), αIIbβ3 integrin (clone MWReg30, PE-conjugated) and activated αIIbβ3 integrin (JON/A-PE, clone Leo.H4) were from Emfret Analytics. Anti-GPIX antibodies were conjugated with AlexaFluor dyes (488, 647) from Life Technologies. Anti-human IL-4R antibody was from R&D Systems. Convulxin (Cvx) was purchased from Kenneth Clemetson (Theodor Kocher Institute, University of Berne, Switzerland). PAR4 activating peptide (PAR4p) was from GL Biochem Inc. Adenosine diphosphate (ADP) and acetylsalicylic acid were from Sigma-Aldrich.
Human subjects
GT patients had been enrolled within the aims of the “Inherited Platelet Disorders Project, Hemorrhagic Diathesis Working Group, Spanish Society for Thrombosis & Hemostasis (SETH). Investigations in this project abided by the Declaration of Helsinki and were approved by the Local Ethical Committees of Hospital Universitario Reina Sofía (Murcia, Spain). All patients and healthy subjects gave their written informed consent.
Mice
The following mouse mutant strains were used in this study: Rasgrp2−/−, Talin1f/f x Pf4-Cre+, hIL-4Rα/GPIbα-Tg and P2ry12−/−, all on a C57BL/6 background and bred in-house. Both male and female mice were used at 8–16 weeks old (20–25 g). All experimental protocols were approved by The University of North Carolina IACUC.
Platelet isolation
Whole blood was obtained via the retro-orbital plexus using heparinized glass capillaries (VWR) and collected into tubes containing 30 IU/mL low molecular weight heparin. Platelet rich plasma (PRP) was obtained by 2 rounds of centrifugation of whole blood at 130 x g and collecting the supernatant with some erythrocytes, followed by 2 rounds of centrifugation at 100 x g to pellet erythrocytes. Platelets were then pelleted by centrifugation at 700 g in the presence of 1 μg/mL PGI2, and the pellet was resuspended in modified Tyrode’s buffer (137 mM NaCl, 12 mM NaHCO3, 2.0 mM KCl, 0.3 mM Na2HPO4, 1 mM MgCl2, 5mM N-2-hydroxyethylpiperazine-N’−2-ethanesulfonic acid, 5 mM Glucose, pH 7.3). Platelets in Tyrode’s buffer were allowed to rest in a 37º water bath for 20–30 minutes prior to transfusion.
Platelet transfusion
For non-thrombocytopenic recipients, endogenous platelets were labeled by I.V. injection of anti-GPIX-AlexaFluor488/647 (2.5 µg/mouse) at least 4 hours prior to platelet transfusion. Platelet counts in Rasgrp2−/− mice were similar to WT mice (1 × 109/mL, ± 1 × 108). Recipient mice received transfusions of WT platelets to achieve circulating counts of 1 or 3 × 108/mL, which were slightly adjusted if the baseline platelet count of the recipient mouse was greater or less than 1 × 109/mL. This transfusion scheme resulted in approximate ratios of 10:1 or 3:1, respectively. For thrombocytopenic recipients, hIL-4Rα/GPIbα-Tg mice were depleted of endogenous platelets via I.V. injection of anti-hIL-4Rα antibody (2 µg/g body weight) at least 4 hours prior to platelet transfusion(28). Thrombocytopenia was confirmed shortly after injection by flow cytometric analysis of whole blood sample. Donor platelets were washed as described above and labeled with 2.5–5 µg/mL anti-GPIX-AlexaFluor488 or anti-GPIX-AlexaFluor647 for 15 minutes at room temperature, washed again, and injected into recipient mice via the retro-orbital plexus. In some experiments, platelets were treated ex vivo with O-sialoglycoprotein endopeptidase (OSGE, 250 μg/mL, 60 mins, 37°C) prior to labeling(32). Cleavage of the 45 kDa N-terminal of GPIbα was confirmed by flow cytometry using an anti-GPIbα antibody (clone Xia.G5) recognizing the N-terminus. For “DAPT” experiments, P2ry12−/− platelets(63) were treated ex vivo with acetylsalicylic acid (2 mM) for 10 minutes at room temperature prior to labeling. In mice receiving two populations of platelets, the two populations were washed after labeling to remove residual antibody and then combined for a single injection. To achieve desired circulating platelet counts, the platelet count/mL was multiplied by 2.5 to estimate the total number of platelets for transfusion (~2 mL blood volume, +50% to account for platelet loss/sequestration). Following transfusion, circulating platelet counts/ratios were confirmed by flow cytometry prior to starting saphenous vein laser injury experiments.
Saphenous vein laser ablation hemostasis model
Saphenous vein laser injury was performed as previously described(25), with modifications. Mice (8–16 weeks of age) were anesthetized by isofluorane inhalation (Veterinary Anesthesia Vaporizer, Dre Veterinary). Hair removal was performed using Nair, the saphenous vein was exposed, and the mouse was placed on the stage on a heating pad. A perfusion drip on the exposed saphenous vein was started and maintained at 37° C via a Sloflo In-line solution heater (SF-28) and single channel heater controller [(Model TC-324B, Warner Instruments) (physiologic salt solution containing mmol/L (132 NaCl, 4.7 KCl, 1.2 MgS04, 2 CaCl2, 18 NaHCO3) pH 7.4, which was bubbled with 5% CO2 / 95% N2 15 minutes prior to the start of the experiment)]. Baseline fluorescence was recorded for 30 seconds using a Zeiss Axio Examiner Z1 upright microscope (Intelligent Imaging Innovations) equipped with a 20x/1 NA water immersion objective lens and a Hamamatsu Orca Flash 4.0 camera (Hamamatsu Photonics). Injury to the endothelium was initiated using an Ablate! photoablation system equipped with an attenuable 532 nm pulse laser (Intelligent Imaging Innovations) and fluorescence intensity was recorded for 270 seconds (SPECTRA X light engine, Lumencor). All data were recorded and analyzed using Slidebook6.0 software (Intelligent Imaging Innovations). Time-to-hemostasis was defined as the time from laser ablation to the visual cessation of blood flow from the injury site. Platelet adhesion at the injury site was determined by quantifying sum fluorescence intensity in the 488 and 647 channels. To directly compare fluorescence intensities between experiments, due to variations in intensity of platelet labeling, all data points were normalized to the intensity at time-to-hemostasis for each individual injury. For some conditions, hemostatic plug formation following laser ablation was imaged in real-time (RT) using spinning disk confocal microscopy (SDC; Yokogawa CSU-W1, equipped with a LaserStack (Intelligent Imaging Innovations) with 488 and 647 laser lines), essentially as described for epifluorescence imaging. Laser ablation was initiated, and RT SDC microscopy was performed in a single plane at or superior to the position of the endothelium for up to 10 minutes. For one mouse, up to 10 injuries were performed along the saphenous vein, beginning distal (upstream) and moving proximal (downstream) so that subsequent injuries were not affected by embolizations from previous injuries. Following real-time imaging, mice were euthanized by isofluorane overdose, and SDC Z-stacks of hemostatic plugs were acquired, from the most intraluminal point of the plug to the most extravascular.
Flow chamber assay
Flow chamber assay was performed essentially as previously described(64). Platelets in heparinized whole blood were labeled with AlexaFluor-conjugated anti-GPIX antibody (2.5 μg/mL) and mixed at desired ratios just prior to starting the experiment. No double-labeling of platelets was observed after mixing, as confirmed by flow cytometry. Collagen (200 μg/mL) was deposited on a glass slide and the PDMS microfluidic device was oriented perpendicularly to the collagen strip. Blood was flowed at arterial shear rates (1600 s−1) for 5 minutes. Platelet adhesion was monitored using a NikonTE300 (Nikon) inverted microscope equipped with a Hamamatsu Orca Flash 4.0 camera and a 100X oil objective and analyzed with NIS Elements software.
Flow cytometry
Flow cytometry was performed essentially as previously described(64). Platelet counts were determined in diluted whole blood using a BD Accuri C6 Flow Cytometer (BD). In samples where platelets were not pre-labeled, blood samples were first incubated with anti-GPIX antibody (2 μg/mL) for 10 minutes. For blood samples from transfused TP Tg mice, all platelets in blood were labeled by incubating with MWReg30-PE (2 μg/mL) for 10 minutes prior to analyzing sample. Transfused platelets were gated on MWReg30-PE intensity, and WT or mutant populations were distinguished by anti-GPIX-AlexaFluor488 or 647 intensity. For determination of platelet integrin activation, diluted whole blood was incubated in Tyrode’s buffer with agonist (Cvx, PAR4p or ADP) for 10 minutes in the presence of 1 mM Ca2+ and 2 μg/mL JON/A-PE. Following activation, samples were diluted in PBS and immediately analyzed. Platelet populations were gated on anti-GPIX-488 or 647 intensity and JON/A-PE intensity was analyzed.
Impact-R
Venous blood was drawn from three healthy volunteers and three patients previously diagnosed with Glanzmann’s Thrombasthenia(65), into buffered 3.2% sodium citrate. Aliquots of these citrated blood samples were saved for in vitro transfusion assays, and the rest of the volume was used for preparation of platelet concentrates using the buffy-coat procedure. Blood samples from controls and GT patients were centrifuged at 1000 x g for 15 mins, and the plasma supernatant and the buffy-coat layer separated into different tubes. After 15 mins resting, the buffy-coat was diluted 1:4 with autologous plasma, centrifuged at 150 x g for 7 mins, and the upper phase containing the majority of platelets was separated. Platelet count in these platelet concentrates from control (PC-C) or GT (PC-GT) (1–2 × 1012/L) was determined using a Sysmex haematological analyser (Sysmex Corporation).
We performed in vitro transfusion by adding to the non-manipulated control blood, variable volumes of PC-C and PC-GT. The mixtures were then tested on the Impact-R™ cone and plate analyzer (Matis Medical Inc.-DANED SA), essentially as originally described(34). In brief, 130 uL blood mixture, at least in duplicate, was placed into a polystyrene well, a Teflon cone was placed on top and rotated (2050/sec) to induce a shear stress that promotes platelet adhesion to the plastic. After washing the well and staining with May-Grünwal solution, the plate surface covered with platelets (% SC) was quantified using an inverted light microscope connected to a camera and image analyzing software that calculates median values from seven images from each well.
Statistics
Results are reported as mean ± standard error of the mean (SEM) and statistical significance was assessed by ANOVA test with post-hoc analysis when appropriate, unless otherwise indicated. A P value less than 0.05 was considered significant.
Supplementary Material
Acknowledgements:
We acknowledge David Paul for expert technical assistance with SDC imaging experiments, Kathryn Poe for assistance with mouse husbandry, Maribel Diaz-Ricart for assistance with human platelet experiments, Jerry Ware for providing the hIL-4Rα/GPIbα mice, Brian Petrich for providing the Talin1f/f mice, Pamela Conley (Portola Pharmaceuticals) for providing P2ry12−/− mice, and especially The C.T. and Nancy Owens Fund for a generous contribution made towards the purchase of the SDC confocal system. MLL and JR are the coordinators of the multicentric project “Functional and Molecular Characterization of Patients with Inherited Platelet Disorders” Spanish Society of Thrombosis and Haemostasis (Hemorrhagic Diathesis Working Group, Spanish Society for Hemostasis and Thrombosis [SETH]).
Funding:
This work was supported by NIH grants 1R35 HL144976-01 to WB and T32HL007149 to RHL. MLL and JR were supported by grants from Instituto de Salud Carlos III and Feder (PI17/01311 & CB15/00055) and Fundación Séneca (19873/GERM/15).
Footnotes
Competing interests: All authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are available in the main text or the supplementary materials.
Supplemental Materials
fig. S1. Integrin activation is similar between normal and transfused platelets.
fig. S2. WT and Rasgrp2−/− platelets adhere at similar rates to the site of injury.
Data File S1. Raw data.
movie S1. TP Tg mouse WT platelets only epifluorescence.
movie S2. TP Tg mouse Rasgrp2−/− platelets only epifluorescence .
movie S3. TP Tg mouse Rasgrp2−/−:WT platelets 5:1 epifluorescence.
movie S4. TP Tg mouse Rasgrp2−/−:WT platelets 2:1 epifluorescence.
movie S5. TP Tg mouse Rasgrp2−/−:WT platelets 2:1 spinning disk confocal Z-stack.
movie S6. TP Tg mouse WT:WT platelets 2:1 spinning disk confocal Z-stack.
movie S7. TP Tg mouse Rasgrp2−/−:WT platelets 2:1 real-time spinning disk confocal.
movie S8. TP Tg mouse WT:WT platelets 2:1 real-time spinning disk confocal.
movie S9. “BSS-like” mouse WT platelets epifluorescence.
movie S10. TP Tg mouse WT:WT-OSGE platelets epifluorescence.
movie S11. “GT-like” mouse WT platelets epifluorescence.
movie S12. “GT-like” mouse WT platelets real-time spinning disk confocal.
movie S13. TP Tg “DAPT”:WT platelets real-time spinning disk confocal.
References
- 1.Savage B, Saldívar E, Ruggeri ZM, Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor, Cell 84, 289–297 (1996). [DOI] [PubMed] [Google Scholar]
- 2.Stegner D, Nieswandt B, Platelet receptor signaling in thrombus formation, J. Mol. Med 89, 109–121 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Lee RH, Stefanini L, Bergmeier W, in Platelets, Michelson AD, Ed. (Elsevier, 2019), pp. 329–348. [Google Scholar]
- 4.Stefanini L, Bergmeier W, RAP1-GTPase signaling and platelet function, J. Mol. Med 94, 13–19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu L, Yang J, Bromberger T, Holly A, Lu F, Liu H, Sun K, Klapproth S, Hirbawi J, Byzova TV, Plow EF, Moser M, Qin J, Structure of Rap1b bound to talin reveals a pathway for triggering integrin activation, Nat. Commun 8, 1744 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Savoia A, Kunishima S, De Rocco D, Zieger B, Rand ML, Pujol-Moix N, Caliskan U, Tokgoz H, Pecci A, Noris P, Srivastava A, Ward C, Morel-Kopp M-C, Alessi M-C, Bellucci S, Beurrier P, de Maistre E, Favier R, Hézard N, Hurtaud-Roux M-F, Latger-Cannard V, Lavenu-Bombled C, Proulle V, Meunier S, Négrier C, Nurden A, Randrianaivo H, Fabris F, Platokouki H, Rosenberg N, HadjKacem B, Heller PG, Karimi M, Balduini CL, Pastore A, Lanza F, Spectrum of the Mutations in Bernard-Soulier Syndrome, Hum. Mutat 35, 1033–1045 (2014). [DOI] [PubMed] [Google Scholar]
- 7.George JN, Caen JP, Nurden A, Glanzmann’s thrombasthenia: the spectrum of clinical disease, Blood 75, 1383–1395 (1990). [PubMed] [Google Scholar]
- 8.Nurden AT, Pillois X, ITGA2B and ITGB3 gene mutations associated with Glanzmann thrombasthenia, Platelets 29, 98–101 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Canault M, Ghalloussi D, Grosdidier C, Guinier M, Perret C, Chelghoum N, Germain M, Raslova H, Peiretti F, Morange PE, Saut N, Pillois X, Nurden AT, Cambien F, Pierres A, van den Berg TK, Kuijpers TW, Alessi M-C, Tregouet D-A, Human CalDAG-GEFI gene ( RASGRP2 ) mutation affects platelet function and causes severe bleeding, J. Exp. Med 211, 1349–1362 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kato H, Nakazawa Y, Kurokawa Y, Kashiwagi H, Morikawa Y, Morita D, Banno F, Honda S, Kanakura Y, Tomiyama Y, Human CalDAG-GEFI deficiency increases bleeding and delays αIIbβ3 activation, Blood 128, 2729–2733 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Lozano ML, Cook A, Bastida JM, Paul DS, Iruin G, Cid AR, Adan-Pedroso R, González-Porras JR, Hernández-Rivas JM, Fletcher SJ, Johnson B, Morgan N, Ferrer-Marin F, Vicente V, Sondek J, Watson SP, Bergmeier W, Rivera J, Novel mutations in RASGRP2, which encodes CalDAG-GEFI, abrogate Rap1 activation, causing platelet dysfunction, Blood 128, 1282–1289 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sevivas T, Bastida JM, Paul DS, Caparros E, Palma-Barqueros V, Coucelo M, Marques D, Ferrer-Marín F, González-Porras JR, Vicente V, Hernández-Rivas JM, Watson SP, Lozano ML, Bergmeier W, Rivera J, Identification of two novel mutations in RASGRP2 affecting platelet CalDAG-GEFI expression and function in patients with bleeding diathesis, Platelets 29, 192–195 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bermejo E, Alberto MF, Paul DS, Cook AA, Nurden P, Sanchez Luceros A, Nurden AT, Bergmeier W, Marked bleeding diathesis in patients with platelet dysfunction due to a novel mutation in RASGRP2, encoding CalDAG-GEFI (p.Gly305Asp), Platelets 29, 84–86 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Westbury SK, Canault M, Greene D, Bermejo E, Hanlon K, Lambert MP, Millar CM, Nurden P, Obaji SG, Revel-Vilk S, Van Geet C, Downes K, Papadia S, Tuna S, Watt C, Freson K, Laffan MA, Ouwehand WH, Alessi MC, Turro E, Mumford AD, Expanded repertoire of RASGRP2 variants responsible for platelet dysfunction and severe bleeding, Blood 130, 1026–1030 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desai A, Bergmeier W, Canault M, Alessi M-C, Paul DS, Nurden P, Pillois X, Jy W, Ahn YS, Nurden AT, Phenotype analysis and clinical management in a large family with a novel truncating mutation in RASGRP2 , the CalDAG-GEFI encoding gene, Res. Pract. Thromb. Haemost 1, 128–133 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Canault M, Saultier P, Fauré S, Poggi M, Nurden AT, Nurden P, Morange PE, Alessi M-CC, Gris J-CC, Peripartum bleeding management in a patient with CalDAG-GEFI deficiency, Haemophilia 23, e533–e535 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Palma-Barqueros V, Ruiz-Pividal J, Bohdan N, Vicente V, Bastida JM, Lozano M, Rivera J, RASGRP2 gene variations associated with platelet dysfunction and bleeding, Platelets 30, 535–539 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Crittenden JR, Bergmeier W, Zhang Y, Piffath CL, Liang Y, Wagner DD, Housman DE, Graybiel AM, CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation, Nat. Med 10, 982–986 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Poon MC, d’Oiron R, Recombinant activated factor VII (NovoSeven) treatment of platelet-related bleeding disorders. International Registry on Recombinant Factor VIIa and Congenital Platelet Disorders Group., Blood Coagul. Fibrinolysis 11 Suppl 1, S55–68 (2000). [PubMed] [Google Scholar]
- 20.T. C. in U. A. to P. R. E. T. Investigators, Effects of Clopidogrel in Addition to Aspirin in Patients with Acute Coronary Syndromes without ST-Segment Elevation, N. Engl. J. Med 345, 494–502 (2001). [DOI] [PubMed] [Google Scholar]
- 21.O’Gara PT, Kushner FG, Ascheim DD, Casey DE, Chung MK, de Lemos JA, Ettinger SM, Fang JC, Fesmire FM, Franklin BA, Granger CB, Krumholz HM, Linderbaum JA, Morrow DA, Newby LK, Ornato JP, Ou N, Radford MJ, Tamis-Holland JE, Tommaso CL, Tracy CM, Woo YJ, Zhao DX, 2013 ACCF/AHA Guideline for the Management of ST-Elevation Myocardial Infarction: Executive Summary, J. Am. Coll. Cardiol 61, 485–510 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Baschin M, Selleng S, Hummel A, Diedrich S, Schroeder HW, Kohlmann T, Westphal A, Greinacher A, Thiele T, Preoperative platelet transfusions to reverse antiplatelet therapy for urgent non‐cardiac surgery: an observational cohort study, J. Thromb. Haemost 16, 709–717 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Nagalla S, Sarode R, Role of Platelet Transfusion in the Reversal of Anti-Platelet Therapy, Transfus. Med. Rev 33, 92–97 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Orsini S, Noris P, Bury L, Heller PG, Santoro C, Kadir RA, Butta NC, Falcinelli E, Cid AR, Fabris F, Fouassier M, Miyazaki K, Lozano ML, Zúñiga P, Flaujac C, Podda GM, Bermejo N, Favier R, Henskens Y, De Maistre E, De Candia E, Mumford AD, Ozdemir GN, Eker I, Nurden P, Bayart S, Lambert MP, Bussel J, Zieger B, Tosetto A, Melazzini F, Glembotsky AC, Pecci A, Cattaneo M, Schlegel N, Gresele P, Bleeding risk of surgery and its prevention in patients with inherited platelet disorders, Haematologica 102, 1192–1203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Getz TM, Piatt R, Petrich BG, Monroe D, Mackman N, Bergmeier W, Novel mouse hemostasis model for real-time determination of bleeding time and hemostatic plug composition, J. Thromb. Haemost 13, 417–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piatt R, Paul DS, Lee RH, McKenzie SE, Parise LV, Cowley DO, Cooley BC, Bergmeier W, Mice Expressing Low Levels of CalDAG-GEFI Exhibit Markedly Impaired Platelet Activation With Minor Impact on Hemostasis, Arterioscler. Thromb. Vasc. Biol 36, 1838–1846 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morowski M, V??gtle T, Kraft P, Kleinschnitz C, Stoll G, Nieswandt B, Only severe thrombocytopenia results in bleeding and defective thrombus formation in mice, Blood 121, 4938–4947 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Boulaftali Y, Hess PR, Getz TM, Cholka A, Stolla M, Mackman N, Owens AP, Ware J, Kahn ML, Bergmeier W, Platelet ITAM signaling is critical for vascular integrity in infammation, J. Clin. Invest 123, 908–916 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bergmeier W, Goerge T, Wang HW, Crittenden JR, Baldwin ACW, Cifuni SM, Housman DE, Graybiel AM, Wagner DD, Mice lacking the signaling molecule CalDAG-GEFI represent a model for leukocyte adhesion deficiency type III, J. Clin. Invest 117, 1699–1707 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanaji T, Russell S, Ware J, Amelioration of the macrothrombocytopenia associated with the murine Bernard-Soulier syndrome, Blood 100, 2102–2107 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Petrich BG, Marchese P, Ruggeri ZM, Spiess S, Weichert RAM, Ye F, Tiedt R, Skoda RC, Monkley SJ, Critchley DR, Ginsberg MH, Talin is required for integrin-mediated platelet function in hemostasis and thrombosis, J. Exp. Med 204, 3103–3111 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bergmeier W, Bouvard D, Eble JA, Mokhtari-Nejad R, Schulte V, Zirngibl H, Brakebusch C, Fässler R, Nieswandt B, Rhodocytin (Aggretin) Activates Platelets Lacking α2β1 Integrin, Glycoprotein VI, and the Ligand-binding Domain of Glycoprotein Ibα, J. Biol. Chem 276, 25121–25126 (2001). [DOI] [PubMed] [Google Scholar]
- 33.Sakurai Y, Hardy ET, Ahn B, Tran R, Fay ME, Ciciliano JC, Mannino RG, Myers DR, Qiu Y, Carden MA, Baldwin WH, Meeks SL, Gilbert GE, Jobe SM, Lam WA, A microengineered vascularized bleeding model that integrates the principal components of hemostasis, Nat. Commun 9, 509 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shenkman B, Savion N, Dardik R, Tamarin I, Varon D, Testing of Platelet Deposition on Polystyrene Surface Under Flow Conditions by the Cone and Plate(let) Analyzer: Role of Platelet Activation, Fibrinogen and von Willebrand Factor, Thromb. Res 99, 353–361 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Lambert MP, What To Do When You Suspect an Inherited Platelet Disorder, ASH Educ. Progr. B 2011, 377–383 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Kaufman RM, Djulbegovic B, Gernsheimer T, Kleinman S, Tinmouth AT, Capocelli KE, Cipolle MD, Cohn CS, Fung MK, Grossman BJ, Mintz PD, O’Malley BA, Sesok-Pizzini DA, Shander A, Stack GE, Webert KE, Weinstein R, Welch BG, Whitman GJ, Wong EC, Tobian AAR, Platelet Transfusion: A Clinical Practice Guideline From the AABB, Ann. Intern. Med 162, 205 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Pisciotto P, Benson K, Hume H, Glassman A, Oberman H, Popovsky M, Hines D, Anderson K, Prophylactic versus therapeutic platelet transfusion practices in hematology and/or oncology patients, Transfusion 35, 498–502 (1995). [DOI] [PubMed] [Google Scholar]
- 38.Schiffer CA, Bohlke K, Delaney M, Hume H, Magdalinski AJ, McCullough JJ, Omel JL, Rainey JM, Rebulla P, Rowley SD, Troner MB, Anderson KC, Platelet Transfusion for Patients With Cancer: American Society of Clinical Oncology Clinical Practice Guideline Update., J. Clin. Oncol 36, 283–299 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Beutler E, Platelet transfusions: the 20,000/microL trigger [see comments], Blood 81 (1993) (available at http://www.bloodjournal.org/content/81/6/1411.long?sso-checked=true). [PubMed] [Google Scholar]
- 40.Davis KB, Slichter SJ, Corash L, Corrected count increment and percent platelet recovery as measures of posttransfusion platelet response: problems and a solution, Transfusion 39, 586–592 (1999). [DOI] [PubMed] [Google Scholar]
- 41.Jennings LK, Wang WC, Jackson CW, Fox CF, Bell A, Hemostasis in glanzmann’s thrombasthenia (GT): Gt platelets interfere with the aggregation of normal platelets, J. Pediatr. Hematol. Oncol 13, 84–90 (1991). [PubMed] [Google Scholar]
- 42.Yun JW, Lee K-O, Jung CW, Oh S-Y, Kim S-H, Choi CW, Kim H-J, Hereditary platelet function disorder from RASGRP2 gene mutations encoding CalDAG-GEFI identified by whole exome sequencing in a Korean woman with severe bleeding., Haematologica , haematol.2019.218487 (2019). [DOI] [PMC free article] [PubMed]
- 43.Levy-Mendelovich S, Levy T, Budnik I, Barg A, Rosenberg N, Seligsohn U, Kenet G, Livnat T, Low Concentrations of Recombinant Factor VIIa May Improve the Impaired Thrombin Generation of Glanzmann Thrombasthenia Patients, Thromb. Haemost 119, 117–127 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Lisman T, Adelmeijer J, Heijnen HFG, de Groot PG, Recombinant factor VIIa restores aggregation of alphaIIbbeta3-deficient platelets via tissue factor-independent fibrin generation., Blood 103, 1720–7 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Cifuni SM, Wagner DD, Bergmeier W, CalDAG-GEFI and protein kinase C represent alternative pathways leading to activation of integrin αIIbβ3 in platelets, Blood 112, 1696–1703 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nechipurenko DY, Receveur N, Yakimenko AO, Shepelyuk TO, Yakusheva AA, Kerimov RR, Obydennyy SI, Eckly A, Léon C, Gachet C, Grishchuk EL, Ataullakhanov FI, Mangin PH, Panteleev MA, Clot Contraction Drives the Translocation of Procoagulant Platelets to Thrombus Surface, Arterioscler. Thromb. Vasc. Biol 39, 37–47 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Ahmad F, Boulaftali Y, Greene TK, Ouellette TD, Poncz M, Feske S, Bergmeier W, Relative contributions of stromal interaction molecule 1 and CalDAG-GEFI to calcium-dependent platelet activation and thrombosis, J. Thromb. Haemost 9, 2077–2086 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Civaschi E, Klersy C, Melazzini F, Pujol-Moix N, Santoro C, Cattaneo M, Lavenu-Bombled C, Bury L, Minuz P, Nurden P, Cid AR, Cuker A, Latger-Cannard V, Favier R, Nichele I, Noris P, Analysis of 65 pregnancies in 34 women with five different forms of inherited platelet function disorders, Br. J. Haematol 170, 559–563 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Al-Battat S, Rand ML, Bouskill V, Lau W, Blanchette VS, Kahr WHA, Rivard GE, Carcao MD, Glanzmann thrombasthenia platelets compete with transfused platelets, reducing the haemostatic impact of platelet transfusions, Br. J. Haematol 181, 410–413 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Tomaiuolo M, Matzko CN, Poventud-Fuentes I, Weisel JW, Brass LF, Stalker TJ, Interrelationships between structure and function during the hemostatic response to injury., Proc. Natl. Acad. Sci. U. S. A 116, 2243–2252 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Welsh JD, Poventud-Fuentes I, Sampietro S, Diamond SL, Stalker TJ, Brass LF, Hierarchical organization of the hemostatic response to penetrating injuries in the mouse macrovasculature, J. Thromb. Haemost 15, 526–537 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fuhrmann J, Jouni R, Alex J, Zöllner H, Wesche J, Greinacher A, Bakchoul T, Assessment of human platelet survival in the NOD/SCID mouse model: technical considerations, Transfusion 56, 1370–1376 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Kulkarni S, Dopheide SM, Yap CL, Ravanat C, Freund M, Mangin P, Heel KA, Street A, Harper IS, Lanza F, Jackson SP, A revised model of platelet aggregation., J. Clin. Invest 105, 783–91 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bergmeier W, Piffath CL, Goerge T, Cifuni SM, Ruggeri ZM, Ware J, Wagner DD, The role of platelet adhesion receptor GPIb far exceeds that of its main ligand, von Willebrand factor, in arterial thrombosis, Proc. Natl. Acad. Sci 103, 16900–16905 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grainger JD, Thachil J, Will AM, How we treat the platelet glycoprotein defects; Glanzmann thrombasthenia and Bernard Soulier syndrome in children and adults, Br. J. Haematol 182, 621–632 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Mazzeffi MA, Lee K, Taylor B, Tanaka KA, Perioperative management and monitoring of antiplatelet agents: a focused review on aspirin and P2Y 12 inhibitors, Korean J. Anesthesiol 70, 379 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Conley PB, Delaney SM, Scientific and therapeutic insights into the role of the platelet P2Y12 receptor in thrombosis., Curr. Opin. Hematol 10, 333–8 (2003). [DOI] [PubMed] [Google Scholar]
- 58.FitzGerald GA, Mechanisms of platelet activation: thromboxane A2 as an amplifying signal for other agonists., Am. J. Cardiol 68, 11B–15B (1991). [DOI] [PubMed] [Google Scholar]
- 59.Shively JA, Gott CL, Jongh DS, The Effect of Storage on Adhesion and Aggregation of Platelets, Vox Sang 18, 204–215 (1970). [DOI] [PubMed] [Google Scholar]
- 60.Cesar JM, Vecino AM, Survival and function of transfused platelets. Studies in two patients with congenital deficiencies of platelet membrane glycoproteins, Platelets 20, 158–162 (2009). [DOI] [PubMed] [Google Scholar]
- 61.Panzer S, Eichelberger B, Koren D, Kaufmann K, Male C, Monitoring survival and function of transfused platelets in Bernard-Soulier syndrome by flow cytometry and a cone and plate(let) analyzer (Impact-R), Transfusion 47, 103–106 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Stolla M, Fitzpatrick L, Gettinger I, Bailey SL, Pellham E, Christoffel T, Slichter SJ, In vivo viability of extended 4°C-stored autologous apheresis platelets, Transfusion 58, 2407–2413 (2018). [DOI] [PubMed] [Google Scholar]
- 63.André P, Delaney SM, LaRocca T, Vincent D, DeGuzman F, Jurek M, Koller B, Phillips DR, Conley PB, P2Y12regulates platelet adhesion/activation, thrombus growth, and thrombus stability in injured arteries, J. Clin. Invest 112, 398–406 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stefanini L, Lee RH, Paul DS, O’Shaughnessy EC, Ghalloussi D, Jones CI, Boulaftali Y, Poe KO, Piatt R, Kechele DO, Caron KM, Hahn KM, Gibbins JM, Bergmeier W, Functional redundancy between RAP1 isoforms in murine platelet production and function, Blood 132, 1951–1962 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sánchez-Guiu I, Antón AI, Padilla J, Velasco F, Lucia JF, Lozano M, Cid AR, Sevivas T, Lopez-Fernandez MF, Vicente V, González-Manchón C, Rivera J, Lozano ML, Functional and molecular characterization of inherited platelet disorders in the Iberian Peninsula: results from a collaborative study, Orphanet J. Rare Dis 9, 213 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.