

Abstract
PURPOSE
Ipilimumab (IPI), in combination with nivolumab (NIVO), is an approved frontline treatment option for patients with intermediate- or poor-risk advanced renal cell carcinoma (aRCC). We conducted a randomized phase II trial to evaluate whether administering IPI once every 12 weeks (modified), instead of once every 3 weeks (standard), in combination with NIVO, is associated with a favorable toxicity profile.
METHODS
Treatment-naïve patients with clear-cell aRCC were randomly assigned 2:1 to receive four doses of modified or standard IPI, 1 mg/kg, in combination with NIVO (3 mg/kg). The primary end point was the proportion of patients with a grade 3-5 treatment-related adverse event (trAE) among those who received at least one dose of therapy. The key secondary end point was 12-month progression-free survival (PFS) in the modified arm compared with historical sunitinib control. The study was not designed to formally compare arms for efficacy.
RESULTS
Between March 2018 and January 2020, 192 patients (69.8% intermediate/poor-risk) were randomly assigned and received at least one dose of study drug. The incidence of grade 3-5 trAEs was significantly lower among participants receiving modified versus standard IPI (32.8% v 53.1%; odds ratio, 0.43 [90% CI, 0.25 to 0.72]; P = .0075). The 12-month PFS (90% CI) using modified IPI was 46.1% (38.6 to 53.2). At a median follow-up of 21 months, the overall response rate was 45.3% versus 35.9% and the median PFS was 10.8 months versus 9.8 months in the modified and standard IPI groups, respectively.
CONCLUSION
Rates of grade 3-5 trAEs were significantly lower in patients receiving modified versus standard IPI. Although 12-month PFS did not meet the prespecified efficacy threshold compared with historical control, informal comparison of treatment groups did not suggest any reduction in efficacy with the modified schedule.
12-weekly ipilimumab, plus nivolumab, is better tolerated than 3-weekly dosing and does not appear to compromise efficacy in mRCC (PRISM trial).
INTRODUCTION
Ipilimumab (IPI) and nivolumab (NIVO), checkpoint inhibitors targeting cytotoxic T-lymphocyte–associated protein 4 (CTLA-4) and PD-1, respectively, are approved in combination as a frontline treatment option for patients with intermediate- or poor-risk advanced renal cell carcinoma (aRCC), as defined by International Metastatic Renal Cell Carcinoma Database Consortium (IMDC) criteria.1 The superiority of the combination over the previous standard of care, the VEGFR-targeted tyrosine kinase inhibitor, sunitinib, was established in the randomized phase III CheckMate 214 study.2,3 IPI was administered at 1 mg/kg (IPI1) and NIVO was administered at 3 mg/kg (NIVO3), once every 3 weeks for four doses, followed by single-agent NIVO.
CONTEXT
Key Objective
This randomized phase II trial was designed to investigate whether, in patients with advanced renal cell carcinoma, modified scheduling of ipilimumab (IPI), in combination with nivolumab, is associated with a favorable toxicity profile in comparison with standard dosing once every 3 weeks.
Knowledge Generated
Giving IPI every 12 weeks for four doses led to a significant reduction in the rate of grade 3-5 treatment-related adverse events. Rates of treatment discontinuation were also in favor of the modified schedule. Although not designed to formally compare arms for efficacy, no clear differences in response rate, progression-free survival, or overall survival were observed.
Relevance (M.A. Carducci)
-
Extended-interval dosing strategies for anti-cytotoxic T-lymphocyte associated protein 4 therapies have the potential to improve patient-reported outcomes by providing flexibility and convenience, while spacing out infusion time. This study suggests these longer dosing strategies can remain efficacious while reducing toxicity experienced by patients with renal cell cancer, much like other studies in lung cancer and melanoma.*
*Relevance section written by JCO Associate Editor Michael A. Carducci, MD.
Dose and scheduling of IPI appear to correlate with treatment safety and tolerability. In the phase I CheckMate 016 study in aRCC, higher rates of toxicity were observed with IPI3+NIVO1 versus IPI1+NIVO3, given once every 3 weeks, on which basis the IPI1+NIVO3 regimen was taken forwards.4 More formal comparison of these dosing regimens was undertaken in patients with metastatic melanoma, in the phase IIIb/IV CheckMate 511 study. IPI1+NIVO3, once every 3 weeks, was again associated with a significantly lower rate of grade 3-5 trAEs compared to IPI3+NIVO1, with similar survival rates at three years.5
Increased interval dosing of IPI has been explored in other settings, suggesting improved tolerability compared with dosing once every 3 weeks. The CheckMate 012 multiarm phase Ib study in patients with non–small-cell lung cancer (NSCLC) included cohorts receiving IPI once every 6 weeks and once every 12 weeks, in combination with NIVO.6 Rates of treatment discontinuation because of trAEs were low (13% and 11%), with encouraging activity, leading to subsequent adoption of the once every 6 weeks regimen. Recently, the KEYNOTE-029 study in patients with metastatic melanoma has explored an alternative IPI dose and schedule in combination with pembrolizumab (anti–PD-1).7 Standard-dose pembrolizumab (200mg, once every 3 weeks), plus 50 mg IPI every 6 weeks, was associated with a grade 3-5 trAE rate of 24%, with antitumor activity above the prespecified threshold of interest.
The PRISM trial was designed to formally establish whether scheduling of IPI once every 12 weeks, in combination with NIVO, was associated with an improved safety profile in comparison with conventional IPI dosing once every 3 weeks in the setting of aRCC. The comparative frequency of adverse events in the two arms was the primary end point.
METHODS
Patients
Adult patients (18 years and older) with untreated, locally advanced, or metastatic clear-cell renal cell carcinoma (RCC), measurable disease as per RECIST version 1.1, and a Karnofsky performance status score of ≥70 and who were belonging to any IMDC risk group were recruited from participating UK sites. IMDC favorable-risk patients were included as the study commenced before the results of CheckMate 214 were available. All patients provided written informed consent. Ethical approval was obtained from the Leeds East Research Ethics Committee (17/YH/0187). Further details of the trial Protocol (online only) were reported previously, including the full list of patient eligibility criteria.8
Study Design and Treatment
PRISM was a multicenter, phase II, parallel-group, randomized controlled trial. The primary end point of the trial was the proportion of participants experiencing a Common Terminology Criteria for Adverse Events (CTCAE; version 5.0) grade 3-5 adverse reaction within the first 12 months of trial treatment. The key secondary end point of the trial was an external comparison against historical progression-free survival (PFS) data associated with sunitinib, included to provide supportive evidence of efficacy.9 Formal comparison with historical data was planned to occur only if the internal comparison of the primary end point achieved statistical significance. The efficacy statistics of the study was designed before the results of Checkmate 214, which is why benchmarking with sunitinib was used.
Participants were registered prospectively and underwent trial-specific assessments of eligibility.8 Eligible participants were individually randomly assigned on a 2:1 basis to receive either modified scheduling or standard scheduling of treatment, respectively. Random assignment was performed centrally by an automated 24-hour system provided by the Leeds Clinical Trials Research Unit (CTRU) using a minimization algorithm incorporating a random element. Minimization factors were the IMDC risk group (favorable/intermediate/poor risk), disease status (metastatic/locally advanced), and nephrectomy status (nephrectomy/no nephrectomy). Treatment allocation was not blinded to participants, medical staff, or trial staff.
Treatment schedules were altered once during the trial after the approval of NIVO dosing once every 4 weeks. Following this amendment, participants randomized to the modified schedule received four doses of combination 3 mg/kg NIVO plus 1 mg/kg IPI once every 12 weeks, with 240 mg maintenance NIVO once every 2 weeks between the first and second combination doses, and 480 mg maintenance NIVO once every 4 weeks between all other combination doses. Single-agent NIVO (480 mg, once every 4 weeks) continued after all combination doses had been administered until disease progression, unacceptable toxicity, or participant choice.
Participants randomized to the standard schedule received four doses of combination 3 mg/kg NIVO plus 1 mg/kg IPI once every 3 weeks, with 480 mg single-agent NIVO once every 4 weeks continuing thereafter, until disease progression, unacceptable toxicity, or participant choice. The Data Supplement (online only) shows all treatment schedules used in the trial for both treatment groups. In alignment with the CheckMate 214 study, only those participants completing their IPI induction phase were permitted to progress to single-agent NIVO maintenance. Participants were permitted to continue treatment beyond first progression on the basis of investigator-assessed clinical benefit, study drug tolerance, and stable performance status.
Trial Outcomes
The primary end point was the proportion of participants experiencing a CTCAE (version 5.0) grade 3-5 adverse reaction within the first 12 months of trial treatment. The key secondary outcome was PFS with the modified schedule, where PFS was calculated from random assignment to first documented evidence of disease progression or death, whichever occurred first. Secondary end points included safety and tolerability (assessed by serious adverse events and treatment compliance), overall response rate (ORR), duration of response, overall survival (OS), and response rate post–first progression.
Health-related quality of life (QoL) was assessed using the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire (QLQ)-C30, Comprehensive Cancer Network Functional Assessment of Cancer Therapy-Kidney Symptom Index (FKSI-19), and EuroQol 5-dimension (EQ-5D-5L) instruments. Given the exploratory nature of the analysis, missing QoL data were not imputed, unless an approach for handling missing data was specified in the appropriate scoring manual. All disease response assessments were graded locally according to RECIST version 1.1 on the basis of computed tomography scans once every 12 weeks. Extended follow-up data were collected 12 months after the final analysis for PFS and OS outcomes. This was performed after the primary analysis to explore the longer-term outcomes for the key groups.
Statistical Analysis
One hundred eighty-nine participants were required to formally assess both the safety and efficacy aspects of the primary objective in a hierarchical testing framework. Specifically, 178 participants would provide an 80% power to detect a clinically relevant reduction in CTCAE grade 3-5 toxicity rate from 40% to 22% with the modified schedule (equivalent to an odds ratio [OR], 0.42) using a two-sided 10% significance level and allowing for 5% attrition. Should the toxicity rate in the control arm be between 30% and 50%, the study would provide 80% power to detect ORs in the range of 0.38 to 0.45; these reductions are deemed clinically relevant. One hundred twenty participants were required in the modified schedule arm to target a minimum clinically relevant hazard ratio of 0.73 compared with historical sunitinib data, corresponding to 50.9% alive and progression-free at 12 months, giving 80% power at the one-sided 5% significance level. Given the 2:1 allocation ratio in favor of the modified schedule, this corresponds to a target sample size of 189 participants allowing for 5% attrition. No formal interim analysis was planned.
Analysis of trial end points was performed using SAS 9.410 by statisticians at Leeds CTRU, and a statistical analysis plan was written before any analyses were undertaken. Analysis was conducted using modified intention-to-treat (mITT) principles for the primary end point and all efficacy end points, meaning that participants were analyzed according to randomized allocation and were included in the analysis, provided that they had received at least one dose of trial treatment. Secondary safety analyses were conducted using the safety population, whereby participants were analyzed according to the treatment they received. Analysis of the safety (primary end point) and efficacy (key secondary end point) components of the primary objective was hierarchical to preserve the power of the trial.
For the primary end point, treatment groups were formally compared by fitting a logistic regression model adjusting for minimization factors. Adjusted ORs, alongside corresponding 90% CIs and P values, are presented. The results for the key secondary end point are based on the lower limit of the one-sided 95% CI for the proportion of patients alive and progression-free at 12 months postrandomization in the modified schedule arm. No formal comparison of PFS was performed between the modified and standard schedule arms; however, PFS has been summarized descriptively for treatment groups, alongside exploratory post hoc hazard ratios, and for IMDC intermediate-/poor-risk subgroups.
Other end points are summarized using appropriate descriptive statistics, alongside appropriate two-sided CIs.
RESULTS
The trial opened to recruitment on March 16, 2018, and completed recruitment on January 15, 2020, randomly assigning 195 participants from 15 sites. Of those, 192 participants formed the mITT population, with 128 in the modified schedule arm and 64 in the standard schedule arm. Three participants did not receive any trial treatment and were excluded. Participant flow is shown in the CONSORT diagram (Fig 1).
FIG 1.

CONSORT flow diagram.
Baseline characteristics for the mITT population were well balanced between treatment groups (Table 1). The majority (133 of 192 [69.3%]) of participants had IMDC intermediate- or poor-risk disease.
TABLE 1.
Baseline Patient Characteristics

Primary Analysis
Overall, 76 of 192 (39.6%) participants experienced a CTCAE grade 3-5 adverse reaction within the first 12 months of trial treatment, with 42 of 128 (32.8%) in the modified schedule and 34 of 64 (53.1%) in the standard schedule. In particular, lower rates of colitis (3.9% v 6.3%), arthralgia (1.6% v 7.8%), serum lipase increase (1.6% v 9.4%), and hypophysitis (0.8% v 3.1%) were observed among patients receiving modified scheduling compared with standard scheduling (Fig 2). The logistic regression model showed a statistically significant estimated OR of 0.43 (90% CI, 0.25 to 0.72; P = .0075) in favor of modified scheduling, after adjusting for minimization factors. The Data Supplement (Table S1) contains adjusted ORs and 90% CIs from the fitted model.
FIG 2.

Key trAEs by severity. CTCAE, Common Terminology Criteria for Adverse Events; trAE, treatment-related adverse event.
Safety, Toxicity, and Tolerability
Rates of treatment discontinuation because of treatment-related toxicity were lower among participants receiving modified scheduling (29 of 128 participants [22.7%]) compared with standard scheduling (25 of 64 participants [39.1%]; unadjusted risk difference: −16.4% [95% CI, −30.4 to −2.4]). The median (IQR) duration of treatment was 209 (105, 406) days and 84 (35, 314) days in the modified and standard schedule arms, respectively. The median (range) number of IPI doses received was 3 (1-4, modified) and 4 (1-4, standard).
Overall, 1,158 trAEs, 87 serious adverse reactions (SARs), and six suspected unexpected serious adverse reactions (SUSARs) were reported in the trial: 756 trAEs, 45 SARs, and four SUSARs in the modified schedule and 402 trAEs, 42 SARs, and two SUSARs in the standard schedule. Key clinical trAEs, by trial arm and CTCAE definition, are presented in Figure 2 alongside the maximum observed CTCAE grade. A plot including all trAEs that occurred in more than 2.5% of patients is presented in the Data Supplement (Fig S1).
Similar numbers and duration of treatment delays were observed between schedules. The number of participants experiencing at least one treatment delay or interruption was 88 of 128 (68.8%) and 37 of 64 (57.8%) for the modified and standard schedule, respectively. The mean (standard deviation [SD]) number of delays per participant was 1.5 (1.66) in the modified schedule arm and 1.4 (1.92) in the standard schedule arm.
Forty-seven deaths were observed among participants randomly assigned to the trial. The primary cause of death was most often related to RCC (modified schedule: 23 of 32 deaths [71.9%], standard schedule: 12 of 15 deaths [80%]). One treatment-related death because of immune-related hepatitis was reported in the modified schedule arm. All remaining deaths were attributed to other causes, including three that involved COVID-19.
Key Secondary Analysis
The median follow-up time at the time of final analysis for PFS was 21 months (95% CI, 17 to 22) using the modified schedule and 22 months (95% CI, 15 to 25) using the standard schedule. Kaplan-Meier curves summarizing PFS by arm are presented in Figure 3A. At 12 months postrandomization, the PFS estimate for the modified schedule was 46.1% (90% CI, 38.6 to 53.2). Therefore, formal comparison of the lower limit of the CI narrowly failed to exclude the historical control rate of 39.7% observed with sunitinib.9
FIG 3.
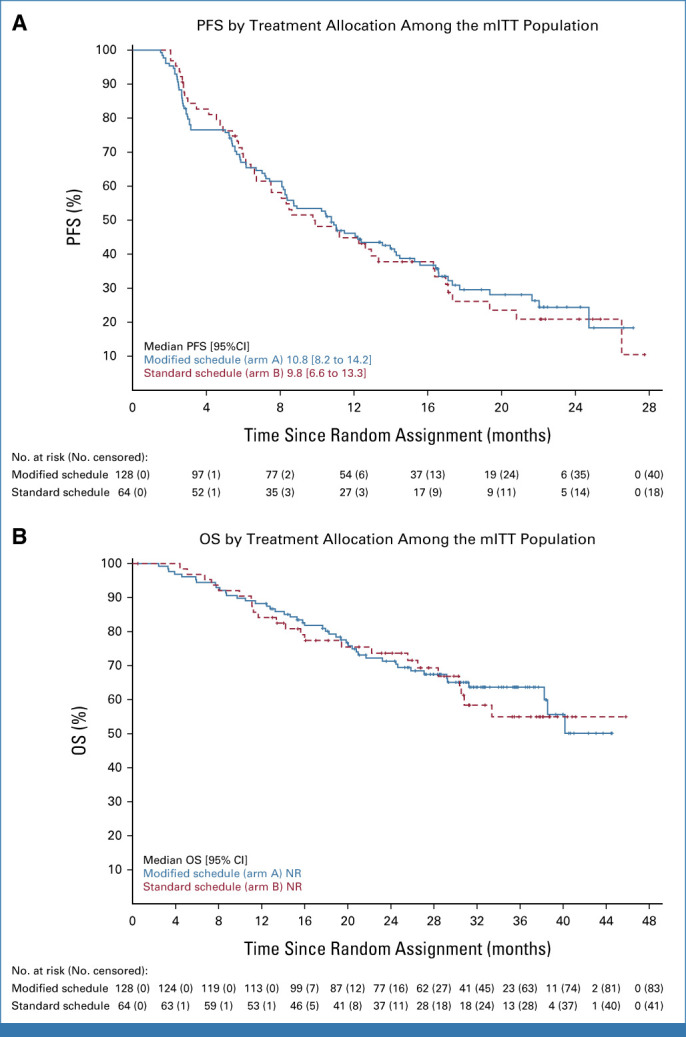
(A) PFS and (B) OS by treatment allocation among the mITT population. mITT, modified intention-to-treat; NR, not reached; OS, overall survival; PFS, progression-free survival.
The standard schedule PFS at 12 months postrandomization was 44.8% (32.1 to 56.7) and appears to be similar to the modified schedule PFS although it is important to recognize that the trial was not powered to detect a difference between arms. Exploratory analysis showed a post hoc unadjusted hazard ratio of 0.95 (95% CI, 0.67 to 1.36). Furthermore, PFS remained similar between arms with extended follow-up of participants, conducted 1 year after the trial follow-up period ended; median follow-up and Kaplan-Meier curves of the extended PFS data are presented in the Data Supplement (Fig S2). PFS by IMDC risk group and PD-L1 expression status (where available) is also available in the Data Supplement (Figs S3 and S4).
ORR and Duration of Response
The proportion of participants achieving a complete or partial response was 45.3% (95% CI, 36.5 to 54.4) with modified scheduling and 35.9% (95% CI, 24.3 to 48.9) with standard scheduling (Table 2). Median duration of response data is also presented in Table 2.
TABLE 2.
Secondary Outcome Measures

Overall Survival
The median follow-up time for OS was 32 months (95% CI, 31 to 34) using the modified schedule and 31 months (95% CI, 28 to 37) using the standard schedule. Kaplan-Meier curves summarizing OS by arm are presented in Figure 3B. The postrandomization OS estimate at 12 months was 88.3% (95% CI, 81.3 to 92.8) using modified scheduling and 84.1% (95% CI, 72.5 to 91.1) using standard scheduling. At 24 months, the OS estimate was 71.3% using modified scheduling and 73.7% using standard scheduling. Median OS was not reached (NR) in either arm. The trial was not designed to compare the two regimens directly. Exploratory analysis showed a post hoc unadjusted hazard ratio of 0.93 (95% CI, 0.56 to 1.54).
IMDC Intermediate- and Poor-Risk Patients
Exploratory Kaplan-Meier curves summarizing PFS and OS for participants with IMDC intermediate- or poor-risk disease by treatment arm are presented in Figure 4A and Figure 4B, respectively. The median PFS was 10.5 months and 8.6 months with modified and standard scheduling, respectively. The 12-month PFS estimates (95% CI) were 43.3% (32.7 to 53.3) in the modified arm and 46.1% (30.7 to 60.1) in the standard arm. The median OS was 38.5 (95% CI, 27.1 to NR) months in the modified arm and NR in the standard arm. The 24-month OS rates were 65.2% and 66.7% in the modified and standard arms, respectively. Among patients with IMDC intermediate- or poor-risk disease, the ORR was 46.7% (95% CI, 36.1 to 57.5) in the modified arm and 40.9% (95% CI, 26.3 to 56.8) in the standard arm (Table 2).
FIG 4.
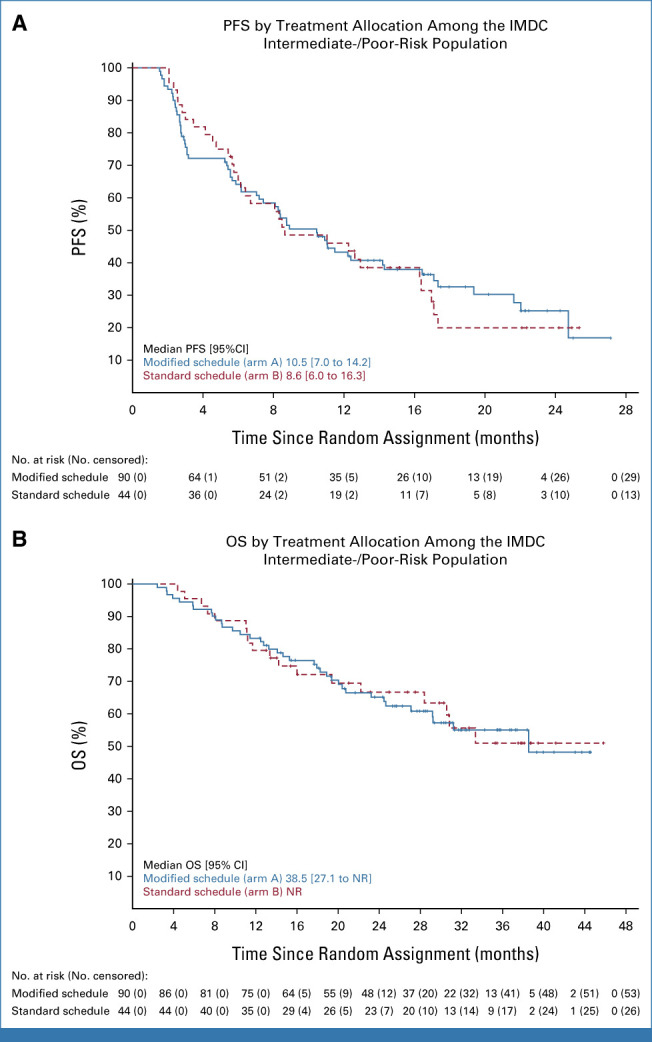
(A) PFS and (B) OS by treatment allocation among the IMDC intermediate-/poor-risk population. IMDC, International Metastatic Renal Cell Carcinoma Database Consortium; NR, not reached; OS, overall survival; PFS, progression-free survival.
Quality of Life
Baseline scores were available for 115 of 128 (89.8%) modified schedule participants and 55 of 64 (85.9%) standard schedule participants. Scores were collected through week 61 although, beyond week 25, only a small number (n ≤ 21) of standard schedule patients completed questionnaires.
QoL, as measured by QLQ-C30 global health status, FKSI-19 total score, and the EQ-5D-5L visual analog scale, did not meaningfully change from baseline at any time point in either arm (Figs 5A-5C). Considering the FKSI GP5 global item “bothered by side effects of treatment,” mean scores were in favor of the modified schedule during the initial 12 weeks of treatment and subsequently in favor of the standard schedule beyond this time point. However, the 95% CI of mean scores was overlapping throughout (Data Supplement, Fig S5). Means (SDs) and corresponding 95% CIs by questionnaire subscales, time point, and arm are available in the Data Supplement (Figs S5 and S6).
FIG 5.
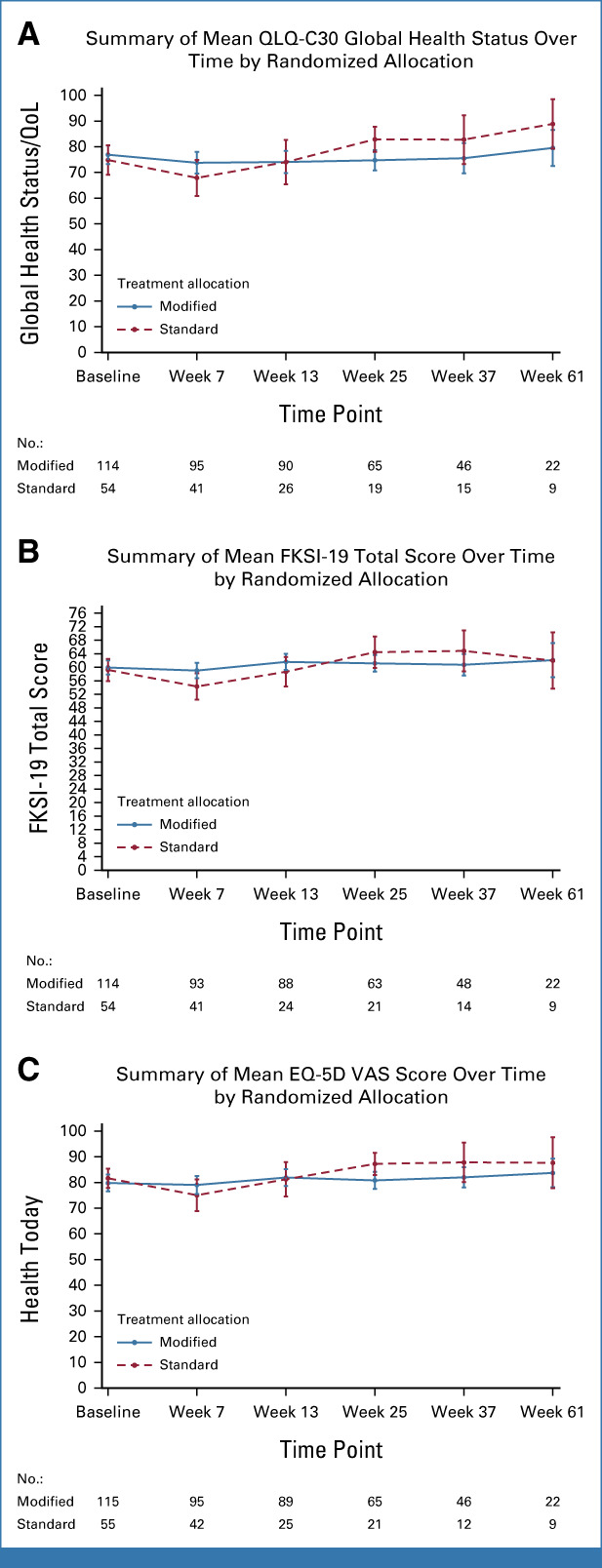
Summaries of mean (A) QLQ-C30 global health status, (B) FKSI-19 total score, and (C) EQ-5D VAS over time, by randomized allocation. EQ-5D, EuroQol 5-dimension; FKSI, Functional assessment of cancer-therapy Kidney Symptom Index; QLQ-C30, Quality of Life Questionnaire-C30; QoL, quality of life; VAS, visual analogue scale.
DISCUSSION
The results of the PRISM study demonstrate that tolerability of IPI + NIVO in the frontline treatment of patients with aRCC can be improved by delivering IPI once every 12 weeks instead of once every 3 weeks. Health-related QoL was generally well maintained using either schedule. Although not designed to formally compare treatment arms for efficacy, no clear differences in ORR, PFS, and OS were observed at a minimum follow-up of 2 years.
Just over half of patients (53.1%) receiving standard scheduling in PRISM experienced a grade 3-5 trAE, which is consistent with the rate (47%) reported in CheckMate 214.3 Rates of treatment discontinuation because of trAE associated with standard IPI were, however, higher in PRISM (39.1%) than in CheckMate 214, which, at 23%, is more akin to that observed with the modified PRISM schedule. The reasons for this difference are uncertain. It is possible, given the now more well-established potential for ongoing benefits beyond treatment discontinuation,11 that a lower threshold to stop treatment was used by PRISM investigators.
Focusing on adverse events rather than efficacy as the primary end point is unusual, but not unprecedented in advanced renal cancer.12 The purpose of PRISM was to establish if there were clear differences in tolerability by altering the drug schedule. If this was the case, and there were also promising efficacy signals, larger randomized phase III trials could be considered. We did not consider large noninferiority trials were justified without preliminary data.
The activity of standard IPI + NIVO in PRISM was broadly in line with previous data.3 A higher proportion of patients had favorable-risk disease (31%) and a lower proportion had previous nephrectomy (63%) in PRISM compared with CheckMate 214 (23% and 82%, respectively), but, otherwise, study populations were similar. The median PFS of 9.8 months among the mITT PRISM population receiving standard IPI sits within the 95% CI (12.4 months [9.8 to 16.5]) of the CheckMate 214 intention-to-treat (ITT) population.3 Among intermediate-/poor-risk patients, the corresponding figures were 8.6 months versus 11.6 months (95% CI, 8.4 to 15.5). The ORRs of 35.9% and 40.9% in this study are comparable with the 39% and 42% ORRs reported in CheckMate 214, when considering ITT and intermediate-/poor-risk patients, respectively.
The opportunity to optimize the dose and schedule of drugs, including immune checkpoint inhibitors, in cancer care to reduce cost, widen access, and improve safety is increasingly being recognized,13 as exemplified by initiatives such as the US Food and Drug Administration’s Project Optimus. This randomized phase II trial serves as an exemplar of such efforts. It does, however, have limitations. The decision to include favorable-risk patients reflects the design of the study before the results of the CheckMate 214 trial, which also included favorable-risk patients, were available. This is also reflected in the choice of single-agent sunitinib to benchmark the activity of the modified IPI schedule. The study did not meet the prespecified efficacy threshold (12-month PFS rate) using the modified schedule on the basis of this comparison. However, when considering both the mITT and the intermediate-/poor-risk subgroup of participants, efficacy data by median PFS, 12-month PFS, and ORR were comparable between PRISM arms and were in line with the data from CheckMate 214. OS rates also remained similar between treatment arms although, with a median follow-up of 32 months, no definite conclusions regarding the impact on longer-term survival can be drawn. The fact that PRISM was not powered to compare treatment arms for efficacy represents a further limitation of our study. Large noninferiority trials would be needed to formally address this, which do not appear justified on the basis of our results, in the opinion of the authors.
It is concerning that patient-reported outcome data in PRISM did not track the trAE data. The reasons for this are unclear. The relationship between adverse events and QoL has been explored previously in aRCC, with inconsistent results.12 Modification to the patient-reported outcome questions to better reflect immune-related toxicity has been suggested.14
Despite the introduction of IPI more than a decade ago, the mechanisms by which the CTLA-4 blockade induces both antitumor responses and trAE remain poorly defined. Intriguingly, however, preclinical studies suggest that CTLA-4–targeting agents that favor regulatory T-cell depletion within the tumor microenvironment, while avoiding peripheral T-cell activation, may be associated with a favorable toxicity profile, potentially paving the way for a new generation of safer and more efficacious anti–CTLA-4 antibodies.15-17
In conclusion, the results of the PRISM trial establish the superior safety of IPI dosing once every 12 weeks compared with once every 3 weeks, in combination with NIVO, in patients with aRCC. Although a formal internal efficacy comparison was not possible, no meaningful differences between treatment arms were observed on the basis of informal comparisons. Our data are consistent with studies in melanoma and NSCLC, suggesting that low dose and/or increased interval dosing of IPI, in combination with anti–PD-1 blockade, can remain efficacious while reducing toxicity experienced by patients.
ACKNOWLEDGMENT
We thank all the patients and clinical study teams who participated in the PRISM trial. We are grateful to the University of Leeds, United Kingdom, for sponsoring the trial and the University of Leeds CTRU for running the study. We thank Geraldine Murden, Hannah Buckley, and Heather Poad at the Leeds CTRU. We are grateful for the support and advice given by the Trial Steering Group (Professor Sarah Danson, Dr Natalie Charnley, Dr Kristian Brock) and the Data Management and Ethics Committee (Dr Hilary Glen, Professor Paul Lorigan and Mr Jamie Stobo). The authors would also like to thank Yorkshire Cancer Research and Bristol Myers Squibb who supported and funded the study.
Naveen S. Vasudev
Consulting or Advisory Role: Pfizer
Speakers’ Bureau: Bristol Myers Squibb, Eisai, Ipsen, EUSA Pharma
Research Funding: Bristol Myers Squibb (Inst)
Travel, Accommodations, Expenses: Ipsen, EUSA Pharma
Gemma Ainsworth
Research Funding: Bristol Myers Squibb (Inst), AstraZeneca (Inst)
Sarah Brown
Research Funding: AstraZeneca (Inst), Janssen (Inst), Bristol Myers Squibb/Celgene (Inst), Adlai Nortye (Inst), GlaxoSmithKline (Inst)
Lisa Pickering
Consulting or Advisory Role: BMS, Eisai, MSD Oncology, Pfizer
Speakers’ Bureau: MSD
Research Funding: NIHR (Inst), Rosetrees Trust (Inst), Kidney and Melanoma Cancer Fund of RMH Charity (Inst)
Tom Waddell
Stock and Other Ownership Interests: The Christie Clinic LLP
Honoraria: Pfizer, Ipsen, Bristol Myers Squibb, EUSA Pharma
Consulting or Advisory Role: Roche, Pfizer, Ipsen, Bristol Myers Squibb, Merck Sharp & Dohme, Eisai Europe
Research Funding: Bristol Myers Squibb (Inst), Pfizer (Inst), Ipsen (Inst), Merck Sharp & Dohme (Inst), Roche (Inst), Eisai (Inst)
Travel, Accommodations, Expenses: EUSA Pharma, Bristol Myers Squibb, Ipsen
Kate Fife
Employment: GlaxoSmithKline (I)
Stock and Other Ownership Interests: GlaxoSmithKline (I)
Honoraria: Eisai, Sanofi, Ipsen, MSD Oncology, EUSA Pharma
Consulting or Advisory Role: Bristol Myers Squibb/Celgene
Research Funding: Merck (Inst), Exelixis (Inst)
Travel, Accommodations, Expenses: Ipsen
Richard Griffiths
Honoraria: Pfizer, Eisai, BMSi, Merck Serono, Ipsen, MSD Oncology
Consulting or Advisory Role: Ipsen, Eisai
Anand Sharma
Consulting or Advisory Role: MSD Oncology, BMS
Speakers’ Bureau: MSD Oncology, EUSA Pharma, Merck Serono, Eisai/MSD, BMS GmbH & Co KG, Janssen Oncology, Pfizer/EMD Serono, Ipsen
Travel, Accommodations, Expenses: MSD, EUSA Pharma, Ipsen
Galina Velikova
Honoraria: Eisai, Pfizer, Novartis
Consulting or Advisory Role: Roche UK, Eisai, Novartis, Sanofi, Pfizer, AstraZeneca, Seagen
Speakers’ Bureau: Novartis
Research Funding: Pfizer (Inst), IQVIA (Inst)
Travel, Accommodations, Expenses: Roche UK, Novartis, Eisai
Other Relationship: University of Leeds
Anthony Maraveyas
Honoraria: Bristol Myers Squibb, Bayer, Pfizer, Boehringer Ingelheim, Sanofi, LEO Pharma
Consulting or Advisory Role: Bristol Myers Squibb, Bayer, Pfizer
Speakers’ Bureau: Bristol Myers Squibb, Bayer
Research Funding: Bristol Myers Squibb (Inst), Bayer
Travel, Accommodations, Expenses: Bayer, EUSA Pharma, Bristol Myers Squibb
Janet Brown
Honoraria: Novartis, Bayer, Amgen, Ipsen, MSD Oncology, Bristol Myers Squibb
Consulting or Advisory Role: Novartis, Bayer, Amgen, Ipsen, MSD Oncology, Bristol Myers Squibb
Speakers’ Bureau: Ipsen, Amgen, MSD
Research Funding: Amgen (Inst), Bayer (Inst)
Travel, Accommodations, Expenses: Ipsen, MSD Oncology, Amgen, Novartis, Bayer
Carmel Pezaro
Honoraria: Amgen, Pfizer, Bayer, Astellas Pharma
Travel, Accommodations, Expenses: Bayer, Ipsen
Mark Tuthill
Honoraria: Eisai
Consulting or Advisory Role: Novartis, Roche, Oxford VacMedix, Gilead Sciences, Seagen
Speakers’ Bureau: Bayer, Seagen, Daiichi Sankyo/Astra Zeneca, Lilly, Gilead Sciences
Research Funding: AstraZeneca (Inst)
Travel, Accommodations, Expenses: Merck, Novartis
Ekaterini Boleti
Honoraria: Ipsen, MSD Oncology, BMS, Pfizer
Consulting or Advisory Role: MSD Oncology, Ipsen, BMS
Travel, Accommodations, Expenses: Ipsen, MSD Oncology
Amit Bahl
Honoraria: Astellas Pharma, Janssen Oncology, Johnson & Johnson/Janssen, Bayer, Ipsen, Pfizer, BMS
Research Funding: Janssen (Inst), Sanofi (Inst), Bayer (Inst)
Bernadett Szabados
Honoraria: Merck, Ellipses Pharma, Ipsen, Roche/Genentech
Travel, Accommodations, Expenses: Roche/Genentech
Rosamonde E. Banks
Research Funding: Randox Laboratories (Inst)
Patents, Royalties, Other Intellectual Property: Patent filed for the use of a novel biomarker aminoacylase-1 in renal transplantation (Inst)
Balaji Venugopal
Honoraria: Pfizer, Bristol Myers Squibb, EUSA Pharma, Eisai, Ipsen, Merck, MSD Oncology
Consulting or Advisory Role: Pfizer/EMD Serono, MSD Oncology, Bristol Myers Squibb, Eisai
Speakers’ Bureau: MSD Oncology, Pfizer, Eisai, Janssen Oncology, Ipsen
Research Funding: Pfizer/EMD Serono (Inst), Calithera Biosciences (Inst), MSD Oncology (Inst), Ipsen (Inst)
Travel, Accommodations, Expenses: Ipsen, EUSA Pharma, Merck Serono
Poulam Patel
Speakers’ Bureau: Britol Myers Squibb (Inst)
Research Funding: Scancell (Inst)
Travel, Accommodations, Expenses: EUSA Pharma
Stefan N. Symeonides
Consulting or Advisory Role: Vaccitech (Inst), Bicycle Therapeutics (Inst), Ellipses Pharma (Inst), EUSA Pharma (Inst), Eisai (Inst), MSD (Inst), Bristol Myers Squibb (Inst), Pfizer/EMD Serono (Inst), MedAnnex (Inst), Boxer Capital (Inst), Duke Street Bio (Inst), Eugit Therapeutics (Inst)
Speakers’ Bureau: Bristol Myers Squibb (Inst), EUSA Pharma (Inst), Ipsen (Inst), Eisai (Inst)
Research Funding: Merck Sharp & Dohme (Inst), Verastem (Inst), Boston Pharmaceuticals (Inst), Sierra Oncology (Inst), Nucana (Inst), BioNTech (Inst), BiolineRx (Inst), Nouscom (Inst), Sapience Therapeutics (Inst), Roche/Genentech (Inst), Incyte (Inst), Scancell (Inst), Medannex (Inst)
Travel, Accommodations, Expenses: Ipsen, Bristol Myers Squibb, MSD, BioNTech, EUSA Pharma
Paul Nathan
Consulting or Advisory Role: AstraZeneca, Bristol Myers Squibb, MSD, Immunocore, Pfizer, Pierre Fabre, Novartis, GlaxoSmithKline, Ipsen, 4SC, Merck
Speakers’ Bureau: Bristol Myers Squibb, Novartis, MSD, Merck
Research Funding: Immunocore (Inst)
Travel, Accommodations, Expenses: Bristol Myers Squibb, MSD, Immunocore
Fiona J. Collinson
Speakers’ Bureau: Bayer
Research Funding: Bristol Myers Squibb, Roche/Genentech (Inst)
Thomas Powles
Honoraria: AstraZeneca, Eisai, Merck, Novartis, Pfizer, Roche, Astellas Pharma, BMS GmbH & Co KG, Exelixis, Incyte, Ipsen, Seagen, Merck Serono, Johnson & Johnson/Janssen, MashupMD
Consulting or Advisory Role: Bristol Myers Squibb, AstraZeneca, Ipsen, Pfizer, Novartis, Seagen, Roche, Exelixis, MSD, Merck Serono, Astellas Pharma, Johnson & Johnson, Eisai, MashupMD, Merck, Incyte
Research Funding: AstraZeneca, Roche, Bristol Myers Squibb, Exelixis, Ipsen, MSD, Novartis, Pfizer, Seagen, Merck Serono, Astellas Pharma, Johnson & Johnson, Eisai
Travel, Accommodations, Expenses: Pfizer, MSD, AstraZeneca, Roche, Ipsen
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
Presented in part at the European Society for Medical Oncology 2021 Congress, Paris, France, September 16-21, 2021.
SUPPORT
Supported by Bristol Myers Squibb (Princeton, NJ) and by Yorkshire Cancer Research (YCR) through the YCR Center for Early Phase Clinical Trials.
CLINICAL TRIAL INFORMATION
ISRCTN95351638
DATA SHARING STATEMENT
Data supporting this work are available on reasonable request. All requests will be reviewed by relevant stakeholders, based on the principles of a controlled access approach. Requests to access data should be made to CTRU-DataAccess@leeds.ac.uk in the first instance.
AUTHOR CONTRIBUTIONS
Conception and design: Naveen S. Vasudev, Thomas Powles, Sarah Brown, Paul Nathan, Galina Velikova, Fiona J. Collinson
Provision of study materials or patients: Naveen S. Vasudev, Lisa Pickering, Tom Waddell, Kate Fife, Richard Griffiths, Anand Sharma, Anthony Maraveyas, Janet Brown, Carmel Pezaro, Mark Tuthill, Ekaterini Boleti, Amit Bhal, Balaji Venugopal, Poulam Patel, Ankit Jain, Stefan N. Symeonides, Paul Nathan, Thomas Powles
Collection and assembly of data: Gemma Ainsworth, Sarah Brown, Eszter Katona, Helen Howard, Rosamonde E. Banks, Joanne Brown, Bernadett Szabados, Richard Griffiths, Anand Sharma, Anthony Maraveyas, Janet Brown, Carmel Pezaro, Mark Tuthill, Ekaterini Boleti, Amit Bhal, Balaji Venugopal, Poulam Patel, Ankit Jain, Stefan N. Symeonides, Paul Nathan, Thomas Powles
Data analysis and interpretation: Naveen S. Vasudev, Gemma Ainsworth, Sarah Brown, Galina Velikova, Lisa Pickering, Tom Waddell, Kate Fife, Bernadett Szabados, Paul Nathan, Stefan N. Symeonides, Fiona J. Collinson, Thomas Powles
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS’ DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Standard Versus Modified Ipilimumab, in Combination with Nivolumab, in Advanced Renal Cell Carcinoma: A Randomized Phase II Trial (PRISM)
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO’s conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Naveen S. Vasudev
Consulting or Advisory Role: Pfizer
Speakers’ Bureau: Bristol Myers Squibb, Eisai, Ipsen, EUSA Pharma
Research Funding: Bristol Myers Squibb (Inst)
Travel, Accommodations, Expenses: Ipsen, EUSA Pharma
Gemma Ainsworth
Research Funding: Bristol Myers Squibb (Inst), AstraZeneca (Inst)
Sarah Brown
Research Funding: AstraZeneca (Inst), Janssen (Inst), Bristol Myers Squibb/Celgene (Inst), Adlai Nortye (Inst), GlaxoSmithKline (Inst)
Lisa Pickering
Consulting or Advisory Role: BMS, Eisai, MSD Oncology, Pfizer
Speakers’ Bureau: MSD
Research Funding: NIHR (Inst), Rosetrees Trust (Inst), Kidney and Melanoma Cancer Fund of RMH Charity (Inst)
Tom Waddell
Stock and Other Ownership Interests: The Christie Clinic LLP
Honoraria: Pfizer, Ipsen, Bristol Myers Squibb, EUSA Pharma
Consulting or Advisory Role: Roche, Pfizer, Ipsen, Bristol Myers Squibb, Merck Sharp & Dohme, Eisai Europe
Research Funding: Bristol Myers Squibb (Inst), Pfizer (Inst), Ipsen (Inst), Merck Sharp & Dohme (Inst), Roche (Inst), Eisai (Inst)
Travel, Accommodations, Expenses: EUSA Pharma, Bristol Myers Squibb, Ipsen
Kate Fife
Employment: GlaxoSmithKline (I)
Stock and Other Ownership Interests: GlaxoSmithKline (I)
Honoraria: Eisai, Sanofi, Ipsen, MSD Oncology, EUSA Pharma
Consulting or Advisory Role: Bristol Myers Squibb/Celgene
Research Funding: Merck (Inst), Exelixis (Inst)
Travel, Accommodations, Expenses: Ipsen
Richard Griffiths
Honoraria: Pfizer, Eisai, BMSi, Merck Serono, Ipsen, MSD Oncology
Consulting or Advisory Role: Ipsen, Eisai
Anand Sharma
Consulting or Advisory Role: MSD Oncology, BMS
Speakers’ Bureau: MSD Oncology, EUSA Pharma, Merck Serono, Eisai/MSD, BMS GmbH & Co KG, Janssen Oncology, Pfizer/EMD Serono, Ipsen
Travel, Accommodations, Expenses: MSD, EUSA Pharma, Ipsen
Galina Velikova
Honoraria: Eisai, Pfizer, Novartis
Consulting or Advisory Role: Roche UK, Eisai, Novartis, Sanofi, Pfizer, AstraZeneca, Seagen
Speakers’ Bureau: Novartis
Research Funding: Pfizer (Inst), IQVIA (Inst)
Travel, Accommodations, Expenses: Roche UK, Novartis, Eisai
Other Relationship: University of Leeds
Anthony Maraveyas
Honoraria: Bristol Myers Squibb, Bayer, Pfizer, Boehringer Ingelheim, Sanofi, LEO Pharma
Consulting or Advisory Role: Bristol Myers Squibb, Bayer, Pfizer
Speakers’ Bureau: Bristol Myers Squibb, Bayer
Research Funding: Bristol Myers Squibb (Inst), Bayer
Travel, Accommodations, Expenses: Bayer, EUSA Pharma, Bristol Myers Squibb
Janet Brown
Honoraria: Novartis, Bayer, Amgen, Ipsen, MSD Oncology, Bristol Myers Squibb
Consulting or Advisory Role: Novartis, Bayer, Amgen, Ipsen, MSD Oncology, Bristol Myers Squibb
Speakers’ Bureau: Ipsen, Amgen, MSD
Research Funding: Amgen (Inst), Bayer (Inst)
Travel, Accommodations, Expenses: Ipsen, MSD Oncology, Amgen, Novartis, Bayer
Carmel Pezaro
Honoraria: Amgen, Pfizer, Bayer, Astellas Pharma
Travel, Accommodations, Expenses: Bayer, Ipsen
Mark Tuthill
Honoraria: Eisai
Consulting or Advisory Role: Novartis, Roche, Oxford VacMedix, Gilead Sciences, Seagen
Speakers’ Bureau: Bayer, Seagen, Daiichi Sankyo/Astra Zeneca, Lilly, Gilead Sciences
Research Funding: AstraZeneca (Inst)
Travel, Accommodations, Expenses: Merck, Novartis
Ekaterini Boleti
Honoraria: Ipsen, MSD Oncology, BMS, Pfizer
Consulting or Advisory Role: MSD Oncology, Ipsen, BMS
Travel, Accommodations, Expenses: Ipsen, MSD Oncology
Amit Bahl
Honoraria: Astellas Pharma, Janssen Oncology, Johnson & Johnson/Janssen, Bayer, Ipsen, Pfizer, BMS
Research Funding: Janssen (Inst), Sanofi (Inst), Bayer (Inst)
Bernadett Szabados
Honoraria: Merck, Ellipses Pharma, Ipsen, Roche/Genentech
Travel, Accommodations, Expenses: Roche/Genentech
Rosamonde E. Banks
Research Funding: Randox Laboratories (Inst)
Patents, Royalties, Other Intellectual Property: Patent filed for the use of a novel biomarker aminoacylase-1 in renal transplantation (Inst)
Balaji Venugopal
Honoraria: Pfizer, Bristol Myers Squibb, EUSA Pharma, Eisai, Ipsen, Merck, MSD Oncology
Consulting or Advisory Role: Pfizer/EMD Serono, MSD Oncology, Bristol Myers Squibb, Eisai
Speakers’ Bureau: MSD Oncology, Pfizer, Eisai, Janssen Oncology, Ipsen
Research Funding: Pfizer/EMD Serono (Inst), Calithera Biosciences (Inst), MSD Oncology (Inst), Ipsen (Inst)
Travel, Accommodations, Expenses: Ipsen, EUSA Pharma, Merck Serono
Poulam Patel
Speakers’ Bureau: Britol Myers Squibb (Inst)
Research Funding: Scancell (Inst)
Travel, Accommodations, Expenses: EUSA Pharma
Stefan N. Symeonides
Consulting or Advisory Role: Vaccitech (Inst), Bicycle Therapeutics (Inst), Ellipses Pharma (Inst), EUSA Pharma (Inst), Eisai (Inst), MSD (Inst), Bristol Myers Squibb (Inst), Pfizer/EMD Serono (Inst), MedAnnex (Inst), Boxer Capital (Inst), Duke Street Bio (Inst), Eugit Therapeutics (Inst)
Speakers’ Bureau: Bristol Myers Squibb (Inst), EUSA Pharma (Inst), Ipsen (Inst), Eisai (Inst)
Research Funding: Merck Sharp & Dohme (Inst), Verastem (Inst), Boston Pharmaceuticals (Inst), Sierra Oncology (Inst), Nucana (Inst), BioNTech (Inst), BiolineRx (Inst), Nouscom (Inst), Sapience Therapeutics (Inst), Roche/Genentech (Inst), Incyte (Inst), Scancell (Inst), Medannex (Inst)
Travel, Accommodations, Expenses: Ipsen, Bristol Myers Squibb, MSD, BioNTech, EUSA Pharma
Paul Nathan
Consulting or Advisory Role: AstraZeneca, Bristol Myers Squibb, MSD, Immunocore, Pfizer, Pierre Fabre, Novartis, GlaxoSmithKline, Ipsen, 4SC, Merck
Speakers’ Bureau: Bristol Myers Squibb, Novartis, MSD, Merck
Research Funding: Immunocore (Inst)
Travel, Accommodations, Expenses: Bristol Myers Squibb, MSD, Immunocore
Fiona J. Collinson
Speakers’ Bureau: Bayer
Research Funding: Bristol Myers Squibb, Roche/Genentech (Inst)
Thomas Powles
Honoraria: AstraZeneca, Eisai, Merck, Novartis, Pfizer, Roche, Astellas Pharma, BMS GmbH & Co KG, Exelixis, Incyte, Ipsen, Seagen, Merck Serono, Johnson & Johnson/Janssen, MashupMD
Consulting or Advisory Role: Bristol Myers Squibb, AstraZeneca, Ipsen, Pfizer, Novartis, Seagen, Roche, Exelixis, MSD, Merck Serono, Astellas Pharma, Johnson & Johnson, Eisai, MashupMD, Merck, Incyte
Research Funding: AstraZeneca, Roche, Bristol Myers Squibb, Exelixis, Ipsen, MSD, Novartis, Pfizer, Seagen, Merck Serono, Astellas Pharma, Johnson & Johnson, Eisai
Travel, Accommodations, Expenses: Pfizer, MSD, AstraZeneca, Roche, Ipsen
No other potential conflicts of interest were reported.
REFERENCES
- 1.Heng DYC, Xie W, Regan MM, et al. : Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: Results from a large, multicenter study. J Clin Oncol 27:5794-5799, 2009 [DOI] [PubMed] [Google Scholar]
- 2.Motzer RJ, Escudier B, McDermott DF, et al. : Survival outcomes and independent response assessment with nivolumab plus ipilimumab versus sunitinib in patients with advanced renal cell carcinoma: 42-month follow-up of a randomized phase 3 clinical trial. J Immunother Cancer 8:e000891, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Motzer RJ, Tannir NM, McDermott DF, et al. : Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med 378:1277-1290, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hammers HJ, Plimack ER, Infante JR, et al. : Safety and efficacy of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma: The CheckMate 016 study. J Clin Oncol 35:3851-3858, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lebbé C, Meyer N, Mortier L, et al. : Evaluation of two dosing regimens for nivolumab in combination with ipilimumab in patients with advanced melanoma: Results from the phase IIIb/IV CheckMate 511 trial. J Clin Oncol 37:867-875, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hellmann MD, Rizvi NA, Goldman JW, et al. : Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): Results of an open-label, phase 1, multicohort study. Lancet Oncol 18:31-41, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Long GV, Robert C, Butler MO, et al. : Standard-dose pembrolizumab plus alternate-dose ipilimumab in advanced melanoma: KEYNOTE-029 cohort 1C, a phase 2 randomized study of two dosing schedules. Clin Cancer Res 27:5280-5288, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buckley HL, Collinson FJ, Ainsworth G, et al. : PRISM protocol: A randomised phase II trial of nivolumab in combination with alternatively scheduled ipilimumab in first-line treatment of patients with advanced or metastatic renal cell carcinoma. BMC Cancer 19:1102, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Motzer RJ, Hutson TE, Cella D, et al. : Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med 369:722-731, 2013 [DOI] [PubMed] [Google Scholar]
- 10.SAS Institute Inc : SAS® 9.4 Statements: Reference. Cary, NC, SAS Institute, 2013 [Google Scholar]
- 11.Regan MM, Jegede OA, Mantia CM, et al. : Treatment-free survival after immune checkpoint inhibitor therapy versus targeted therapy for advanced renal cell carcinoma: 42-Month results of the CheckMate 214 trial. Clin Cancer Res 27:6687-6695, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Escudier B, Porta C, Bono P, et al. : Randomized, controlled, double-blind, cross-over trial assessing treatment preference for pazopanib versus sunitinib in patients with metastatic renal cell carcinoma: PISCES study. J Clin Oncol 32:1412-1418, 2014 [DOI] [PubMed] [Google Scholar]
- 13.Hirsch I, Goldstein DA, Tannock IF, et al. : Optimizing the dose and schedule of immune checkpoint inhibitors in cancer to allow global access. Nat Med 28:2236-2237, 2022 [DOI] [PubMed] [Google Scholar]
- 14.Bergerot CD, Malhotra J, Bergerot P, et al. : Patients’ perceptions regarding the relevance of items contained in the Functional Assessment of Cancer Therapy Kidney Symptom Index-19. Oncologist 28:494-500, 2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du X, Liu M, Su J, et al. : Uncoupling therapeutic from immunotherapy-related adverse effects for safer and effective anti-CTLA-4 antibodies in CTLA4 humanized mice. Cell Res 28:433-447, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong MMY, Maleki Vareki S: Addressing the elephant in the immunotherapy room: Effector T-cell priming versus depletion of regulatory T-cells by anti-CTLA-4 therapy. Cancers (Basel) 14:1580, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gan X, Shan Q, Li H, et al. : An anti-CTLA-4 heavy chain-only antibody with enhanced T(reg) depletion shows excellent preclinical efficacy and safety profile. Proc Natl Acad Sci USA 119:e2200879119, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data supporting this work are available on reasonable request. All requests will be reviewed by relevant stakeholders, based on the principles of a controlled access approach. Requests to access data should be made to CTRU-DataAccess@leeds.ac.uk in the first instance.