

Abstract
Under anaerobic conditions and at circumneutral pH, cells of the widely distributed, obligate chemolithoautotrophic bacterium Thiobacillus denitrificans oxidatively dissolved synthetic and biogenic U(IV) oxides (uraninite) in nitrate-dependent fashion: U(IV) oxidation required the presence of nitrate and was strongly correlated with nitrate consumption. This is the first report of anaerobic U(IV) oxidation by an autotrophic bacterium.
In situ microbial reductive immobilization of radionuclides in aquifers and saturated soils is a remedial approach that has been the subject of considerable research effort over the past decade. The essence of this approach is that many radionuclides of concern are redox active and are less soluble in their reduced form and thus can be immobilized in aquifers via microbially mediated reduction under anaerobic conditions. For example, U(VI) in the form of UO22+ and its complexes is relatively soluble in water, whereas the mineral uraninite (UO2), typically formed by U(VI)-reducing bacteria (5, 10), has very low solubility. Since the earliest reports of direct microbial reduction of uranium by Geobacter metallireducens, Shewanella oneidensis (formerly Shewanella putrefaciens), and Desulfovibrio desulfuricans (9-11), other species have also been shown to have this capability. However, recent studies have suggested that microbially mediated U(IV) oxidation could complicate efforts at long-term reductive immobilization. In 2002, two articles (4, 14) reported that uranium reoxidation can occur anaerobically in the presence of nitrate, which is a common cocontaminant with uranium at U.S. Department of Energy sites (13). Finneran et al. (4) showed that nitrate-grown, but not Fe(III)-grown, cells of G. metallireducens carried out nitrate-dependent U(IV) oxidation in anaerobic cell suspensions amended with UBr4, a soluble form of U(IV). Senko et al. (14) observed nitrate-dependent uranium solubilization during push-pull (in situ) field studies and invoked an indirect, microbial mechanism [namely, abiotic oxidation of U(IV) by intermediates of bacterial nitrate reduction, such as nitrite]. Although they invoked an indirect mechanism, Senko et al. (14) could not rule out direct microbial oxidation of U(IV) on the basis of their data.
In this article, it is reported that the widely distributed, obligate chemolithoautotrophic bacterium Thiobacillus denitrificans, known for its ability to couple the oxidation of various S- and Fe(II)-containing electron donors with denitrification (8, 15), is capable of anaerobic, nitrate-dependent oxidative dissolution of synthetic and biogenic U(IV) oxides. T. denitrificans has relevance to certain uranium-contaminated sites, as this species (or species with >98% 16S rRNA gene sequence similarity) was found to account for a relatively large proportion of the bacterial community from an open-pit uranium mine (representing ∼24% of the 16S rRNA gene clones analyzed [16]). This is the first report of anaerobic, nitrate-dependent U(IV) oxidation by an autotrophic bacterium, although the aerobic oxidation of U4+ (aqueous) by the autotrophic bacterium Acidithiobacillus ferrooxidans (formerly Thiobacillus ferrooxidans) at pH 1.5 was reported by DiSpirito and Tuovinen (3).
All experiments described in this article were performed at 30°C under strictly anaerobic conditions in an anaerobic glove box (Coy Laboratory Products, Inc., Grass Lake, Mich.) with a nominal gas composition of 80% N2, 10% CO2, and 10% H2. The glass and plastic materials used to contain or manipulate the cultures were allowed to degas in the glove box for at least several days before use. T. denitrificans (strain ATCC 25259, obtained from the American Type Culture Collection) was cultivated in anaerobic growth medium (pH ∼7) that included the following compounds added at the indicated concentrations: Na2S2O3 · 5H2O (20 mM), NH4Cl (18.7 mM), KNO3 (20 mM), KH2PO4 (14.7 mM), NaHCO3 (30 mM), MgSO4 · 7H2O (3.25 mM), FeSO4 · 7H2O (0.08 mM), CaCl2 · 2H2O (0.05 mM), and anaerobic and sterile vitamin, trace element, and selenite-tungstate solutions prepared as described by Widdel and Bak (18) (stock solutions 1, 4, 6, 7, and 8). Anaerobic techniques used in the preparation of growth medium and stock solutions are described elsewhere (1). Highly purified water (18 MΩ resistance) obtained from a Milli-Q UV Plus system (Millipore, Bedford, Mass.) was used to prepare the growth medium and all other aqueous solutions described in this article.
For U(IV) oxidation experiments, T. denitrificans cells in late exponential phase (∼108 cells/ml) were harvested anaerobically by centrifugation in sealed polycarbonate bottles and washed once in anaerobic resuspension buffer. The composition of the resuspension buffer was similar to that of the growth medium with the following exceptions: it contained no Na2S2O3 · 5H2O, CaCl2 · 2H2O, or KNO3, the KH2PO4 concentration was reduced to 1.5 mM [to effectively preclude the formation of soluble U(IV)-phosphate complexes, as indicated by geochemical modeling with the PHREEQC computer program (12)], and the FeSO4 · 7H2O concentration was reduced to 0.0075 mM [to preclude the possibility that iron could markedly enhance U(IV) oxidation, as has been documented in studies with G. metallireducens involving FeSO4 concentrations of 5 to 6 mM (4)]. For cell suspension experiments, the resuspension buffer was amended with additional compounds as described below.
Assays for U(IV) oxidation were performed in 25-ml serum bottles sealed with butyl rubber stoppers and containing either 5 or 6 ml of liquid culture, depending on the experiment. Positive controls to assess the batch-specific denitrification activity of T. denitrificans cells were carried out in the growth medium (i.e., with thiosulfate as the electron donor) and received the same inoculum (in terms of number and concentration of cells) as cultures assayed for U(IV) oxidation. Dissolved uranium was determined according to the following steps: (i) passage of samples through 0.2-μm-pore-size syringe filters under anaerobic conditions, (ii) immediate centrifugation of the filtrate (∼20,000 × g, 4°C, 4 min) to minimize the possibility that colloidal U(IV) could be included in the analysis (although colloidal material was never visible in the filtrates), (iii) addition of 50 (or 500) μl of the supernatant to 4,950 (or 4,500) μl of 0.32 N HNO3 containing thallium (10 μg/liter) as an internal standard, and (iv) analysis of the resulting solution by inductively coupled plasma-mass spectrometry (ICP-MS). The filtration step resulted in minimal sorptive loss of U(VI), as indicated by a test in which uranyl acetate dissolved in resuspension buffer had a U concentration of 37.41 ± 0.19 μM (mean ± standard deviation) before filtration and 37.00 ± 0.22 μM after filtration. ICP-MS analysis, which was conducted with a quadrupole, single-collector instrument (model 4500; Agilent Technologies, Palo Alto, Calif.), attained precision within 5% and accuracy of >95% using standards traceable to the National Institute of Standards and Technology. The underlying assumption of this analytical method, namely, that dissolved uranium would be solely in the +VI oxidation state under the experimental conditions, was founded on geochemical modeling with the PHREEQC computer program (12) and was tested for selected samples by comparing results of ICP-MS analysis to those of kinetic phosphorescence analysis (KPA) (2), which is specific to U(VI). The concentrations of nitrate and nitrite were determined by ion chromatography (model DX 500; Dionex Corporation, Sunnyvale, Calif.) with micromembrane suppression and electrochemical conductivity detection. Protein concentrations were determined with a Coomassie dye-protein binding colorimetric method (Pierce Biotechnology, Rockford, Ill.) after hydrolysis of the cells in 0.5 N NaOH at 100°C for 10 min; bovine serum albumin was used as the standard.
T. denitrificans cells catalyzed oxidative dissolution of U(IV) oxide in anaerobic resuspension buffer containing ca. 3 mM nitrate and ca. 1 mmol of synthetic U(IV) oxide per liter (Fig. 1A). To confirm that the dissolved uranium that appeared over time was truly oxidized to U(VI), two active samples collected on day 17 were subjected to KPA. For both samples, the ICP-MS and KPA results agreed within 4%, thereby confirming oxidative dissolution. Live (without nitrate) and sterile controls indicated that oxidative dissolution was dependent on nitrate and was catalyzed by active T. denitrificans cells: the increase in dissolved uranium in the live (without nitrate) and sterile controls was <3% of that observed in the active cultures (Table 1). The live controls differed from the active cultures only in that the controls contained no nitrate; the sterile controls differed from the active cultures only in that the cells were autoclaved (anaerobically) before addition to the bottles. In this experiment, U(IV) oxide was produced under strictly anaerobic conditions by adding 0.1 mmol of uranyl acetate dihydrate to 30 ml of sterile, degassed Milli-Q water and then adding 1 mmol of sodium dithionite that had been stored in the glove box. The precipitate was washed three times with sterile, degassed Milli-Q water. The washed U(IV) oxide pellet, which had a slurry consistency, was added to serum bottles with a sterile spatula.
FIG. 1.

Anaerobic, oxidative dissolution of two different synthetic U(IV) oxides by washed cell suspensions of T. denitrificans: (A) freshly prepared U(IV) oxide with a slurry consistency and (B) aggregated grains of U(IV) oxide that had dried under anaerobic conditions. Datum points represent the averages of duplicate suspensions.
TABLE 1.
Summary of experiments testing nitrate-dependent U(IV) oxidation by T. denitrificans
| U(IV) oxide form | Net U(IV) dissolved (μM)a
|
Portion of U(IV) dissolved (%)b | Positive-control denitrification activityc | ||
|---|---|---|---|---|---|
| Active culture | Sterile control | Live control without nitrate | |||
| Synthetic, slurryd | 42 | 0.92 | 0.18 | 4 | 1.2 |
| Synthetic, driede | 0.067 | 0.0027 | <0 | 0.02 | 1.2 |
| Biogenic UO2f | 39 | 15 | 13 | 22 | 3.3 |
Net U(IV) dissolved is defined as the final U concentration minus the initial U concentration. Values represent the averages of duplicate cultures for synthetic U(IV) oxides or of triplicate cultures for biogenic uraninite.
Approximate fraction of initial U(IV) that was dissolved in the active cultures, expressed as a percentage. The amounts of initial U(IV) present were approximately 1, 0.4, and 0.18 mmol/liter, respectively, in the experiments with slurry (synthetic), dried (synthetic), and biogenic U(IV) oxide.
In micromoles of electron equivalents per minute. Positive controls contained thiosulfate as the electron donor rather than a U(IV) oxide (see text). Protein content ranged from approximately 1.1 to 1.3 mg.
See Fig. 1A. Net U(IV) dissolved data are calculated for day 17.
See Fig. 1B. Net U(IV) dissolved data are calculated for day 20.
See Fig. 2A. Net U(IV) dissolved data are calculated for day 14.
When the U(IV) oxide synthesis procedure was modified by allowing the slurry to dry for 5 days in an anaerobic glove box and form aggregated particles, results indicating nitrate-dependent U(IV) oxidation by T. denitrificans (Fig. 1B) were qualitatively similar to those depicted in Fig. 1A. However, the rate of U(IV) oxidation for the dried U(IV) oxide was <0.2% of that observed for the U(IV) oxide slurry (Table 1 and Fig. 1). Furthermore, the percentage of total U(IV) that was oxidatively dissolved with the dried U(IV) oxide was 2 to 3 orders of magnitude lower than the percentage for the U(IV) slurry (Table 1). This difference cannot be attributed to batch-to-batch differences in the activity of T. denitrificans cells, as the total denitrification activities of the positive controls for the experiments represented in Fig. 1 agreed within 5% (Table 1). It is possible that the differences in U(IV) oxidation could be explained by greater mass transfer limitations or lower specific surface area for the dried U(IV) oxides. Unfortunately, reliable physical characterization of the materials was precluded by the vulnerability of the U(IV) oxides to rapid oxidation in air and the possibility of associated changes in physical properties.
Additional studies performed with biogenic uraninite (UO2) also clearly indicated nitrate-dependent U(IV) oxidation by T. denitrificans (Fig. 2A), as active cultures produced significantly more dissolved U(VI) than sterile or live controls (P ≪ 0.01 by t test). However, the background concentration of dissolved uranium was higher and the differences between active and control cultures were less pronounced than for synthetic U(IV) oxides (Fig. 2A and Table 1). Biogenic uraninite, which was graciously provided by J. Fredrickson (Pacific Northwest National Laboratory), was generated with H2-oxidizing suspensions of S. putrefaciens CN32 (6) and has been characterized by X-ray diffraction (5). In the present study, biogenic UO2 was dispensed from a 7.5-mmol/liter stock solution in anoxic water to attain a final concentration of approximately 0.18 mmol/liter.
FIG. 2.

Production of dissolved uranium (A) and consumption of nitrate (B) during anaerobic, oxidative dissolution of biogenic uraninite by washed cell suspensions of T. denitrificans. Datum points represent the averages of triplicate suspensions, and error bars represent 1 standard deviation (error bars are plotted for all datum points but are obscured by the symbol in some cases).
Nitrate and nitrite data were collected for suspensions with biogenic uraninite. Nitrite was never detected (detection limit, ca. 10 μM) during the experiment depicted in Fig. 2. Thus, there is no evidence to suggest that abiotic reaction of dissolved nitrite with uranium accounted for the observed net oxidation of 39 μM U(IV), which would have required from 13 to 78 μM nitrite, depending on whether nitrite were reduced to ammonium or nitrogen monoxide, respectively. Notably, nitrite was also not detected in the positive control for this experiment, which consumed 13 mM nitrate over a 2-h period. Both U(IV) oxidation and nitrate consumption slowed over time (Fig. 2), and in fact, these processes were strongly correlated over time (r2 = 0.98) (Fig. 3). Despite this strong linear correlation, it is clear that U(IV) could not have served as the predominant electron donor for denitrification, as the net amount of U(IV) oxidized (39 μM) is <2% of the amount that would be required to denitrify the 0.96 mM nitrate consumed. In fact, if the entire amount of added U(IV) had been oxidized in conjunction with denitrification, it would only have accounted for 7.5% of the observed nitrate consumption.
FIG. 3.

Correlation between net U(IV) oxidation and nitrate consumption during the oxidative dissolution of biogenic uraninite by washed cells of T. denitrificans (experiment represented in Fig. 2). Datum points represent the averages of triplicate suspensions. A linear regression fit is plotted.
Hydrogen (present in the glove box atmosphere to drive the oxygen-consuming catalyst) is the only electron donor that could have accounted for nitrate consumption [note that Fe(II), present at 7.5 μM, could account for <0.2% of the observed denitrification]. An initial concentration of 1.8% H2 in the headspace of active cultures (which is lower than the nominal composition but generally consistent with the H2 monitor in the glove box) would account for reduction of ca. 1 mM nitrate to N2 and could explain why nitrate consumption effectively ceased by day 14 (i.e., the primary electron donor was depleted). Further information about the relationship of H2 oxidation to nitrate consumption and U(IV) oxidation was obtained from additional experimental trials in which the headspace of suspensions was rigorously purged and replaced with an H2-free, anaerobic mixture of 90% N2-10% CO2. These studies indicated that, in contrast to results for suspensions with H2, oxidative dissolution of biogenic uraninite was not significantly different in nitrate-amended active cultures compared to both sterile and live (without nitrate) controls (P ≫ 0.05 by t test) and that nitrate consumption was markedly greater in the presence of H2 than in its absence (data not shown).
Whether nitrate-dependent U(IV) oxidation is coupled to energy conservation in T. denitrificans and G. metallireducens cannot be determined from existing data. The overall reaction
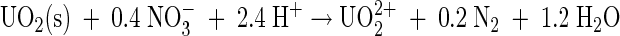 |
is thermodynamically favorable (ΔGo′ = −65 or −93 kJ/mol for crystalline or amorphous UO2, respectively, based on data from references 7 and 17), although the reduction potential of the UO22+/UO2(s) couple is relatively high (E0′ = 0.41 or 0.26 V for crystalline or amorphous UO2, respectively) and would thus require a relatively high-potential electron transport carrier (possibly a c-type cytochrome). If U(IV) oxidation were coupled to nitrate reduction to nitrite rather than denitrification, it would yield little free energy (E0′ = 0.43 V for the NO3−/NO2− couple). The strong correlation between U(IV) oxidation and nitrate consumption (Fig. 3) cannot be used to definitively conclude that these processes were coupled for energy conservation, as U(IV) oxidation only accounted for <2% of nitrate consumption and ceased when nitrate was still available. These observations, along with the lack of significant U(IV) oxidation in the absence of H2, suggest that U(IV) oxidation by T. denitrificans may require ongoing nitrate reduction coupled to another electron donor. Notably, U(IV) oxidation also accounted for a small portion of nitrate consumption (no more than ca. 10%) during nitrate-dependent U(IV) oxidation by the heterotroph G. metallireducens in cell suspension experiments (4), although it is not clear what electron donor drove nitrate reduction in that system. The exact nature of the nitrate dependence of U(IV) oxidation, for example, whether proteins involved in nitrate reduction have any direct role in U(IV) oxidation, remains to be determined.
Acknowledgments
I thank B. Esser (Lawrence Livermore National Laboratory) for valuable discussions and, in combination with S. Szechenyi, for performing ICP-MS analyses of uranium, and I thank J. Fredrickson and D. Kennedy (Pacific Northwest National Laboratory) for graciously providing biogenic uraninite and KPA analyses of U(VI) in selected samples.
This work was performed under the auspices of the U.S. Department of Energy by the University of California, Lawrence Livermore National Laboratory, under contract W-7405-Eng-48.
REFERENCES
- 1.Beller, H. R., A. M. Spormann, P. K. Sharma, J. R. Cole, and M. Reinhard. 1996. Isolation and characterization of a novel toluene-degrading, sulfate-reducing bacterium. Appl. Environ. Microbiol. 62:1188-1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brina, R., and A. G. Miller. 1992. Direct detection of trace levels of uranium by laser-induced kinetic phosphorimetry. Anal. Chem. 64:1413-1418. [Google Scholar]
- 3.DiSpirito, A. A., and O. H. Tuovinen. 1982. Uranous ion oxidation and carbon dioxide fixation by Thiobacillus ferrooxidans. Arch. Microbiol. 133:28-32. [Google Scholar]
- 4.Finneran, K. T., M. E. Housewright, and D. R. Lovley. 2002. Multiple influences of nitrate on uranium solubility during bioremediation of uranium-contaminated subsurface sediments. Environ. Microbiol. 4:510-516. [DOI] [PubMed] [Google Scholar]
- 5.Fredrickson, J. K., J. M. Zachara, D. W. Kennedy, M. C. Duff, Y. A. Gorby, S. W. Li, and K. M. Krupka. 2000. Reduction of U(VI) in goethite (α-FeOOH) suspensions by a dissimilatory metal-reducing bacterium. Geochim. Cosmochim. Acta 64:3085-3098. [Google Scholar]
- 6.Fredrickson, J. K., J. M. Zachara, D. W. Kennedy, C. Liu, M. C. Duff, D. B. Hunter, and A. Dohnalkova. 2002. Influence of Mn oxides on the reduction of uranium(VI) by the metal-reducing bacterium Shewanella putrefaciens. Geochim. Cosmochim. Acta 66:3247-3262. [Google Scholar]
- 7.Grenthe, I., J. Fuger, R. J. Lemire, A. B. Muller, C. Nguyen-Trung, and H. Wanner. 1992. Chemical thermodynamics of uranium. Nuclear Energy Agency, Organisation for Economic Co-operation and Development, Paris, France.
- 8.Kelly, D. P., and A. P. Wood. 2000. Confirmation of Thiobacillus denitrificans as a species of the genus Thiobacillus, in the β-subclass of the Proteobacteria, with strain NCIMB 9548 as the type strain. Int. J. Syst. Evol. Microbiol. 50:547-550. [DOI] [PubMed] [Google Scholar]
- 9.Lovley, D. R., and E. J. P. Phillips. 1992. Bioremediation of uranium contamination with enzymatic uranium reduction. Environ. Sci. Technol. 26:2228-2234. [Google Scholar]
- 10.Lovley, D. R., and E. J. P. Phillips. 1992. Reduction of uranium by Desulfovibrio desulfuricans. Appl. Environ. Microbiol. 58:850-856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lovley, D. R., E. J. P. Phillips, Y. A. Gorby, and E. R. Landa. 1991. Microbial reduction of uranium. Nature (London) 350:413-416. [Google Scholar]
- 12.Parkhurst, D. L., and C. A. J. Appelo. 1999. User's guide to PHREEQC (version 2)—a computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations. U.S. Geological Survey Water-Resources Investigations Report 99-4259. U.S. Geological Survey, Reston, Va.
- 13.Riley, R. G., and J. M. Zachara. 1992. Nature of chemical contamination on DOE lands and identification of representative contaminant mixtures for basic subsurface science research. Subsurface Science Program, Office of Energy Research, U.S. Department of Energy, Washington, D.C.
- 14.Senko, J. M., J. D. Istok, J. M. Suflita, and L. R. Krumholz. 2002. In-situ evidence for uranium immobilization and remobilization. Environ. Sci. Technol. 36:1491-1496. [DOI] [PubMed] [Google Scholar]
- 15.Straub, K. L., M. Benz, B. Schink, and F. Widdel. 1996. Anaerobic, nitrate-dependent microbial oxidation of ferrous iron. Appl. Environ. Microbiol. 62:1458-1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suzuki, Y., S. D. Kelly, K. M. Kemner, and J. F. Banfield. 2003. Microbial populations stimulated for hexavalent uranium reduction in uranium mine sediment. Appl. Environ. Microbiol. 69:1337-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thauer, R. K., K. Jungermann, and K. Decker. 1977. Energy conservation in chemoautotrophic anaerobic bacteria. Bacteriol. Rev. 41:100-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Widdel, F., and F. Bak. 1992. Gram-negative mesophilic sulfate-reducing bacteria, p. 3352-3378. In A. Balows, H. G. Trüper, M. Dworkin, W. Harder, and K.-H. Schleifer (ed.), The prokaryotes. Springer-Verlag, New York, N.Y.