

Abstract
A number of oxygenated monoterpenes present at low concentrations in plant oils have anticarcinogenic properties. One of the most promising compounds in this respect is (−)-perillyl alcohol. Since this natural product is present only at low levels in a few plant oils, an alternative, synthetic source is desirable. Screening of 1,800 bacterial strains showed that many alkane degraders were able to specifically hydroxylate l-limonene in the 7 position to produce enantiopure (−)-perillyl alcohol. The oxygenase responsible for this was purified from the best-performing wild-type strain, Mycobacterium sp. strain HXN-1500. By using N-terminal sequence information, a 6.2-kb ApaI fragment was cloned, which encoded a cytochrome P450, a ferredoxin, and a ferredoxin reductase. The three genes were successfully coexpressed in Pseudomonas putida by using the broad-host-range vector pCom8, and the recombinant converted limonene to perillyl alcohol with a specific activity of 3 U/g (dry weight) of cells. The construct was subsequently used in a 2-liter bioreactor to produce perillyl alcohol on a scale of several grams.
The production of (−)-perillyl alcohol from l-limonene is of interest because of the limited availability of (−)-perillyl alcohol in nature and its proven anticarcinogenic properties; phase II trials to evaluate perillyl alcohol for the treatment of breast, pancreatic, and colorectal cancer are in progress (37). The only microbial enzyme system described thus far that transforms limonene to perillyl alcohol was found in Bacillus stearothermophilus BR388. However, this enzyme system is not sufficiently regiospecific; significant quantities of carveol, carvone, and terpineol are also produced (25). Removal of these side products is difficult as their boiling points and hydrophobicities are almost identical, and expensive purification methods (for example, chromatography) would be required to obtain sufficiently pure perillyl alcohol. Therefore, the industrial production of perillyl alcohol with this Bacillus enzyme system is not attractive. The conversion of limonene to perillic acid by a Pseudomonas putida strain expressing a cymene monooxygenase was described by Mars et al. (21) and could be interesting as perillyl alcohol is likely to be an intermediate in the production of perillic acid. Other literature concerning limonene biotransformations was reviewed recently (5).
The approach that we used to find strains capable of regiospecific hydroxylation of limonene consisted of screening a collection of 1,800 bacterial strains grown on a range of relatively reduced substrates, such as toluene, naphthalene, and various alkanes. Using this approach, we anticipated that we would find oxygenases involved in catabolic pathways that would accept l-limonene as a substrate. Previous work has demonstrated that many catabolic oxygenases accept a wide range of unnatural substrates. Toluene dioxygenases, for example, have been shown to oxygenate more than 100 substrates (reference 11 and references therein). Another example is the P. putida GPo1 alkane hydroxylase, which hydroxylates a wide range of aliphatic, alicyclic, and alkyl-substituted compounds (33). More recently, a Sphingomonas isolate was shown to contain a soluble alkane hydroxylase that is able to hydroxylate or epoxidate a wide range of (hetero)alicyclic compounds (17, 18). Other enzymes that oxidize alkanes include the nonheme iron monooxygenases related to the soluble and particulate methane monooxygenases, as well as cytochrome P450 alkane hydroxylases found in yeasts and some bacteria (34). The yeast cytochrome P450 alkane hydroxylases have not been extensively tested with substrates other than alkane or alkane metabolites, while the bacterial cytochrome P450 alkane hydroxylases have not been cloned and characterized yet, with the exception of an Acinetobacter enzyme, the first member of a new family of cytochrome P450 enzymes, CYP153 (20).
Our screening analysis yielded two major results. First, we found a range of toluene and naphthalene dioxygenases that hydroxylate d-limonene exclusively in the 6 position, yielding pure (+)-trans-carveol (6). Second (as described in the present paper and in a recent patent application [8]), we found a number of alkane-degrading organisms that hydroxylate both enantiomers of limonene in the 7 position. The best strain in this respect was Mycobacterium sp. strain HXN-1500, which was described previously in a study focusing on alkane hydroxylases in high-G+C-content gram-positive bacteria (35). One of the findings was that HXN-1500 does not contain a membrane-bound alkane hydroxylase gene. Since this strain was not suitable for a large-scale process, we cloned the oxygenase responsible for limonene hydroxylation (a cytochrome P450 enzyme belonging to the CYP153 family) and expressed its components in P. putida.
MATERIALS AND METHODS
Bacterial strains.
A total of 137 alkane-degrading strains were obtained from strain collections or were isolated from a range of natural and artificial environments. The sources and references for 18 strains have been described previously (30). Twenty other strains (mainly rhodococci) that were isolated and partially characterized as described previously were a gift from Susanne Schorcht (28, 35). Fifteen strains were isolated from a tricking-bed air filter in Stuttgart, Germany, and were characterized by Gram staining, morphology, metabolic capabilities, fatty acid analysis, and 16S rRNA gene sequencing (26, 35). Three strains were a gift from Bioclear, Groningen, The Netherlands. Eighty-one additional alkane-degrading strains were isolated from soil, water, sewage sludge, and tree leaves based on their ability to grow with octane as the sole carbon and energy source.
Other strains used in this study are listed in Table 1. Luria-Bertani (LB) broth (27) and E2 medium (16) supplemented with carbon sources or antibiotics were used throughout this study. MT trace elements (16) were added to minimal media. All cultures were grown aerobically at 30°C. For growth on n-alkanes, petri dishes containing E2 medium were incubated at 30°C with the n-alkanes supplied through the vapor phase. In the case of C5 to C10 n-alkanes the compounds were supplied by placing an open Erlenmeyer flask with the alkane in a sealed container holding the petri dishes; for longer n-alkanes (up to C16) the compounds were supplied by placing a Whatman 3MM filter disk with 200 μl of the n-alkane in the lid of the petri dish. Recombinants were cultured at 130 rpm and 30°C in baffled Erlenmeyer flasks with E2 medium and with 0.5% (vol/vol) n-alkanes as the carbon sources.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant phenotype and/or genotype | Source or reference |
|---|---|---|
| Strains | ||
| E. coli DH10B | Cloning strain | Gibco BRL |
| E. coli GEc137(pGEc47ΔB) | DH-1 thi fadR | 29 |
| P. putida GPo12(pGEc47ΔB) | P. putida GPo1 cured of the OCT plasmid | 29 |
| Plasmids | ||
| pGEc47ΔB | Contains all genes necessary for growth on n-alkanes except an alkane hydroxylase gene | 29 |
| pCom8 | Broad-host-range expression vector with PalkB, GmroriT alkS | 31 |
| pCom8-PFR1500 | P450 operon in pCom8 | This study |
| pCR2.1-TOPO | PCR cloning vector | Invitrogen |
| TOPO-HXN-1500 | pCR2.1-TOPO with HXN-1500 PCR fragment | This study |
| pKKPalk | E. coli expression vector with PalkB, Apr | 31 |
| pZErO2.1 | Cloning vector, Kmr | Invitrogen |
| pZErO2.1-Apa6.1 | pZErO2.1 with 6.1-kb ApaI fragment | This study |
Miniaturized screening.
Multiple strains stored in one 96-well microtiter plate at −80°C were sampled simultaneously without thawing the bulk cultures by using a spring-loaded 96-pin replicator (Kühner, Basel, Switzerland) as previously described (7) and were transferred to a regular sterile polystyrene microtiter plate (type 3072; Costar, Cambridge, Mass.); each well (working volume, 350 μl) contained 180 μl of a solidified mineral medium agar (2% [wt/vol]) without a C source. The inoculated microtiter plate was placed in a desiccator together with a beaker of water and a 50-ml beaker containing 10 ml of a 1:1:1:1:1 (vol/vol/vol/vol/vol) mixture of hexane, octane, decane, dodecane, and hexadecane. The lid on top was kept 2 mm from the wells in order to allow for a sufficient supply of gaseous alkanes to the wells. After 7 days of growth at the ambient temperature (28°C), the cell mass that developed on the agar surface was harvested as follows. First, 110 μl of potassium phosphate buffer (50 mM, pH 7.0) was added to each well. Repeated lateral movement of the spring-loaded replicator in the wells resulted in suspension of a large part of the cell mass. The suspensions were subsequently transferred by using a 12-channel multipipette and wide-orifice tips to a microtiter plate with 0.5-ml conical wells (Maxi-plaque; Polylabo, Geneva, Switzerland). The microtiter plate was centrifuged for 15 min at 4,000 rpm in an Eppendorf type 5403 centrifuge. After disposal of the supernatant, the cells were resuspended in 100 μl of a buffer (50 mM potassium phosphate buffer [pH 7.0] containing 100 mM glucose) by repeated filling and emptying of wide-orifice pipette tips by using a 12-channel multipipette. Subsequently, 2 μl of optically pure d-limonene or l-limonene (Fluka, p.a.) was added to each well, and the wells were closed by using a sandwich cover consisting of a pierced layer of soft silicone combined with a rigid polypropylene plate (7). The microtiter plate was incubated for 2 h at 25°C with orbital shaking at 300 rpm and an amplitude of 5 cm. The microtiter plate was then centrifuged for 15 min at 2,000 × g. Fifty microliters of the supernatant without the remaining d-limonene phase was transferred into a half-area microtiter plate (Costar type 3696; Corning, Corning, N.Y.) by using a 12-channel multipipette.
Analysis by high-performance liquid chromatography-MS.
An analysis was performed by using a high-performance liquid chromatograph (Agilent 1100 series) equipped with a diode array detector and a mass detector in series. Solvent 1 was acetonitrile-methanol (1:1, vol/vol) containing 0.1% formic acid. Solvent 2 was water-0.1% formic acid. Solvents 1 and 2 were used at a 60:40 (vol/vol) ratio for 10 min, followed by a steep gradient to 90% solvent 1, with a total run time of 13.2 min in a Machery Nagel C18 column (70 by 3 mm) equipped with an 8-mm-long precolumn of the same material. The settings of the mass spectrometer (MS) were as follows: APCI mode; positive ionization; fragmenter voltage, 50 V; gas temperature, 350°C; vaporizer temperature, 375°C; drying gas (N2) flow rate, 4 liters min−1; nebulizer pressure, 50 lb/in2; capillary voltage, 2,000 V; corona current, 6 μA. The MS was run in the selected ion mode set to detect the following masses (relative dwell times are indicated in parentheses): 135 (40%), 137, (5%), 151, (10%), 153 (7%), and 176 (15%). Perillic acid did not give a good MS signal at any mass but could be accurately quantified by the diode array detector signal at 225 nm. The characteristic UV spectrum was helpful in confirming the presence of perillic acid. In all cases, authentic standards were used to determine retention times, UV-visible spectra, and mass spectra.
Cultivation of Mycobacterium sp. strain HXN-1500.
Mycobacterium sp. strain HXN-1500 was grown at 30°C and pH 7.0. For growth of HXN-1500 on solid media, 0.1% ethanol was added to 0.5× Evans medium (9) or n-octane was supplied through the vapor phase by incubating petri dishes in a sealed container together with an open Erlenmeyer flask containing n-octane. For growth in liquid cultures, 0.1% ethanol or 1% n-octane was added to the medium. A 2.1-liter KLF 2000 bioreactor (Bioengineering, Wald, Switzerland) controlled by Biologics software (Bioengineering) was inoculated with two 100-ml Erlenmeyer flask cultures to an optical density at 450 nm (OD450) of 0.1. The stirrer speed was set at 550 rpm, and the airflow was set at 0.25 liter/min. The carbon source was supplied by passing the incoming air through a washing flask containing n-octane. To prevent foaming, polypropylene glycol P2000 was added at regular intervals. Cells were harvested by centrifugation at 4°C for 30 min at 2,000 × g. Each pellet was resuspended in 50 mM sodium phosphate buffer and centrifuged again. The pellets were stored at −40°C.
Purification of limonene-hydroxylating enzyme.
About 30 to 50% of the total cell protein was released from Mycobacterium sp. strain HXN-1500 cells by two passages through a 4-ml (working volume) French press cell operated at 1,000 lb/in2. The crude cell extract was centrifuged at 150,000 × g for 1 h to remove intact cells, cell debris, and total membranes, and the supernatant was fractionated by ammonium sulfate precipitation. Ammonium sulfate was added to a concentration of 40% at 0°C. After 10 min of stirring, the solution was centrifuged for 20 min at 50,000 × g, and the pellet was discarded. Subsequently, ammonium sulfate was added to a concentration of 60%, and the preparation was stirred for 10 min and centrifuged as described above. The supernatant was discarded, and the pellet containing the target enzyme was redissolved in 25% ammonium sulfate. The 48-kDa protein associated with limonene hydroxylation activity was purified further by hydrophobic interaction chromatography on a phenyl Sepharose column by using a 25 to 0% ammonium sulfate gradient in 25 mM Tris-HCl (pH 6.8). The 48-kDa protein eluted near the end of the gradient. Anion-exchange chromatography on a Source Q15 column with a 0 to 0.5 M NaCl gradient in 25 mM Tris-HCl (pH 7.0) was used as a polishing step. The best fractions were subjected to Edman degradation for N-terminal sequencing. During purification the activity of the fractions (formation of perillyl alcohol from limonene) was monitored by liquid chromatography-MS (see below).
Limonene hydroxylase activity assay.
To determine limonene hydroxylation activity, 100 μl of protein extract was incubated with 1 μl of l-limonene and 5 μl of a 5 mM NADH solution. Incubation was carried out in an Eppendorf thermomixer at 30°C and 1,400 rpm. The reaction was stopped at appropriate times by adding 100 μl of acetonitrile. After centrifugation (15 min, 15,000 × g), samples were stored at −20°C before analysis by liquid chromatography-MS as described above. Because HXN-1500 ferredoxin and ferredoxin reductase were not available, the assay was not optimized for specific activity.
CO difference spectra.
To quantitate cytochrome P450 in P. putida GPo12(pGEc47ΔB)(pCom8-PFR1500), cells were harvested by centrifugation at 4°C and resuspended in 100 mM potassium phosphate buffer (pH 7.5) containing 2 mM EDTA, 20% glycerol, 1.5 mM dithiothreitol, 0.4% Triton X-100, and 1 mM phenylmethylsulfonyl fluoride. The cells were disrupted by one passage through a French press cell (Thermo Electron Corporation) at 10,000 lb/in2. Cell debris and a large part of the membranes were removed by centrifugation at 130,000 × g for 10 min. The soluble fraction was then used to measure the CO-reduced P450 absorption with a UV-visible spectrophotometer (Varian type CARY3). Extracts were diluted in the same buffer, and the CO-reduced difference spectra were measured as described by Omura and Sato (24). To calculate the concentration of P450, an extinction coefficient of 91 mM−1 cm−1 was used.
Molecular genetics.
Restriction enzymes, T4 DNA ligase, and dideoxynucleotides were obtained from Roche Molecular Biologicals and were used as specified by the supplier. Oligonucleotides were synthesized by Microsynth, Balgach, Switzerland. Chromosomal DNA was isolated from strain HXN-1500 by the method of van Soolingen et al. (36). The 16S rRNA gene of HXN-1500 was amplified with amplification primers 16F27 and 16R1525 and was sequenced with primers 16F355 and 16R519 (14, 15). Because our standard 16R1488 primer (15) did not work for HXN-1500, we developed a new sequence primer, 16R1470 (TTCCGGTACGGCTACCTTGTTACGACTT). This primer worked well for HXN-1500.
A 339-bp internal gene fragment of the CYP153 gene was amplified with primers P450fw1 (GTSGGCGGCAACGACACSAC) and P450rv3 (GCASCGGTGGATGCCGAAGCCRAA). Taq DNA polymerase was obtained from Promega. The PCR product was separated from other fragments and primers on a 1% agarose gel by using Tris-borate-EDTA buffer. The 339-bp band was cut out from the gel, isolated by electroelution (27), and cloned in pCR2.1-TOPO. Both strands of the inserts were sequenced with a Li-Cor 4000L sequencer by using IRD800-labeled primers −40 forward (AGGGTTTTCCCAGTCACGACGTT) and −40 reverse (GAGCGGATAACAATTTCACACAGG) (MWG-Biotech) and an Amersham Thermosequenase cycle sequencing kit.
To obtain the complete CYP153 gene and flanking DNA, the cloned CYP153 fragment was used as a probe in Southern blotting to identify restriction fragments that were the appropriate sizes. An enriched gene bank containing 5.5- to 7.0-kb chromosomal ApaI fragments was constructed. The fragments were isolated from a preparative gel by electroelution, ligated between the appropriate sites of pZErO-2, and transformed into Escherichia coli DH10B (Gibco). Transformants containing the desired fragment were identified by colony blotting. Both strands of the insert were sequenced by using clones made with an EZ:TN <TET-1> kit (Epicentre) and IRD800-labeled primers described in the manual of the kit.
The CYP153 operon consisting of a P450 gene, a ferredoxin gene, and a ferredoxin reductase gene was amplified with primers HXN-1500-op-FW2 (CAATTGGAAATGACGGTGGCCGCCAGCGACGCGAC) and HXN-1500-op-RV2 (AAGCTTCTAATGTTGTGCAGCTGGTGTCCGTACGA). These primers introduced a MunI restriction site three bases upstream of the start codon of the P450 gene and a HindIII restriction site downstream of the operon (the restriction sites are underlined). The 2.9-kb fragment was cloned in pCR2.1-TOPO, sequenced to verify that no mutations had been introduced by the PCR, and recloned as a MunI-HindIII fragment between the EcoRI and HindIII sites of pCom8 (31). The forward primer was designed to place the pCom8 ribosome binding site (RBS) six bases upstream of the ATG start codon of the first gene in the operon (AAAAATTGGAGAATTGGAAATG; the RBS and ATG start codon are indicated by boldface type, and the sequence created by ligation of the MunI and EcoRI sites is underlined). The final construct, pCom8-PFR1500, was introduced into E. coli GEc137(pGEc47ΔB) by electroporation (4) and into P. putida GPo12(pGEc47ΔB) by triparental mating with E. coli HB101(pRK2013) as the helper strain (3). Transconjugants were selected on E2 medium containing the appropriate antibiotics. E. coli strains or transformants harboring plasmids were grown with appropriate antibiotics (12.5 μl of tetracycline per ml for pGEc47ΔB, 10 μg of gentamicin per ml for pCom8 in E. coli, and 25 to 100 μg of gentamicin per ml for pCom8 in P. putida).
Nucleotide and amino acid sequences were analyzed and compared by using LASERGENE Navigator from DNASTAR (Madison, Wis.). Nucleotide and amino acid sequences were compared with the EMBL, Swiss-Prot, and GenBank databases by using BLAST (1). BLAST searches were carried out at the National Center for Biotechnology Information.
Bioconversions.
P. putida GPo12(pGEc47ΔB)(pCom8-PFR1500) was grown in E2 medium containing MT trace metals, gentamicin (50 μg/ml), and n-octane as the sole carbon source to an OD450 of approximately 1. Cells were harvested by centrifugation for 10 min at 4,500 × g. The supernatant was discarded, and the cells were suspended in 50 mM potassium phosphate buffer (pH 7.0) at a concentration of 1 mg (dry weight) per ml. The assays were performed in 5-ml (total volume) mixtures at 25°C with agitation with a magnetic stirrer at 1,000 rpm in 40-ml headspace vials with polytetrafluoroethylene-coated silicon septum caps. l-Limonene or (−)-perillyl alcohol was added to a concentration of 2 mM from 1 M stock solutions in ethanol. Samples (1 ml) were taken with a syringe. The samples were prepared for gas chromatography (GC) analysis as follows. One milliliter of ethyl acetate was added to a sample in a Pyrex tube with a Teflon cap, was thoroughly mixed with a vortex mixer, and was centrifuged for 5 min at 3,000 × g. The ethyl acetate phase was transferred to a fresh tube, and Na2SO4 was added to extract water from the ethyl acetate and was removed by centrifugation for 3 min at 3,000 × g. Then 300 to 400 μl was transferred to GC vials and analyzed with a 25-m Optima 5 column (Macherey Nagel, Oensingen, Switzerland). The oven was programmed with a temperature gradient from 100 to 140°C at a rate of 10°C/min and from 140 to 280°C at a rate of 20°C/min.
For large-scale bioconversions, P. putida GPo12(pGEc47ΔB)(pCom8-PFR1500) was grown in a 2.1-liter KLF 2000 bioreactor with 1 liter of E2 medium with n-octane as the sole carbon source (provided through the gas phase by passing air through a washing flask filled with n-octane and feeding liquid n-octane at a rate of 0.5 to 5 ml h−1). At an OD450 of 30, a second phase consisting of 500 ml of bis(2-ethylhexyl)phthalate and 50 ml l-limonene was added (time zero in Fig. 6). Incubation was continued for 72 h at 30°C with mechanical stirring at 1,500 rpm and an average feed rate of 1 ml of octane h−1. The culture was aerated at a rate of 0.3 liter min−1.
FIG. 6.

Biotransformation of limonene with P. putida GPo12(pGEc47ΔB)(pCom8-PFR1500) at the 1.5-liter scale. Symbols: ♦, perillyl alcohol concentration in the organic phase; ▪, perillic acid concentration in the water phase; ×, n-octane feed rate.
Nucleotide sequence accession numbers.
The accession number of the Mycobacterium sp. strain HXN-1500 rRNA gene sequence is AJ457057, and the accession number of the 6.2-kb ApaI fragment containing the ahpGHI operon is AJ783967.
RESULTS
To identify a biocatalyst for the production of perillyl alcohol from limonene, we screened 1,800 isolates, mainly hydrocarbon degraders. After preliminary analysis of the results, we focused on 137 alkane-degrading bacterial strains. For 10 of these strains perillyl alcohol was the sole product, while 33 strains produced mixtures of perillyl alcohol and perillic acid. Traces of perillyl aldehyde were found in only a few cases. The remaining 94 strains did not produce detectable amounts of oxidation products at the 7 position. Most of the positive strains for which taxonomic data were available belonged to the CNM group (pleomorphic high-G+C-content gram positive bacteria), while only a few of the gram-negative strains produced significant amounts of the product. The best strain with respect to the specific activity for perillyl alcohol formation was Mycobacterium sp. strain HXN-1500, which converted d-limonene to (+)-perillyl alcohol at a rate of 13 U g (dry weight)−1 and converted l-limonene to (−)-perillyl alcohol at a rate of 10 U g (dry weight)−1.
Properties of HXN-1500.
HXN-1500 was originally isolated from a trickling-bed bioreactor (26, 35). On solid media this gram-positive strain formed bright yellow, smooth, convex, round colonies at temperatures up to 37°C. HXN-1500 cells are nonmotile coccoid short rods. The 16S rRNA gene sequence of HXN-1500 was closely related to but not identical to those of Mycobacterium frederiksbergense DSM 44346 (99.5%) (38) and Mycobacterium neoaurum ATCC 25795 (99.0%) (32). Therefore, we designated the organism Mycobacterium sp. strain HXN-1500. On solid LB medium, HXN-1500 showed very weak growth, like many other nocardioforms. Linear alkanes ranging from n-hexane to n-dodecane were good growth substrates, while longer alkanes also supported growth, but the cell densities were low. Ethanol and, to a lesser extent, acetate supported growth as well. However, the limonene-hydroxylating activity was significantly decreased when ethanol was used as the carbon source or when yeast extract was added to the minimal medium. As the highest limonene hydroxylation activities were obtained with n-octane-grown cells, we attempted to cultivate HXN-1500 on a 2-liter scale with n-octane added directly to the medium. However, this was quite problematic as the cells became very hydrophobic, entered the n-octane layer, and formed clumps that could not be resuspended. It was possible to grow cells in suspension if n-octane was supplied through the vapor phase, but the growth rates and final cell densities were low. In view of the difficulties of cultivating HXN-1500 and the fact that this bacterium must be considered a biosafety class II organism, we set out to clone and express the enzyme responsible for limonene hydroxylation in a more suitable host.
Purification and N-terminal sequencing of the limonene hydroxylase.
As only octane-grown cells convert limonene to perillyl alcohol, it seemed likely that an alkane hydroxylase is responsible for the bioconversion. Therefore, we attempted to amplify homologs of AlkB, the membrane-bound alkane hydroxylase of P. putida GPo1, from HXN-1500 as described previously (30, 35). However, as this approach was not successful, we decided to purify the limonene-hydroxylating enzyme and clone the encoding genes using protein sequence data.
Octane-grown cells were disrupted by French press treatment, and after ultracentrifugation of cell extracts the activity was found in the supernatant. The activity was reasonably stable at 0°C, with a half-life of about 6 h, but at room temperature the activity was rapidly lost. The pH optimum was close to pH 7.0, while activity could hardly be detected at pH 5.5 or 8.0. Almost all of the activity (93%) could be recovered in a 40 to 60% ammonium sulfate fraction, and comparison of this fraction with a similar fraction from ethanol-grown cells clearly showed strong induction of a 48-kDa protein in n-octane-grown cells. Subsequent hydrophobic interaction chromatography and anion-exchange chromatography yielded almost pure 48-kDa protein associated with limonene hydroxylase activity (Fig. 1). During purification, the specific activities varied strongly for fractions that had similar amounts of the 48-kDa protein when they were analyzed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis. As many hydroxylases are multicomponent systems, this was attributed to the (partial) loss of one or more hydroxylase components (data not shown).
FIG. 1.
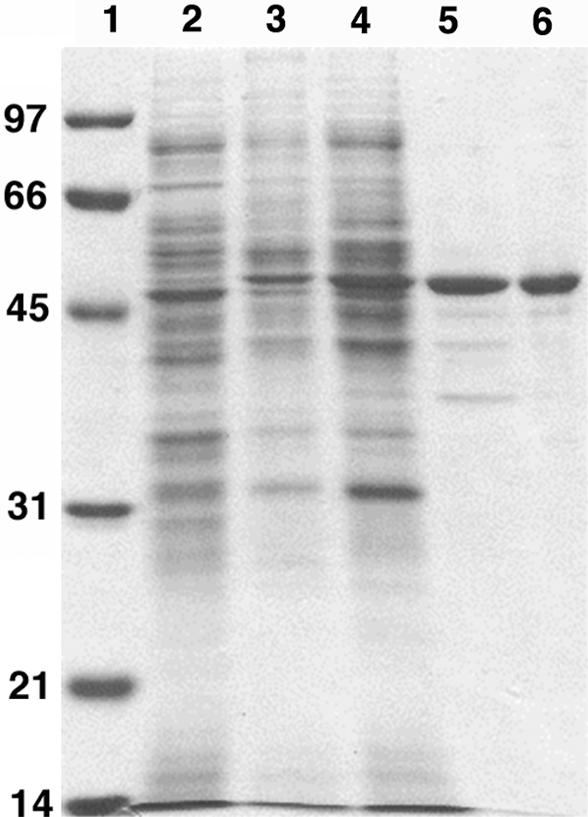
Purification of the limonene-hydroxylating activity. Proteins were separated on a 12% SDS-polyacrylamide gel. Approximately 10 μg of protein was loaded in each lane. Lane 1, marker proteins (kDa); lane 2, cell extract of HXN-1500 grown on ethanol as the C source; lane 3, cell extract of HXN-1500 grown on n-octane; lane 4, 40 to 60% ammonium sulfate fraction; lane 5, best fraction after hydrophobic interaction chromatography; lane 6, best fraction after anion-exchange chromatography.
Fractions containing the 48-kDa protein were red-brown. A visible light absorption spectrum showed a major peak at 418 nm and minor peaks at 355, 540, and 570 nm, which is very similar to the absorption spectra of cytochrome P450 proteins. The best fraction from the anion-exchange chromatography step was sequenced by Edman degradation, which resulted in a 30-amino-acid sequence, (M/T)EMTVAASDATNAAYGMALED(F/I)DVANPVLF (underlining indicates that the signals were weak or ambiguous).
Cloning and sequencing of genes encoding the limonene hydroxylase activity.
Several oligonucleotides were designed based on the N-terminal sequence, with the aim of amplifying an 85-nucleotide gene segment corresponding to the N terminus. However, these primers did not yield PCR products that were the correct length. Comparison of the N-terminal sequence with the sequence databases yielded clear sequence identity to the N terminus of a putative cytochrome P450 encoded by the Caulobacter crescentus genome sequence (level of identity, 52%). The Caulobacter sequence in turn showed high full-length homology (>50% sequence identity) to the recently described sequence of the hexane hydroxylase of Acinetobacter calcoaceticus EB014, the first described member of the CYP153 family (20). In a multiple alignment of the A. calcoaceticus EB104 sequence and its homologs encoded by the C. crescentus and Bradyrhizobium japonicum genomes, the oxygen-binding stretch in the I helix (GGNDTTRN) and the sequence ending with the heme-binding cysteine (HLSFGFGIHRC) were found to be perfectly conserved. Primers based on the N-terminal sequence combined with primers based on the two conserved sequences again did not yield the expected products. However, forward and reverse primers based only on the two conserved peptide motifs did yield a PCR product, which was cloned and sequenced. The encoded peptide had close to 70% sequence identity to the EB104 hexane hydroxylase and 74% sequence identity to the C. crescentus P450 sequence, indicating that we had cloned a fragment of the target gene or at least a close homolog of the target gene. Subsequently, the cloned PCR fragment was used as a probe in Southern blots to select a suitable restriction fragment for cloning. Restriction enzyme ApaI yielded a 6.2-kb fragment, which was cloned and sequenced. Analysis of the sequence for open reading frames (ORFs) showed that a 1,260-bp ORF encoded a P450 homolog, the N-terminal sequence of which was identical to the sequence of the limonene-hydroxylating enzyme except for S26, which was close to the end of the N-terminal sequence. The N-terminal methionine appeared to have been processed.
Sequence analysis.
Due to the high full-length sequence identity with the EB014 hexane hydroxylase, the HXN-1500 cytochrome P450 can be classified as a member of the CYP153 family, and it was designated CYP153A6 (22). It exhibits about 28% sequence identity to CYP119 and P450terp, whose three-dimensional structures have been determined. The CYP153 ORF was preceded by an RBS (AAGGGA) 5 nucleotides upstream of the ATG start codon. The calculated molecular mass (46,675.5 Da) was close to the value obtained by SDS-polyacrylamide gel electrophoresis (48 kDa). A 47,407.7-Da peptide encoded immediately downstream of the P450 gene was most closely related to the EB104 ferredoxin reductase encoded downstream of the EB104 hexane hydroxylase (47% sequence identity) and to putidaredoxin, the ferredoxin reductase of P450cam (44%). The start codon of the reductase gene was not preceded by a recognizable RBS and overlapped the stop codon of the P450 gene, which is indicative of translational coupling. A third ORF 21 nucleotides downstream of the reductase gene encoded a protein that was most closely related to a C. crescentus ferredoxin (57%) and to the A. calcoaceticus EB104 ferredoxin encoded immediately upstream of the CYP153A1 gene (45%). Six nucleotides upstream of the predicted start codon, an RBS (AAGGA) was found. The gene arrangement (cytochrome P450, ferredoxin reductase, ferredoxin) (Fig. 2) and the sequence similarities indicated that we cloned an operon encoding all three components of a typical bacterial class I cytochrome P450 system. We propose the following designations for the ORFs: CYP153A6 P450 gene, ahpG; reductase gene, ahpH; and ferredoxin gene, ahpI (alkane hydroxylase P450).
FIG. 2.

Organization of cytochrome P450 operon and flanking DNA. The open reading frames are indicated by arrows. The scale is in bases.
Functional heterologous expression.
To obtain functional expression of the HXN-1500 cytochrome P450 system, we cloned the complete operon in pCom8, a broad-host-range expression vector for E. coli and Pseudomonas based on the PalkB promoter (31). The resulting construct was transferred to E. coli GEc137(pGEc47ΔB) and P. putida GPo12(pGEc47ΔB). Both of these host strains contain all genes necessary for growth on n-octane except a functional alkane hydroxylase gene and can be complemented by membrane-bound alkane hydroxylases expressed from pCom8 or related vectors (29). While the E. coli recombinant did not grow on n-octane, the P. putida recombinant grew slowly on n-octane, with a doubling time in liquid cultures of about 15 h. Several rounds of cultivation resulted in a decrease in the doubling time of the recombinant to 3 h. Sequencing of the pCom8 insert did not reveal any changes. However, the phenotype of the recombinant (faster growth on n-octane) was stable after passage in LB medium. Retransformation of plasmids obtained from fast-growing recombinants in P. putida GPo12(pGEc47ΔB) resulted in recombinants with intermediate growth rates. We subsequently tested other n-alkanes as growth substrates for the P. putida recombinant. N-Heptane supported slightly faster growth than n-octane, while other alkanes, ranging from n-pentane to n-undecane, supported growth with lower growth rates (Fig. 3).
FIG. 3.

Growth rates of P. putida GPo12(pGEc47ΔB)(pCom8-PFR1500) on n-alkanes. C5, n-pentane; C6, n-hexane; C7, n-heptane; C8, n-octane; C9, n-nonane; C10, n-decane; C11, n-undecane.
Protein gels indicated that significant levels of expression were obtained in P. putida, but only if the recombinants were grown with n-octane as the C source. Cultivation of the P. putida recombinant on citrate and subsequent induction of the P450 operon with the nonmetabolizable inducer dicyclopropylketone (DCPK) or n-octane never yielded high expression levels or measurable limonene hydroxylation activity (Fig. 4 and data not shown).
FIG. 4.
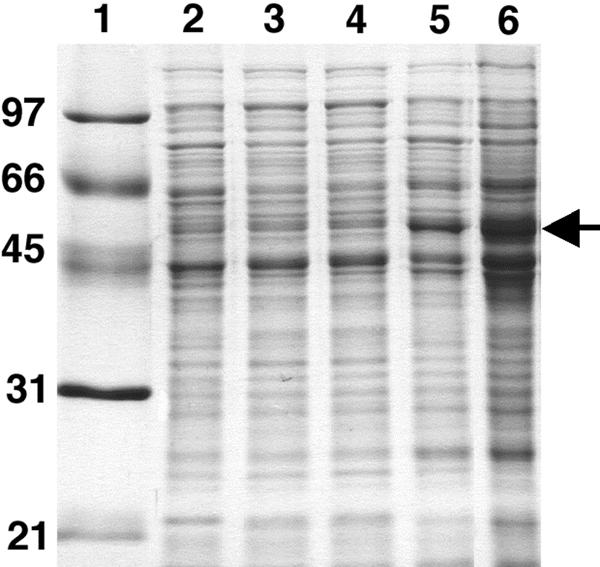
Expression of CYP153A6 in P. putida GPo12(pGEc47ΔB)(pCom8-PFR1500). Proteins were separated on a 12% SDS-polyacrylamide gel. Approximately 10 μg of protein was loaded in each lane. Lane 1, marker proteins (kDa); lane 2, P. putida GPo12(pGEc47ΔB) grown on citrate; lane 3, P. putida GPo12(pGEc47ΔB)(pCom8-PFR1500) grown on citrate; lane 4, same recombinant induced with DCPK; lane 5, same recombinant grown on n-octane; lane 6, crude cell extract of P. putida recombinant used to record the CO difference spectrum. The CYP153A6 band is indicated by the arrow.
To confirm the expression of the P450 component in n-octane-grown P. putida GPo12(pGEc47ΔB), CO difference spectra were examined. A cell extract with a protein concentration of 7 mg ml−1 (Fig. 5) gave a difference in the A450 of 0.2, which corresponded to a calculated P450 concentration of 2.2 μM, 0.1 mg ml−1, or 1.5% of the protein in the cell extract. Based on an estimated level of expression of the oxygenase component of 5% (wt/wt) of the total protein (Fig. 4), this indicates that only about one-third of the P450 protein could be detected in the CO difference spectrum.
FIG. 5.

CO difference spectrum of a P. putida GPo12(pGEc47ΔB)(pCom8-PFR1500) cell extract.
Conversion of limonene to perillyl alcohol.
E. coli and P. putida recombinants containing pGEc47ΔB and pCom8-PFR1500 were tested for conversion of limonene to perillyl alcohol. In the case of the E. coli recombinant, a small amount of perillyl alcohol was formed only if the P450 genes were present and induced, and the specific activity was 0.1 U g−1 (data not shown). In the case of P. putida, perillyl alcohol was produced with a specific activity of 3 U g (dry weight) of cells−1, but only when the recombinant was grown with n-octane as the C source. Induction of citrate-grown cells with DCPK or octane did not result in measurable conversion of l-limonene to perillyl alcohol.
The small-scale experiments showed that limonene by itself was relatively toxic to the P. putida recombinant, and production of perillyl alcohol was limited to concentrations of 1 to 2 mM. Therefore, we set up a bioreactor experiment with 1 liter of E2 medium in a 2-liter bioreactor. The recombinant strain was grown on n-octane to a cell density of approximately 10 g liter−1. Subsequently, an organic phase consisting of 500 ml of bis(2-ethylhexyl)phthalate and 50 ml of l-limonene was added (time zero in Fig. 6), and the production of perillyl alcohol and perillic acid was monitored by GC analysis. The n-octane feed rate was set at a level that supported limited growth of the recombinant, without accumulation of n-octane in the organic phase, and was varied occasionally to determine the response of the culture (based on oxygen consumption and n-octane levels in the organic phase). In 75 h, perillyl alcohol was produced at a concentration of 6.8 g liter−1 in the organic phase [bis(2-ethylhexyl)phthalate] or 2.3 g liter−1 calculated for the entire bioreactor contents. The water phase contained about 0.14 g of perillic acid liter−1 (Fig. 6), and the final cell density was 15 g liter−1.
DISCUSSION
A screening project to identify hydrocarbon-degrading bacteria that are able to hydroxylate limonene in the 7 position yielded almost exclusively high-G+C-content gram-positive alkane degraders. Conversely, almost one-third of the alkane degraders were able to hydroxylate limonene, which shows that preselection for a rather common metabolic capability can greatly facilitate a screening process. In fact, a previous screening program for hydroxylation of N-substituted pyrrolidines led to the same conclusion: many alkane degraders possess alkane hydroxylases with a wide substrate range and excellent regio- and stereoselectivity (17, 19).
The most active strain in our screening for perillyl alcohol production (strain HXN-1500, a close relative of M. frederiksbergense and M. neoaurum) produced only a little perillic acid; however, it was difficult to use as a biocatalyst and must be considered a biosafety class II organism. To circumvent these problems and to characterize the enzyme system responsible for the bioconversion, we isolated the limonene-hydroxylating enzyme and used N-terminal sequence data to clone the encoding gene. This yielded an operon consisting of three genes encoding all three components of a class I cytochrome P450 belonging to the CYP153 family (20). The physiological function of the cytochrome P450 is most likely hydroxylation of alkanes, as HXN-1500 produced the enzyme only when it was grown on n-octane. Furthermore, as the heterologously expressed enzyme allowed a P. putida recombinant to grow on linear alkanes and no subterminal oxidation products, such as 2- or 3-alkanols or alkanones, were detected, the alkane hydroxylation takes place exclusively at terminal positions. This contrasts strongly with mutants of Bacillus megaterium CYP102A1 (P450-bm3) and P. putida CYP101 (P450cam) that hydroxylate alkanes at high rates but with low specificities (2, 12). Comparison of these mutants with CYP153 enzymes may well reveal the basis of the specificity for terminal methyl groups of these enzymes. Interestingly, members of the other enzyme class known for its ability to hydroxylate at the terminal methyl groups, the integral membrane alkane hydroxylases, do not hydroxylate limonene to perillyl alcohol. This suggests that the CYP153 enzyme might provide a substrate range that is complementary to that of the integral membrane alkane hydroxylases.
We chose P. putida to express the HXN-1500 cytochrome P450 alkane hydroxylase for several reasons. First, it is the first gram-negative soil organism that has been certified as a safe strain by the Recombinant DNA Advisory Committee (23). Second, it is well suited for biocatalysis as specific isolates are quite solvent resistant, and the strain which we used is easy to handle in bioreactors. Third, unlike E. coli, various natural P. putida isolates express cytochrome P450 enzymes at high levels (e.g., P450cam). Fourth, we previously constructed recombinant P. putida strains which allowed selection for growth on n-octane or other n-alkanes if alkane hydroxylases were successfully expressed. Functional heterologous expression of cytochrome P450 systems is generally considered difficult, and very often CO difference spectra and in vitro reconstitution with isolated components are the only evidence of successful expression. Most expression experiments are carried out with E. coli, which produces small amounts of heme and requires addition of α-levulinic acid. We previously used a broad-host-range expression system to obtain functional expression in Pseudomonas strains of membrane-bound alkane hydroxylases from a wide range of gram-negative α-, β-, and γ-proteobacteria and high-G+C-content gram-positive bacteria (29). Here, we found that by using the same vector-host combination the HXN-1500 cytochrome P450 system could be functionally expressed in P. putida at levels that allow the host to grow on n-octane with doubling times of 3 h, which corresponds to an alkane hydroxylase activity of 10 to 20 U g (dry weight) of cells−1. It is important to note that these growth rates were obtained only after prolonged selection for faster growth on alkanes. We have not yet investigated the nature of the mutations in pCom8 and the P. putida GPo12 chromosome that allow higher functional levels of expression. However, we have obtained similar results with other CYP153 enzymes (data not shown). This shows (i) that specific features of our expression system are not optimal for expression of CYP153 enzymes and (ii) that these features can be optimized by a selection process. The selected improvements could include higher heme levels, optimized heme insertion in the apoprotein, and higher copy numbers. Further study of these improvements could provide a general method for optimizing the expression of cytochrome P450 enzymes.
Because the growth rate of the P. putida recombinant on n-octane has yet to reach the physiological limits (the membrane-bound alkane hydroxylase AlkB allows doubling times of less than 1.5 h), the strategy described above allows further selection (e.g., in continuous cultures) for better functional expression. CO difference spectra indicate that only 30% of the expressed peptide in n-octane-grown cells contains heme, suggesting that part of the improvement might come from better loading of the holoenzyme with heme. However, the addition of α-levulinic acid did not increase the growth rate of the recombinant on n-octane (data not shown).
Another interesting observation is the fact that optimal levels of expression were obtained only when the recombinant P. putida strain was grown on alkanes, not when the same recombinant (after growth on n-octane) was grown on citrate and induced with alkanes or other compounds, such as dicyclopropylketone. Microheterogeneity in the population used to inoculate the cultures could not be observed; all cells plated on minimal media and fed with n-octane developed into colonies. This indicates that genetic differences do not play a role. Possibly, there is a delicate balance between CYP153 apoprotein synthesis and heme synthesis, which is achieved only in cells that are actively growing on n-octane. Irrespective of explanations for the high levels of expression due to selection and growth on n-octane, the (functional) expression levels observed in this work are a significant improvement over the levels observed for other P450 enzymes in host strains, such as E. coli strains. Therefore, we are now testing P. putida for expression of other cytochrome P450 enzymes, using the same approach.
The threefold-lower limonene hydroxylation activity of the recombinant strain relative to the wild-type Mycobacterium strain might well be due to limiting rates of substrate uptake or transport and/or product export, an issue that is presently under investigation. In previous studies, the outer membrane of gram-negative bacteria (in particular, the lipopolysaccharide layer) was found to be an effective permeability barrier for monoterpenes (10, 13). Nevertheless, the P. putida recombinant described in this paper allows production of perillyl alcohol in practical amounts, as demonstrated by the bioreactor experiment (Fig. 6). Further work should show whether the levels obtained here can be exceeded by facilitating uptake or by using alternative host strains and whether the process can be scaled up. We consider this is a promising proof of principle for large-scale production, which paves the way for industrial production of this compound starting from l-limonene.
Acknowledgments
We thank Martina Röthlisberger for DNA sequencing, René Brunisholz for N-terminal sequencing, and Karl-Heinrich Engesser for supplying Mycobacterium sp. strain HXN-1500.
This research was supported in part by the Swiss Priority Program in Biotechnology of the Swiss National Science Foundation (project 5002-037023).
REFERENCES
- 1.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 2.Bell, S. G., E. Orton, H. Boyd, J. A. Stevenson, A. Riddle, S. Campbell, and L. L. Wong. 2003. Engineering cytochrome P450cam into an alkane hydroxylase. Dalton Trans. 2003:2133-2140. [Google Scholar]
- 3.Ditta, G., S. Stanfield, D. Corbin, and D. R. Helinski. 1980. Broad host range DNA cloning system for gram-negative bacteria: construction of a gene bank of Rhizobium meliloti. Proc. Natl. Acad. Sci. USA 77:7347-7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dower, W. J., J. F. Miller, and C. W. Ragsdale. 1988. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 16:6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duetz, W. A., H. Bouwmeester, J. B. van Beilen, and B. Witholt. 2003. Biotransformation of limonene by bacteria, fungi, yeasts and plants. Appl. Microbiol. Biotechnol. 61:269-277. [DOI] [PubMed] [Google Scholar]
- 6.Duetz, W. A., A. H. M. Fjallman, S. Ren, C. Jourdat, and B. Witholt. 2001. Biotransformation of d-limonene to (+) trans-carveol by toluene-grown Rhodococcus opacus PWD4 cells. Appl. Environ. Microbiol. 67:2829-2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duetz, W. A., L. Rüedi, R. Hermann, K. O'Connor, J. Büchs, and B. Witholt. 2000. Methods for intense aeration, growth, storage, and replication of bacterial strains in microtiter plates. Appl. Environ. Microbiol. 66:2641-2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duetz, W. A., B. Witholt, and C. Jourdat. September 2002. Process for the preparation of perillyl alcohol. European patent 1,236,802. [Google Scholar]
- 9.Evans, C. T. G., D. Herbert, and D. W. Tempest. 1970. The continuous cultivation of microorganisms. 2. Construction of a chemostat. Methods Microbiol. 2:277-327. [Google Scholar]
- 10.Fontanille, P., and C. Larroche. 2003. Optimization of isonovalal production from alpha-pinene oxide using permeabilized cells of Pseudomonas rhodesiae CIP 107491. Appl. Microbiol. Biotechnol. 60:534-540. [DOI] [PubMed] [Google Scholar]
- 11.Gibson, D. T., and R. E. Parales. 2000. Aromatic hydrocarbon dioxygenases in environmental biotechnology. Curr. Opin. Biotechnol. 11:236-243. [DOI] [PubMed] [Google Scholar]
- 12.Glieder, A., E. T. Farinas, and F. H. Arnold. 2002. Laboratory evolution of a soluble, self-sufficient, highly active alkane hydroxylase. Nat. Biotechnol. 20:1135-1139. [DOI] [PubMed] [Google Scholar]
- 13.Griffin, S. G., S. G. Wyllie, and J. L. Markham. 2001. Role of the outer membrane of Escherichia coli AG100 and Pseudomonas aeruginosa NCTC 6749 and resistance/susceptibility to monoterpenes of similar chemical structure. J. Essent. Oil Res. 13:380-386. [Google Scholar]
- 14.Hauben, L., L. Vauterin, J. Swings, and E. R. B. Moore. 1997. Comparison of 16S ribosomal DNA sequences of all Xanthomonas species. Int. J. Syst. Bacteriol. 47:328-335. [DOI] [PubMed] [Google Scholar]
- 15.Karlson, U., D. F. Dwyer, S. W. Hooper, E. R. B. Moore, K. N. Timmis, and L. D. Eltis. 1993. Two independently regulated cytochromes P-450 in a Rhodococcus rhodochrous strain that degrades 2-ethoxyphenol and 4-methoxybenzoate. J. Bacteriol. 175:1467-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lageveen, R. G., G. W. Huisman, H. Preusting, P. E. F. Ketelaar, G. Eggink, and B. Witholt. 1988. Formation of polyester by Pseudomonas oleovorans: the effect of substrate on the formation and composition of poly-(R)-3-hydroxyalkanoates and poly-(R)-3-hydroxyalkenoates. Appl. Environ. Microbiol. 54:2924-2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li, Z., and D. Chang. 2004. Recent advances in regio- and stereoselective biohydroxylation of non-activated carbon atoms. Curr. Org. Chem. 8:1647-1658. [Google Scholar]
- 18.Li, Z., H.-J. Feiten, D. Chang, W. A. Duetz, J. B. van Beilen, and B. Witholt. 2001. Preparation of (R)- and (S)-N-protected-3-hydroxypyrrolidines by hydroxylation with Sphingomonas sp. HXN-200, a highly active, regio-and stereoselective, and easy to handle biocatalyst. J. Org. Chem. 66:8424-8430. [DOI] [PubMed] [Google Scholar]
- 19.Li, Z., H.-J. Feiten, J. B. van Beilen, W. Duetz, and B. Witholt. 1999. Preparation of optically active N-benzyl-3-hydroxypyrrolidine by enzymatic hydroxylation. Tetrahedron Asymmetry 10:1323-1333. [Google Scholar]
- 20.Maier, T., H.-H. Foerster, O. Asperger, and U. Hahn. 2001. Molecular characterization of the 56-kDa CYP153 from Acinetobacter sp. EB104. Biochem. Biophys. Res. Commun. 286:652-658. [DOI] [PubMed] [Google Scholar]
- 21.Mars, A. E., J. P. L. Gorissen, I. van den Beld, and G. Eggink. 2001. Bioconversion of limonene to increased concentrations of perillic acid by Pseudomonas putida GS1 in a fed-batch reactor. Appl. Microbiol. Biotechnol. 56:101-107. [DOI] [PubMed] [Google Scholar]
- 22.Nelson, D. R., L. Koymans, T. Kamataki, J. J. Stegeman, R. Feyereisen, D. J. Waxman, M. R. Waterman, O. Gotoh, M. J. Coon, R. W. Estabrook, I. C. Gunsalus, and D. W. Nebert. 1996. P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics 6:1-42. [DOI] [PubMed] [Google Scholar]
- 23.Nelson, K. E., C. Weinel, I. T. Paulsen, R. J. Dodson, H. Hilbert, V. dos Santos, D. E. Fouts, S. R. Gill, M. Pop, M. Holmes, L. Brinkac, M. Beanan, R. T. DeBoy, S. Daugherty, J. Kolonay, R. Madupu, W. Nelson, O. White, J. Peterson, H. Khouri, I. Hance, P. C. Lee, E. Holtzapple, D. Scanlan, K. Tran, A. Moazzez, T. Utterback, M. Rizzo, K. Lee, D. Kosack, D. Moestl, H. Wedler, J. Lauber, D. Stjepandic, J. Hoheisel, M. Straetz, S. Heim, C. Kiewitz, J. Eisen, K. N. Timmis, A. Dusterhoft, B. Tummler, and C. M. Fraser. 2002. Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ. Microbiol. 4:799-808. [DOI] [PubMed] [Google Scholar]
- 24.Omura, T., and R. Sato. 1964. The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J. Biol. Chem. 239:2370-2378. [PubMed] [Google Scholar]
- 25.Oriel, P. J., S. Savithiry, and H. C. Chang. November 1997. Process for the preparation of monoterpenes using bacterium containing recombinant DNA. U.S. patent 5,688,673. [Google Scholar]
- 26.Plaggemeier, T. 2000. Elimination der schwer wasserlöslichen Modellabluftinhaltsstoffe n-Hexan und Toluol in Biorieselbettverfahren. Ph.D. thesis. Universität Stuttgart, Stuttgart, Germany.
- 27.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 28.Schorcht, S. 1998. Mikrobiologische und molekularbiologische Charakterisierung alkanabbauender Bakteriengemeinschaften. Ph.D. thesis. Universität Bremen, Bremen, Germany.
- 29.Smits, T. H. M., S. B. Balada, B. Witholt, and J. B. van Beilen. 2002. Functional analysis of alkane hydroxylases from gram-negative and gram-positive bacteria. J. Bacteriol. 184:1733-1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smits, T. H. M., M. Röthlisberger, B. Witholt, and J. B. van Beilen. 1999. Molecular screening for alkane hydroxylase genes in gram-negative and gram-positive strains. Environ. Microbiol. 1:307-318. [DOI] [PubMed] [Google Scholar]
- 31.Smits, T. H. M., M. A. Seeger, B. Witholt, and J. B. van Beilen. 2001. New alkane-responsive expression vectors for E. coli and Pseudomonas. Plasmid 46:16-24. [DOI] [PubMed] [Google Scholar]
- 32.Stahl, D. A., and J. W. Urbance. 1990. The division between fast- and slow-growing species corresponds to natural relationships among the myobacteria. J. Bacteriol. 172:116-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Beilen, J. B., J. Kingma, and B. Witholt. 1994. Substrate specificity of the alkane hydroxylase of Pseudomonas oleovorans GPo1. Enzyme Microb. Technol. 16:904-911. [Google Scholar]
- 34.van Beilen, J. B., Z. Li, W. A. Duetz, T. H. M. Smits, and B. Witholt. 2003. Diversity of alkane hydroxylase systems in the environment. Oil Gas Sci. Technol. 58:427-440. [Google Scholar]
- 35.van Beilen, J. B., T. H. M. Smits, L. G. Whyte, S. Schorcht, M. Röthlisberger, T. Plaggemeier, K.-H. Engesser, and B. Witholt. 2002. Alkane hydroxylase homologues in gram-positive strains. Environ. Microbiol. 4:676-682. [DOI] [PubMed] [Google Scholar]
- 36.van Soolingen, D., P. E. W. de Haas, P. W. M. Hermans, and J. D. A. van Embden. 1994. DNA-fingerprinting of Mycobacterium tuberculosis, p. 196-205. In Bacterial pathogenesis, part A, vol. 235. Academic Press, San Diego, Calif. [DOI] [PubMed]
- 37.Wagner, K.-H., and I. Elmadfa. 2003. Biological relevance of terpenoids. Ann. Nutr. Metab. 47:95-106. [DOI] [PubMed] [Google Scholar]
- 38.Willumsen, P., U. Karlson, E. Stackebrandt, and R. M. Kroppenstedt. 2001. Mycobacterium frederiksbergense sp. nov., a novel polycyclic aromatic hydrocarbon-degrading Mycobacterium species. Int. J. Syst. Evol. Microbiol. 51:1715-1722. [DOI] [PubMed] [Google Scholar]