

Abstract
Plasminogen (Plg), the zymogen of plasmin (Plm), is a glycoprotein involved in fibrinolysis and a wide variety of other physiological processes. Plg dysregulation has been implicated in a range of diseases. Classically, human Plg is categorized into two types, supposedly having different functional features, based on the presence (type I) or absence (type II) of a single N-linked glycan. Using high-resolution native mass spectrometry, we uncovered that the proteoform profiles of human Plg (and Plm) are substantially more extensive than this simple binary classification. In samples derived from human plasma, we identified up to 14 distinct proteoforms of Plg, including a novel highly stoichiometric phosphorylation site at Ser339. To elucidate the potential functional effects of these post-translational modifications, we performed proteoform-resolved kinetic analyses of the Plg-to-Plm conversion using several canonical activators. This conversion is thought to involve at least two independent cleavage events: one to remove the N-terminal peptide and another to release the active catalytic site. Our analyses reveal that these processes are not independent but are instead tightly regulated and occur in a step-wise manner. Notably, N-terminal cleavage at the canonical site (Lys77) does not occur directly from intact Plg. Instead, an activation intermediate corresponding to cleavage at Arg68 is initially produced, which only then is further processed to the canonical Lys77 product. Based on our results, we propose a refined categorization for human Plg proteoforms. In addition, we reveal that the proteoform profile of human Plg is more extensive than that of rat Plg, which lacks, for instance, the here-described phosphorylation at Ser339.
Keywords: plasma proteins, plasminogen, proteoform profiling, plasmin, plasminogen phosphorylation
Graphical Abstract
Highlights
-
•
Plasminogen (Plg) is typically subdivided, based on N-glycosylation, into type I and type II.
-
•
Proteoform analysis revealed ∼16 forms of Plg, due to glyco- and phosphomodifications.
-
•
Conversion of Plg to plasmin was found to involve an intermediate step.
-
•
Activation of Plg can be monitored using mass spectrometry.
In Brief
We have identified a novel intermediate step of plasminogen (Plg) conversion to plasmin by monitoring Plg activation with native mass spectrometry. A putative phosphorylation site of Plg was also identified and placed on Ser339 and found to be highly occupied. The proteoform characterization of Plg using mass spectrometry allowed us to divide Plg not into two but eight types. These novel annotated characteristics of Plg can help to better understand the Plg–plasmin system.
Plasmin (Plm) is a circulating protease involved in various physiological processes (1, 2). Plm is mostly known for its role in fibrinolysis, but it is also implicated in cell adhesion, cell migration, cell–cell signaling, tumor development, ovulation, and wound healing (1, 3). Proteolytically active Plm results through a complex activation mechanism from its corresponding proprotein, plasminogen (Plg) (3). Plg is a 791-residue circulatory liver–derived glycoprotein of approximately 90 kDa. The balance between Plm and Plg plays a key role in hemostasis, and imbalance can lead to various diseases and conditions (4, 5, 6). For example, overexpression of Plm can lead to excessive bleeding. Conversely, Plm deficiency can induce thrombosis and improper wound healing (7). Moreover, Plm can activate other enzymes like collagenases and component system factors (8, 9, 10). Generally, hemostasis depends on the balance between Plm and Plg promotion and inhibition, relying on different activators and inhibitors. The two primary activators are the urokinase and tissue Plg activators (termed uPa and tPa, respectively). Notably, recombinant tPa is clinically used in response to embolic or thrombotic stroke because of its role in the Plm–Plg system, catalyzing the conversion of Plg to Plm (11).
The most abundant circulating form of Plg, Glu-Plg, contains the entire amino acid sequence and is comprised of seven domains: the Pan-apple domain (PAp), five consecutive kringle domains (KR1–5), and the serine protease domain (12). The five KR domains are essential for activating and binding lysine residues (13). In addition, various interdomain interactions, such as between Lys and Arg residues from the PAp and KR domains, have been shown to preserve a closed conformation of Plg (12). The proteolytic processing of Glu-Plg into Plm is thought to occur via two steps (Fig. 1). The main activation step itself occurs through the cleavage of the active site (Arg560–Val561) by Plm, tPa, or uPa when the activation loop is exposed (5). In return, active Plm can cleave tPa and uPa into their two-chain form, which has a significantly increased affinity for converting Plg into Plm (14). In addition, Glu-Plg can undergo the proteolytic removal of the first 77 amino acids (cleaving between Lys77 and Lys78), forming Lys-Plg (15). This step, described as Plm-induced preactivation, is thought to generate a 10- to 20-fold more readily cleaved form of Plg (Plm) than Glu-Plg, related to the fact that Lys-Plg has a more open conformation (3, 16). It is thought that Lys-Plg is found only at the site of activation in vivo (13, 17), that is, at cell surfaces and does not circulate (13).
Fig. 1.

Schematic overview of Plg/Plm structures and putative post-translational modifications. Activation of full-length Glu-Plg occurs by the cleavage of the activation site at R561. The conversion of Glu-Plg to Lys-Plg occurs through the cleavage of an N-terminal peptide. Plg and thus also Plm can harbor up to two core O-glycans (S249 and T346) and one complex N-glycan (N289). The N-glycan site is not always occupied, which has led to the classical distinction between type I (occupied) and type II Plg/Plm. Amino acids are numbered without the signal peptide. Plg, plasminogen; Plm, plasmin.
Plg possesses several glycosylation sites of high occupancy (Fig. 1). Traditionally, Plg (and thus also Plm) is categorized into two types (type I and type II), based on the occupancy of these glycosylation sites, which have been described to also give rise to distinctive function and localization (18). Plg type I contains an N-glycan at Asn289 in the KR3 domain (19, 20), whereas this N-glycosylation site is not occupied in Plg type II. The N-glycan is believed to cause steric hindrance, leading to a slower activation of Plg type I by uPa or tPa than type II (12). The binding of certain protein interactors is also distinct for Plg type I or II. For example, the streptococcal protein PAM has been reported to have an >10-fold higher affinity for Plg type II over type I (21). When nonglycosylated, Asn289 in Plg type II forms key interactions with the serine protease domain (17). In addition, Plg type I is thought to be more prominent in intravascular fibrinolysis and type II to have a greater affinity for cell surfaces (22). In addition, both Plg types have a fully occupied O-glycan site at position Thr346 and a partly occupied O-glycan site at Ser249 (between KR3 and KR4) (19, 20, 23). The number of possible Plg proteoforms that arise from the combinations of proteolytic processing and glycan occupancy as described previously results putatively in a diverse palette of potential Plg proteoforms. This heterogeneity could be further exacerbated by the variety of different glycan structures presented for each individual glycosylation site.
Given its central role in fibrinolysis, the activation of Plg to Plm has been the focus of extensive research over several decades (5), using various experimental methods such as nuclear magnetic resonance spectroscopy (20), gel electrophoresis (24), and affinity/enzymatic assays (25). A general drawback of these techniques is that they cannot easily distinguish different proteoforms, yielding only ensemble-averaged information over the entire proteoform profile. While Plg type I and type II can be separated by, for example, ion-exchange chromatography (26), this does also not resolve individual Plg (glyco)proteoforms (27, 28). As a direct consequence, the effects of the inherent microheterogeneity of Plg on its structure and function have so far been poorly explored.
High-resolution native mass spectrometry (native MS) (29) is an analytical technique that is well suited to dissect proteoforms. Since the introduction of high-resolution Orbitrap mass spectrometers, native MS has enabled the characterization of intact macromolecular assemblies (30), complex glycosylation features (31, 32), and many other aspects, such as structural heterogeneity or protein interactions (33). In a native MS experiment, intact proteins are introduced into the mass spectrometer under nondenaturing conditions without a proteolysis step. Under these conditions, any post-translational modifications (PTMs) present on the protein are readily detected as a shift in protein mass (29). Because of this, native MS excels at revealing unexpected PTMs that could be overlooked if they are not included in search criteria for peptide-centric (bottom–up) MS approaches (34). An additional benefit to native MS is that if a protein contains multiple PTMs, the combinatorial profile of different PTMs is simultaneously detected, in contrast to bottom–up proteomics where this information is often lost (35). The unique information obtained by native MS can be combined with the localization and quantification of PTMs by bottom–up MS into a single analytical approach to greatly improve confidence in identified PTMs (36, 37).
Here, we use native MS to characterize the entire proteoform ensemble of pooled human serum–derived Plg and plasma-derived Plg from individual donors. Moreover, we apply this native MS–based methodology to probe the proteoform-level conversion kinetics of Plg under activating conditions. Our data reveal several novel details within the Plg proteoform landscape that have previously escaped characterization, with implications on the appropriate characterization of Plg subtypes and their associated structural and functional consequences.
Experimental Procedures
Chemicals and Materials
Human pooled serum–derived Plg was purchased from Sigma–Aldrich. Individual donor plasma–derived Plg was purified via a three-step protocol (17). Plasma was acquired from five healthy donors recruited to participate (human ethics approval: 34706, Monash University). Recombinant tPa, uPa, and rat serum–derived Plg were kindly provided by Ruby Law (Monash University). The bacterial staphylokinase (SAK) was kindly provided by Suzan Rooijakkkers (UMCU Utrecht). All chemicals and enzymes used, that is, ammonium acetate (AMAC), Tris(2-carboxyethyl)phosphine (TCEP), neuraminidase (sialidase) from Arthrobacter ureafaciens, and peptide-N-glycosidase F from Elizabethkingia meningoseptica were purchased from Sigma–Aldrich. Phosphatase was acquired from New England Biolabs.
Activation of Plg
Per activation experiment, 7 μl of 1 μg/μl of Plg was buffer exchanged six times as described later at 4 °C and immediately kept on ice. The activator of choice was also buffer exchanged six times and kept on ice. The concentration of Plg and activator was determined using a Nanodrop spectrophotometer (Thermo Scientific), and concentrations were adjusted using AMAC to achieve 1 μg/μl of Plg. The measurement at time point 0 min was always taken before the activator was added. The time of activation was recorded from the moment that activator was added to Plg at a final molar ratio of 1:250, activator:Plg. Upon adding the activator, the mixture was shortly vortexed, spun down in a benchtop spinner, and incubated at 25 °C in a thermos-shaker. Activation with uPa was performed and measured in triplicate. Activation with SAK and tPa was performed once.
Native MS Analysis
Protein fractions were buffer exchanged into 150 mM AMAC (pH 7.5) by ultrafiltration with a 10 kDa cutoff filter. Sialidase was used to remove sialic acid residues from several studied proteins, and peptide-N-glycosidase F was used to cleave off N-glycans. Deglycosylation was performed in PBS or 50 mM ammonium bicarbonate. Enzyme-treated samples were also buffer exchanged prior to native MS analysis. Protein samples were analyzed on a modified Exactive Plus Orbitrap instrument with an extended mass range (Thermo Fisher Scientific) as previously described (30) using an m/z range of 500 to 15,000. Voltage offsets on the transport multipoles and ion lenses were tuned to achieve optimal transmission of the protein ions. Nitrogen was used in the higher-energy collisional dissociation (HCD) cell at a gas pressure of 6 to 8 × 10−10 bar. Spray voltage was set to 1.3 kV. Source fragmentation and collision energy were optimized for the analysis of each protein. Source temperature was set to 250 °C. Acquisition time per transient was set to obtain a resolution (at m/z 200) of 30,000. The instrument was mass calibrated using cesium iodide.
Data Analysis
For native MS data analysis, accurate masses of proteoforms were extracted by deconvolution of the raw native MS spectra to zero-charge spectra using UniDec software (version 5.0.1) (38). Resulting data were manually crosschecked with raw spectra. Analysis of PTM composition after deconvolution was done manually. Glycan structures and other PTMs were deduced based on reported glycosylation, glycopeptide data, and removal of sialic acids and glycans. Phosphorylation was confirmed by dephosphorylation. For calculations, average masses were used, namely hexose/mannose/galactose (162.1424 Da), N-acetylglucosamine (NAc, 291.26 Da), fucose (146.14 Da), neuraminic acid (NeuAc, 291.26 Da), and phosphorylation (79.89 Da). Symbols and text nomenclature are based on the recommendations of the Consortium for Functional Glycomics (39). Quantification of all Plg and Plm proteoforms was done using relative intensities.
Peptide-Centric MS Analysis
For PTM identification and quantification, protein samples of two donors were digested in solution by adding 5 μg in 200 mM Tris and reducing with 50 mM TCEP for 30 min at 60 °C. After cooling to room temperature, samples were alkylated with 200 mM chloroacetamide. This reaction was quenched with 50 mM TCEP, and sodium deoxycholate was added to 1%. Protein samples were digested for 4 h using trypsin at an enzyme-to-protein ratio of 1:100, then (optionally) overnight with Glu-C enzyme at a ratio of 1:75, at 37 °C. All digests were desalted using a protocol as previously described (40) using OASIS plates (Waters), dried, and dissolved in 2% fatty acid (FA) prior to LC–MS analysis. Peptides of protein samples (100 ng) were separated and analyzed using an Ultimate HPLC nanoflow system coupled to an Exploris Orbitrap mass spectrometer (both Thermo Fisher Scientific) as previously described (40). In short, analysis was performed in positive ion mode using electrospray ionization. MS1 scans were obtained in a mass range from m/z 375 to 1600 at a resolution of 60,000. Data-dependent MS2 acquisition was at a resolution of 15,000 with a mass range from m/z 40 to 2500. Fragmentation was HCD and electron transfer HCD. Raw data were searched manually to identify glycan fragment ions and confirm results obtained by data interpretation with Byonic software (version 4.3.4; Protein Metrics, Inc) using the sequence of human Plg (P00747) as described in UniProt, version 2 (1989). Digestion parameters included the enzyme of choice (trypsin or Glu-C) with two missed cleavages permitted. In all searches, mass tolerance for precursor ions was set to 10 ppm and for fragment ions to 20 ppm. All annotated peptides were filtered with 1% false discovery rate (FDR) and a log probability of >1.50 at the peptide level. Modifications included in the search were human N- and O-glycans, Cys alkylation, Met- and Try-oxidation, and S/T phosphorylation. Quantification was performed with Skyline (version 20.2) (41) on the tryptic digestion of one of the donors (female 2). Identified peptides can be found in Supplemental Table S3. Peptides selected for quantification and their abundances can be found in Supplemental Table S4. Spectral simulation was performed using R.
For further identification of the phosphorylation site, an additional peptide-centric MS analysis was performed. Here, 50 μl of protein aliquot was digested with sequence grade Glu-C at 1:50 enzyme/protein ratio overnight. The digested peptide mixture was acidified with 1 μl formic acid and subjected to LC–MS/MS. Peptides were separated on a Waters Acquity BEH peptide C18 column (100 mm × 2.1 mm, 1.7 μm). The mobile phases consisted of 0.1% FA as phase A and 90% acetonitrile and 0.1% FA as phase B. A gradient program with a flow rate of 125 μl/min was applied as follows: 2.5% B for 2.5 min, a linear increase to 60% B for 67.5 min, an increase to 95% B for 5 min and holding for 10 min, and then re-equilibrated to the starting conditions for 5 min. Injection volume was 10 μl. Data-dependent MS2 acquisition was at a range of 300 to 2000 m/z on a high-resolution TOF, followed by MS/MS of 10 ion candidates per cycle with rolling CE. Other settings were as follows: 5500 V spray voltage, 300 °C source temperature, ion source gas 1 and 2 set at 35, curtain gas set at 15, and declustering potential set at 150 V. Phosphorylation sites were identified using the MASCOT search add-on in Analyst software, version 1.6 (Sciex) with phosphorylation as dynamic modification. Digestion parameters included the enzyme of choice (Glu-C) with two missed cleavages permitted. Data were searched against the sequence of human Plg (as described in the UniProt human proteome database, January 2020), using a 20 ppm precursor ion tolerance for total protein-level analysis and 0.04 Da fragment ion tolerance. Peptide-spectrum matches were identified, quantified, and filtered to a 1% peptide FDR and then collapsed further to a final protein-level FDR of 1%. Mass spectra of phosphorylated and dephosphorylated peptides were manually curated in PeakView software, version 2.2 (Sciex).
Phosphoprotein gel Analysis
Phosphorylation using recombinant Fam20C (R&D Systems) and dephosphorylation using Lambda protein phosphatase (New England BioLabs) of native Plg was performed as recommended by the manufacturer. Phosphoprotein stain was performed using Pro-Q diamond phosphoprotein gel stain (ThermoFisher Scientific).
Phylogenetic Analysis
The 250 most correlating sequences to human Plg were downloaded after a BLAST search. Using the msa (multiple sequence aligner) package (42), all sequences were aligned with the human sequence. Amino acid positions of interest of all 250 organisms were manually compared with human, and the relative conservation was calculated. The included sequences can be found in Supplemental Table S5 (UniProt accession numbers).
Experimental Design and Statistical Rationale
In this study, we used commercially available Plg, from pooled serum, to identify PTMs naturally present in populations. We confirmed the presence of identified PTMs with the analysis of plasma-derived Plg from individual donors (n = 5). These donors were manually and visually compared with pooled serum–derived Plg without statistical analysis. Activation kinetics were measured on a high-resolution Orbitrap as a technical triplicate under controlled conditions (temperature, reaction time, preparation time, mixing, and volumes). Proteoform intensities of the triplicates were averaged to create a dataset in which proteoform-specific intensities could be compared. Duplicate peptide-centric proteomic studies were performed to corroborate our findings from native MS analysis.
Results
The Proteoform Landscape of Human Plg
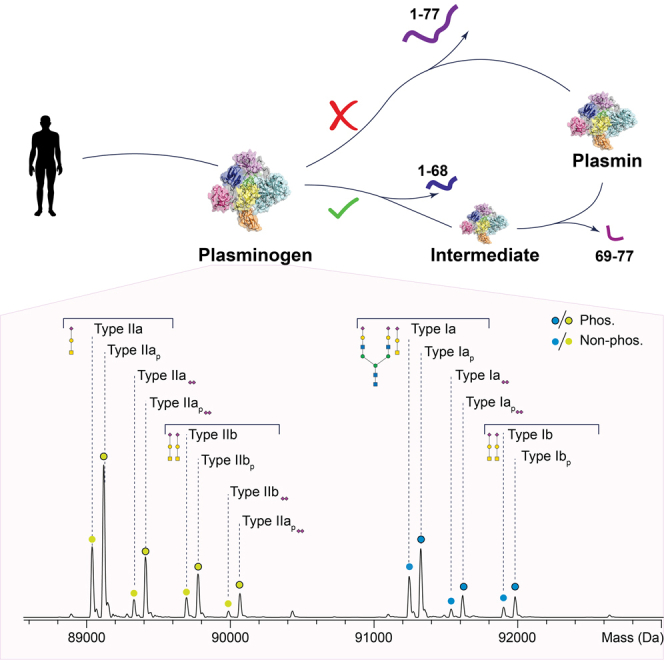
To determine the diversity of the proteoform landscape of Plg, we first analyzed human Plg purified from pooled serum using native MS (Fig. 2A). The charge state series for Plg ranged from +18 to +21 at around 4200 to 5200 m/z (Supplemental Fig. S1). Following charge-state deconvolution, we observed around 14 distinct masses, ranging from 89,038.2 to 92,269.0 Da. The sequence-predicted protein backbone mass of 88,384.4 Da (88,432.4–48 Da from 24 disulfide bonds) was not observed. Instead, the observed mass difference between the lowest measured mass and the amino acid sequence backbone mass of Plg agrees well with Plg harboring the expected (fully occupied) O-glycan at Thr346 (20), composed of galactose, N-acetylgalactosamine, and one NeuAc (GalNAcGalNeuAc, 656.6 Da). Therefore, the lowest mass peak in the mass spectrum can be annotated as Plg type II.
Fig. 2.

Annotation and quantification of the proteoform profile of hPlg.A, deconvoluted mass spectrum of Plg reveals at least 14 annotated Plg glycoproteoforms being either phosphorylated (75%) or nonphosphorylated (25%). The O-glycan at Thr346 is always occupied, whereas the O-glycan at S249 is partly occupied. B, quantification and comparison of type I/II distribution and the phosphorylation ratio and Ser249 O-glycosylation occupancy in six samples originating from serum of five individuals and one pooled serum–derived Plg. To support our annotation of the seven unique glycoforms including the sialylation, we treated Plg with sialidase (Supplemental Fig. S2B) and observed Plg type I to lose the mass of 3× NeuAc, resulting from the removal of two NeuAc from the N-glycan and one NeuAc from the O-glycan. For both types of Plg, sialidase treatment simplified the proteoform profile into four glycoforms, one or two O-glycans, one N-glycan, and their respective phosphorylated proteoforms. From our annotations, we also conclude that one O-glycan site is a 100% occupied. hPlg, human plasminogen; NeuAc, NeuAc, neuraminic acid.
The native MS spectrum of Plg revealed two similar proteoform profiles, one starting at a mass around of 89.5 kDa and the other around 91.5 kDa. Plg is reported to be both N- and O-glycosylated, with the glycoform type I/II distinction based on the presence/absence of an N-glycan (19). Comparing the masses revealed that each peak of the first proteoform profile has a paired peak in a second proteoform profile shifted by 2206.1 Da, an exact fit of a complex N-glycan composed of 4× N-acetylhexosamine, 5× hexose, and 2× N-acetyl-NeuAc (HexNAc4Hex5NeuAc2). Based on known biosynthetic pathways for human serum glycosylation, this structure most likely represents a fully sialylated biantennary complex N-glycan. Consequently, we annotated the most abundant peak representing Plg type II, with one occupied O-glycan site and the N-glycosylation site Asn289 unoccupied. Additional proteoform peaks of Plg represented mass differences of +291.7 and +656.8 Da with an additional +291.5 Da on Plg type II. The mass shifts are induced by the addition of NeuAc or GalNAcGalNeuAc O-glycans, in line with our expectations as Plg is known to be able to harbor up to two O-glycans (23). On Plg type I, we annotated a +291.5 and +646.5 Da mass shift as a NeuAc and the addition of a second O-glycan. As the NeuAc is seen both on Plg type I and type II, we concluded that this is caused by sialylation of the Gal or GalNAc of an already monosialylated core 1 type O-glycan. To annotate the most abundant mass peak, we looked at the prevalent doublet peaks that seemed to be consistently present throughout the native MS spectrum. Each pair of peaks in the spectrum differed in mass by ∼80 Da. From this pattern, we hypothesized that a major fraction of Plg is phosphorylated. To confirm this, we performed native MS after treatment with a phosphatase (Supplemental Fig. S2A), after which the higher mass and putatively phosphorylated proteoform of each doublet pair indeed disappeared.
hPlg Harbors a Novel Highly Occupied Phosphorylation Site
Our native MS results hint that 75% of Plg, purified from pooled serum, is phosphorylated (Fig. 2B). Phosphorylation is not a very widespread and abundant PTM for serum proteins, with only a few of them known to be phosphorylated (43). To confirm that the observed phosphorylation is not a unique occurrence, we analyzed Plg derived from the plasma of five different donors, in which phosphorylation was observed in every individual with comparable prevalence (Supplemental Fig. S3). To pinpoint the identity of the phosphorylation site, we performed peptide-centric bottom–up analysis of Plg. Tryptic digestion of Plg yielded a 37-residue glycopeptide (330IPSCD SSPVS TEQLA PTAPP ELTPV VQDCY HGDGQ SYR367) containing both the expected Thr346 O-glycan and the putative 80 Da phosphorylation. Initial site localization on the tryptic peptide (SPVSTEQLAPTAPPELTPVV) containing both the O-glycan and the phosphorylation (Supplemental Fig. S4A) using standard HCD- and electron transfer HCD–based approaches was not successful, which we hypothesized was due to the close proximity of the O-glycan and the phosphorylation negatively impacting their fragmentation behaviors. Therefore, we also performed bottom–up analysis utilizing GluC as an alternative protease in an attempt to sequester these two modifications onto different peptides. This analysis successfully localized the O-glycan site to be as expected on Thr346 and the phosphorylation site on a 15 AA peptide containing the STE motif but not the O-glycan site. First, the MS1 spectrum of a 24 AA peptide (327YCKIP SCDSS PVSTE QLAPT APPE351) provided a smaller phosphorylated peptide containing the O-glycan. This placed both modifications somewhere on SPVSTEQLAPTAPPE. Furthermore, two peptides were measured that contained either only the phosphate moiety or the O-glycan (Supplemental Fig. S4B). A 9 AA peptide (342QLAPT APPE351) localized the O-glycan on Thr346, and a 15 AA peptide (327YCKIP SCDSS PVSTE342) carried the phosphorylation. We were able to pinpoint the phosphorylation on Ser339 from the fragmentation spectra of the peptide YCKIPSCDSSPVSTE (Supplemental Fig. S4C). Interestingly, Ser339 is found within an SxE motif, a known recognition sequence for the prevalent serum protein kinase FAM20c (43, 44). Using recombinant FAM20c, we confirmed that Plg is indeed a target of this kinase in vitro (Supplemental Fig. S5). Further investigation is necessary to determine whether FAM20c is the true Plg kinase in vivo.
Considering this very high occupancy (even higher than the partially occupied O-glycan), it is somewhat surprising that Plg phosphorylation has remained largely unrecognized in the recent Plg literature. Barlati et al. (45) proposed phosphotyrosine and phosphothreonine on the basis of immunoblotting assays in 1995, whereas Wang et al. (46) explicitly ruled out these modifications and instead proposed a near-stoichiometric quantity of phosphoserine at Ser578 on the basis of MALDI-TOF MS1 spectra of Plg following cyanogen bromide digestion. More recently, the possibility of Plg phosphorylation was previously noted in a study of serum proteoforms, although no site identification was performed (47). By contrast, we observed a ∼75% occupancy of phosphoserine at Ser339, with no evidence of phosphorylation at any other site, on the combined basis of native MS- and MS2-validated bottom–up proteomics. We noted that Ser578 is buried within the catalytic domain of Plg and is not surface exposed, making it a less likely target for phosphorylation.
Next, using bottom–up proteomics data, we quantified the PTMs of Plg in a sample purified from an individual donor (Supplemental Fig. S6A). We confirmed a near 100% occupancy of the Thr346 O-glycan site. The N-glycosylation site Asn289 exhibited partial occupancy with complex, mostly disialylated biantennary glycans as also annotated in the native MS spectrum (Fig. 2). Interestingly, the additional O-glycosylation site Ser249 appeared to be only infrequently occupied and rarely disialylated. When the proteoform profile of Plg is reconstructed using the bottom–up data described previously, an excellent agreement to the experimentally observed native MS profiles, indicating that we have identified and localized PTMs of all significantly abundant proteoforms of Plg (Supplemental Fig. S6B). Overall, our combined native MS and bottom–up MS data demonstrate that Plg is far more diverse than the current “type I” and “type II” categorization. Rather, we annotate at least eight prominent apparent types: type Ia, IIa, and I/IIb (with Ser249 O-glycan). Each type can then be phosphorylated, leading to Iap, Ibp, IIap, and IIbp. In addition, each type can contain an extra sialic acid placed on an O-glycan. For completeness, we provide annotation for each type and PTM in the Supplemental Table S1, including their respective masses.
Plg Activation from a Proteoform Perspective
It is well established that glycosylation differences between Plg type I and type II affect the activation rate and tissue specificity of Plg. Type I is known to have a lower affinity for cells than type II and lower activation rates in the presence of uPa and tPa, but type I seemingly works faster on fibrin clots (18, 48, 49). Given the wide variety of Plg proteoforms that we describe, we hypothesized that these differences could be attributed to specific proteoforms. As each Plg proteoform is resolved in mass, we used native MS to monitor the in vitro activation and transformation of Plg to Plm using uPa as a representative activator, exploring the effect that the various PTMs may have on the rate (Fig. 3). Upon incubation with uPa, several new proteoforms appeared in the deconvoluted native mass spectra (Fig. 3A, Supplemental Fig. S7).
Fig. 3.

Activation of Plg with uPa reveals a novel step-wise processing.A, monitoring by native mass spectrometry, the species formed following the addition of uPa to Plg (at a ratio of 1:250) reveals a step-wise N-terminal cleavage, starting with the release of Glu1–Arg67, followed by Met68–Lys77. The canonical Lys-Plm product is formed only later in time, when compared with the product that released Glu1–Arg67. B, annotated proteoform profile of all species, including Plg, the novel intermediate and Lys-Plm. C, schematic of the stepwise activation of Plg. Plg, plasminogen; uPa, urokinase plasminogen activator.
Because the mechanism of Plg activation has been so extensively studied (6), we expected to observe formation of several specific proteoforms. Specifically, the expected mass changes following activation of Plg are (1) a +18.0 Da mass shift because of the hydrolysis of the active site peptide bond (Arg560–Val561) and (2) a −8790.0 Da mass shift because of cleavage of the N-terminal activation peptide, covering the first 77 amino acids. Instead, after 10 min of incubation with uPa, we observed a species harboring a mass difference of −7652.5 Da, 1119 Da lighter than the expected Glu1–Lys77 N-terminal cleavage. Based on the sequence and structure of Plg, we noted that cleavage of the first 68 amino acids of the N-terminal peptide (including two disulfide bonds and the active site hydrolysis) should lead to a mass shift of 7653.6 Da, nicely in agreement with the experimental data. Incubating Plg longer with uPa, a second species with the same proteoform profile appeared corresponding to a mass shift of −8771.7 Da compared with Plg, or −1119.2 Da when compared with the newly identified intermediate species. This final mass loss corresponds to the full cleavage of the first 77 amino acids, thus forming the well-described Glu1–Lys77 product. To confirm that N-terminal cleavage is initiated with the cleavage of the first 68 AAs and not co-occurring with the full 77 AA cleavage, we next monitored the formation of the N-terminal cleaved Plg-derived peptides throughout incubation (Supplemental Fig. S9, Supplemental Table S2). Observed and annotated peptides included Glu1–Arg68, Met69–Lys77, Met69–Lys78, and further digested fragments thereof, but never Glu1–Lys77 or Glu1–Lys78. These data indicate that the activation is tightly coupled to the new intermediate N-terminal cleavage product, happening either very shortly before or after. To some degree, Lys78-Plm was observed, where the N-terminal peptide was cleaved at a consecutive lysine at site 78 rather than Lys77 (Fig. 3B). Over the whole course of Plg activation, we thus observed the active site hydrolysis (+18 Da) that forms Plm and the cleavage of the N-terminal peptide that converts Glu-Plg/Plm to Lys-Plg/Plm. As such, we categorized the reaction products as Glu-Plg (inactive, unreacted Plg), Int-Plm (hydrolyzed Glu-Plg, cleaved at Arg68), and Lys-Plm (canonically cleaved, active Plm). While Plg contains 24 disulfide pairs, we see no evidence of breakage of any of these bonds to facilitate the activation process, either in the activation peptide (Supplemental Fig. S8) or the produced Plm (Supplemental Table S1). However, we cannot rule out, for example, transient and/or substoichiometric quantities of disulfide disruption.
Thus, the newly described intermediate species corresponds to the N-terminal cleavage at Arg68, including active site hydrolysis. We also observed that this intermediate cleavage step precedes the canonical 77 AA N-terminal cleavage, suggesting a step-wise conversion of Glu-Plg to Lys-Plm. We performed similar experiments with the activators tPa and SAK, a bacterial activator of Plg (Supplemental Fig. S9). With all three activators, we observed this intermediate cleavage step at Arg68 to precede the canonical cleavage at Lys77.
Proteoform-Resolved Activation Kinetics
The annotation of the full proteoform profile (Fig. 3B) enables the monitoring of apparent reaction rates of individual proteoforms upon activation by uPa. We particularly investigated the effect of Plg phosphorylation and O-glycosylation on the activation. We grouped proteoforms to PTMs of interest and used the relative abundances of corresponding proteoforms over the course of Plg activation. As such, we were able to determine the relative abundances of groups of proteoforms (i.e., type I, II or (non)phosphorylated) and even individual proteoforms over time (Supplemental Fig. S10).
We first compared the relative abundances of the sum of all proteoforms categorized as either Plg type I or type II, that is, in the presence or the absence of the N-glycan (Fig. 4A). Assuming the activation path to follow: Glu-Plg → Lys-Int → Lys-Plm. we observed that the first step (to Lys-Int) seems equally fast for both types. However, Plg type I is converted faster to Lys-Plm than type II. Next, we analyzed differences in the proteoforms of Plg in the presence or the absence of phosphorylation (Fig. 4B) but found no substantial differences in their activation rates. No substantial differences were also found in the absence/presence of the second O-glycan (Ser249) (Fig. 4C). In summary, of all PTMs present in human Plg, only the presence or the absence of the N-glycan seems to affect the conversion of Glu-Plg to Lys-Plm substantially in our in vitro assays, with the phosphorylation and additional O-glycosylation having no measurable effect.
Fig. 4.

Plg activation rates in vitro are affected by its N-glycosylation but not by its phosphorylation and O-glycosylation.A, monitored relative abundances (measured by native MS) of type I and type II Plg show a difference in the rate of formation of Lys-Plm, whereby the first step to Lys-Int seems not to be rate dependent. B and C, the presence/absence of phosphorylation and the additional O-glycan (Ser249) on human Plg have no substantial effect on the activation of Plg. Pie charts show the average relative abundance of proteoform corresponding to the indicated PTM categories (measured by native MS). Plg, plasminogen; PTM, post-translational modification.
Phylogenetic Analysis of Plg, Conservation of PTM, and Cleavage Sites
Phosphorylation is uncommon for plasma proteins, especially when compared with glycosylation (43). To follow up the highly abundant phosphorylation observed here for human Plg, we next queried whether this Ser339 site is a conserved feature. Therefore, we aligned the sequences of Plg proteins from ∼250 mammalian species, and queried conservation of the human phosphosite, and the N- and O-glycosylation sites. The human pSxE motif (43) around Ser339 was conserved on only ∼40% of the queried species. The Ser249 O-glycosylation site, which in humans is (less frequently) occupied, was conserved on ∼35% of the sequences. Surprisingly, the conservation of the fully occupied O-glycan site at Thr346 was only conserved on ∼15% of the sequences. Moreover, the N-glycosylation site at N289, responsible for the canonical Plg type I–type II classification in human Plg, is conserved in just around half of the sequences. In contrast, the active site Arg561 was found to be 100% conserved. Interestingly, both the canonical N-terminal cleavage site and the here-reported novel intermediate N-terminal cleavage site were practically fully conserved (99.2% and 98.8%, respectively).
These data suggest two things. First, the activation mechanism, at least with respect to the active site hydrolysis and the N-terminal cleavage of Glu-Plg is a highly conserved process and occurs in similarly in all queried mammals. In addition, this means it is highly likely that the cleavage of the N terminus will also occur stepwise in most mammals. Second, most PTMs, including the type-defining N-glycosylation, are not a highly conserved feature of Plg. It is therefore likely that, based on the (lack of) conservation of the glycosylation sites and the STE motif, Plg from other mammals may not necessarily portray the same spectral complexity (and amount of proteoforms) as human Plg. To demonstrate this point, we also purified rat serum–derived Plg, which according to its sequence does not conserve the human SxE motif and the O-glycosylation sites but harbors the N-glycosylation site. Indeed, the native MS spectrum of rat serum–derived Plg was found to be displaying a much simpler proteoform profile (Fig. 5), revealing the presence of an N-glycan, but there was no evidence for O-glycosylation or phosphorylation. Hence, we stipulate that phosphorylation on human Plg represents a relatively recent feature in evolution.
Fig. 5.

Conservation of PTMs of Plg between rat and human. Comparison of the native mass spectrum of rat Plg (red) with the native mass spectrum of human Plg (blue) with the mass of the first peak of human Plg aligned with that of rat Plg. Compared with human Plg, the proteoform profile of rat serum–derived Plg looks simple, lacking the human O-linked glycosylation and phosphorylation. The distribution over type I and II seems similar between human and rat Plg. Plg, plasminogen; PTM, post-translational modification.
Discussion
By using high-resolution native MS, we here monitored full proteoform profiles of human Plg, following its activation. Our analysis revealed novel features of Plg activation and Plg PTMs. By monitoring the Plg activation in vitro by native MS, we could dissect proteoform-specific N-terminal processing and confirmed differences in activation rate between canonical Plg type I and II. In contrast, we did not observe substantial differences in N-terminal processing between proteoforms being differentially O-glycosylated and/or phosphorylated. We propose that the degree of complexity incurred by PTMs warrants the annotation of more Plg types, based on their N- and O-glycosylation and phosphorylation status. We reveal here that the conversion of Glu-Plg to Lys-Plm occurs differently than thus far anticipated, namely via a two-step process. First, an intermediate cleavage at Arg67 creates Int-Plg, which is further cleaved at Lys77 to form Lys-Plm.
Unequivocally, our work revealed the conversion of Glu-Plg/Plm to Lys-Plm to occur strictly via a two-step mechanism. Although Plg activation has been studied extensively, only once has the cleavage of the N-terminal peptide at Arg67 been described, albeit not as a feature in Plg activation. The conversion of Glu-Plg to modified forms was described as already reported by Wiman et al. (50), occurring either through cleavage of the Arg67 to 68Met or Lys77 to 78Lys bonds. Holvoet et al. (51) focus on the Lys-Plg intermediate form using an antibody that bound an epitope, which is not exposed in Glu-Plg. It appears that the focus on Lys-Plg in the literature has overshadowed the existence of the here described methionine-cleaved intermediate. As described by Fredenburgh (52) in 1992, Lys-Plg is a significant intermediate in the activation of Glu-Plg during fibrinolysis. Perhaps the well-known lysine-binding capability of Plm and Plg can play a role in this (53) as well as the fact that the small change in mass (1.1 kDa) would be hard to detect when techniques such as gel electrophoresis would be employed. Here, owing to its “what-you-see-is-what-you-get” overview of proteoform profiles by native MS, we were able to distinguish the two N-terminal fragments and conclude based on the species abundances that this step happens strictly in a consecutive fashion. The step-wise cleavage is also supported by reported structures of Plg. After analysis of a previously published structure for Plg (17), it is clear that the canonical cleavage site (Lys77) is still buried within the PAp domain even when Plg is in an open conformation (Fig. 6, C–E). Access to Lys77 requires removal of the first 68 AA, which are exposed in an open conformation and leaves the Lys77 cleavage site exposed.
Fig. 6.

Plasminogen (Plg) phosphorylation and activation.A and B, X-ray crystal structure of human Plg (Protein Data Bank ID: 4DUR) in the closed conformation. Plg, a seven-domain protein, from the N terminus, Pan-apple domain (PAp, blue), kringles 1 to 5 domain (KR1–5, magenta, yellow, orange, red, and green, respectively), and the serine protease domain (SP, cyan). B, the phosphorylated S339 is located at the loop (residues 334–356) connecting KR3 and KR4. In the crystal structure, residues 335 to 344 cannot be accurately modeled based on the electronic map suggesting this region is flexible. Here, it is represented by a dotted line with the phosphorylated S339 modeled. In the closed conformation, this loop is near the activation loop R561|V562 (red sticks). C, in the closed conformation, in the first cleavage site R68|M69 (red sticks), the P1 residue R68 binds to KR4 via the main chain with W427 (yellow sticks) and the side chain with D411 and D413 (yellow sticks) from the lysine-binding site. In the second cleavage site K77|K78 (red sticks), the P1 residue K77 is buried within the PAp domain, and the P1 residue K78 bonds with D175 of KR2 (yellow sticks). As a result, when in the closed position, both sites are predicted to be inaccessible. D, in the open conformation, the first cleavage site becomes accessible, whilst the second cleavage site remains restricted by the PAp domain. E, upon cleavage of the first site, the second cleavage site becomes exposed.
Next, one may wonder why the here reported highly abundant phosphorylation of human Plg has been overlooked so far. We attribute this to the close proximity, in sequence, of the highly occupied Thr346 O-glycan site and the highly occupied phosphorylation site Ser339. MS2 fragmentation of the tryptic peptide harboring both an O-glycan and a phosphorylation may be efficient, hampering proper annotation when using standard tryptic digests and HCD approaches. In contrast, the facile detection of phosphorylation via native MS accents the advantages and complementarity of protein-centric over peptide-centric proteomics approaches. Nevertheless, using peptide-centric MS, we could confirm the presence and location of the Ser249 O-glycosylation (23) albeit by using alternative proteases.
The close proximity between the O-glycan at Thr346 and the phosphorylation at Ser339 site imply some potential crosstalk between these two PTMs. The position of Ser339 close to the activation loop implies that Plg phosphorylation may impact the rate of Plg activation, although we did not observe differences between phosphorylated and nonphosphorylated Plg by activation with uPa. The exact role of Plg phosphorylation therefore remains unknown but could be related to tissue specificity (localization), cell and protein binding, receptor interaction and substrate recognition, and changes in Plg/Plm conformation (43, 54). Further work, for example by comparing wildtype Plg versus Ser339Ala and/or Thr346Ala mutants in vivo, will be necessary to fully delineate these intricacies. Moreover, given that this phosphorylation site is not that well conserved between species, one may wonder whether it has a critical role.
SAK, one of the other activators tested in this study, is secreted by group A Streptococcus, an exclusively human pathogen. SAK binds to and activates closed Plg (55). Here too, it remains to be further investigated if there are any differences in the rate of Plg activation by streptokinase from different clusters (56). Elucidating the role of Plg/Plm phosphorylation would therefore be a significant advancement of our understanding of the Plg/Plm system.
In conclusion, in this work, we reveal the presence of novel PTMs in human Plg and determine the occupancy of all these sites in plasma-derived Plg from several donors. We show that these features are omnipresent in the studied donors. Of all these PTMs (N-glycosylation, O-glycosylation, and phosphorylation), only the presence/absence of the N-glycosylations seems to have an effect on the activation of Plg. In the activation of Plg, our data refine the activation mechanism of Plg, describing a new initial cleavage, preceding the known cleavage step. Overall, monitoring the proteoform profiles of plasma proteins is a strong approach to monitor in-depth and quantitively the presence/absence of PTMs, which is combination with peptide-centric approaches can be used to assign the sites, and nature of the modifications attached in detail. As such PTMs may fine tune the interactions and function of plasma proteins, these methods can contribute to an improved understanding of for instance hemostasis.
Data availability
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (57) partner repository with the dataset identifier PXD042225. Reviewer username: reviewer_pxd042225@ebi.ac.uk; password: RZzDSXwz
Supplemental data
This article contains supplemental data.
Conflict of interest
The authors declare no competing interests.
Acknowledgments
We acknowledge Suzan Rooijakkers and Carla Gosselaar-De Haas (UMC Utrecht) for supplying recombinant SAK. We thank Adam Quek and Qing He for the purification and characterization of human plasminogen; and Bruce Kemp for the critical comments. We acknowledge support from the Netherlands Organization for Scientific Research funding the Netherlands Proteomics Centre through the X-omics Road Map program (project no.: 184.034.019) and support from the National Health and Medical Research Council of Australia (grant no.: APP1172593).
Funding and additional information
D. A. T. C., V. F., and A. J. R. H. acknowledge further support by the Netherlands Organization for Scientific Research Satin 731.017.202 and ENPPS.LIFT.019.001 grants. J. C. W. acknowledges the Senior Principal Research Fellowship support from the National Health and Medical Research Council of Australia and the previous Federation Fellowship support from the Australian Research Council.
Author contributions
J. C. W., R. H. P. L., and A. J. R. H. conceptualization; A. J. R. H., D. A. T. C., V. Y., D. Y., G. W., G. L., C. L., and R. H. P. L. methodology; D. A. T. C., V. Y., T. C., V. F., D. Y., G. W., G. L., C. L., and A. J. R. H. formal analysis; D. A. T. C. and V. Y. investigation; D. A. T. C. and V. Y. writing–original draft; J. C. W. and R. H. P. L. writing–review & editing; R. H. P. L. and A. J. R. H. writing–review & editing; J. C. W. and A. J. R. H. funding acquisition.
Contributor Information
Ruby H.P. Law, Email: ruby.law@monash.edu.
Albert J.R. Heck, Email: a.j.r.heck@uu.nl.
Supplementary Data
References
- 1.Li W.Y., Chong S.S., Huang E.Y., Tuan T.L. Plasminogen activator/plasmin system: a major player in wound healing? Wound Repair Regen. 2003;11:239–247. doi: 10.1046/j.1524-475x.2003.11402.x. [DOI] [PubMed] [Google Scholar]
- 2.Chana-Muñoz A., Jendroszek A., Sønnichsen M., Wang T., Ploug M., Jensen J.K., et al. Origin and diversification of the plasminogen activation system among chordates. BMC Evol. Biol. 2019;19:27. doi: 10.1186/s12862-019-1353-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keragala C.B., Medcalf R.L. Plasminogen: an enigmatic zymogen. Blood. 2021;137:2881–2889. doi: 10.1182/blood.2020008951. [DOI] [PubMed] [Google Scholar]
- 4.Longstaff C., Kolev K. Basic mechanisms and regulation of fibrinolysis. J. Thromb. Haemost. 2015;13(Suppl 1):S98–105. doi: 10.1111/jth.12935. [DOI] [PubMed] [Google Scholar]
- 5.Katz J.M., Tadi P. StatPearls. StatPearls Publishing; Treasure Island (FL: 2022. Physiology, plasminogen activation. [PubMed] [Google Scholar]
- 6.Baker S.K., Strickland S. A critical role for plasminogen in inflammation. J. Exp. Med. 2020;217 doi: 10.1084/jem.20191865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kolev K., Longstaff C. Bleeding related to disturbed fibrinolysis. Br. J. Haematol. 2016;175:12–23. doi: 10.1111/bjh.14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Milner J.M., Elliott S.F., Cawston T.E. Activation of procollagenases is a key control point in cartilage collagen degradation: interaction of serine and metalloproteinase pathways. Arthritis Rheum. 2001;44:2084–2096. doi: 10.1002/1529-0131(200109)44:9<2084::AID-ART359>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 9.Barthel D., Schindler S., Zipfel P.F. Plasminogen is a complement inhibitor. J. Biol. Chem. 2012;287:18831–18842. doi: 10.1074/jbc.M111.323287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leung L.L., Morser J. Plasmin as a complement C5 convertase. EBioMedicine. 2016;5:20–21. doi: 10.1016/j.ebiom.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gravanis I., Tsirka S.E. Tissue-type plasminogen activator as a therapeutic target in stroke. Expert Opin. Ther. Targets. 2008;12:159–170. doi: 10.1517/14728222.12.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aĭsina R.B., Mukhametova L.I. [Structure and functions of plasminogen/plasmin system] Bioorg. Khim. 2014;40:642–657. doi: 10.1134/s1068162014060028. [DOI] [PubMed] [Google Scholar]
- 13.Rijken D.C., Sakharov D.V. Basic principles in thrombolysis: regulatory role of plasminogen. Thromb. Res. 2001;103:S41–S49. doi: 10.1016/s0049-3848(01)00296-1. [DOI] [PubMed] [Google Scholar]
- 14.Urano T., Castellino F.J., Suzuki Y. Regulation of plasminogen activation on cell surfaces and fibrin. J. Thromb. Haemost. 2018;16:1487–1497. doi: 10.1111/jth.14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cesarman-Maus G., Hajjar K.A. Molecular mechanisms of fibrinolysis. Br. J. Haematol. 2005;129:307–321. doi: 10.1111/j.1365-2141.2005.05444.x. [DOI] [PubMed] [Google Scholar]
- 16.Miles L.A., Parmer R.J. Plasminogen receptors: the first quarter century. Semin. Thromb. Hemost. 2013;39:329–337. doi: 10.1055/s-0033-1334483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Law R.H., Caradoc-Davies T., Cowieson N., Horvath A.J., Quek A.J., Encarnacao J.A., et al. The X-ray crystal structure of full-length human plasminogen. Cell Rep. 2012;1:185–190. doi: 10.1016/j.celrep.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 18.Hajjar K. Saunders Co; Philadelphia, PA: 2003. Hematology of Infancy and Childhood. [Google Scholar]
- 19.Hayes M.L., Castellino J.F. Carbohydrate of the human plasminogen variants. I. Carbohydrate composition, glycopeptide isolation, and characterization. J. Biol. Chem. 1979;254:8768–8771. [PubMed] [Google Scholar]
- 20.Marti T., Schaller J., Rickli E.E., Schmid K., Kamerling J.P., Gerwig G.J., et al. The N- and O-linked carbohydrate chains of human, bovine and porcine plasminogen. Species specificity in relation to sialylation and fucosylation patterns. Eur. J. Biochem. 1988;173:57–63. doi: 10.1111/j.1432-1033.1988.tb13966.x. [DOI] [PubMed] [Google Scholar]
- 21.De Oliveira D.M., Law R.H., Ly D., Cook S.M., Quek A.J., McArthur J.D., et al. Preferential acquisition and activation of plasminogen glycoform II by PAM positive group A streptococcal Isolates. Biochemistry. 2015;54:3960–3968. doi: 10.1021/acs.biochem.5b00130. [DOI] [PubMed] [Google Scholar]
- 22.Mori K., Dwek R.A., Downing A.K., Opdenakker G., Rudd P.M. The activation of type 1 and type 2 plasminogen by type I and type II tissue plasminogen activator. J. Biol. Chem. 1995;270:3261–3267. doi: 10.1074/jbc.270.7.3261. [DOI] [PubMed] [Google Scholar]
- 23.Pirie-Shepherd S.R., Stevens R.D., Andon N.L., Enghild J.J., Pizzo S.V. Evidence for a novel O-linked sialylated trisaccharide on Ser-248 of human plasminogen 2. J. Biol. Chem. 1997;272:7408–7411. doi: 10.1074/jbc.272.11.7408. [DOI] [PubMed] [Google Scholar]
- 24.Müllertz S. Different molecular forms of plasminogen and plasmin produced by urokinase in human plasma and their relation to protease inhibitors and lysis of fibrinogen and fibrin. Biochem. J. 1974;143:273–283. doi: 10.1042/bj1430273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kok P., Nilsson T. Assay characteristics and fibrin affinity of plasminogen activators of the intrinsic fibrinolytic system. Thromb. Res. 1986;41:197–209. doi: 10.1016/0049-3848(86)90229-x. [DOI] [PubMed] [Google Scholar]
- 26.Hayes M.L., Castellino F.J. Carbohydrate of the human plasminogen variants. III. Structure of the O-glycosidically linked oligosaccharide unit. J. Biol. Chem. 1979;254:8777–8780. [PubMed] [Google Scholar]
- 27.Lee H.S., Qi Y., Im W. Effects of N-glycosylation on protein conformation and dynamics: protein Data Bank analysis and molecular dynamics simulation study. Sci. Rep. 2015;5:8926. doi: 10.1038/srep08926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wandall H.H., Nielsen M.A.I., King-Smith S., de Haan N., Bagdonaite I. Global functions of O-glycosylation: promises and challenges in O-glycobiology. FEBS J. 2021;288:7183–7212. doi: 10.1111/febs.16148. [DOI] [PubMed] [Google Scholar]
- 29.Tamara S., den Boer M.A., Heck A.J.R. High-resolution native mass spectrometry. Chem. Rev. 2022;122:7269–7326. doi: 10.1021/acs.chemrev.1c00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rose R.J., Damoc E., Denisov E., Makarov A., Heck A.J. High-sensitivity orbitrap mass analysis of intact macromolecular assemblies. Nat. Methods. 2012;9:1084–1086. doi: 10.1038/nmeth.2208. [DOI] [PubMed] [Google Scholar]
- 31.Čaval T., Heck A.J.R., Reiding K.R. Meta-heterogeneity: evaluating and describing the diversity in glycosylation between sites on the same glycoprotein. Mol. Cell. Proteomics. 2021;20 doi: 10.1074/mcp.R120.002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Čaval T., Tian W., Yang Z., Clausen H., Heck A.J.R. Direct quality control of glycoengineered erythropoietin variants. Nat. Commun. 2018;9:3342. doi: 10.1038/s41467-018-05536-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu D., Robinson C.V. Understanding glycoprotein structural heterogeneity and interactions: insights from native mass spectrometry. Curr. Opin. Struct. Biol. 2022;74 doi: 10.1016/j.sbi.2022.102351. [DOI] [PubMed] [Google Scholar]
- 34.Savaryn J.P., Catherman A.D., Thomas P.M., Abecassis M.M., Kelleher N.L. The emergence of top-down proteomics in clinical research. Genome Med. 2013;5:53. doi: 10.1186/gm457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leney A.C., El Atmioui D., Wu W., Ovaa H., Heck A.J.R. Elucidating crosstalk mechanisms between phosphorylation and O-GlcNAcylation. Proc. Natl. Acad. Sci. U. S. A. 2017;114:E7255–e7261. doi: 10.1073/pnas.1620529114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lössl P., Brunner A.M., Liu F., Leney A.C., Yamashita M., Scheltema R.A., et al. Deciphering the Interplay among Multisite phosphorylation, interaction dynamics, and conformational transitions in a tripartite protein system. ACS Cent. Sci. 2016;2:445–455. doi: 10.1021/acscentsci.6b00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deslignière E., Diemer H., Erb S., Coliat P., Pivot X., Detappe A., et al. A combination of native LC-MS approaches for the comprehensive characterization of the antibody-drug conjugate trastuzumab deruxtecan. Front. Biosci. (Landmark Ed.) 2022;27:290. doi: 10.31083/j.fbl2710290. [DOI] [PubMed] [Google Scholar]
- 38.Marty M.T., Baldwin A.J., Marklund E.G., Hochberg G.K.A., Benesch J.L.P., Robinson C.V. Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to Polydisperse ensembles. Anal. Chem. 2015;87:4370–4376. doi: 10.1021/acs.analchem.5b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neelamegham S., Aoki-Kinoshita K., Bolton E., Frank M., Lisacek F., Lütteke T., et al. Updates to the Symbol nomenclature for glycans guidelines. Glycobiology. 2019;29:620–624. doi: 10.1093/glycob/cwz045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luijkx Y.M.C.A., Henselijn A.J., Bosman G.P., Cramer D.A.T., Giesbers K.C.A.P., van 't Veld E.M., et al. Detection of bacterial α-l-Fucosidases with an ortho-quinone methide-based probe and mapping of the probe-protein adducts. Molecules. 2022;27:1615. doi: 10.3390/molecules27051615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Henderson C.M., Shulman N.J., MacLean B., MacCoss M.J., Hoofnagle A.N. Skyline Performs as well as Vendor software in the Quantitative analysis of serum 25-Hydroxy Vitamin D and Vitamin D binding Globulin. Clin. Chem. 2018;64:408–410. doi: 10.1373/clinchem.2017.282293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bodenhofer U., Bonatesta E., Horejš-Kainrath C., Hochreiter S. msa: an R package for multiple sequence alignment. Bioinformatics. 2015;31:3997–3999. doi: 10.1093/bioinformatics/btv494. [DOI] [PubMed] [Google Scholar]
- 43.Tagliabracci V.S., Wiley S.E., Guo X., Kinch L.N., Durrant E., Wen J., et al. A single kinase generates the majority of the secreted phosphoproteome. Cell. 2015;161:1619–1632. doi: 10.1016/j.cell.2015.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tagliabracci V.S., Engel J.L., Wen J., Wiley S.E., Worby C.A., Kinch L.N., et al. Secreted kinase phosphorylates extracellular proteins that regulate biomineralization. Science. 2012;336:1150–1153. doi: 10.1126/science.1217817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barlati S., De Petro G., Bona C., Paracini F., Tonelli M. Phosphorylation of human plasminogen activators and plasminogen. FEBS Lett. 1995;363:170–174. doi: 10.1016/0014-5793(95)00312-w. [DOI] [PubMed] [Google Scholar]
- 46.Wang H., Prorok M., Bretthauer R.K., Castellino F.J. Serine-578 is a major phosphorylation locus in human plasma plasminogen. Biochemistry. 1997;36:8100–8106. doi: 10.1021/bi970328d. [DOI] [PubMed] [Google Scholar]
- 47.Cramer D.A.T., Franc V., Caval T., Heck A.J.R. Charting the proteoform landscape of serum proteins in individual donors by high-resolution native mass spectrometry. Anal. Chem. 2022;94:12732–12741. doi: 10.1021/acs.analchem.2c02215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takada Y., Makino Y., Takada A. Glu-plasminogen I and II: their activation by urokinase and streptokinase in the presence of fibrin and fibrinogen. Thromb. Res. 1985;39:289–296. doi: 10.1016/0049-3848(85)90224-5. [DOI] [PubMed] [Google Scholar]
- 49.Gonzalez-Gronow M., Gawdi G., Pizzo S.V. Tissue factor is the receptor for plasminogen type 1 on 1-LN human prostate cancer cells. Blood. 2002;99:4562–4567. doi: 10.1182/blood.v99.12.4562. [DOI] [PubMed] [Google Scholar]
- 50.Wiman B., Collen D. Molecular mechanism of physiological fibrinolysis. Nature. 1978;272:549–550. doi: 10.1038/272549a0. [DOI] [PubMed] [Google Scholar]
- 51.Holvoet P., Lijnen H.R., Collen D. A monoclonal antibody specific for Lys-plasminogen. Application to the study of the activation pathways of plasminogen in vivo. J. Biol. Chem. 1985;260:12106–12111. [PubMed] [Google Scholar]
- 52.Fredenburgh J.C., Nesheim M.E. Lys-plasminogen is a significant intermediate in the activation of Glu-plasminogen during fibrinolysis in vitro. J. Biol. Chem. 1992;267:26150–26156. [PubMed] [Google Scholar]
- 53.Smith J.H., Morris J.P., Chibber B.A., Castellino F.J. The role of the lysine binding sites of human plasminogen in the fibrinogen stimulated rate of active site formation in the streptokinase-plasminogen equimolar complex. Thromb. Res. 1984;34:499–506. doi: 10.1016/0049-3848(84)90254-8. [DOI] [PubMed] [Google Scholar]
- 54.Seok S.H. Structural Insights into protein regulation by phosphorylation and substrate recognition of protein Kinases/Phosphatases. Life (Basel) 2021;11:957. doi: 10.3390/life11090957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang X., Lin X., Loy J.A., Tang J., Zhang X.C. Crystal structure of the catalytic domain of human plasmin complexed with streptokinase. Science. 1998;281:1662–1665. doi: 10.1126/science.281.5383.1662. [DOI] [PubMed] [Google Scholar]
- 56.McArthur J.D., Cook S.M., Venturini C., Walker M.J. The role of streptokinase as a virulence determinant of Streptococcus pyogenes--potential for therapeutic targeting. Curr. Drug Targets. 2012;13:297–307. doi: 10.2174/138945012799424589. [DOI] [PubMed] [Google Scholar]
- 57.Perez-Riverol Y., Bai J., Bandla C., García-Seisdedos D., Hewapathirana S., Kamatchinathan S., et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022;50:D543–D552. doi: 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (57) partner repository with the dataset identifier PXD042225. Reviewer username: reviewer_pxd042225@ebi.ac.uk; password: RZzDSXwz