

Abstract
Chronic hepatitis B virus (HBV) infection remains a global health problem due to the lack of treatments that prevent viral rebound from HBV covalently closed circular (ccc)DNA. In addition, HBV DNA integrates in the human genome, serving as a source of hepatitis B surface antigen (HBsAg) expression, which impairs anti-HBV immune responses. Cytosine base editors (CBEs) enable precise conversion of a cytosine into a thymine within DNA. In this study, CBEs were used to introduce stop codons in HBV genes, HBs and Precore. Transfection with mRNA encoding a CBE and a combination of two guide RNAs led to robust cccDNA editing and sustained reduction of the viral markers in HBV-infected HepG2-NTCP cells and primary human hepatocytes. Furthermore, base editing efficiently reduced HBsAg expression from HBV sequences integrated within the genome of the PLC/PRF/5 and HepG2.2.15 cell lines. Finally, in the HBV minicircle mouse model, using lipid nanoparticulate delivery, we demonstrated antiviral efficacy of the base editing approach with a >3log10 reduction in serum HBV DNA and >2log10 reduction in HBsAg, and 4/5 mice showing HBsAg loss. Combined, these data indicate that base editing can introduce mutations in both cccDNA and integrated HBV DNA, abrogating HBV replication and silencing viral protein expression.
Keywords: MT: RNA/DNA Editing, HBV, cccDNA, base editing, HBsAg, therapeutics, gene editing, lipid nanoparticles
Graphical abstract
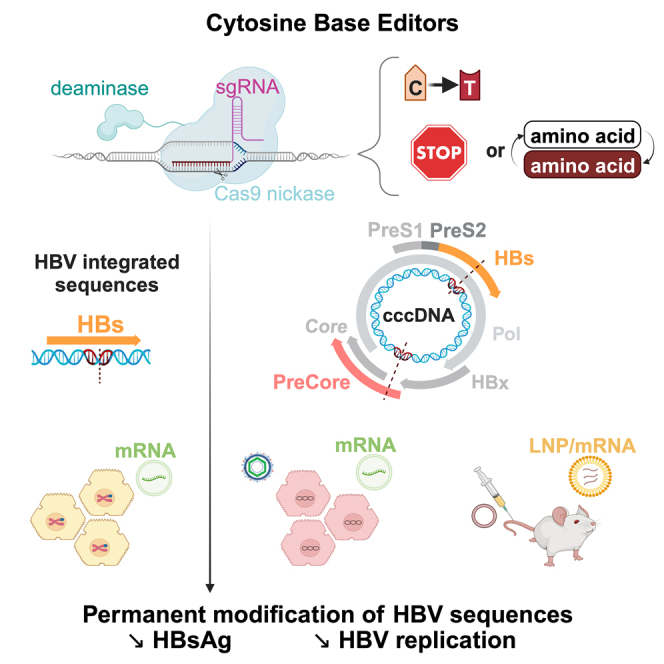
Zoulim and colleagues demonstrate in cell culture and in preclinical mouse models that hepatitis B virus (HBV) replication and antigen production can be durably abrogated by the use of base editing technology to introduce permanent mutations in the HBV genome, opening new perspectives toward a cure for chronic HBV infection.
Introduction
Chronic hepatitis B (CHB) remains a global health problem with more than 250 million people infected with hepatitis B virus (HBV) worldwide.1,2,3 It is estimated that 30% of patients develop hepatocellular carcinoma and cirrhosis, which leads to 800,000 deaths per year.4,5 The HBV genome is maintained in the hepatocyte nucleus as a 3.2-kb episomal covalently closed circular DNA (cccDNA), which is the source of continuous viral replication.6,7 Although standard-of-care antivirals (nucleoside or nucleotide analogs [NAs]) efficiently inhibit viral replication, they do not affect cccDNA, which persists in the liver during the lifetime of a patient, preventing CHB from being cured.5 In addition to being a persistent reservoir of cccDNA, HBV DNA sequences are known to integrate into the human genome. These copies of integrated HBV DNA can serve as a template for up to 80% of HBs gene transcripts in the late phase of infection.8 The resulting expression of hepatitis B surface antigen (HBsAg) impairs host immune responses against the virus and contributes to the persistence of HBV.8,9,10 Therefore, novel classes of drugs that could inactivate both cccDNA and integrated HBV DNA are needed to enable a cure for CHB.7
Gene editing technologies have the potential to directly target and inactivate both of the aforementioned viral DNA species. Although nuclease gene editing strategies have been shown to reduce cccDNA levels within in vitro and in vivo models,11,12 one concern with this approach is the generation of double-strand break (DSB) intermediates.13 Given that HBV DNA can integrate at multiple locations in the human genome within a single cell, gene editing with a nuclease targeting an HBV DNA sequence could simultaneously generate DSBs at multiple genomic loci, leading to undesirable genomic rearrangements.14,15 Unlike nucleases, base editors perform a chemical reaction, deamination, thus converting one nucleotide into another without the need for a DSB intermediate.16,17 Base editing enables multiple edits in a single cell with high efficacy and minimal genomic rearrangements, compared to CRISPR-Cas9.18 Cytosine base editors (CBEs) convert cytosine into thymine, enabling gene silencing through the introduction of the stop codons, an approach that previously showed promise for targeting HBV DNA in cell models.16,19,20 Although previous studies19,20 demonstrated base editing of HBV sequences and silencing of viral gene expression, the models that were used were exclusively in vitro systems unsuitable to assess base editing on an established cccDNA pool characteristic of CHB.
In this study, we have identified a combination of two guide RNAs (gRNAs), that, when paired with a CBE, inactivated both cccDNA and integrated HBV DNA in relevant HBV cell models, including HBV-infected HepG2-NTCP, HBV-infected primary human hepatocytes (PHHs), as well as HepG2.2.15 and PLC/PRF/5 cell lines with artificially and naturally integrated HBV DNA, respectively. Furthermore, for the first time, we have shown durable antiviral efficacy, including HBsAg loss, in vivo in the HBVcircle mouse model using lipid nanoparticulate (LNP) delivery of base editing reagents (mRNA/gRNA). Combined with a thorough evaluation of gRNA-dependent off-target effects, these data advance our understanding of the potential of base editing to enable a functional cure for chronic HBV infection.
Results
HBV gRNA design and screen
HBV genotype D (subgenotype ayw) was used for gRNA design because this is a common genotype used in many cellular and animal models of HBV infection, and therefore, extensive sequence data are available.21 Two different strategies were used to design gRNAs. In the first strategy, we used the Benchling CRISPR tool22 to identify 33 gRNAs that, when paired with a prototypical CBE, BE4, would be expected to introduce stop codons in the four main HBV genes: Polymerase, HBs, (Pre)Core, and X. In the second strategy, we identified the gRNAs exhibiting high target site conservation across HBV isolate sequences in the HBVdb database23 that, in combination with the cytosine base editor, were predicted to introduce missense mutations in HBV genes (Tables S1 and S2) if C:G to T:A substitutions were made within the editing window of BE4. To test the editing efficacy of the gRNAs, we generated HEK293T cell lines with integrated HBV DNA sequences. Briefly, HEK293T cells were transduced with a lentivirus carrying HBV DNA sequences 2309–1622 (HBs, Pol) or 1176–2451 (X, Core). The resulting cell lines were transfected with the two plasmids encoding base editing reagents: BE4 and a gRNA. The gRNAs were then ranked based on two factors: (1) the rate of editing outcomes encoding stop codons or missense mutations as assessed by next-generation sequencing (NGS) and (2) the conservation across HBV genotypes (Tables S1 and S2).
Base editing with selected gRNAs suppresses HBV viral parameters in de novo infected HepG2-NTCP cells
We examined the effect of BE4 with the six selected gRNAs (Figure S1A) on HBV parameters in HBV-infected HepG2 cells stably expressing the hNTCP receptor (i.e., HepG2-NTCP cells).24 RNA transfection methods have proven to be more efficient than DNA transfection and less toxic for cells.25 Therefore, RNA transfection was chosen to test selected gRNAs in HBV-infected HepG2-NTCP cells. Protocols were optimized to efficiently deliver BE4 mRNA and gRNAs to study the effect of editing on the replicative ability of de novo established cccDNA (Figures 2A and S1B).26 Levels of extracellular HBsAg and HBe antigen (HBeAg) were measured in supernatants by ELISA, and total HBV DNA and 3.5-kb RNA were quantified intracellularly using quantitative PCR (qPCR) and quantitative reverse transcription-PCR (qRT-PCR), respectively. Several of these treatment groups exhibited a reduction in selected HBV viral markers (Figures S1C–S1F). In particular, gRNAs g37 (designed to introduce a stop codon in the HBs gene) and g40 (designed to introduce a stop codon in the Precore gene) (Figure 1A) drastically reduced HBsAg and HBeAg, respectively (Figures 2B, 2C, S1C, and S1D). Interestingly, inhibition of total HBV DNA and 3.5-kb RNA upon treatment with g37 and g40 was also observed (Figures 2D, 2E, S1E, and S1F). Furthermore, to increase the suppression of all four viral parameters, we combined the two gRNAs (g37 + g40) and successfully achieved the reduction of HBsAg, HBeAg, total HBV, and 3.5-kb RNA (Figures 2B–2E). The reduction in 3.5-kb RNA was confirmed by northern blot, along with a reduction in 2.4-kb and 2.1-kb HBs mRNAs, although the effect was less pronounced for these shorter HBV mRNA species (Figure S2A). At the intracellular protein level, a decrease in all three HBs isoforms (L, M, and S) upon g37 + g40 treatment was demonstrated by western blot (Figure S2B).
Figure 2.

Effect of CBE and gRNAs g37, g40, or their combination on HBV extracellular and intracellular parameters in HepG2-NTCP cells
(A) Experimental scheme. A protocol similar to Figure S1B was used with a few modifications. All of the samples were collected at 15 dpi. (B and C) Extracellular HBsAg and HBeAg were measured by ELISA. (D) Total HBV DNA was quantified by quantitative (qPCR) from DNA extracted from cell lysates. (E) Total cellular RNA was extracted and HBV 3.5-kb RNA levels were quantified by quantitative reverse transcription-PCR (qRT-PCR). Data were normalized to PCSK9 control gRNA targeting Proprotein convertase subtilisin/kexin type 9 (gene unrelated to HBV). (F) cccDNA level was assessed by qPCR on the DNA samples pretreated with ExoI/III in HepG2-NTCP. (G) Level of the C>T functional editing that leads to the introduction of the stop codons in HBs and Precore genes, assessed by NGS on ExoI/III-treated cccDNA samples from HepG2-NTCP, as well as PCSK9 (assessed on total DNA). Data are represented as mean ± SEM for n = 4 to 6 replicates.
Figure 1.

cccDNA organization with the location of the 2 selected gRNAs g37 and g40 and gRNA/targeted sequences
The edited nucleotides and resulting amino acid changes are highlighted in red. CBE/g37 editing of the targeted cytosines C7 and/or C8 leads to the introduction of the stop codon in HBs; CBE/g40 editing of the cytosine C8 and/or C9 results in the stop codon in the Precore gene.
HBV DNA intermediates (e.g., cccDNA, protein-free-relaxed circular [rc]DNA, rcDNA) share high sequence similarity, and gRNA/BE4 complexes could target any of these DNA species, if the recognition sequence is present as double-stranded DNA (dsDNA). We therefore investigated the direct impact of gRNA/BE4 editing on cccDNA by pretreating infected cells with 2′,3′-dideoxy-3′-thiacytidine (3TC, or lamivudine), which reduced the amount of viral DNA replicative intermediates with respect to cccDNA at the time of base editing (Figure S3A). Similar to the 3TC untreated condition (as shown in Figure 2), a reduction in viral parameters was observed after the transfection with BE4 and g37, g40, or (g37 + g40) (Figures S3B–S3F), and no difference in BE4 expression was observed in 3TC-untreated or -treated conditions (Figure S3G). These results strongly suggest that the reduction in viral replicative parameters is a consequence of base editing direct effect on cccDNA.
We next assessed cccDNA levels by two methods: cccDNA-specific qPCR on the total DNA samples treated with ExoI/III nucleases (Figures 2F and S3H) and Southern blot analysis on Hirt extracted samples followed by ExoI/III digestion (Figure S4A) to decrease the levels of HBV replicative intermediates other than cccDNA.27 Densitometry analysis of Southern blot bands and qPCR consistently showed no difference in cccDNA levels in edited samples.
We further performed NGS on total DNA samples treated with ExoI/III nucleases to assess cccDNA editing rates that led to the introduction of the Stop codons in HBs and Precore genes in the presence or absence of 3TC. Treatment of HBV-infected HepG2-NTCP cells with BE4/g37 led to a C7T edit, successfully introducing W156Stop amino acid change in the HBs gene. Similarly, treatment with BE4/g40 led to a C8T edit, resulting in a W28Stop change in the Precore gene. Multiplexing BE4 and gRNAs g37 + g40 led to the editing of both C7T (HBs) and C8T (Precore) sites (Figure 2G). High levels of editing at all of the positions were maintained in the case of the combinatorial treatment with 3TC (Figure S3I). NGS on the Hirt extracted samples followed by ExoI/III digestion that were analyzed by Southern blotting (Figure S4A) also confirmed a very high editing efficiency in the presence or absence of 3TC (48%–70% C7T for the HBs gene and 60%–70% for the Precore gene) (Figure S4B).
Taken together, these data demonstrate that BE4 mediates the inhibition of HBV replication and viral antigen production in infected HepG2-NTCP cells. Combination with 3TC showed that this inhibition is maintained in the context of reduced replicative intermediates, suggesting direct base editing and introduction of stop codons in cccDNA.
Antiviral efficacy of base editing in HBV-infected PHHs
PHHs isolated from chimeric mouse liver allow the maintenance of hepatocyte differentiation and HBV replication for up to 30 days.28,29 These PHHs were infected with HBV for at least 4 days to allow the generation of a stable pool of cccDNA before transfection. Two consecutive transfections with mRNA and gRNA (days 5 and 12 after HBV infection) were performed to increase efficacy. After the last transfection, PHHs were kept in culture for 2 weeks, and the experiment terminated 25 days postinfection (Figure 3A). Consistent with the data in HepG2-NTCP cells, transfection with BE4 and individual gRNAs led to a reduction in the respective viral markers, whereas the combination of BE4 with both g37 and g40 resulted in a simultaneous reduction in HBsAg, HBeAg, intracellular HBV total DNA, and 3.5-kb RNA (Figures 3B and S5A). The albumin expression level was similar in the control and treatment conditions, confirming that PHH functionality was not compromised by HBV-targeting gRNAs (Figure S5B). We have further assessed extracellular HBV DNA load in the supernatant over the course of the experiment. In this case, the treatment with BE4/(g37 + g40) was compared to the temporary 3TC treatment. As expected, 3TC efficiently reduced HBV replication; however, after treatment discontinuation, HBV DNA levels increased, indicating viral rebound. Contrary to 3TC, there was no rebound in the case of treatment with BE4/(g37 + g40) (Figure 3C), suggesting that base editing efficiently reduces HBV replication and prevents viral rebound.
Figure 3.

Antiviral efficacy of base editing in HBV-PHH
(A) Experimental scheme. (B) Multiplexing the 2 gRNAs with BE4 simultaneously reduces HBsAg, HBeAg, 3.5-kb RNA, and total HBV DNA. Viral parameters assessed at the end of the experiment, day 25 postinfection. BE4 with the PCSK9 gRNA was used as a control for normalizing the data. (C) HBV replication assessed by HBV DNA qPCR in PHH supernatant. Discontinuation of 3TC leads to HBV rebound, whereas base editing prevents viral rebound. (D) cccDNA level was assessed by qPCR on the DNA samples pretreated with ExoI/III. (E) Level of the C>T functional editing that leads to the introduction of the stop codons in HBs and Precore genes, assessed by NGS on ExoI/III treated cccDNA samples as well as PCSK9 (assessed on total DNA). Data are represented as mean ± SEM for n = 3.
BE4 protein expression from the transfected mRNA was transient and could not be detected by 24 h posttransfection (Figure S5C), suggesting that the observed antiviral efficacy resulted from permanent changes in HBV cccDNA rather than transcriptional interference of the BE4 protein with the viral genome.
Similar to HepG2-NTCP, the cccDNA level did not change in HBV-infected PHHs after treatment with the base editing reagents (Figure 3D). In PHHs, two transfections with BE4/(g37 + g40) resulted in 59% cccDNA editing at the g37 site (HBs) and 81% cccDNA editing at the g40 site (Precore) (Figure 3E).
The expression of cellular deaminases is known to generate uracils within cccDNA that are processed by uracil glycosylase into abasic sites, ultimately leading to cccDNA degradation.30 Although CBEs may also generate uracil intermediates within cccDNA, BE4 contains a uracil glycosylase inhibitor (UGI) domain, which inactivates base excision repair and thus increases the efficiency of the base editing.16 To assess whether base editing could promote uracil glycosylase-induced cccDNA degradation, we transfected the BE4 base editor without UGI (BE4_noUGI) with gRNAs 37 and 40 in HBV-infected PHHs. Similar to BE4, BE4_noUGI with g37 or g40 reduced HBV viral parameters and enabled robust cccDNA editing, but it did not affect cccDNA levels (Figures S5D and S5E). This result supports the concept that the antiviral efficacy of base editing primarily functions through the introduction of nucleotide changes within cccDNA and not through any changes in cccDNA stability, at least under tested PHH in vitro experimental conditions.
We have further assessed the editing and antiviral efficacy of the next-generation cytosine base editors BE4-PpAPOBEC1 and CBE-T. They display high on-target activity, but at the same time exhibit minimal guide-independent off-target effects associated with cytosine deamination on cellular RNA and genomic DNA.31,32 Two transfections with the combination of gRNAs (g37 + g40) and one of the editors (BE4, BE4-PpAPOBEC1, or CBE-T) were performed in HBV-infected PHHs. Both BE4-PpAPOBEC1 and CBE-T were efficient in reducing the four assessed viral markers: HBsAg, HBeAg, 3.5-kb RNA, and HBV DNA (Figure S6A). BE4-PpAPOBEC1 and CBE-T were less efficient than BE4 in editing both HBs (46% and 36%, respectively) and Precore (62% and 28%). The level of editing correlated with the level of HBsAg and HBeAg reduction (Figures S6A and S6B). These results show the broad applicability of different CBEs to directly edit the HBV genome and inhibit viral replication and antigen production.
CBE inhibits HBsAg expression from integrated HBV in vitro
Integrated HBV DNA is a source of HBsAg, which could represent most of the antigen production in late stages of CHB.8,33,34 Therefore, we next examined whether HBs targeting gRNA, g37, and the combination (g37 + g40) were able to suppress the expression of HBsAg from integrated HBV genomes. For this, we used HepG2.2.15 cells harboring artificially integrated replication competent dimeric HBV genomes,35 which were transfected with BE4 mRNA and g37 (Figure 4A). These cells were treated with 3TC to reduce the abundance of HBV DNA replicative intermediates that could compete as base editor substrates. On day 6 after transfection, a strong decline in extracellular and intracellular HBsAg protein levels was observed in HepG2.2.15 cells treated with BE4 and g37 or (g37 + g40) (Figures 4B and S7). Base editing efficiency was assessed by NGS of extracted genomic DNA, which revealed 60% C-to-T editing at the HBs target site in the samples treated with either g37 or (g37 + g40) (Figure 4D). Consistent with the results in HBV-infected HepG2-NTCP, targeting the integrated Precore gene with g40 resulted in a reduction in the extracellular HBeAg level in HepG2.2.15 cells, along with ∼70% C8T edit introducing the premature stop codon in Precore (Figures 4C and 4D).
Figure 4.

Anti-HBV efficacy of CBE and gRNAs in HBV-integrated cell lines
(A) Schematic representation of the protocol used for HepG2.2.15 cells. All of the samples were collected 6 days posttransfection (dpt). (B and C) Extracellular HBsAg and HBeAg levels were assessed in the supernatant of the cells treated with 3TC 4 days before the transfection with base editing reagents. (D) Level of the C>T functional editing was assessed by NGS on the purified DNA. (E) Schematic representation of the protocol used for PLC/PRF/5 cells. (F) At 6 dpt, extracellular HBsAg was measured in the supernatants of the cells transfected with g37-PLC (g37 adapted for genotype A HBs targeting site within PLC/PLF/5 cells). (G) The level of the C>T editing on the HBs targeting site of g37-PLC was assessed by NGS. All of the ELISA data were normalized to the BE4/PCSK9 (control gRNA) condition. Error bars indicate SEM of 4 or 6 replicates.
In addition, PLC/PRF/5 cells36,37 were used to test the efficiency of CBE to inactivate HBsAg from replication-incompetent naturally integrated HBV genotype A DNA sequences (Figure 4E). Upon Sanger sequencing, we observed a mismatch in the g37 binding site in the genotype A HBs gene compared to the ayw genotype D of HepG2.2.15. Therefore, a gRNA compatible with the HBV sequence in PLC/PRF/5, represented as g37-PLC, was designed. Similar to the effect of g37 in HepG2.2.15, g37-PLC reduced the amount of secreted HBsAg (Figure 4F). A rate of 45% editing was sufficient to observe a robust anti-HBs effect in this cellular model (Figure 4G). We thus demonstrated that the introduction of stop codons by cytosine base editing inhibits HBsAg expression produced from the integrated HBV sequences in cell lines with either artificially or naturally integrated HBV DNA.
Base editing leads to sustained reduction of HBV viral markers in vivo
To test antiviral efficacy of base editing in vivo, we used the HBV minicircle mouse model. This in vivo model supports persistent HBV replication and expression of viral antigens resulting from hydrodynamic injection (HDI) with a cccDNA-like plasmid.38 Four weeks after HDI, the mice secreting HBsAg were organized into four groups. Hepatic delivery of base editing reagents was achieved via the systemic administration of the LNPs formulated with mRNA encoding BE4 and control PCSK9 gRNA39 or HBV-targeting gRNAs (g37 + g40). Mice received one intravenous injection of LNPs; after the injection, serum HBsAg, HBV DNA, and HBeAg levels were assessed weekly. Entecavir (ETV)-treated mice received antiviral alone orally for 2 weeks, and then the treatment was discontinued (Figure 5A). Six weeks after the beginning of the treatment, we detected more than 2log10 mean serum HBsAg reduction in the mice treated with BE4/(g37 + g40); 4 out of 5 mice showed HBsAg reduction below the limit of detection (Figures 5B, S8A, and S8B). Treatment with HBV-targeting base editing reagents further led to a sustained reduction in serum HBV DNA with no HBV viral rebound observed, compared to the ETV group, in which serum HBV DNA was reduced following administration, but rebounded after ETV treatment was discontinued (Figures 5C, S8C, and S8D). Two weeks after treatment, loss of expression of the viral marker HBeAg was observed in all of the mice that received HBV-targeting base editing reagents (Figures 5D, S8E, and S8F). Six weeks after the beginning of treatment, the study was terminated, and total HBV DNA level was assessed in mice livers. Compared to controls, there was a decrease in the HBV DNA amount in BE4/gRNAs(37 + 40)-treated mice (Figure 5E). NGS showed that in vivo BE4/g37 introduced stop codon W156∗ in the HBs gene with approximately 30% efficacy; BE4/g40 introduced stop codon W28∗ in the Precore gene with 42% efficacy (Figure 5F). Taken together, this is the first demonstration of LNP-mediated delivery of base editing reagents targeting HBV sequences, showing a sustained reduction in HBV parameters in vivo.
Figure 5.

LNP-mediated delivery of base editor and HBV-targeting gRNAs leads to sustained reduction of viral markers in HBV minicircle mouse model
(A) Experimental scheme. (B) Seven weeks after the injection (day 0), mice injected with BE4/(g37 + g40) showed > 2log10 mean serum HBsAg reduction; 4/5 mice injected with HBV-specific LNPs showed HBsAg reduction below the limit of detection. (C) HBV replication is reduced in ETV-treated mice and then rebounds when the treatment is discontinued at day 14 (positive control). Base editing treated mice showed up to 3log10 sustained reduction in serum HBV DNA with no rebound observed. (D) Two weeks after the beginning of treatment, all mice treated with BE4/(g37 + g40) showed HBeAg below the limit of detection. (E) Total HBV DNA levels were assessed on the DNA purified from mice liver at the end of the experiment. (F) Editing was assessed on the DNA purified from mice liver by NGS. The data are represented as mean ± SEM, n = 4 or 5 mice per group.
Off-target editing assessment
To evaluate gRNA-dependent off-target effects, we performed RNase H-dependent amplification and sequencing (rhAmpSeq) analysis40,41 of the in silico predicted off-target sites on the DNA samples derived from HBV-infected PHH transfected with the gRNAs (g37 + g40) and cytosine base editors BE4, BE4-PpAPOBEC1, and CBE-T. Briefly, guide-dependent off-target candidates are identified in silico by running Cas-OFFinder42 on the GRCh38/hg38 reference genome using the protospacer and NRR PAM specificity as input. Candidate off-target sites were stratified by their genomic location and PAM sequence. All of the sites with ≤3 mismatches to the on-target locus were included in the rhAmpSeq panel for evaluation in edited cells. Candidate off-target sites with 4–7 mismatches from the on-target that overlapped annotated exons or were within 100 bp of an exon were also included. In this study, we have screened 499 and 685 potential off-targets in rhAmp-seq panels of g40 and g37, respectively. Each base in a candidate off-target site was compared between treated and untreated samples. A Fisher’s exact test was used to generate a significance score for the enrichment of off-target edits in treated cells when compared to untreated cells.
The number of the identified off-target sites correlated with the level of on-target editing (BE4 > BE4-PpAPOBEC1 > CBE-T) (Table S3). Each off-target site was further characterized by its genomic location (Table S4). Standard BE4 base editor with g37 and g40 resulted in 19 and 7 off-target sites, respectively. The majority of the off-target sites were located in the noncoding regions of the genome, namely intronic and intergenic regions, except for the two sites: [g37] in the BCL7A gene (nonsynonymous mutation with 0.45% off-target editing) and [g40] in the long noncoding RNA gene AC131254.1. Next-generation CBE BE4-PpAPOBEC1 exhibited a more advantageous off-target profile (8 sites with g37 and 1 site with g40). CBE-T treatment was associated with the lowest off-target editing: HBs/Pol-targeting g37 yielded two off-target sites in the noncoding regions of the genome, and none were identified for the Precore-targeting g40.
Discussion
Currently available NA therapies, approved for CHB patients, suppress HBV replication but do not target HBV cccDNA or HBs expression from the integrated DNA sequences and hence do not cure CHB. The persistence of highly stable HBV cccDNA in infected hepatocytes leads to viral relapse upon the discontinuation of therapy, and therefore lifelong NA treatment is necessary. Undoubtedly, there is an urgent need for HBV therapeutics that target the HBV genomic reservoir (cccDNA) and integrated HBV DNA to avoid the need for long-term treatment.1,5,7
In recent years, several research groups applied CRISPR-Cas9 nuclease technology to eliminate intrahepatic HBV genomes.11,13,26 However, this approach suffers from several limitations. For example, DSBs generated upon dsDNA cleavage by wild-type Cas9 endonuclease can lead to host genomic instability. Moreover, upon multiplexing, shorter episomal cccDNA variants could be generated that remain transcriptionally active.26 In this study, we demonstrate the potential of base editing technology to irreversibly silence cccDNA and integrated HBV DNA, which was associated with the reduction/loss of the viral markers.
We have examined the utility of cytosine base editors (BE4) for the inactivation of HBV cccDNA and integrated DNA templates. To do this, we screened 40 different gRNAs and identified a combination of 2 that enabled simultaneous reduction of all 4 tested HBV markers (HBsAg, HBeAg, HBV DNA, and 3.5-kb RNA) in HepG2-NTCP, as well as in PHHs. We further showed that BE4/(g37 + g40)-mediated editing of HBV genomes reduced not only HBV replication in HepG2-NTCP and PHHs but also HBsAg expression in HepG2.2.15 and PLC/PRF/5 cells, which harbor artificially and naturally integrated HBV DNA, respectively.
The high rates of base editing observed in samples from which HBV DNA replicative intermediates were removed using nucleases digestion27 suggest that base editing antiviral efficacy is mediated by the editing of cccDNA. To further test this hypothesis, infected HepG2-NTCP cells were treated with 3TC, which decreases the abundance of replicative intermediates, leaving predominantly cccDNA as a substrate for base editing. The reduction in viral parameters and even higher editing rates under this experimental condition demonstrates that cccDNA can be directly targeted by BE4. Furthermore, in clinical trials novel anti-HBV therapeutics are often added to the standard regimen of approved NA antivirals, such as 3TC.1 Therefore, it is encouraging that base editing efficacy in a relevant HBV cell model was not compromised when combined with 3TC treatment.
Two previous studies have shown the potential of cytosine base editing to target HBV genomes; however, these studies used the lentiviral-based delivery of base editors.19,20 Lentiviral transduction with the base editing reagents performed before infection does not allow the establishment of cccDNA, and therefore has limited relevance to CHB. Here, we demonstrated the delivery of gRNAs and an mRNA encoding a cytosine base editor in infection models with an established cccDNA pool, including in vivo in an HBV mouse model.
As a delivery method, we have used LNPs. This delivery strategy enables transient protein expression from short-lived mRNA,43 limiting exposure of the genome to the gene/base editor, which is associated with a more favorable off-target profile.44,45,46 In this study, we examined for the first time in vivo efficacy of the base editing approach for inhibiting viral replication in an HBV minicircle mouse model. This immunocompetent mouse model supports high levels of HBV replication and persistence. Contrary to HBV AAV mice, HBV replication in this model is driven directly from the cccDNA-like viral genomes. Hence, it is more physiologically relevant to test the efficacy of cccDNA targeting drugs or cccDNA-related processes.38 Furthermore, HBV replication recovery after the discontinuation of ETV showed that HBVcircle mice enabled rebound of the virus under the analyzed experimental conditions, indicating the relevance of the model. Contrary to ETV, the LNP-mediated hepatocellular delivery of BE4 mRNA and (g37 + g40) led to a sustained reduction of not only serum HBV DNA but also secreted HBsAg in mouse serum for 6 weeks after LNP injection. For secreted HBeAg, we observed a drastic reduction below the limit of detection at an earlier time point (2 weeks postinjection). This dramatic reduction in viral antigens was accompanied by a partial editing of the cccDNA pool (30% and 40% for g37 and g40 target regions, respectively), which could be explained by the fact that editing would be targeting preferentially the transcriptionally active cccDNA molecules, which are more accessible to the editor.47 Because HBsAg and HBeAg play important roles in establishing and maintaining chronic infection through immunomodulatory effects, this approach could have additional benefits by restoring a more active immune environment in patients.48 The sustained reductions in viral markers after a single administration of base editing reagents in this model reinforces the unique and potentially advantageous mechanism of gene editing relative to other modalities such as small interfering RNA or antisense oligonucleotides, which require repeated administrations.49,50
Due to the overlapping nature of the HBV genome, nonsense mutation in an open reading frame (ORF) can introduce missense mutations in another ORF and/or affect a regulatory region important for viral gene expression/replication. For example, base editing with g40 not only results in W28∗ in precore but also introduces mutations into the epsilon encoding region. Because epsilon plays an important role in pregenomic RNA (pgRNA) encapsidation,51 it is possible that mutating this site affects pgRNA packaging and reduces HBV replication, as observed in the present study. Similarly for g37, the W156∗ mutation in HBs also corresponds to a G500N amino acid substitution in the polymerase. Although the effect of this mutation on polymerase function has not been reported, mutating G500 could influence rcDNA synthesis because it is located in the RT domain of the polymerase. A previous report by Melegari et al.. showed that a mutant carrying F501L (located next to G500) is replication defective.52 In any case, the enveloped HBV nucleocapsids would not be released from the cells treated with g37 + g40 in the absence of HBs proteins.
Our most surprising result was the reduction of 3.5-kb RNA (pgRNA) levels with g37 and g40 in vitro. We do not have a clear explanation for this observation, but it could be due to the impact of the mutations on viral transcription or transcript stability/degradation via nonsense-mediated RNA decay, as previously suggested for HBs RNA.20 This warrants further studies exploring in-depth mechanistic insights. However, from the therapeutic point of view, this decrease in pgRNA is beneficial because it would contribute to reduced HBV DNA replication.
Although we have generated promising results in various models with prototypical cytosine base editor BE4, we have also demonstrated the antiviral efficacy of gRNAs (g37 + g40) in combination with the next-generation cytosine base editors BE4-PpAPOBEC1 and CBE-T.32 Compared with BE4, these editors have markedly reduced rates of guide-independent off-target editing in mammalian cells,31 which makes them more promising for potential therapeutic application.
Thorough evaluation of potential off-target activity is an important aspect of gene (base) editing drug discovery. Two previously published studies addressing base editing for silencing HBV genes assessed up to 11 potential off-target sites for the investigated gRNAs.19,20 In particular, the top 3 predicted off-target sites were evaluated for gRNA gS8,19 which has the same sequence as g37 used in our experiments. In this study, using the rhAmpSeq CRISPR analysis system,40,41 we were able to perform a much more thorough assessment; 685 potential off-target sites were evaluated for g37 and 499 off-target sites were evaluated for g40. The BE4 base editor was associated with a significant number of off-target sites: 19 in the case of g37 and 7 in the case of g40. BE4-PpAPOBEC1 and CBE-T had a more favorable off-target profile, while maintaining robust editing and antiviral activity in the HBV-PHH cell system. A full assessment of the consequences of the detected off-target edits would be needed before making a decision on the clinical use of a particular base editor with the gRNAs (g37 + g40).
Taken together, our findings show that a nonviral vector can deliver cytosine base editing reagents capable of efficiently and irreversibly silencing cccDNA and integrated HBV DNA sequences in relevant in vitro and in vivo systems. These data improve our understanding of the potential of the base editing to cure HBV and contribute to our knowledge of the molecular mechanism of action by which base editing can serve as an effective antiviral.
Materials and methods
Generation of HEK293T-lenti-HBV cell lines and transfection with the base editors (DNA format)
Two lentiviral plasmids containing partial HBV DNA sequences 2309–1622 (HBs, Pol) or 1176–2451 (X, Core) were cloned and further used for the lentiviral production. The resulting lentiviruses were transduced into HEK293T cells to generate the cell lines containing a single partial HBV DNA sequence per cell. HEK293T-lenti-HBV cell lines were transfected with the plasmid encoding BE4 (750 ng) and a plasmid encoding gRNA (250 ng) in a 48-well plate using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s protocol. gRNA sequences are mentioned in Tables S1 and S2. The g37 sequence was also reported by Yang et al. in 2020.19
Hepatoma cell line culture
HepG2-NTCP and HepG2.2.15 cells were cultured in DMEM supplemented with l-glutamine (Gibco) sodium pyruvate (Gibco), 5% fetal calf serum (Fetalclone II), 100 U/mL penicillin (Gibco), and 100 μg/mL streptomycin (Gibco) at 37°C and 5% CO2, all provided by Life Technologies. A total of 5 μg/mL puromycin (InvivoGen, Toulouse, France) and 400 μg/mL G418 (Eurobio Scientific, Les Ulis, France) were also added for HepG2-NTCP and HepG2.2.15 cells, respectively. For PLC/PRF/5 cells, Eagle’s Minimum Essential Medium (American Type Culture Collection) was used as a base medium with 10% heat-inactivated fetal bovine serum (FBS).
HBV infection and BE4/gRNA transfection of HepG2-NTCP
HepG2-NTCP cells were seeded at 105/cm2 in complete DMEM growth medium. From the next day onward, cells were maintained in 2.5% DMSO (Merck-Sigma-Aldrich) containing medium to enhance HBV infection.53 After 72 h, cells were infected with HBV inoculum at a MOI of 1000 (using polyethylene glycol [PEG] 4%). For Figure 2A, the infected cells were replated at 6 dpi. The next day, these cells were transfected with BE4-encoding mRNA and gRNA (ratio 2:1) using Lipofectamine Messenger MAX (Life Technologies). In experiments using a combination of two gRNAs, the mRNA:g37:g40 ratio was adjusted to 2:0.5:0.5. At 15 dpi, supernatants were collected for assessing extracellular HBV parameters and cells were harvested for measuring intracellular parameters and DNA base editing. For 3TC-treated cells as shown in Figure S3A, 10 μM 3TC was added at 4 days postinfection (dpi), cells were replated at 6 dpi, and 3TC was maintained until 15 dpi (Merck-Sigma-Aldrich). For Figure S1, cells were replated at 2 dpi, transfection was performed the next day, and samples were collected at 14 dpi.
PHHs maintenance, infection, and transfection
Plated PHHs isolated from chimeric mouse liver were purchased from PhoenixBio.28,29 PHHs were cultured at a concentration of 350,000 cells/well in a 24-well plate. Infection media was prepared using dHCGM/FBS with PEG 4% and HBV at MOI 500. Cells were incubated with 500 μL infection media for 20–24 h and washed the next day 3 times with dHCGM/FBS media. A final media change with dimethyl sulfoxide-supplemented hepatocyte clonal growth medium (dHCGM)/FBS was done after the 3 washes to complete the infection protocol and cells were maintained at 37°C and 5% CO2, with media changes every 72 h. Infected cells were transfected with BE4 encoding mRNA (600 ng) and gRNA (200 ng) (ratio 3:1) in each well of a 24-well plate using Lipofectamine Messenger MAX (Invitrogen) mixed with Opti-MEM media (Gibco). Cells were incubated with transfection reagents for 16–18 h. The media (dHCGM/FBS) was changed the next day.
BE4/gRNA transfection of HepG2.2.15 and PLC/PRF/5 cells
PLC/PRF/5 or 3TC pretreated HepG2.2.15 cells were transfected with BE4 mRNA and gRNA (ratio 2:1). At day 6 posttransfection, culture supernatants and cells were collected for the detection of HBV antigens and DNA base editing, respectively.
Immunoblotting
Transfected HepG2-NTCP and HepG2.2.15 cells were washed with PBS (Eurobio Scientific) and lysed with radioimmunoprecipitation assay buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.5, 1 mM EDTA pH 8.0, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS, protease inhibitor cocktail [Roche]) for 30 min at 4°C followed by centrifugation at 12,000 × g to remove cell debris. Protein concentrations were measured using a BCA assay kit (Life Technologies). Equal amount of total protein was subjected to SDS-PAGE using 4%–20% mini-PROTEAN TGX stain-Free Precast Gel or 3%–8% Criterion XT Tris-Acetate (BioRad Laboratories). Immunodetection was done using anti-HBs (Abbott H166 mouse monoclonal), anti-Ku80 (ab119935, Abcam), and anti-Cas9 (C15310258 Diagenode) primary antibodies followed by incubation with horseradish peroxidase conjugated secondary antibodies. Signals were detected using Bio-Rad Clarity Western ECL and the Chemidoc XRS (Bio-Rad Laboratories).
Quantification of total intracellular HBV DNA and HBV RNA
Total cellular DNA and RNA were extracted using the Epicentre MasterPure kit (Lucigen) and the Nucleospin RNA kit (Macherey-Nagel), respectively. qPCR was performed as described earlier.10 Briefly, total HBV DNA was quantified using TaqMan PCR Pa03453406_s1(Life Technologies). cccDNA amplification was performed on ExoI/III-treated samples (to degrade genomic DNA and incomplete double-stranded circular rcDNA intermediate species) using specific primers (forward: 5′CCGTGTGCACTTCGCTTCA3′; reverse: 5′GCACAGCTTGGAGGCTTGA3′; probe: 5′(6FAM)CATGGAGACCACCGTGAACGCCC[BBQ (BlackBerry® Quencher)]). Serial dilutions of an HBV plasmid served as quantification standard. Human β-globin amplification (TaqMan Assay ID: Hs00758889_s1) was performed for the internal normalization of total HBV DNA or cccDNA. Using primers, 3.5-kb RNA quantification was done (forward: 5′ GGAGTGTGGATTCGCACTCCT3′; reverse: 5′AGATTGAGATCTTCTGCGAC3′); probe: [6FAM]AGGCAGGTCCCCTAGAAGAAGAACTCC[BBQ], and normalized to β-glucuronidase (GUSb) (TaqMan Assay ID: Hs99999908_m1). qPCRs were set up in an Applied Biosystems QuantStudio 7 machine.
PHH and extracellular HBV DNA assessment
A total of 5 μL PHH supernatant was mixed with 45 μL of the buffer containing 40 μg/mL salmon sheared DNA (Invitrogen) in 10 mM Tris, pH 8. Samples were boiled at 95°C for 15 min and kept on ice for qPCR preparation. HBV DNA qPCR was performed with Universal PCR Master Mix (Applied Biosystems) and DNA oligonucleotide probe ES70 (/56FAM/ccgtgtgca/ZEN/cttcgcttcacctctgc/3IABkFQ) and the primers ES72 (CCGTCTGTGCCTTCTCATCTG), and ES73 (AGTCCAAGAGTCCTCTTATGTAAGACCTT).
Detection of HBV antigens
HBsAg and HBeAg were detected in cell supernatants by ELISA using the chemiluminescence immunoassay kit from Autobio Diagnostic according to the manufacturer’s instructions.
Southern blot analysis
Southern blotting was performed as described earlier using the International Coalition to Eliminate HBV harmonized protocol.27,54 Briefly, total DNA was extracted using the Hirt extraction protocol followed by treatment with Exo I/III. All of the samples were quantified by Qubit. Mitochondrial NADH dehydrogenase (ND2, TaqMan Assay ID: Hs02596874_g1, Life Technologies) levels were quantified by qPCR and used to normalize loading. Samples were separated on 1.2% agarose gel in 1× Tris-acetate EDTA buffer at 15 V. Depurination followed by denaturation and neutralization was performed in gel before transferring to a nylon membrane with 20× saline-sodium citrate buffer using a Whatman TurboBlotter. DNA was crosslinked to the membrane by ultraviolet light at 120 mJ/cm2. The membrane was hybridized overnight at 55°C with DNA probes. The hybridized signal was amplified using the QuantiGene Singleplex Assay kit (Life Technologies) and detected by a ChemiDoc imager.
Northern blot analysis
Total RNA was extracted using the TRI reagent (Molecular Research Center) protocol following recommendation from the manufacturer and quantified using Nanodrop One. Total RNA, 10 μg, was mixed with glyoxal, denatured (50°C, 1 h) and resolved for 5 h at 60 mV with phosphate buffer recircularization on a 1.2% agarose gel, after which an RNA integrity profile was assessed by a ChemiDoc imager. After RNA transfer on a Hybond-N+ membrane and crosslinking (2 h, 80°C), HBV RNAs were detected using DIG-labeled probes and DIG Wash and Block Buffer Set (Merck-Sigma-Aldrich).
NGS of DNA
DNA samples were sequenced by NGS (Illumina MiSeq platform), and sequencing reads were analyzed to obtain editing rates, as described in the methods of Packer et al.55
Animal care and treatments
All animal care and procedures were carried out according to the relevant National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee and the Office of Laboratory Animal Research at Charles River Accelerator and Development Laboratory. C3H male mice of 5–6 weeks of age were purchased from Charles River Laboratories. HBV minicircle DNA was injected into C3H mice using hydrodynamic delivery as described.38 Four weeks later, HDI mice were assessed for HBsAg and organized into the four groups for further treatment, as indicated in Figure 5. Serum was collected every 7 days from the submandibular vein and used to assess the levels of HBsAg. Lipid nanoparticles were diluted in sterile 1× Tris-buffered saline and intravenously injected via tail vein. Animals were euthanized 42 days post-LNP injection.
LNP formulations
The base editor mRNA and gRNA were coformulated at a weight ratio 1:1 in LNPs. For LNPs containing a combination of two gRNAs, the mRNA:g37:g40 ratio was adjusted to 1:0.5:0.5. The formulations were generated by mixing an aqueous solution of the RNA (pH 4.0) with the four lipid components, a proprietary ionizable lipid, dioleoylphosphatidylethanolamine, cholesterol, and DMG-PEG2000, in ethanol solution. The two solutions were mixed in the microfluidics device from Precision Nanosystems. The LNPs were dialyzed overnight against 1× Tris-buffered saline at 4°C, further concentrated in 100,000 molecular weight cutoff Amicon Ultra centrifugation tubes (Millipore Sigma) and subsequently filtered through 0.2-μm filters (Pall Corporation). Particle size was assessed using the Malvern Panalytical Zetasizer. Endotoxin was measured using the Pierce Chromogenic Endotoxin Quant Kit (Thermo Fisher Scientific) following the manufacturer’s protocol.
Off-target site identification with rhAmpSeq
DNA from edited and untreated cells was extracted with the PureLink Genomic DNA Mini Kit (Thermo Fisher Scientific) following the manufacturer’s protocol. The extracted genomic DNA was amplified with custom rhAmpSeq panels (Integrated DNA Technologies), and sequencing libraries were prepared using the rhAmpSeq Library Kit (Integrated DNA Technologies). Sequencing libraries were sent to the Novogene Corporation and were sequenced on a Novoseq S4 (Illumina) to a target depth of 50,000 sequencing reads per candidate off-target site per sample.
Sequencing reads were preprocessed to trim low-quality base calls. Paired end reads were subsequently stitched to create consensus reads with adjusted base-quality scores, and those stitched reads were aligned to the human reference genome. Frequencies of base calls at each position in all candidate off-target sites were calculated from the read alignments and compared across treated and untreated samples. An odds ratio quantifying the enrichment of each observed variant in the treated samples was calculated, and a Fisher’s exact test was used to assess statistical significance.
Data and code availability
Data that underlie the reported results will be made available upon request 3 months after publication for a period of 5 years after the publication date.
Acknowledgments
We acknowledge Inserm U1052 for discussions and P. Huchon for technical assistance. The graphical abstract was created with BioRender.com. F.Z. received public grants overseen by the French National Research Agency (ANR) as part of the second “Investissements d’Avenir” program (reference no. ANR-17-RHUS-0003), the European Union (grant EU H2020-847939-IP-cure-B), and LabEx DEvweCAN (ANR-10-LABX-61). The work was performed in Lyon, France and in Cambridge, MA.
Author contributions
Conceptualization: E.M.S., M.G.M., E.C., A.K., M.S.P., B.T., F.G., F.Z., and G.C. Formal analysis: E.M.S., M.G.M., E.C., A.K., S.D., D.L., C.-Y.C., J.R.D., L.A.B., and M.S.P. Funding acquisition: B.T., F.G., F.Z., and G.C. Investigation: E.M.S., M.G.M., E.C., A.K., S.D., D.L., C.-Y.C., J.R.D., L.A.B., M.S.P., L.S.S., S.K., and L.Y. Methodology: E.M.S., M.G.M., E.C., A.K., D.L., C.-Y.C., J.R.D., L.A.B., M.S.P., L.S.S., S.K., and L.Y. Supervision: B.T., F.G., and F.Z. Visualization: E.M.S., M.G.M., E.C., A.K., and B.T. Writing – original draft: E.M.S., M.G.M., E.C., A.K., B.T., and M.S.P. Writing – review & editing: all authors.
Declaration of interests
E.M.S., S.D., D.L., C.-Y.C., J.R.D., L.A.B., M.S.P., G.C., L.S.S., S.K., L.Y., and F.G. are employees and shareholders of Beam Therapeutics. This work was funded by Beam Therapeutics, which is developing base editing therapeutics and has filed patent applications on this work. F.Z. received consulting fees from Aligos, Antios, Assembly, Gilead, and GSK; F.Z. and B.T. received research funding to INSERM from Assembly Biosciences, Beam Therapeutics, and Janssen.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2023.102112.
Contributor Information
Michael S. Packer, Email: mpacker@beamtx.com.
Fabien Zoulim, Email: fabien.zoulim@inserm.fr.
Supplemental information
References
- 1.Roca Suarez A.A., Testoni B., Zoulim F. HBV 2021: New therapeutic strategies against an old foe. Liver Int. 2021;41(Suppl 1):15–23. doi: 10.1111/liv.14851. [DOI] [PubMed] [Google Scholar]
- 2.Fanning G.C., Zoulim F., Hou J., Bertoletti A. Therapeutic strategies for hepatitis B virus infection: towards a cure. Nat. Rev. Drug Discov. 2019;18:827–844. doi: 10.1038/s41573-019-0037-0. [DOI] [PubMed] [Google Scholar]
- 3.WHO Hepatitis B. 2023. https://www.who.int/news-room/fact-sheets/detail/hepatitis-b
- 4.Revill P.A., Penicaud C., Brechot C., Zoulim F. Meeting the Challenge of Eliminating Chronic Hepatitis B Infection. Genes. 2019;10:260. doi: 10.3390/genes10040260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Revill P.A., Chisari F.V., Block J.M., Dandri M., Gehring A.J., Guo H., Hu J., Kramvis A., Lampertico P., Janssen H.L.A., et al. A global scientific strategy to cure hepatitis B. Lancet. Gastroenterol. Hepatol. 2019;4:545–558. doi: 10.1016/S2468-1253(19)30119-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wei L., Ploss A. Mechanism of Hepatitis B Virus cccDNA Formation. Viruses. 2021;13:1463. doi: 10.3390/v13081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinez M.G., Boyd A., Combe E., Testoni B., Zoulim F. Covalently closed circular DNA: The ultimate therapeutic target for curing HBV infections. J. Hepatol. 2021;75:706–717. doi: 10.1016/j.jhep.2021.05.013. [DOI] [PubMed] [Google Scholar]
- 8.Podlaha O., Wu G., Downie B., Ramamurthy R., Gaggar A., Subramanian M., Ye Z., Jiang Z. Genomic modeling of hepatitis B virus integration frequency in the human genome. PLoS One. 2019;14 doi: 10.1371/journal.pone.0220376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma Z., Zhang E., Gao S., Xiong Y., Lu M. Toward a Functional Cure for Hepatitis B: The Rationale and Challenges for Therapeutic Targeting of the B Cell Immune Response. Front. Immunol. 2019;10:2308. doi: 10.3389/fimmu.2019.02308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lebossé F., Testoni B., Fresquet J., Facchetti F., Galmozzi E., Fournier M., Hervieu V., Berthillon P., Berby F., Bordes I., et al. Intrahepatic innate immune response pathways are downregulated in untreated chronic hepatitis B. J. Hepatol. 2017;66:897–909. doi: 10.1016/j.jhep.2016.12.024. [DOI] [PubMed] [Google Scholar]
- 11.Ramanan V., Shlomai A., Cox D.B.T., Schwartz R.E., Michailidis E., Bhatta A., Scott D.A., Zhang F., Rice C.M., Bhatia S.N. CRISPR/Cas9 cleavage of viral DNA efficiently suppresses hepatitis B virus. Sci. Rep. 2015;5 doi: 10.1038/srep10833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorsuch C.L., Nemec P., Yu M., Xu S., Han D., Smith J., Lape J., van Buuren N., Ramirez R., Muench R.C., et al. Targeting the hepatitis B cccDNA with a sequence-specific ARCUS nuclease to eliminate hepatitis B virus in vivo. Mol. Ther. 2022;30:2909–2922. doi: 10.1016/j.ymthe.2022.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez M.G., Smekalova E., Combe E., Gregoire F., Zoulim F., Testoni B. Gene Editing Technologies to Target HBV cccDNA. Viruses. 2022;14:2654. doi: 10.3390/v14122654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leibowitz M.L., Papathanasiou S., Doerfler P.A., Blaine L.J., Sun L., Yao Y., Zhang C.-Z., Weiss M.J., Pellman D. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet. 2021;53:895–905. doi: 10.1038/s41588-021-00838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A·T to G·C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diorio C., Murray R., Naniong M., Barrera L., Camblin A., Chukinas J., Coholan L., Edwards A., Fuller T., Gonzales C., et al. Cytosine base editing enables quadruple-edited allogeneic CART cells for T-ALL. Blood. 2022;140:619–629. doi: 10.1182/blood.2022015825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Y.-C., Chen Y.-H., Kao J.-H., Ching C., Liu I.-J., Wang C.-C., Tsai C.-H., Wu F.-Y., Liu C.-J., Chen P.-J., et al. Permanent Inactivation of HBV Genomes by CRISPR/Cas9-Mediated Non-cleavage Base Editing. Mol. Ther. Nucleic Acids. 2020;20:480–490. doi: 10.1016/j.omtn.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou H., Wang X., Steer C.J., Song G., Niu J. Efficient silencing of hepatitis B virus S gene through CRISPR-mediated base editing. Hepatol. Commun. 2022;6:1652–1663. doi: 10.1002/hep4.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rajoriya N., Combet C., Zoulim F., Janssen H.L.A. How viral genetic variants and genotypes influence disease and treatment outcome of chronic hepatitis B. Time for an individualised approach? J. Hepatol. 2017;67:1281–1297. doi: 10.1016/j.jhep.2017.07.011. [DOI] [PubMed] [Google Scholar]
- 22.Mohr S.E., Hu Y., Ewen-Campen B., Housden B.E., Viswanatha R., Perrimon N. CRISPR guide RNA design for research applications. FEBS J. 2016;283:3232–3238. doi: 10.1111/febs.13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayer J., Jadeau F., Deléage G., Kay A., Zoulim F., Combet C. HBVdb: a knowledge database for Hepatitis B Virus. Nucleic Acids Res. 2013;41:D566–D570. doi: 10.1093/nar/gks1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Y., Qi Y., Peng B., Li W. NTCP-Reconstituted In Vitro HBV Infection System. Methods Mol. Biol. 2017;1540:1–14. doi: 10.1007/978-1-4939-6700-1_1. [DOI] [PubMed] [Google Scholar]
- 25.Zou S., Scarfo K., Nantz M.H., Hecker J.G. Lipid-mediated delivery of RNA is more efficient than delivery of DNA in non-dividing cells. Int. J. Pharm. 2010;389:232–243. doi: 10.1016/j.ijpharm.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez M.G., Combe E., Inchauspe A., Mangeot P.E., Delberghe E., Chapus F., Neveu G., Alam A., Carter K., Testoni B., Zoulim F. CRISPR-Cas9 Targeting of Hepatitis B Virus Covalently Closed Circular DNA Generates Transcriptionally Active Episomal Variants. mBio. 2022;13 doi: 10.1128/mbio.02888-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allweiss L., Testoni B., Yu M., Lucifora J., Ko C., Qu B., Lütgehetmann M., Guo H., Urban S., Fletcher S.P., et al. Quantification of the hepatitis B virus cccDNA: evidence-based guidelines for monitoring the key obstacle of HBV cure. Gut. 2023;72:972–983. doi: 10.1136/gutjnl-2022-328380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamasaki C., Kataoka M., Kato Y., Kakuni M., Usuda S., Ohzone Y., Matsuda S., Adachi Y., Ninomiya S.i., Itamoto T., et al. In vitro evaluation of cytochrome P450 and glucuronidation activities in hepatocytes isolated from liver-humanized mice. Drug Metabol. Pharmacokinet. 2010;25:539–550. doi: 10.2133/dmpk.dmpk-10-rg-047. [DOI] [PubMed] [Google Scholar]
- 29.Winer B.Y., Huang T.S., Pludwinski E., Heller B., Wojcik F., Lipkowitz G.E., Parekh A., Cho C., Shrirao A., Muir T.W., et al. Long-term hepatitis B infection in a scalable hepatic co-culture system. Nat. Commun. 2017;8:125. doi: 10.1038/s41467-017-00200-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lucifora J., Xia Y., Reisinger F., Zhang K., Stadler D., Cheng X., Sprinzl M.F., Koppensteiner H., Makowska Z., Volz T., et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science. 2014;343:1221–1228. doi: 10.1126/science.1243462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu Y., Leete T.C., Born D.A., Young L., Barrera L.A., Lee S.-J., Rees H.A., Ciaramella G., Gaudelli N.M. Cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity. Nat. Commun. 2020;11:2052. doi: 10.1038/s41467-020-15887-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lam D.K., Feliciano P.R., Arif A., Bohnuud T., Fernandez T.P., Gehrke J.M., Grayson P., Lee K.D., Ortega M.A., Sawyer C., et al. Improved cytosine base editors generated from TadA variants. Nat. Biotechnol. 2023;41:686–697. doi: 10.1038/s41587-022-01611-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wooddell C.I., Yuen M.-F., Chan H.L.-Y., Gish R.G., Locarnini S.A., Chavez D., Ferrari C., Given B.D., Hamilton J., Kanner S.B., et al. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci. Transl. Med. 2017;9:eaan0241. doi: 10.1126/scitranslmed.aan0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meier M.-A., Calabrese D., Suslov A., Terracciano L.M., Heim M.H., Wieland S. Ubiquitous expression of HBsAg from integrated HBV DNA in patients with low viral load. J. Hepatol. 2021;75:840–847. doi: 10.1016/j.jhep.2021.04.051. [DOI] [PubMed] [Google Scholar]
- 35.Sells M.A., Chen M.L., Acs G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc. Natl. Acad. Sci. USA. 1987;84:1005–1009. doi: 10.1073/pnas.84.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alexander J.J., Bey E.M., Geddes E.W., Lecatsas G. Establishment of a continuously growing cell line from primary carcinoma of the liver. S. Afr. Med. J. 1976;50:2124–2128. [PubMed] [Google Scholar]
- 37.Ishii T., Tamura A., Shibata T., Kuroda K., Kanda T., Sugiyama M., Mizokami M., Moriyama M. Analysis of HBV Genomes Integrated into the Genomes of Human Hepatoma PLC/PRF/5 Cells by HBV Sequence Capture-Based Next-Generation Sequencing. Genes. 2020;11:661. doi: 10.3390/genes11060661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan Z., Zeng J., Yu Y., Xiang K., Hu H., Zhou X., Gu L., Wang L., Zhao J., Young J.A.T., Gao L. HBVcircle: A novel tool to investigate hepatitis B virus covalently closed circular DNA. J. Hepatol. 2017;66:1149–1157. doi: 10.1016/j.jhep.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 39.Chadwick A.C., Wang X., Musunuru K. In Vivo Base Editing of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) as a Therapeutic Alternative to Genome Editing. Arterioscler. Thromb. Vasc. Biol. 2017;37:1741–1747. doi: 10.1161/ATVBAHA.117.309881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dobosy J.R., Rose S.D., Beltz K.R., Rupp S.M., Powers K.M., Behlke M.A., Walder J.A. RNase H-dependent PCR (rhPCR): improved specificity and single nucleotide polymorphism detection using blocked cleavable primers. BMC Biotechnol. 2011;11:80. doi: 10.1186/1472-6750-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muller A., Sullivan J., Schwarzer W., Wang M., Park-Windhol C., Klingler B., Matsell J., Hostettler S., Galliker P., Duman M., et al. High-efficiency base editing for Stargardt disease in mice, non-human primates, and human retina tissue. bioRxiv. 2023 doi: 10.1101/2023.04.17.535579. Preprint at. [DOI] [Google Scholar]
- 42.Bae S., Park J., Kim J.-S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Finn J.D., Smith A.R., Patel M.C., Shaw L., Youniss M.R., van Heteren J., Dirstine T., Ciullo C., Lescarbeau R., Seitzer J., et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018;22:2227–2235. doi: 10.1016/j.celrep.2018.02.014. [DOI] [PubMed] [Google Scholar]
- 44.Chen F., Alphonse M., Liu Q. Strategies for nonviral nanoparticle-based delivery of CRISPR/Cas9 therapeutics. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020;12 doi: 10.1002/wnan.1609. [DOI] [PubMed] [Google Scholar]
- 45.Paunovska K., Loughrey D., Dahlman J.E. Drug delivery systems for RNA therapeutics. Nat. Rev. Genet. 2022;23:265–280. doi: 10.1038/s41576-021-00439-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu B., Javidi-Parsijani P., Makani V., Mehraein-Ghomi F., Sarhan W.M., Sun D., Yoo K.W., Atala Z.P., Lyu P., Atala A. Delivering SaCas9 mRNA by lentivirus-like bionanoparticles for transient expression and efficient genome editing. Nucleic Acids Res. 2019;47:e44. doi: 10.1093/nar/gkz093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y., Li Y., Zai W., Hu K., Zhu Y., Deng Q., Wu M., Li Y., Chen J., Yuan Z. HBV covalently closed circular DNA minichromosomes in distinct epigenetic transcriptional states differ in their vulnerability to damage. Hepatology. 2022;75:1275–1288. doi: 10.1002/hep.32245. [DOI] [PubMed] [Google Scholar]
- 48.Revill P., Testoni B., Locarnini S., Zoulim F. Global strategies are required to cure and eliminate HBV infection. Nat. Rev. Gastroenterol. Hepatol. 2016;13:239–248. doi: 10.1038/nrgastro.2016.7. [DOI] [PubMed] [Google Scholar]
- 49.Vaillant A. Oligonucleotide-Based Therapies for Chronic HBV Infection: A Primer on Biochemistry, Mechanisms and Antiviral Effects. Viruses. 2022;14:2052. doi: 10.3390/v14092052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hui R.W.-H., Mak L.-Y., Seto W.-K., Yuen M.-F. RNA interference as a novel treatment strategy for chronic hepatitis B infection. Clin. Mol. Hepatol. 2022;28:408–424. doi: 10.3350/cmh.2022.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kramvis A., Kew M.C. Structure and function of the encapsidation signal of hepadnaviridae. J. Viral Hepat. 1998;5:357–367. doi: 10.1046/j.1365-2893.1998.00124.x. [DOI] [PubMed] [Google Scholar]
- 52.Melegari M., Scaglioni P.P., Wands J.R. Hepatitis B virus mutants associated with 3TC and famciclovir administration are replication defective. Hepatology. 1998;27:628–633. doi: 10.1002/hep.510270243. [DOI] [PubMed] [Google Scholar]
- 53.Yan R., Zhang Y., Cai D., Liu Y., Cuconati A., Guo H. Spinoculation Enhances HBV Infection in NTCP-Reconstituted Hepatocytes. PLoS One. 2015;10 doi: 10.1371/journal.pone.0129889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.ICE-HBV Southern Blot. 2022. https://ice-hbv.org/protocol/a-sensitive-and-rapid-southern-blot-assay-based-on-branched-dna-technology-for-the-detection-of-hbv-dna-in-cell-culture-and-liver-tissue-samples/
- 55.Packer M.S., Chowdhary V., Lung G., Cheng L.-I., Aratyn-Schaus Y., Leboeuf D., Smith S., Shah A., Chen D., Zieger M., et al. Evaluation of cytosine base editing and adenine base editing as a potential treatment for alpha-1 antitrypsin deficiency. Mol. Ther. 2022;30:1396–1406. doi: 10.1016/j.ymthe.2022.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data that underlie the reported results will be made available upon request 3 months after publication for a period of 5 years after the publication date.