

Abstract
Diverse factors including metabolism, chromatin remodeling, and mitotic kinetics influence development at the cellular level. These factors are well known to interact with the circadian transcriptional-translational feedback loop (TTFL) after its emergence. What is only recently becoming clear, however, is how metabolism, mitosis, and epigenetics may become organized in a coordinated cyclical precursor signaling module in pluripotent cells prior to the onset of TTFL cycling. We propose that both the precursor module and the TTFL module constrain cellular identity when they are active during development, and that the emergence of these modules themselves is a key lineage marker. Here we review the component pathways underlying these ideas; how proliferation, specification, and differentiation decisions in both developmental and adult stem cell populations are or are not regulated by the classical TTFL; and emerging evidence that we propose implies a primordial clock that precedes the classical TTFL and influences early developmental decisions.
Keywords: circadian, cell cycle, epigenetics, metabolism, stem cell, development
1. COUPLING OF THE CIRCADIAN CLOCK TO METABOLISM, EPIGENETIC STATE, AND CELL DIVISION
1.1. The Molecular Circadian Clock in Mammals
The mammalian circadian clock drives an oscillation of 24-hour period in diverse physiological functions, including epigenetic modification, metabolism, and cell division, in the whole body. Each cell possesses an intrinsic circadian clock driven by transcriptional-translational feedback loops (TTFLs) composed of circadian clock genes (Balsalobre et al. 1998, Yamazaki 2000). At the center of the TTFLs are two basic helix-loop-helix transcription factors: CLOCK and BMAL1 (ARNTL) (Figure 1a). These proteins heterodimerize, then bind to E-box enhancer elements and transactivate associated genes, driving gene expression that includes their negative regulators Period (PER) and Cryptochrome (CRY). Once the CLOCK:BMAL1 heterodimer forms a complex with PER and CRY proteins, the CLOCK:BMAL1 heterodimers dissociate from DNA, ending transactivation (Chiou et al. 2016). A series of phosphorylation and ubiquitination events as well as other checkpoints regulates the kinetics of PER and CRY nuclear reentry and competence to associate with CLOCK:BMAL1 (Patke et al. 2020). The latency of interactions between the positive CLOCK:BMAL1 and the negative CRY-PER arms contributes to the 24-hour period of the core TTFL loop.
Figure 1.
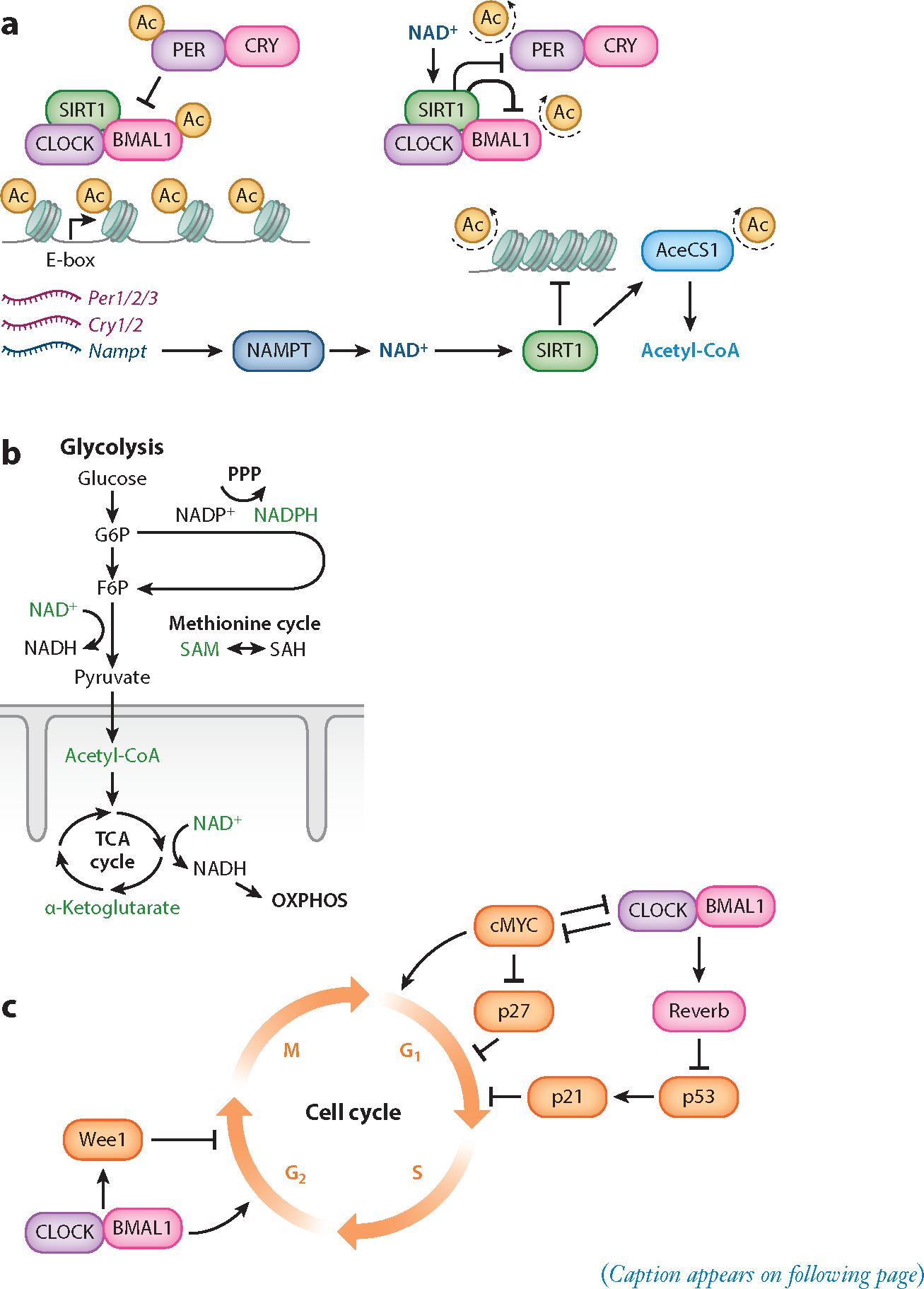
Circadian clock interaction with metabolism, cell cycle, and chromatin remodeling. (a) The core circadian TTFL is composed of the heterodimer CLOCK:BMAL1, which drives the transcription of their repressors PER1/2/3 and CRY1/2; these repressors dimerize in the cytoplasm and translocate to the nucleus to repress CLOCK:BMAL1. CLOCK:BMAL1 also drives the rhythmic accumulation of NAD+ by driving the transcription of the gene encoding the protein NAMPT, the rate-limiting enzyme in the NAD+-salvage pathway. The cyclic synthesis of NAD+ corresponds to the rhythmic activity of NAD+-dependent HDACs including SIRT1. Rhythms in SIRT1 activity over 24-hour periods also contribute to the cyclic synthesis of the universal acetyl donor acetyl-CoA through the enzyme AceCS1, which is activated by deacetylation. (b) Major catabolic pathways conserved across living cells include glycolysis, the TCA cycle, and OXPHOS. Each of these pathways produces metabolites that interact with circadian rhythms through the production of metabolites that act as substrates for posttranslational modifications or can activate protein-modifying enzymes (labeled in green). Among anabolic pathways, the PPP and the methionine cycle generate metabolites that interact with circadian rhythms. (c) Prominent connections of the circadian clock (purple) with the cell cycle (orange), emphasizing the checkpoints that are especially important for their coupling in stem cells. Abbreviations: Ac, acetyl; AceCS1, acetyl-CoA synthetase 1; CRY, Cryptochrome; F6P, fructose 6-phosphate; G6P, glucose 6-phosphate; HDAC, histone deacetylase; NAD+, nicotinamide adenine dinucleotide; NADP+, nicotinamide adenine dinucleotide phosphate; NAMPT, nicotinamide phosphoribosyltransferase; OXPHOS, oxidative phosphorylation; PER, Period; PPP, pentose phosphate pathway; SAH, S-adenosylhomocysteine; SAM, S-adenosyl methionine; TCA, tricarboxylic acid; TTFL, transcriptional-translational feedback loop.
The cell-autonomous circadian clock also contains secondary loops that serve to stabilize 24-hour transcriptional rhythms. A prominent example, particularly in development, is modulation by NR1 family transcription factors, which include transactivating RORα/β and repressive REVERBα/β family members (Ueda et al. 2005). These proteins compete for binding to RORE cis elements that regulate expression of clock genes, including Bmal1 and the Reverbs, making these family members’ interactions a self-contained regulatory loop that directly modulates BMAL1 expression to influence the core TTFL.
1.2. Circadian Clock Interaction with Metabolism, Chromatin Remodeling, and the Cell Cycle
While the composition of the circadian TTFL is relatively consistent, its coupling to inputs and outputs from other cellular signaling pathways varies substantially across cell types. Major pathways commonly integrated with the circadian TTFL, with functions that vary throughout and exert important influences upon developmental lineage restriction, include cellular metabolism, epigenetics, and the cell cycle (Figure 1).
1.2.1. Metabolism.
The primary function of circadian rhythms is to synchronize behavior and physiology with the diurnal rhythm of Earth rotating around its own axis as it revolves around the Sun. Perhaps the core function of this arrangement is to align the sleep/wake cycle of each organism with the solar cycle to maximize the opportunity to acquire available nutrients from its environment. It is therefore no surprise that extensive coupling exists between circadian rhythms and cellular metabolism throughout both development and adult function. For example, the abundance of ~50% of metabolites produced within the mouse liver is regulated by circadian clocks (Krishnaiah et al. 2017). These cyclically regulated metabolites include cofactors for chromatin-modifying enzymes such as nicotinamide adenine dinucleotide (NAD+) and S-adenosyl methionine (SAM), suggesting that chromatin structure is dynamically regulated in tune with circadian rhythms through cellular metabolism (Krishnaiah et al. 2017).
Cellular metabolism includes the set of pathways through which molecules are broken down into smaller units to generate energy (catabolism) and through which biomolecules such as fatty acids, nucleotides, and amino acids are synthesized from smaller units (anabolism). The primary catabolic pathways present within all living cells include glycolysis, the tricarboxylic acid (TCA) cycle, and oxidative phosphorylation (OXPHOS). Together these pathways combine to break down glucose, generating crucial metabolites including most of the adenosine triphosphate (ATP) used by the cell to provide energy for biochemical reactions. Glycolysis, the TCA cycle, and OXPHOS each exhibit reciprocal interaction with circadian rhythms by requiring TTFL-expressed enzymes and generating metabolites involved in the modification of both chromatin and core clock proteins (Figure 1b, Table 1).
Table 1.
Clock-regulated metabolites that participate in chromatin remodeling contribute to cellular differentiation prior to clock development
| Metabolite | Metabolic pathway | Chromatin modification | Circadian interaction | Stem cell fate change |
|---|---|---|---|---|
| Acetyl-CoA | Glycolysis Tricarboxylic acid (TCA) cycle | Histone acetylation | Activity of acetyl-CoA synthetase 1 (AceCS1) requires cyclic acetylation dependent on a functional circadian clock and the NAD+-dependent deacetylase SIRT1 (Sahar et al. 2014). Catabolism of glucose to acetyl-CoA depends on ATP-citrate lyase (ACLY); ACLY protein levels are cyclic in the mouse liver (Mauvoisin et al. 2014). |
Maintenance of pluripotency (Moussaieff et al. 2015) |
| α-Ketoglutarate | TCA cycle | Histone demethylation DNA demethylation | Inhibits JumonjiC domain–histone demethylase 1a (JARIDla), which associates with CLOCK:BMAL1 to facilitate Per2 transcription (DiTacchio et al. 2011). | Maintenance of pluripotency (Carey et al. 2015) |
| Nicotinamide adenine dinucleotide (NAD+) | Glycolysis | Histone deacetylation | In mouse liver, 24-hour cycling rhythm is exhibited (Krishnaiah et al. 2017). CLOCK:BMAL1 drives transcription of Nampt, a crucial enzyme for NAD+ production (Nakahata et al. 2009, Ramsey et al. 2009). |
Acquisition and maintenance of pluripotency (Calvanese et al. 2010, Y. Lee et al. 2012, Lees et al. 2020, Tang et al. 2014) |
| S-adenosyl methionine (SAM) | One-carbon metabolism (SAM and methionine cycle) | Histone methylation DNA methylation | In mouse liver, 24-hour cycling rhythm is exhibited (Krishnaiah et al. 2017). Accumulation of SAM by-product SAH (S-adenosyl homocysteine) hinders transmethylation and elongates circadian period (Fustin et al. 2013). |
Maintenance of pluripotency (Shiraki et al. 2014, Shyh-Chang et al. 2013) |
1.2.1.1. Interaction of glycolysis with circadian rhythms through NAD+ and glucose catabolism.
Glycolysis is a sequence of cytosolic oxidation-reduction (redox) reactions that converts glucose into two pyruvate molecules, while also generating two net ATP and reducing two NAD+ molecules to NADH. While glycolysis generates fewer net ATPs than OXPHOS, this pathway can function in a low-oxygen environment such as the postimplantation uterus and certain adult stem cell niches (Fischer & Bavister 1993, Simsek et al. 2010).
In particular, NAD+ is a key metabolite for interactions among metabolism, chromatin remodeling, and the circadian clock (Figure 1a). NAD+ shows robust diurnal rhythms in vitro and in vivo (Bellet et al. 2013, Nakahata et al. 2009, Ramsey et al. 2009), and operates as a cofactor for the sirtuins, a family of class III histone deacetylases (HDACs). NAD+ itself influences the circadian TTFL through the SIRT1-dependent deacetylation of PER2, a CLOCK:BMAL1 inhibitor (Asher et al. 2008). In turn, the circadian TTFL controls NAD+ levels through the NAD+ biosynthetic salvage pathway, in which nicotinamide is converted into the NAD+ precursor β-nicotinamide mononucleotide. This reaction is catalyzed by the rate-limiting enzyme nicotinamide phosphoribosyltransferase (NAMPT), whose expression is driven by the direct binding of CLOCK:BMAL1 to E-boxes in the Nampt promoter (Nakahata et al. 2009, Ramsey et al. 2009). While formation of fructose-1,6-bisphosphate upstream of NAD+ reduction is the rate-limiting step of glycolysis, increasing the concentration of NAD+ is sufficient to increase ATP production in the cytosol (Pitelli et al. 2011). Thus, a transcriptional-enzymatic feedback loop controls NAD+ biosynthesis and links circadian rhythms to the glycolytic catabolism of glucose.
Additionally, the availability of glucose itself contributes to circadian TTFL function by providing derivatives that serve as substrates for posttranslational modifications to core clock machinery. For example, CLOCK, BMAL1, and PER2 can be posttranslationally modified through the addition of O-linked N-acetylglucosamine (GlcNAc).This modification, catalyzed by the enzyme O-GlcNAc transferase (OGT), changes the activity of each clock protein (Kaasik et al. 2013, Li et al. 2013). Liver-specific OGT ablation dampens Bmal1 oscillation.
1.2.1.2. Interaction of the TCA cycle with circadian rhythms through acetyl-CoA and α-ketoglutarate.
When oxygen is available, pyruvate (primarily generated by glycolysis) is transported to the inner mitochondrial matrix and converted into acetyl-CoA to fuel the TCA cycle. In this pathway, acetyl-CoA produced from pyruvate or through fatty acid oxidation is further oxidized to generate NADH and FADH2. In addition to its catabolic function, several partially oxidized TCA cycle intermediates can be extracted to serve as building blocks for anabolic processes, including lipid, amino acid, and nucleotide biosynthesis as well as posttranslational protein modifications (reviewed by Boroughs & DeBerardinis 2015; see also Wellen et al. 2009). In this way the TCA cycle represents a central hub of energy metabolism, in which many pathways involved in central carbon metabolism, chromatin remodeling, and circadian rhythms intersect. For example, TCA cycle intermediate α-ketoglutarate inhibits JumonjiC domain–histone demethylase (JARID), which associates with CLOCK:BMAL1 to facilitate Per2 transcription (DiTacchio et al. 2011).
Acetyl-CoA, which interacts with circadian rhythms in part by providing acetyl groups as substrates for chromatin remodeling, exists in two separate pools in the cell: a mitochondrial pool that fuels the TCA cycle and a nuclear/cytosolic pool (reviewed by Albaugh et al. 2011). The nuclear/cytosolic pool, which is responsible for protein acetylation and fatty acid synthesis, is produced by two enzymes: ATP-citrate lyase (ACLY) and acetyl-CoA synthetase 1 (AceCS1). ACLY uses citrate produced during the TCA cycle as a substrate for the production of acetyl-CoA and is subject to circadian regulation. ACLY protein levels are cyclic in the liver (Mauvoisin et al. 2014), and ACLY activity controls global histone acetylation depending on glucose availability (Wellen et al. 2009).
Notably, AceCS1 is also subject to circadian regulation through NAD+. To produce acetyl-CoA, AceCS1 uses acetate produced physiologically by alcohol metabolism and histone deacetylation as a substrate (reviewed by Shimazu et al. 2010). The deacetylation of Lys-661 on AceCS1 by SIRT1 leads to the activation of AceCS1 (Hallows et al. 2006). The deacetylation of AceCS1 is cyclic, and its rhythmicity requires both a functional circadian clock and the NAD+-dependent deacetylase SIRT1 (Sahar et al. 2014). Thus, circadian rhythms directly link interconnected metabolic pathways to chromatin modification by dynamically regulating levels of acetyl-CoA produced through both ALCY and AceCS1 activity. In turn, intermediate metabolites produced by the TCA cycle such as α-ketoglutarate act as signaling molecules that reinforce the core circadian TTFL.
1.2.1.3. Interaction of OXPHOS with circadian rhythms through the NAD+-dependent activation of SIRT1/3.
OXPHOS is the primary catabolic pathway in mature eukaryotic cells for generating ATP, and this pathway plays a critical role in maintaining bioenergetic homeostasis by linking glycolysis, the TCA cycle, and fatty acid oxidation with ATP synthesis. NADH and FADH2, often from the TCA cycle, shuttle electrons to transmembrane protein complexes along the inner mitochondrial membrane that comprise the electron transport chain (ETC). The flow of electrons through these complexes pumps protons from the inner mitochondrial matrix to the outer matrix, creating a high electrochemical gradient. ATP is synthesized through ATP synthase, which uses the energy produced from transporting protons back to the inner mitochondrial matrix to catalyze adding a phosphate group to adenosine diphosphate (ADP).
OXPHOS is indirectly regulated by circadian rhythms, in part through NAD synthesis and thus the diurnal deacetylation of ETC Complex I through the activity of NAD+-dependent HDACs. The reversible acetylation of Complex I inhibits its function, driving its rhythmic activity in a human hepatocyte cell line (HepG2 cells) (Cela et al. 2016). Complex I function peaks in phase with NAMPT and SIRT1/3 expression, implicating the circadian NAD+-salvage pathway as a key mediator of OXPHOS (Cela et al. 2016). Additionally, approximately 38% of mitochondrial proteins oscillate in abundance throughout the day in a PER1/2-dependent manner, including several rate-limiting enzymes contributing to the TCA cycle and ETC (Neufeld-Cohen et al. 2016). In turn, OXPHOS appears to directly regulate the core circadian clock by influencing BMAL1 expression. Disrupting OXPHOS either through pharmacological inhibition or the depletion of mitochondrial DNA dysregulates BMAL1 expression in HepG2 cells (Scrima et al. 2016). Together, these observations suggest that OXPHOS and circadian rhythms reciprocally interact through the production of metabolites that act as substrates for posttranslational modifications of metabolic enzymes and core circadian machinery alike.
1.2.1.4. Anabolism: pentose phosphate pathway (NADPH) and one-carbon metabolism (SAM).
Among anabolic pathways responsible for synthesizing biomolecules, the pentose phosphate pathway (PPP) and one-carbon metabolism each exhibit reciprocal regulation with the circadian clock through metabolite signaling pathways. The PPP uses the glycolysis intermediate glucose 6-phosphate to generate NADPH and ribose 5-phosphate, a precursor for nucleotide synthesis. Signaling from the PPP through production of the redox cofactor NADPH is an important regulator of transcriptional oscillations (Rey et al. 2016). Inhibiting NADPH production by the PPP remodels circadian gene expression in human osteosarcoma (U2OS) cells and is sufficient to alter rhythmic behavior and tissue clocks in Drosophila (Rey et al. 2016). However, whether metabolic oscillations of NADPH persist without a functional circadian TTFL is an area of future study that will be crucial to determining the full extent of interaction between circadian rhythms and the PPP.
Circadian rhythms are also influenced by cyclical methionine production, which is part of a broader set of transformations known as one-carbon metabolism. Methionine production generates S-adenosylmethionine (SAM) as an intermediate metabolite that interacts with circadian rhythms through its function as a methyl-donor cofactor. During transmethylation, SAM donates its methyl group to acceptor molecules to generate S-adenosylhomocysteine (SAH).SAM exhibits diurnal cycling in mouse liver, and the accumulation of SAH elongates the circadian period by inhibiting the m6A-RNA methylation of core clock transcripts including Clock, Bmal1, and Per1/2 (Fustin et al. 2013, Krishnaiah et al. 2017). Because SAH acts as a competitive inhibitor of SAM-dependent-transmethylation, the SAM/SAH ratio within the cell (known as the methylation potential) therefore dynamically regulates the circadian period by titrating the rate of m6A-dependent mRNA degradation (reviewed by Carmel & Jacobsen 2011; see also Fustin et al. 2013).
1.2.2. Chromatin remodeling.
In eukaryotes, the storage and accessibility of genomic DNA is regulated at the level of chromatin, the DNA–protein complex that packages nuclear DNA. Chromatin is predominantly arranged into repeated arrays of nucleosomes composed of histone octamers. Due to their positive charge and the accessibility of their flexible N-terminal tails, histones are ideally suited to package DNA while acting as a platform for reversible, chemical posttranslational modifications that alter chromatin structure and function. These post-translational modifications include combinations of acetylation, methylation, phosphorylation, and ubiquitination events that durably encode physiological and environmental information and in turn modulate the extent to which genome elements are either accessible or compacted depending on cellular needs (reviewed by Bannister & Kouzarides 2011, Smith & Shilatifard 2010). Specific cyclic chromatin transitions associated with circadian TTFL transcription events occur on a genome-wide scale (Masri & Sassone-Corsi 2010). For example, CLOCK:BMAL1 acts as a pioneer transcription factor capable of directly initiating chromatin remodeling, and rhythmic CLOCK:BMAL1 binding to E-box motifs drives circadian chromatin transitions through transcription of the ancillary clock protein DBP (Menet et al. 2014, Ripperger & Schibler 2006). In addition, the circadian TTFL indirectly contributes to chromatin remodeling through interaction with metabolic pathways that generate intermediate products capable of serving as substrates for chromatin modifications (Figure 1a,b).
1.2.2.1. Histone acetylation and deacetylation.
The acetylation of histone lysines is catalyzed by histone acetyltransferases (HATs) and requires acetyl-CoA as the acetyl-donating cofactor (reviewed by Sabari et al. 2017). Several proteins involved in the addition and removal of acetyl groups to histones are associated with the core circadian TTFL. CLOCK acts in concert with HATs such as p300, CBP (CREB-binding protein), and the CBP-associated factor PCAF (Curtis et al. 2004, Etchegaray et al. 2003, Lee et al. 2010, Takahata et al. 2000). CLOCK:BMAL1 inhibitors balance these modifications by recruiting HDACs: PER recruits SIN3A-HDAC1, and CRY1 associates with the complex SIN3B-HDAC1/2 (Duong et al. 2011, Naruse et al. 2004).
In particular, the NAD+-dependent class III HDACs (sirtuins) represent a central node linking metabolism, chromatin remodeling, and circadian rhythms. SIRT1, SIRT3, and SIRT6 have each been functionally linked to circadian control, and they modulate cyclic outputs in response to metabolic cues across different subcellular compartments (reviewed by Sassone-Corsi 2016). While SIRT3 is localized to mitochondria and controls fatty acid oxidation and intermediary metabolism, SIRT1 and SIRT6 directly contribute to circadian patterns of histone deacetylation (Masri et al. 2013, 2014; Peek et al. 2013). SIRT6, which is the only sirtuin localized exclusively to chromatin, interacts directly with CLOCK:BMAL1 (Masri et al. 2014, Tennen et al. 2010). SIRT6 controls circadian chromatin recruitment of SREBP-1, cyclically regulating genes implicated in fatty acid and cholesterol metabolism (Masri et al. 2014). SIRT1 is localized to both the nucleus and cytoplasm and deacetylates histones and nonhistone proteins, including the clock proteins BMAL1 and PER2 (Asher et al. 2008, Hirayama et al. 2007). Through deacetylation of other histone-modifying enzymes such as methyltransferase MLL1 and metabolic enzymes such as AceCS1, SIRT1 positions circadian control of NAD+ production as a principal regulator of epigenetic and metabolic function (Aguilar-Arnal et al. 2013, Sahar et al. 2014).
1.2.2.2. Histone methylation and demethylation.
Histone methylation reactions, which are catalyzed mostly on lysine residues by histone methyltransferases, are also capable of influencing circadian TTFL function. For example, the histone H3 lysine K4 methyltransferase MLL1 (mixed-lineage leukemia 1) interacts with CLOCK:BMAL1 and is needed for the proper regulation of circadian transcription (Katada & Sassone-Corsi 2010). The activating trimethylation of H3K4 has been linked to clock control, and it seems to be essential to permit circadian chromatin transitions that promote clock gene expression (Ripperger & Schibler 2006). Conversely, the repressive trimethylation of H3K27 is clock controlled at the Per1 promoter through a mechanism that involves the methyltransferase EZH2 (Etchegaray et al. 2006). All histone methyltransferases require SAM generated from methionine metabolism as the methyl-donor cofactor. Thus, in addition to its influence on m6A-RNA methylation pathways, methionine metabolism interacts with circadian TTFL function through the SAM-dependent histone methylation.
The removal of methyl modifications is also critical for cyclic chromatin remodeling. However, while several histone demethylases associate with CLOCK:BMAL1, the mechanism through which histones are catalytically demethylated at clock genes remains unclear and is further complicated by the interactions among other chromatin-modifying enzymes. For example, lysine-specific demethylase 1a (LSD1) is an FAD (flavin adenine dinucleotide)-dependent histone demethylase that targets H3K4 and H3K9 and undergoes circadian phosphorylation when complexed with CLOCK:BMAL1 to promote the activation of Per2 (Nam et al. 2014, Shi et al. 2004). However, the loss of circadian phosphorylation on LSD1 does not appear to affect H3K4 or H3K9 methylation but instead inhibits the acetylation of H3K9 (Nam et al. 2014). The mechanism through which LSD1 affects histone acetylation may be related to its association with SIRT1, but how H3K4 and H3K9 undergo circadian demethylation remains unclear (Mulligan et al. 2011). Another histone demethylase implicated in clock function is JARID1a. JARID1a also associates with CLOCK:BMAL1 to facilitate Per2 transcription, but similarly to LSD1 its activity increases histone acetylation by inhibiting HDAC1 independent of demethylation (DiTacchio et al. 2011). Thus, understanding how circadian histone demethylation is achieved will likely require further study of the interactions among chromatin-modifying enzymes.
1.2.3. The cell cycle.
The cell cycle consists of the cell growth (G1), DNA replication (S), and cell division (G2/M) phases. Often the G1/S transition, when signals ranging from growth factors to DNA damage are integrated to determine entry into S phase or exit to a noncycling quiescent phase (G0), is rate determining for cell division. However, the G2/M transition can also become rate determining under conditions of rapid proliferation in which the G1 duration is shortened early in embryonic development (Figure 2) (Van Oudenhove et al. 2016). The circadian TTFL can exert control over both of these major checkpoints. Increasing the flexibility of this relationship, modeling approaches suggest that circadian TTFL coupling can drive ultradian (<24-hour period) or infradian (>24-hour period) harmonics of the circadian period in cell division in addition to the classical 24-hour rhythm, depending upon what intracellular coupling mechanisms and extracellular signaling are present (Almeida et al. 2020, El Cheikh et al. 2014, Feillet et al. 2014, Gupta et al. 2016, Kang et al. 2008, Traynard et al. 2016). Conversely, the cell cycle has also been proposed to entrain the circadian TTFL, and cyclin-dependent kinase signaling can regulate the circadian clock, suggesting that TTFL/cell cycle interactions are reciprocal (Bieler et al. 2014, Ou et al. 2019, Paijmans et al. 2016, Yan & Goldbeter 2019).
Figure 2.
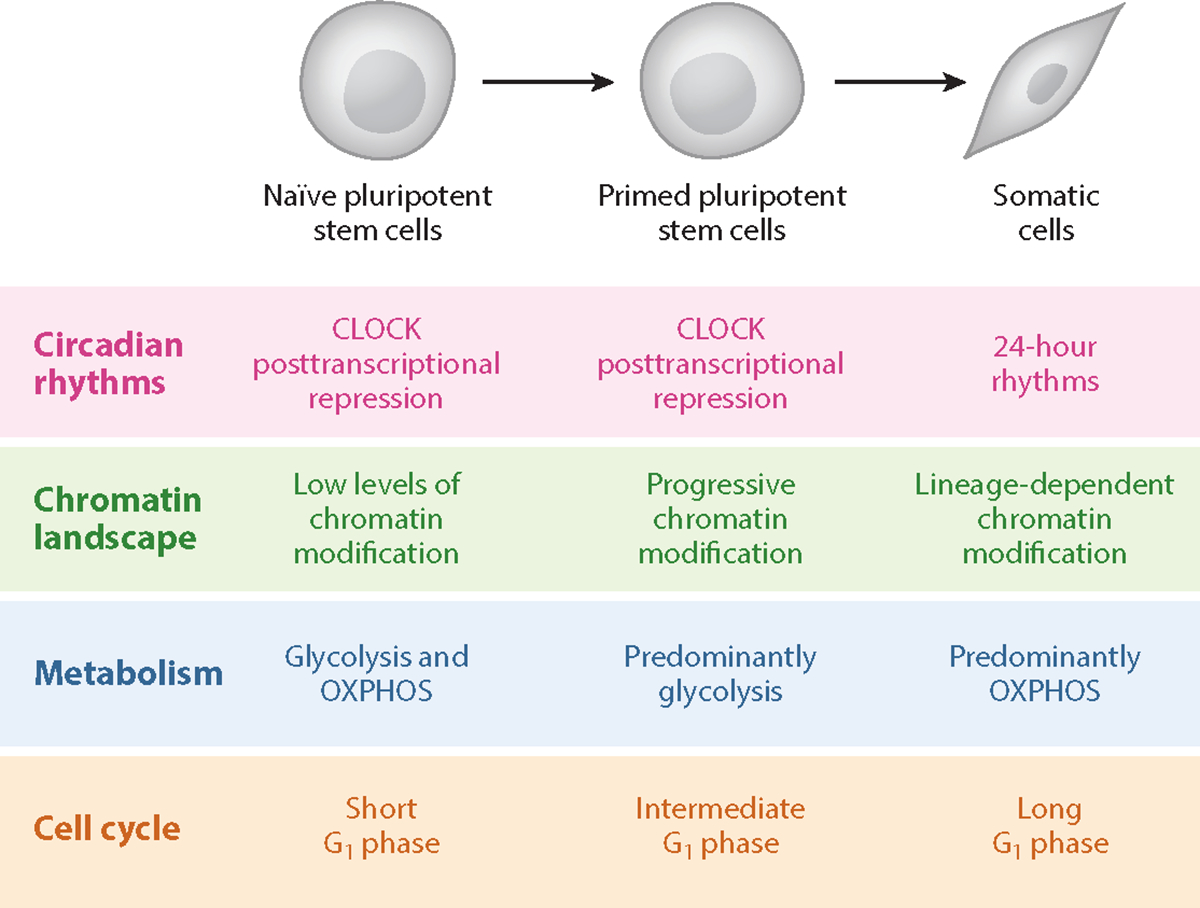
Pluripotent stem cell differentiation and clock development involves sequential changes to cellular metabolism and chromatin remodeling. Distinct phases of pluripotency can be captured in vitro and have characteristic epigenetic, metabolic, and cell cycle profiles. Both naïve and primed pluripotent ESCs lack circadian gene expression, and the core TTFL protein CLOCK is posttranscriptionally repressed. Naïve ESCs have very little chromatin modification across the epigenetic landscape, and progressive DNA methylation is associated with sequential lineage commitment. Naïve ESCs are able to perform OXPHOS, glycolysis, and fatty acid oxidation, whereas primed ESCs rely primarily on glycolysis to generate ATP. Differentiated cells rely primarily on OXPHOS. Abbreviations: ESC, embryonic stem cell; OXPHOS, oxidative phosphorylation; TTFL, transcriptional-translational feedback loop.
Perhaps the most prominent TTFL influence on the cell cycle is the BMAL1-driven, REVERBα suppression of p53 and p21 expression. This in turn gates the cyclin-dependent kinase (CDK) activity necessary for G1-to-S progression, among other cell cycle checkpoints (Figure 1c) (Fu & Kettner 2013, Gérard & Goldbeter 2012, Gréchez-Cassiau et al. 2008, Karimian et al. 2016). More recently, the TTFL has also been shown to exert control on the cell cycle downstream of the CDKs through posttranslational modification of the retinoblastoma protein (Lee et al. 2019).
Another salient example is c-MYC, which accelerates the cell cycle by promoting the G1/S transition in spite of p27-dependent CDK inhibition and promotes the cyclin expression needed for progression through the S phase (Figure 1c) (Pérez-Roger et al. 1997). The CLOCK:BMAL1 complex and c-MYC inhibit one another’s activity by a startling array of mechanisms, and this balance is disrupted by jet lag (Altman et al. 2015; Fu et al. 2002, 2005; Lee et al. 2019; Repouskou & Prombona 2016; Shostak et al. 2016). The TTFL/c-MYC balance is of particular interest in stem cells, given the role of c-MYC in pluripotency (Cartwright 2005, Takahashi & Yamanaka 2006). Other mechanisms coupling the circadian TTFL to the G1/S transition include the clock-regulated expression of cyclins and Wnt signaling components, among other factors (Fu & Kettner 2013).
The classical mechanism coupling the circadian TTFL to the G2/M checkpoint is CLOCK:BMAL1 control of Wee1 expression, which inhibits the initiation of mitosis by the cyclin B1/CDK1 complex (Figure 1c) (Matsuo 2003). More recently, the TTFL regulation of cyclin B1 expression itself has been shown to be relevant (Farshadi et al. 2019). Meanwhile the Timeless protein, while arguably nonessential for TTFL function in mammals, mediates the reciprocal regulation of the TTFL and the cell cycle G2/M checkpoint via Chk signaling (Engelen et al. 2013, Neilsen et al. 2019, Ünsal-Kaçmaz et al. 2005, Yang et al. 2010).
2. CIRCADIAN CLOCKS IN DEVELOPMENT
Embryonic development is a complex process with numerous decision points that any given cell must make to produce differentiated tissues, all of which first derive from pluripotent source material. Although most somatic cells are equipped with an intrinsic circadian clock, the TTFL arises gradually during embryonic development. Most data suggest that the core circadian feedback loops are completely absent in gametes and very early fertilized zygotes, with the TTFL being induced and progressively strengthened as lineage choice becomes more defined (Alvarez et al. 2003, Amano et al. 2009, Morse et al. 2003, Umemura et al. 2017). Thus, the circadian TTFL is completely absent in pluripotent stem cells (PSCs), and these are often used as a model for clock suppression and ontogeny in the context of development (reviewed by Umemura & Yagita 2020; see also Dolatshad et al. 2010, Yagita et al. 2010). At what point the TTFL is initially induced as pluripotency is lost; how metabolic, epigenetic, and cell cycle factors contribute to this shift; and the possible role of the TTFL in early lineage commitment remain unclear.
2.1. Circadian Clocks in Gametes
In both sexes, gametes lack an intrinsic circadian TTFL, and circadian regulation of their biology appears to occur non–cell autonomously. In male mammals, the protein expression of clock genes in testis germ cells is largely stable across the day; this may arise from a loss of transcriptional rhythms, as some studies find stable transcript expression (Alvarez et al. 2003, Morse et al. 2003), or from a lack of posttranscriptional circadian control, as others report circadian or ultradian rhythms in transcript levels despite unchanging protein expression (Bebas et al. 2009, Liu et al. 2007). Histological approaches show stable PER1 expression in developing sperm cells that is controlled by the CREM transcription factor rather than by classical clock control (Alvarez et al. 2003, Morse et al. 2003). Most rhythmic clock gene expression in the male reproductive system is confined to extragametic structures, and poor sperm production in Bmal1−/− mice is attributable to defects in rhythmic testosterone production by Leydig cells of the testes rather than to cell-autonomous defects in sperm (Alvarez et al. 2008, Bebas et al. 2009). While spermatogenesis is circadian regulated, this regulation thus seems to be entirely mediated by extracellular signaling from surrounding clock-competent tissues (Rienstein et al. 1998).
A similar situation prevails in the ovary in female mammals (reviewed in Sellix & Menaker 2010). Unfertilized oocytes have static clock gene expression, with especially high levels of Bmal1, Clock, and Cry1 transcripts that are likely maternally derived and may play a role in maintaining oocyte transcriptional repression (Amano et al. 2009, Ko et al. 2000). The ovary as a whole has inducible circadian TTFL function coupled to luteinizing and gonadotrophin hormone release, but this rhythm is localized in various support cell populations of the ovary and is excluded from quiescent and developing ova (Fahrenkrug et al. 2006, He et al. 2007, Karman & Tischkau 2006, Yoshikawa et al. 2009). A subset of these support cells’ clocks gates the circadian timing of ovulation. Deletion of Bmal1 in theca cells, but not in granulocytes, abolishes rhythmic sensitivity to luteinizing hormone exposure, impairing the normal circadian control of ovulation timing via hormone release (Mereness et al. 2016).
2.2. Circadian Clocks in Embryonic and Induced Pluripotent Stem Cells
While some core clock proteins such as BMAL1 are constitutively expressed early in embryonic development, circadian expression patterns only begin following the metabolic shift from glycolysis to OXPHOS in PSCs transitioning out of pluripotency (Umemura et al. 2019). During differentiation, CLOCK protein expression appears to be a rate-limiting factor for circadian TTFL network maturation (Umemura et al. 2017). CLOCK expression is suppressed posttranscriptionally in PSCs, and this suppression gradually subsides during embryonic development concomitant with an increased amplitude of circadian molecular oscillation (Figure 2) (Umemura et al. 2017). The primary mechanism of posttranscriptional repression of Clock mRNA appears to be Dicer/Dgcr8-dependent microRNA (miRNA)-mediated repression, as knockout of either Diceror Dgcr8 led to early-onset CLOCK expression in mouse embryonic stem cells (mESCs) (Umemura et al. 2017). Despite early CLOCK expression, however, Dicer−/− and Dgcr8−/− mESCs failed to demonstrate circadian rhythms even after DIV28, far beyond the previously observed milestone of their development (Umemura et al. 2017). In this study the lack of circadian oscillation in these cells was ascribed to their failure to properly differentiate, highlighting the crucial role of the transition from pluripotency in the development of the circadian clock. In line with this idea, the disruption of cellular differentiation by genetic and epigenetic perturbations [such as DNA methyltransferase (Dnmt1, Dnmt3a, and Dnmt3b) knockouts or c-MYC overexpression] prevents the development of circadian rhythms in mESCs (Figure 1) (Umemura et al. 2014, 2017).
In fact, the reprogramming of somatic cells using Yamanaka factors [OCT3/4, KLF4, SOX2, and c-MYC (Takahashi & Yamanaka 2006)] to produce human induced PSCs (hiPSCs) abolishes the established circadian rhythm, further demonstrating that circadian clock development is tightly coupled with cellular differentiation (Yagita et al. 2010). In the early stages of reprogramming, Yamanaka factors induce cell proliferation, which increases mitochondrial respiration and shuttles glucose to the PPP to meet the demand for increased nucleotide synthesis (Hawkins et al. 2016). Peaks in cell proliferation and OXPHOS contribute to reactive oxygen species (ROS) accumulation and hypoxia-inducible factor 1α activation, which drives the metabolic switch toward glycolytic energy production (Hawkins et al. 2016). Together these observations lay the foundation for a paradigm through which the dynamic changes in cellular metabolism, chromatin remodeling, and the cell cycle that occur during cell fate transition are intertwined with ontogeny of the circadian clock.
In the mouse embryo, tissues such as the heart and liver begin to demonstrate synchronous circadian rhythms at E13 (Dolatshad et al. 2010, Inada et al. 2014, Landgraf et al. 2015). By E14.5, synchronous circadian rhythms are observable in the mouse suprachiasmatic nucleus (SCN), the region of the hypothalamus responsible for aligning circadian rhythms across peripheral tissues (Carmona-Alcocer et al. 2018). The whole-body circadian clock also emerges gradually during human development. Circadian rhythms in body temperature are present in newborn infants, but circadian rhythms in the sleep/wake cycle only stabilize after about six weeks of life (Kleitman & Engelmann 1953, Rivkees 2003). The gradual development of the mouse and human circadian clocks is recapitulated in vitro: The differentiation of PSCs drives the cell-autonomous circadian oscillation of clock gene expression only after 14 days in vitro (DIV14) in mouse PSCs and after DIV42 in human PSCs (with robust circadian rhythms only observable in human PSCs after DIV90) (reviewed by Umemura & Yagita 2020; see also Umemura et al. 2019, Yagita et al. 2010).
2.3. Differentiation of Pluripotent Stem Cells by Changes to Metabolites That Regulate Chromatin Structure and Eventually Interact with Circadian Rhythms
Pluripotency is the ability of a stem cell to differentiate into any of the three primary germ layers: ectoderm, mesoderm, and endoderm. These three layers can give rise to all cell types of the adult body. Rather than a single fixed stage, pluripotency encompasses a gradient of cellular phenotypes from naïve PSCs of the inner cell mass in early blastocysts to primed PSCs of early postimplantation epiblasts (Figure 2) (reviewed by Weinberger et al. 2016). Although pluripotency exists transiently in vivo, several stages can be stably recapitulated in vitro (Evans & Kaufman 1981).In addition to the ability to give rise to all somatic lineages, naïve PSCs are able to incorporate into a developing blastocyst to create chimeric embryos and use both glycolysis and OXPHOS to generate ATP. In contrast, primed PSCs retain pluripotency, but unlike naïve PSCs they are incapable of integrating into a developing blastocyst to form chimeric embryos and are predominantly glycolytic. Although the mouse naïve-to-primed PSC transition is accompanied by increased glycolytic activity, in human PSCs this increase is not observed (Gu et al. 2016, Sperber et al. 2015, Zhou et al. 2012). However, during differentiation both mouse and human primed PSCs undergo a metabolic shift toward using OXPHOS as the primary source of ATP generation (Diano & Horvath 2012, Zhang et al. 2011).
Metabolites appear to play a significant role in regulating cell fate and development, including PSC differentiation. This regulation occurs primarily through the modification of chromatin that contributes to gene expression (reviewed by Mathieu & Ruohola-Baker 2017, Tsogtbaatar et al. 2020). Interestingly, many of the metabolites that contribute to the maintenance of stem cell pluripotency also influence the function of core circadian machinery following the development of circadian rhythms in differentiating cells (Table 1). This crucial link between cell fate acquisition and circadian TTFL function underlies the potential of metabolic chromatin remodeling pathways to drive the overall trajectory of development with progressive circadian entrainment as a central conductor. For example, entrainment to circadian feeding/fasting cycles triggers islet maturation in human ESCs (hESCs) by inducing the cyclic synthesis of energy metabolism and insulin secretion effectors (Alvarez-Dominguez et al. 2020). After entrainment, hESC-derived islets gain persistent chromatin changes and rhythmic insulin responses with a raised glucose threshold, a hallmark of functional maturity (Alvarez-Dominguez et al. 2020). As such, exploring how metabolites influence cell fate acquisition will provide crucial insights into how cellular differentiation might drive the initiation of circadian rhythms.
2.3.1. The TCA cycle (acetyl-CoA and α-ketoglutarate).
As a central pathway linking chromatin remodeling, glycolysis, and OXPHOS, the TCA cycle is well positioned to influence the metabolic shifts that drive the transition from naïve to primed pluripotency and ultimately the differentiation of primed PSCs to multipotent primordial germ cells. For example, the TCA cycle intermediate α-ketoglutarate promotes naïve stem cell pluripotency through the elevated demethylation of histones and DNA (Carey et al. 2015, Tischler et al. 2019). However, following the metabolic shift toward glycolytic dependence in the transition to primed pluripotency, α-ketoglutarate functions to promote primordial germ cell differentiation by supporting oxidative metabolism (TeSlaa et al. 2016, Tischler et al. 2019). Blocking acetyl-CoA production by the TCA cycle causes a loss of pluripotency in hESCs, while preventing acetyl-CoA consumption delays differentiation (Moussaieff et al. 2015). During the first 24hours of mESC differentiation, a metabolic switch takes place in which pyruvate becomes fully oxidized in mitochondria, leading to acetyl-CoA deprivation, the loss of histone acetylation, and the subsequent loss of pluripotency marker expression (Moussaieff et al. 2015). Together these observations demonstrate that crucial phases of cell fate transition are driven by metabolic shifts that alter substrate availability for chromatin remodeling.
2.3.2. Glycolysis (NAD+).
The connection between acetyl-CoA deprivation and histone acetylation in the maintenance of pluripotency implicates the NAD+-dependent HDAC SIRT1, which promotes acetyl-CoA production through the deacetylation of AceCS1 and directly participates in histone deacetylation. Indeed, the activity of SIRT1, which has been shown to be crucial for reprogramming, regulates mESC pluripotency (Calvanese et al. 2010, Y. Lee et al. 2012, Tang et al. 2014). Interestingly, the downregulation of SIRT2 and the upregulation of SIRT1 are together a molecular signature of primed human PSCs; these proteins regulate primed pluripotency through distinct mechanisms (Cha et al. 2017). NAD+ also promotes the cell fate transition from primed to naïve pluripotency in hESCs through the acquisition and maintenance of a bivalent metabolism (Lees et al. 2020). Increasing intracellular NAD+ levels in hESCs stimulates mitochondrial metabolism while suppressing glycolytic metabolism. This bivalent metabolism has been shown to coincide with increased pluripotency marker expression and decreased global H3K27 trimethylation, suggesting a potential link to SAM metabolism and methylation potential (Lees et al. 2020).
2.3.3. The methionine cycle (SAM).
SAM levels are crucial during the naïve-to-primed hESC transition, in which the epigenetic landscape changes through increased H3K27 trimethylation (Sperber et al. 2015). H3K27 trimethylation regulates key signaling pathways important for the metabolic changes necessary for early human development (Sperber et al. 2015, Z. Xu et al. 2016). These trimethyl modifications are generated by Polycomb repressive complex 2, which among other proteins includes the methyltransferase EZH2 (Vizán et al. 2015). Methionine and SAM are required for hESC self-renewal through the methylation of other histone targets, and the depletion of SAM leads to reduced H3K4 trimethylation and defective maintenance of the hESC state (Shiraki et al. 2014). In naïve stem cells, SAM is consumed by the metabolic enzyme nicotinamide N-methyltransferase, limiting its availability and thus maintaining low levels of histone methylation (Sperber et al. 2015). SAM, the universal donor of methyl groups for histone modifications, is therefore a key regulator both for promoting the primed pluripotent state of ESCs and for regulating their differentiation.
2.3.4. Core clock gene function prior to differentiation.
The observation that CLOCK expression is posttranscriptionally suppressed in mESCs prior to differentiation demonstrates that CLOCK function is not required for either PSC self-renewal or the transition from naïve to primed pluripotency (Umemura et al. 2017). Interestingly, however, Clock knockout in mESCs appears to decrease proliferation and increase apoptosis, potentially indicating a function of CLOCK in PSCs undergoing the first steps of cellular differentiation (Lu et al. 2016).
In contrast to CLOCK, BMAL1 is robustly expressed in both mouse and human PSCs, and several recent reports have demonstrated diverse functions of this transcription factor prior to the development of circadian rhythms. For example, loss of BMAL1 inhibits glycolytic metabolism in mouse PSCs and appears to disrupt the ability of these cells to exit pluripotency (Ameneiro et al. 2020). Bmal1-knockout mESCs express higher levels of Nanog and exhibit altered expression of pluripotency-associated signaling pathways (Ameneiro et al. 2020). Furthermore, Bmal1-knockout mESCs display deficient multilineage cell differentiation capacity during the formation of teratomas and gastrula-like organoids (Gallardo et al. 2020). Considering the clock-independent functions of BMAL1 that have been observed both prior to circadian clock development and in adult function, it will be interesting to explore the factors that dynamically regulate the extent to which BMAL1 functions independently of the core circadian TTFL within single cells (Lipton et al. 2015).
3. CIRCADIAN CLOCKS IN ADULT STEM CELLS
3.1. Neural Stem Cells
Most ectodermally derived neural stem cells (NSCs) that populate the developing nervous system, radiating neurons and glia outward from the ventricles, are transient. What NSCs remain in the adult nervous system are canonically localized to specific niches within the telencephalon that have relatively high baseline levels of neurogenesis: the subgranular zone and dentate gyrus of the hippocampus as well as the subventricular zones (SVZs) of the lateral ventricles. Most research on circadian control of neurogenesis has focused on these traditional niches. More recent studies also suggest broader circadian control of inducible adult NSCs in ventral forebrain-derived structures including the retina and hypothalamus (Figure 3).
Figure 3.
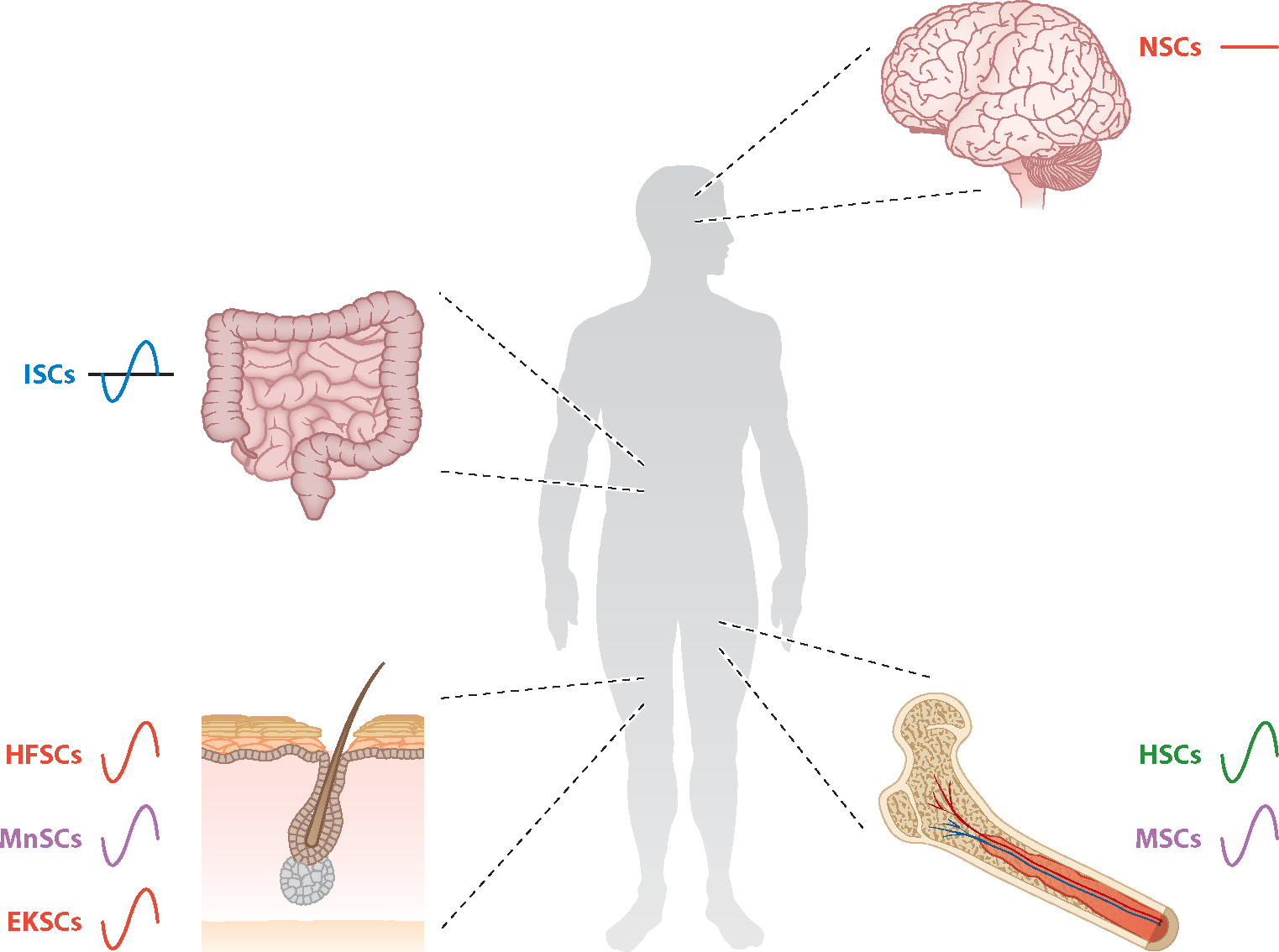
Adult stem cells derived from each germ lineage exhibit different degrees of circadian function. Four body parts (brain, intestine, bone marrow, and skin) containing adult stem cell niches are shown. Each stem cell type is color coded by developmental lineage: ectoderm (red), neural crest (purple), mesoderm (green), or endoderm (blue). The lines adjacent to each stem cell type indicate either the presence (sine wave) or absence (straight line) of the circadian TTFL in those cells. Abbreviations: EKSC, epidermal keratinocyte stem cell; HFSC, hair follicle stem cell; HSC, hematopoetic stem cell; ISC, intestinal stem cell; MnSC, melanocyte stem cell; MSC, mesenchymal stem cell; NSC, neural stem cell; TTFL, transcriptional-translational feedback loop.
3.1.1. Hippocampal neural stem cells.
Prolific neurogenesis in hippocampal NSC niches underpins the region’s impressive plasticity, which is essential for its role in learning and memory formation. Proliferating hippocampal NSCs in vivo express BMAL1 and PER2 (Borgs et al. 2009, Bouchard-Cannon et al. 2013) and lack PER1 (Gilhooley et al. 2011). However, most circadian gene expression analyses purporting to show either rhythmic (Bouchard-Cannon et al. 2013) or constitutive (Borgs et al. 2009) clock gene expression in hippocampal NSCs only looked broadly at the hippocampal domains in question without colocalization with markers of NSC fate, rendering the experiments uninformative. In the only direct test (Malik et al. 2015a) of whether hippocampal NSCs have an autonomous clock to date, PER1::Luc rhythms were detected in hippocampal NSC neurosphere cultures shortly after, but not before, the induction of differentiation. While the possibility of a PER1-independent clock in undifferentiated hippocampal NSCs deserves further scrutiny, the preponderance of available evidence currently suggests that they lack an autonomous clock.
Most studies (reviewed in Draijer et al. 2019), particularly later ones with more refined methodologies, agree that hippocampal NSC proliferation is circadian across vertebrates, with peaks in the G1/S transition in the morning, S phase early in the evening, and mitosis overnight. In young mice constitutively null for Bmal1, Per2, or Reverbα, hippocampal NSCs are arrhythmic and hyperproliferative, demonstrating that clock-controlled cues regulate their mitosis (Ali et al. 2015, Bouchard-Cannon et al. 2013, Schnell et al. 2014). In particular, multiple clock components, including BMAL1 and PER2, seem to gate entry of quiescent hippocampal NSCs into mitosis, while BMAL1 serves the additional role of regulating the number of divisions an active progenitor undergoes (Bouchard-Cannon et al. 2013). When clock control of these NSCs’ cell cycles is compromised, hyperproliferation exacerbates the rundown of the finite proliferative capacity of hippocampal NSCs, leading to decreased hippocampal neurogenesis with age in Bmal1−/− mice in vivo (Ali et al. 2015, Encinas et al. 2011). Similar results have been reported in culture. Young adult Per2−/− hippocampal NSCs yield larger neurospheres with increased percentages of mature neurons at the expense of undifferentiated NSCs (Borgs et al. 2009). In contrast, older Bmal1−/− and Cry1/2−/− hippocampal NSCs, and Reverbβ−/− forebrain NSCs of unreported age, generate smaller, more differentiated neurospheres (Malik et al. 2015a, Shimozaki, 2018). In sum, the niche circadian clock seems to antagonize hippocampal NSC proliferation and prevent their premature exhaustion.
Most mechanisms proposed to couple the clock to hippocampal NSC mitosis are cell autonomous. Modeling based on Bmal1 and Per2 loss-of-function phenotypes has suggested an inhibitor of cyclin-D/CDK4–6 as a key clock-regulated mediator of the cell cycle (Bouchard-Cannon et al. 2013). This model is consistent with rhythmic p20 expression at the G1/S transition and the successful delay of cell cycle progression by a selective CDK4–6 inhibitor in zebra fish NSCs (Figure 1c) (Akle et al. 2017). Separately, REVERBα is proposed to suppress hippocampal NSC proliferation by downregulating expression of the FABP7 fatty acid–binding protein (Schnell et al. 2014). Presumably, currently unidentified clock-controlled extracellular signaling couples the niche circadian TTFL to these cell-autonomous proximal regulators of NSC mitosis.
Bmal1−/− hippocampal NSCs tend to overproduce astrocytes at the expense of neurons both in vivo and in neurosphere culture, suggesting that BMAL1 has a proneural effect on neuron/glial fate choice in differentiating progenitors that is independent of the clock regulation of mitosis (Ali et al. 2015, Malik et al. 2015a). However, Per2−/− neurospheres from P10 mouse pups yield a higher percentage of neurons with no change in glial yields, while Cry1/2−/− neurospheres from 5- to 8-month-old adult mice yield an increase in astrocyte production with no change in neuron yield (Borgs et al. 2009, Malik et al. 2015a). Given the age confound, Per and Cry mutants’ apparent effects on the neuron/glia fate decisions of NSCs seem likely to simply reflect the overproliferation and depletion of NSCs, as hippocampal NSCs produce more glia at the end of their useful lives as progenitors (Encinas et al. 2011). Thus, BMAL1 has a bona fide proneural influence on the fate of individual hippocampal NSCs’ daughter cells independent of the TTFL, while the circadian clockwork as a whole indirectly maintains the neurogenic capacity of the hippocampal niche by limiting the NSCs’ rate of proliferation over developmental time.
3.1.2. Subventricular zone neural stem cells of the lateral ventricles.
High baseline neurogenesis in the SVZ provides replacement cells to the olfactory bulb, where exposure to aerosolized environmental toxins necessitates continuous turnover of mature cells throughout the life span. Similarly to the hippocampus, SVZ NSCs express some clock genes in arrhythmic or very weakly rhythmic patterns that are rapidly induced into rhythmic expression upon exposure to differentiation cues (Kimiwada et al. 2009, Malik et al. 2015b). Of some concern, PER1::Luc is again the only sensor used to directly demonstrate this in live-imaged SVZ neurospheres. Per1 has among the weakest and least rhythmic expression among clock genes in undifferentiated SVZ NSCs in the Kimiwada et al. (2009) study, while some other clock genes including Per2 appear to be potentially circadian in their expression, albeit with very low amplitude. Thus, while available evidence argues against the presence of a cell-autonomous TTFL in SVZ NSCs, this case further emphasizes the need for an attempt to discern whether PER1-independent rhythms are present in undifferentiated NSCs of both major telencephalic neurogenic niches.
Unlike in the hippocampus, the timing of SVZ NSC proliferation may be uncoupled from the circadian clock, with the G2/M phase transition occurring equally across the day and no data currently available for other stages of the cell cycle (Tamai et al. 2008). However, Bmal1−/− SVZ NSCs in 3-month-old mice show reduced proliferation in vivo (Ali et al. 2019), while Bmal1 and Clock RNAi reduce the production of mature neurons but not astrocytes from 6-week-old SVZ-derived neurospheres (Kimiwada et al. 2009). These data are consistent with the rundown of the SVZ NSCs with age in the absence of Bmal1, as well as a role of BMAL1 in promoting neural fate in SVZ daughter cells. Thus, SVZ NSCs are probably regulated by the circadian clock more similarly to hippocampal NSCs than is suggested by the currently available data directly examining this question.
3.1.3. Ventral forebrain neural stem cells.
The retina and hypothalamus of the mammalian brain develop from a common compartment that segregates prior to developmental neurogenesis, producing distinct transient NSC pools that yield neurons and glia with dramatically different morphology and function to serve their respective visual and organism-wide homeostatic functions (reviewed in Bedont et al. 2015, Byerly & Blackshaw 2009, Cepko 2014). Among the more obvious legacies of this shared heritage are Müller glia of the retina and tanycytes of the tuberal hypothalamic third ventricle, which are distinct populations of Rax(+) radial glia-like NSCs. While to our knowledge circadian regulation of these niches’ neurogenesis has not been studied directly, the robust roles of the retina and hypothalamus in organismal circadian rhythmicity suggest that such regulation is extremely likely, and considerable circumstantial evidence supports this viewpoint.
Tanycytes are traditionally divided into lateral α1/2 and ventral β1/2 subpopulations by morphology (Rodriguez et al. 2005). Tanycytes regulate such aspects of homeostasis as feeding and metabolism, seasonality, and hormone release, both directly and by generating daughter cells that integrate into the relevant hypothalamic nuclei. While the sources of hypothalamic neurogenesis are hotly debated, studies (reviewed in Yoo & Blackshaw 2018) have characterized two distinct tanycytic neurogenic niches that contribute at least some of these cells in mammals. β2-Tanycytes of the hypothalamic proliferative zone (HPZ) form a diet-responsive niche that is primarily neurogenic in adolescent and young adult mice (Haan et al. 2013, D. Lee et al. 2012). In contrast, α-tanycytes are robustly proliferative in a growth factor–regulated manner as late as 16 months of age in mice (Chaker et al. 2016, Pérez-Martín et al. 2010, Robins et al. 2013). Likely as a consequence of their extended proliferation, α- but not β-tanycyte pools run down with aging in mice in a manner reminiscent of hippocampal NSCs (Chaker et al. 2016, Encinas et al. 2011). In contrast to the telencephalic neurogenic niches, the tanycytes of the third ventricle have robustly rhythmic clock gene expression. The transcript-level expression of clock genes tends to be either across the entire tanycyte population or enriched in the HPZ (Lein et al. 2007, Yasuo et al. 2008). However, close visual inspection of in-slice sample images from the only protein-level data available suggests that the PER2::Luc signal is rhythmic in both the nonproliferative β1 and adult-quiescent β2 populations, with much stronger amplitude and overall higher expression in the former than in the latter. The relative strength of the α-tanycyte rhythm in these images is difficult to make out due to low resolution and strong signal in the adjoining parenchyma, but these cells appear to less robustly express PER2::Luc than the adjacent hypothalamic nuclei and may not have active autonomous clocks (Guilding et al. 2009). While further study is warranted, it is tempting to speculate that, at least for the HPZ, the observed intermediate rhythms in clock gene expression contribute to their progressive quiescence as mice mature into adulthood.
Such a role would be consistent with the developmentally related Müller glia of the retina. While these cells are constitutively proliferative throughout the life span in nonmammalian vertebrates, including zebra fish, they are generally minimally proliferative or nonproliferative in adult mammals (Bernardos et al. 2007, Das et al. 2006). What is less clear is whether Müller glia retain any residual neurogenic capacity in mammals in response to injury. While bipolar cell and rod production sourced from Müller glia has been reported after injury in rat retina, it is unclear whether mammalian Müller glia are capable of acting as NSCs supporting full retinal regeneration, which is seen after injury in less complex vertebrates (Ooto et al. 2004, Thummel et al. 2008). Adult Müller glia have a robust autonomous circadian clock (L. Xu et al. 2016). In addition, intriguingly, transcript expression of Per, Cry, and Reverb inhibitors of Bmal1 is higher, while positive regulators of Bmal1 are generally lower in Müller glia compared to those in retinal progenitors, and retinal progenitor–like changes in many of these genes’ expression occur in Müller glia injured with N-methyl-d-aspartate (NMDA) (Lin et al. 2019). Phototoxic insult has somewhat similar effects on Müller glia, suppressing the expression of clock genes, including Bmal1 and Per1, which are central to their clockwork (Sifuentes et al. 2016, L. Xu et al. 2016). Much as with hypothalamic tanycytes, this hints that the circadian clock may hinder dedifferentiation and neurogenesis of Müller glia in the mammalian retina. The potential for the application of latent Müller glia neurogenic capacity to regenerative medicine is a topic of considerable translational interest (reviewed in Lahne et al. 2020), making circadian manipulation as a possible tool for modifying this capacity a topic of particular interest.
3.2. Epithelial Stem Cells
The hair follicle is a useful case study of a relatively well-characterized, mixed population of stem cell populations hailing from two distinct developmental lineages: hair follicle stem cells (HFSCs) derived from the ectoderm proper and melanocyte stem cells (MnSCs) derived from the neural crest (Figure 3). These stem cells provide the fuel for a lifelong cycle of anagen (growth), catagen (degeneration), and telogen (quiescent) stages in the hair follicle as a whole. Commingled HFSCs and MnSCs are housed in a relatively quiescent state in a bulge adjoining each hair follicle and, at some stages of the growth cycle, in a more proliferative primed state in secondary hair germ; the hair germ in turn produces more-differentiated transient amplifying cells (TACs) that actually generate most of the terminal cell types incorporated into the hair follicle. In physiological conditions, the interfollicular skin is replenished by a physically separate population of epidermal keratinocyte stem cells (EKSCs); these cells proliferate at the basal layers of the skin and migrate upward as they first differentiate into TACs and then give rise to terminally differentiated cells, similar to the process in the hair follicle (reviewed in Hsu et al. 2014) (Figure 3).
3.2.1. Hair follicle stem cells.
HFSCs are impressively multipotent, with the capacity to contribute TACs to supplement epidermal keratocyte replenishment of the interfollicular skin after wounding (Ito et al. 2005) and produce numerous ectodermal and mesodermal lineages under laboratory conditions (reviewed in Najafzadeh 2015). Despite their malleable fate, the overall population of HFSCs has a robust intrinsic circadian clock even during the quiescent telogen phase (Janich et al. 2011, Lin et al. 2009, Plikus et al. 2013). The expression of HFSC clock genes tracks with the expression of key regulators of their proliferation and differentiation; similarly, HFSC subpopulations collected simultaneously and parsed by clock amplitude unequally express differentiation factors (Janich et al. 2011, Lin et al. 2009). Meanwhile, across time, higher HFSC clock amplitude as anaphase begins correlates with self-renewal and outer root sheath fates in daughter cells, while primed germ progenitors are produced by HFSCs oscillating at lower amplitude in late catagen (Ito et al. 2004, Janich et al. 2011, Rompolas et al. 2013). More lineage-committed TAC progeny have even higher amplitude rhythms than HFSCs during anaphase, demonstrating a progressive increase in clock amplitude during differentiation within the hair follicle niche (Plikus et al. 2013). Together, this suggests a role for the HFSC and TAC clocks in setting the terminal fates of their progeny.
The circadian clock plays a well-established role in driving the progression of HFSCs through the proliferative, refractory, and competent cell cycle states necessary for cyclical hair growth (Janich et al. 2011, Plikus et al. 2011). Clock amplitude in individual HFSCs positively correlates with the expression of pro-proliferative markers, including the Wnt and BMP-inhibitor pathways (Janich et al. 2011). Many of these signaling genes have Bmal1-competent E-boxes in their enhancers, and K14Cre-mediated Bmal1 deletion in HFSCs reduces these pro-proliferative genes’ expression and impedes cells’ proliferative capacity (Janich et al. 2011). This is consistent with gene expression changes in global Bmal1-null skin that specifically upregulate the inhibitory cell cycle regulator Cdkn1a, likely secondary to a dysregulated ROR/REVERBα balance (Lin et al. 2009). Global Period1/2 deletion enhances the HFSC proliferative capacity, supporting an interpretation of the Bmal1 proliferation phenotypes as clock effects (Janich et al. 2011).
Clock-null HFSC proliferation defects would, at first blush, then be expected to hinder hair growth. Indeed, a stalled first anagen delays initial hair growth in global Bmal1−/− and Clock−/− mice (Lin et al. 2009). However, K14Cre-mediated Bmal1 deletion in HFSCs fails to phenocopy first anagen delay (Geyfman et al. 2012, Janich et al. 2011), suggesting that Bmal1 dependence of the first anagen is a local niche effect on the HFSCs. Alternatively, the pacing of the first anagen may be set not by the HFSCs but by the more-differentiated TAC progenitor population. TACs contribute most cells directly incorporated into the hair follicle; these cells have the timing but not the overall throughput of their mitosis disrupted by K14Cre-driven Bmal1 or global Cry1/2 deletion (Geyfman et al. 2012, Plikus et al. 2013, Rompolas et al. 2013).
Surprisingly, tamoxifen-gated Bmal1 deletion in adulthood accelerates hair regrowth after depilation, suggesting that the clock inhibits hair growth later in life (Yang et al. 2016). Intriguingly, these results could reflect gating of the clock by pro–hair growth effects of light. Light signaling conveyed through a retina → SCN → sympathetic relay promotes hair regrowth after manual depilation via accelerated anagen entry (Fan et al. 2018). While the authors did not demonstrate a relationship between this light promotion of hair regrowth and gating by either the HFSC or TAC clocks, a relationship seems plausible.
3.2.2. Melanocyte stem cells.
MnSCs were recently demonstrated to have a cell-autonomous clock (Buhr et al. 2019). Human melanocyte phenotypes including dendritic outgrowth are modified by Bmal1 and Per2 short hairpin RNA (shRNA) injected into the intact hair follicle but not into differentiated melanocytes in culture, possibly reflecting a clock role in MnSC differentiation (Hardman et al. 2015). MnSCs also express neuropsin, allowing local entrainment of their rhythms by light (Buhr et al. 2019). One intriguing possibility is that local clock entrainment in MnSCs tweaks melanocyte proliferation and differentiation to help implement pigmentation changes dependent upon light dosage. Indeed, transcriptomics in snowshoe and mountain hare skin demonstrates seasonal changes not only in pigmentation gene expression but also in Wnts, other HFSC/MnSC proliferation signals, and visual transduction genes (Ferreira et al. 2020). However, the sympathetic signaling that mediates the SCN-dependent light responsiveness of HSFCs also appears to regulate age-related graying through MnSC β-adrenergic receptors (Fan et al. 2018, Zhang et al. 2020). While the MnSC clock itself is insensitive to SCN light cues (Buhr et al. 2019), whether internal and/or external light sensitivity guides other aspects of MnSC adaptation to light is unclear.
3.2.3. Epidermal keratinocyte stem cells.
Cultured human EKSCs have robust cell-autonomous clocks that drive five daily peaks of EKSC gene expression: the first three peaks of (a) Reverbα/β, (b) Per1/3, and (c) Per2+Cry2 correlate with distinct modules of genes required for EKSC differentiation (Janich et al. 2013). Administering Ca2+ or BMP to EKSCs during these peaks exerts a stronger differentiation response than at other times, and forcibly overexpressing Per1 or Per2 forces differentiation of EKSCs (Janich et al. 2013). Both Clock−/− and Per2−/− mice exhibit skewing of luminal and epithelial cell fates present in mammary tissue, supporting a role for the clock in guiding EKSC differentiation (Casey et al. 2016, McQueen et al. 2018). EKSC clock expression peaks of (d) Cry1 and (e) Bmal1 associate with modules controlling DNA repair and cell division (Janich et al. 2013). Constitutive Bmal1−/− and Clock−/− mutants, as well as K14Cretargeted Bmal1 deletion, result in the hyperproliferation of murine EKSCs, suggesting that the clock’s effect on mitosis in this lineage is inhibitory (Casey et al. 2016, Geyfman et al. 2012).
3.3. Bone Marrow Stem Cells
Much like the hair follicle, the bone marrow presents another useful case of distinct adult stem cell lineages colocalized within a common niche: mesoderm-derived hematopoietic stem cells (HSCs) and mesenchymal stem cells (MSCs) derived from the neural crest (Isern et al. 2014) (Figure 3). HSCs give rise to the multipotent progenitors and common lymphocyte progenitors required for diverse white and red blood cell production. In service of this function, HSCs are unusual in being able to leave their niche, trafficking out into the blood and sites important for immune function such as the spleen and thymus, all while retaining the ability to home back into the bone marrow for maintenance and self-renewal (Kumar et al. 2019). Within the bone marrow, HSCs occupy a heavily vascularized stromal niche in close proximity to the MSCs that is distinct from the endosteal compartment housing more lineage-restricted progenitor cells (Ding & Morrison 2013). The MSCs maintain conditions suitable for HSC self-renewal and help to regulate HSC trafficking in addition to their own role as stem cells for adipogenesis (fat), osteogenesis (bone), and chondrogenesis (cartilage) (reviewed in Murray et al. 2014; see also Méndez-Ferrer et al. 2010).
3.3.1. Hematopoietic stem cells.
An HSC-enriched fraction of blood and tissues is obtainable by flow cytometry. Furthermore, host bone marrow HSCs can be readily ablated and replaced with donor HSCs, allowing researchers to easily distinguish cell-autonomous and non-cell-autonomous effects. These tools were leveraged to show that HSCs have a functional circadian TTFL (Garrett & Gasiewicz 2006, Puram et al. 2016, Tsinkalovsky et al. 2006). Indeed, Clock1a expression is an instructive positive regulator of hematopoietic system formation in zebra fish development via control of rhythmic Smad3 expression involved in the nodal induction of mesoderm (Bian et al. 2017). Clock similarly regulates Smad3 expression in 3T3 cells, suggesting that an already-functional circadian TTFL may be essential for HSC production in mammalian development (Bian et al. 2017).
Circadian rhythms in HSC number and associated colony-forming activity have generally been reported in the blood and spleen (García-García et al. 2019, Golan et al. 2018, Kollet et al. 2013, Lucas et al. 2008, Méndez-Ferrer et al. 2008, Stenzinger et al. 2019), as well as by most studies in bone marrow (Garrett & Gasiewicz 2006, Kollet et al. 2013, Singh et al. 2011; but see also Stenzinger et al. 2019). Both rhythms and overall levels of HSCs are perturbed by circadian disruptions, including constant light, jet lag, and most clock gene deletions examined, often but not always in a non-cell-autonomous manner (Bhatwadekar et al. 2013, Kollet et al. 2013, Méndez-Ferrer et al. 2008, Singh et al. 2011, Stenzinger et al. 2019; but see also Bhatwadekar et al. 2017, Cao et al. 2017, Lucas et al. 2008, Wang et al. 2016). Contradictory reports in the literature likely stem from multiple sources, including a tendency to infer circadian rhythmicity from two daily time points; HSC numbers reflecting the integration of HSC cell division and specification dynamics with HSC migration dynamics; and the modulation of all of these factors by aging. We explore these interactions at length below.
Cell division in whole bone marrow of mammals, ranging from mice to humans, definitively occurs on a circadian rhythm (Aardal & Laerum 1983, Bourin et al. 2002, Clark & Korst 1969, Scheving et al. 1978, Smaaland et al. 1992). However, multipotent and lineage-committed progenitors contribute to this, and the rhythmic nature of HSC cell division is unclear. A human CD34+ population enriched for HSCs reportedly displays circadian DNA replication (Abrahamsen et al. 1998). Meanwhile, the percentage of murine LSK HSCs in quiescence varies in more of a 12-hour ultradian pattern, with peaks in HSC differentiation/egress at ZT5 and self-renewal at ZT17 reported to be times of low HSC quiescence (Garrett & Gasiewicz 2006, Golan et al. 2018). Theonly study we found reporting no time-of-day difference in DNA replication of LSK HSCs in blood, spleen, and bone marrow looked only at ZT2 and ZT14 (Stenzinger et al. 2019), time points with similar HSC quiescence rates that predictably generated a false negative result (Garrett & Gasiewicz 2006). Thus, time of day does seem to affect HSC cell division, although more direct studies examining LSK HSC cell division with finer circadian time courses are warranted. Bmal1−/− HSCs show no defects either in colony formation in vitro or in bone marrow renewal in competition with wild-type HSCs in vivo (Ieyasu et al. 2014), suggesting that the cell-autonomous TTFL is not directly coupled to the cell cycle in HSCs. However, perturbing systemic circadian rhythms by administering an aryl hydrocarbon receptor agonist appears to shorten the HSC cell cycle (Garrett & Gasiewicz 2006), while clock-controlled extracellular cues such as the release of the chemokine CXCL12 and the cytokine TNF-α regulate HSC proliferation (Golan et al. 2018, Méndez-Ferrer et al. 2008, Nie et al. 2008, Stenzinger et al. 2019). Thus, circadian control seems to be primarily exerted non–cell autonomously on the HSC cell cycle. That said, the chemokine receptor CXCR4 is probably expressed in a temporally regulated manner in HSCs (ZT5 > ZT13 but ZT2 = ZT14), and adult CXCR4 deletion with ROSACreER increases HSC proliferation in a cell-autonomous manner that is likely dependent on cyclin D1 (Lucas et al. 2008, Nie et al. 2008, Stenzinger et al. 2019). This suggests that HSCs gate their responsiveness to external cell cycle control through the temporal regulation of CXCR4 expression.
HSC migration closely cooperates with HSC cell cycle kinetics to set the hematopoietic proliferative capacity of various tissues. The circadian control of HSC egress from and homing back to bone marrow is primarily mediated non–cell autonomously by clock-controlled sympathetic/parasympathetic release of the neurotransmitters norepinephrine and acetylcholine, as well as by rhythmic corticosteroid and melatonin release (García-García et al. 2019, Golan et al. 2018, Méndez-Ferrer et al. 2008, Stenzinger et al. 2019). These long-range cues converge on other cells present in the bone marrow niche that regulate HSC trafficking through local release of additional signals, including rhythmic expression of the cytokine CXCL12 by MSCs and TNF-α by leukocytes (Golan et al. 2018; Kollet et al. 2013; Méndez-Ferrer et al. 2008, 2010; Stenzinger et al. 2019). This extrinsic circadian control of HSC trafficking is again likely gated by temporal variations in the HSC expression of CXCR4 (Lucas et al. 2008, Stenzinger et al. 2019).
Interestingly, considerable evidence suggests an age-related transition from extracellular circadian control to cell-autonomous HSC TTFL control in setting daughter cells’ specification and differentiation, perhaps related to a withdrawal of norepinephrinergic innervation and the associated displacement of HSCs from their normal niche in aged mice (Maryanovich et al. 2018). Younger constitutive Bmal1−/− mice have lower HSC cell counts in bone marrow, defects in lymphocyte cell numbers, and inefficient differentiation in lymphoid B-cell lineages, all of which appear to be mediated by non-cell-autonomous cues (Puram et al. 2016, Sun et al. 2006). Complementing these findings, young adult constitutive Cry1/2−/− mice have more differentiated B cells, while 4-month-old constitutive Per2−/− mice have an increase in HSCs in younger adult marrow that is depleted by 12 months of age, suggesting unsustainable hyperproliferation with inadequate self-renewal in young mouse HSCs (Bhatwadekar et al. 2013, Cao et al. 2017). While Bmal1−/− HSCs show normal colony formation in vitro and MxCre>Bmal1-deletion in HSCs has no impact on the HSC content of the bone marrow of younger mice, TekCre>Bmal1-deletion in HSCs results in a loss of bone marrow HSCs and colony-forming capacity by 8–10 months of age, suggesting a failure of self-renewal (Bhatwadekar et al. 2017, Ieyasu et al. 2014, Puram et al. 2016). Conversely, very old 20-month Per2−/− HSCs of the lymphoid-biased lineage have cell autonomously enhanced self-renewal, likely due to a role for PER2 in driving specific DNA repair pathways that inhibit proliferation (Wang et al. 2016). Interestingly, the contrast of Wang et al. (2016) with the Bhatwadekar et al. (2013) results in Per2−/− mice implies a reversal of PER2’s effects on HSC self-renewal at a very advanced age in mice. This may be precipitated by the withdrawal of circadian sympathetic signaling and other changes in the niche that have occurred by 20 months of age (Maryanovich et al. 2018).
3.3.2. Neural crest mesenchymal stem cells.
MSCs have a cell-autonomous circadian TTFL (Boucher et al. 2016, Huang et al. 2009,Wu et al. 2008).The TTFL’s effects on the MSC cell cycle have not been heavily studied.While Clock or Per2 RNA interference (RNAi) has surprisingly been reported to increase the proportion of quiescent MSCs in culture (Boucher et al. 2016), Reverbα overexpression has a similar effect (He et al. 2015). At present, there are insufficient reports to confidently determine the relationship between the TTFL and the cell cycle in MSCs.
In contrast, numerous studies have examined the effects of circadian clock components in the specification and differentiation of MSC daughter cells into the fat and bone lineages, motivated in no small part by mutant mouse phenotypes. Bmal1−/−, Clock−/−, and Per3−/− mice are all prone to obesity and/or high percentage of fat by weight (Costa et al. 2011, Guo et al. 2012,Turek 2005). Meanwhile, Bmal1−/− and MSC-targeting Prx1Cre>Bmal1-deletion mice have progressive loss of bone density over the life span that is driven in part by mildly deficient osteogenic potential along with high turnover (Samsa et al. 2016, Tsang et al. 2019).
BMAL1’s role in suppressing adiposity may be clock independent. While Bmal1 shRNA enhances adipogenesis in 10T1/2 MSC-like cells, both Clock and Per2 shRNA actually reduce the adipogenic potential of MSCs, likely at least in part through the downregulation of Wnt pathway components downstream of Bmal1 (Boucher et al. 2016, Guo et al. 2012). In addition, although Per3 overexpression blocks and Per3 deletion enhances MSC adipogenesis, the core clock was unaffected (Costa et al. 2011). Given the glucocorticoids used to stimulate the adipogenic differentiation of MSCs, which can directly bind the Per3 promoter to regulate Per3 expression, PER3 effects on adipogenesis may not be clock dependent (Costa et al. 2011, Pico et al. 2016). However, the circadian clock may play an instructive role later in adipocyte differentiation. For example, REVERBα expression is highly dynamic throughout pre-adipocyte differentiation in a manner that is likely regulated by both the TTFL and rhythmic degradation (DeBruyne et al. 2015, Wang & Lazar 2008).
Bmal1’s role in promoting osteogenesis, in contrast, most likely reflects a clock-dependent function. Complementing the modest osteogenic deficiency of Prx1Cre>Bmal1-deleted MSCs, the overexpression of Bmal1 in MSCs promotes osteogenesis in culture (He et al. 2013, Huang et al. 2020, Tsang et al. 2019). Conversely, knocking down the negative arm components Cry2 or Per2 in MSCs promotes osteogenesis, while the overexpression of Reverbα suppresses osteogenesis (He et al. 2015, Tang et al. 2020, Zhuo et al. 2018). Much as for adipogenesis, the transcriptional control of Wnt pathway components seems to be an important factor coupling the circadian TTFL to MSC osteogenesis (He et al. 2013, 2015). Interestingly, aged MSCs become less proliferative, less osteogenic, and more adipogenic while downregulating BMAL1 and upregulating REVERBα (Chen et al. 2012, Maryanovich et al. 2018, Yu et al. 2011). This suggests that degradation of the circadian TTFL may be relevant for maladaptive age-related changes in MSCs. Finally, two shRNA studies (Boucher et al. 2016, Zhuo et al. 2018) have reported a handful of results inconsistent with the rest of the literature: increased osteogenesis under Bmal1 shRNA and no effect on osteogenesis with Clock or Per2 shRNA. At best guess, these anomalous results reflect insufficient knockdown and/or off-target effects that complicate the shRNAs’ effects on osteogenesis.
3.4. Intestinal Stem Cells
Regrettably little is known about circadian control of adult stem cells of endodermal lineage, but of these, the intestinal niche is best studied (Figure 3). Intestinal stem cells (ISCs) reside within crypts at the base of the lumen. Signals—prominently including Wnts, especially from Paneth cells—regulate their division. Their differentiating daughters migrate into the intestinal lumen, where they constantly turn over intestinal epithelium damaged by the harsh digestive role of the intestine. The rate of this basal turnover becomes elevated in cases of unusual injury or inflammation, resulting in increased cell division (reviewed in Clevers et al. 2014, Parasram & Karpowicz 2020).
Whether ISCs possess an intrinsic TTFL in mammals is unclear. The intestine as a whole rhythmically expresses clock genes (Balakrishnan et al. 2010, Xu et al. 2017). However, the only study to closely examine mammalian ISC-specific rhythms with PER2::mCherry reported relatively flat expression (though still with a nonzero ~1.5X amplitude) from individual ISCs and low-amplitude, rapidly damping rhythms from organoids cultured in ISC-enriching conditions (Matsu-ura et al. 2016). In contrast, studies of invertebrate ISCs in Drosophila melanogaster reveal a clear cell-autonomous TTFL (Karpowicz et al. 2013, Parasram et al. 2018). Whether this reflects a bona fide evolutionary loss of ISC TTFL activity somewhere in the vertebrate clade, or whether mammalian ISCs simply have weak and/or PER2-independent TTFL activity that Matsu-ura et al. (2016) failed to detect, is of considerable interest for understanding the possible role of TTFL induction in early lineage specification (see Section 3).
In either case, the ISC cell cycle is regulated non–cell autonomously by circadian rhythms in a largely conserved manner. The mammalian ISC cell cycle runs on a 12-hour ultradian harmonic coordinated by circadian TTFL-controlled Wnt, TNF-α, and c-Jun N-terminal kinase (JNK) signaling (Matsu-ura et al. 2016, Stokes et al. 2017). These cues set the cell cycle period by gating the G1/S checkpoint, at least in part by regulating p21 expression (Figure 1c) (Matsu-ura et al. 2016, Stokes et al. 2017). While the G1/S transition seems to be generally rate determining in mammalian ISCs, systemic Per1/2 is also required for a proper exit from the S phase, likely from enhanced Wee1 expression that implies BMAL1 as a mediator (Figure 1c) (Matsuo 2003, Pagel et al. 2017). Drosophila ISCs show rhythmically increased proliferation on a circadian 24-hour period in response to injury, which relies upon both the ISC TTFL and circadian signaling from differentiated enterocytes (Karpowicz et al. 2013). As in mammals, Wnt and Hippo/JNK signaling is prominent among the extracellular clock-controlled cues involved (Karpowicz et al. 2013, Parasram et al. 2018).
To what extent circadian rhythms regulate specification and differentiation decisions after ISC cell division is unclear. Bmal1−/− intestine has no obvious morphological defects under physiological conditions, and while Per1/2−/− intestine is deficient in Paneth and goblet cells, this defect could be accounted for by elevated apoptosis (Pagel et al. 2017, Stokes et al. 2017). This question has not been directly asked in the fruit fly, although interestingly, the differentiated enteroen-docrine lineage (which has some secretory function loosely similar to that of Paneth and goblet cells) and enterocyte lineage are distinguished by the deactivation and maintenance, respectively, of circadian TTFL activity (Parasram et al. 2018). We thus suspect circadian involvement in ISC cell fate decisions but definitive determination of this awaits further research.
4. TEMPORAL REGULATION OF LINEAGE COMMITMENTTHROUGHOUT DEVELOPMENT BY METABOLIC RHYTHMS EARLY AND THE CLASSICAL CIRCADIAN TRANSCRIPTIONAL-TRANSLATIONAL FEEDBACK LOOP LATER
At fertilization, de novo clock gene expression is blocked for the first several, rapid cleavage divisions of the cell cycle, and clock gene expression steadily drops (Amano et al. 2009, Ko et al. 2000). However, temporal cues are available, as fertilization coincides with an initial burst of ROS that develops an oscillatory pattern coupled to cell division, as shown in Xenopus (Han et al. 2018). As cell division proceeds, the developing organism first forms a dense, solid ball of cells at the morula stage, which then restructures into a hollow blastula by the approximately 100-cell stage. Somewhere in this morulation/blastulation time frame, the transcription of the embryo’s own clock genes begins in mammals (Amano et al. 2009, Ko et al. 2000). Once the blastula successfully implants itself in the uterine lining, the hollow structure of the blastula allows involution to occur as division continues, forming a layered structure known as the gastrula. The inner layers form the primitive streak, the precursor of mesoderm and endoderm, while the outer layer goes on to become the ectoderm (Aires et al. 2018).
4.1. Possible Requirement for a Primordial Metabolic Clockfor Mesoendodermal Specification
A pair of recent papers (Ameneiro et al. 2020, Gallardo et al. 2020) beautifully demonstrate that while Bmal1−/− ESCs are resistant to differentiation in general, they are particularly poor substrates for primitive streak formation. In both adherent and gastruloid conditions compatible with multilineage differentiation,Bmal1−/− ESCs produced cells with much lower expression of mesodermal and endodermal lineage markers, while ectodermal lineage markers were relatively unaffected. This is consistent with data obtained in zebra fish, in which constitutive Clock1a knockout causes a selective defect in the formation of mesodermal lineages due to a loss of the rhythmic Smad3 expression required to transduce promesoderm nodal signals (Bian et al. 2017). Clock is able to similarly regulate Smad3 expression in mammalian cell culture (Bian et al. 2017).
Furthermore, Bmal1 may be epistatic to the effects of ROS accumulation on the mesoendodermal specification of ESCs. ESCs experiencing elevated ROS under differentiating conditions typically yield progeny enriched for the mesoendodermal lineage (Ji et al. 2010). Nonetheless, elevated ROS resulting from reduced glycolysis and enhanced OXPHOS in Bmal1−/− ESCs fails to prevent the ectodermal enrichment of their progeny (Ameneiro et al. 2020). One tempting hypothesis for this disconnect is the possibility that ROS timing makes the difference; while constant levels of ROS inhibit the mesodermal cardiovascular differentiation of ESCs, pulsatile ROS actually promotes such differentiation (Sauer & Wartenburg 2005). Indeed, ROS cycles in early dividing embryos are rhythmically phase locked to the cell cycle (Han et al. 2018).
The cell cycle also provides a potential means for stem cells to reorganize their metabolic cycles upon mesoendodermal specification. A Wee1-dependent mitotic pause occurs during the selection of mesodermal and endodermal fates in hESCs, providing sustained exposure to the S/G2/M cell cycle phases that are relatively conducive to selecting these lineages over ectoderm (Pauklin & Vallier 2013,Van Oudenhove et al. 2016). This pause is particularly important for the formation of endoderm, having more subtle effects on mesodermal differentiation (Van Oudenhove et al. 2016). Because mitosis freezes transcription, it is a signal that has been proposed to mediate entrainment of the TTFL oscillator by the cell cycle (Paijmans et al. 2016, Yan & Goldbeter 2019). But the slow induction of circadian TTFL cycling in PSC culture, as well as the late appearance of cell-autonomous rhythms in mesodermal tissues such as the heart sometime after E9.5, suggests that a cell-autonomous circadian oscillator is likely not yet present in the gastrula (Umemura et al. 2017, 2019; Yagita et al. 2010).
However, the cell cycle pause during mesoendodermal specification could instead serve as an instructive cue for reorganizing rhythmic metabolism. In addition to transcriptional arrest, mitochondrial OXPHOS is transiently increased during the G2/M transition to provide adequate energy to complete mitosis (Wang et al. 2014). Blocking either this cell cycle–coupled OXPHOS enhancement or the concomitant ROS production arrests the cell cycle at G2/M (Han et al. 2018, Wang et al. 2014). Timing of the G2/M pause in gastrulation may therefore be related to the uterine implantation that precedes it, which results in decreased uterine oxygen content and thus an overall increase in glycolysis and decrease in OXPHOS that presumably raises the barrier to mitosis (Fischer & Bavister 1993, Houghton et al. 1996). However, one outstanding question is why this pause only occurs in the newly specified mesoendoderm.
One possibility is that the extended pause allows an opportunity for the more extensive epigenetic remodeling of the chromatin required for ESCs to avoid defaulting into the ectodermal lineage (see Section 4.2). Consistent with this hypothesis, DNA methylation profiling of ESCs and precursor cells across the three germ layers has revealed that mesoderm precursors undergo 35% more promoter DNA methylation than ectodermal precursors (Isagawa et al. 2011).Broader profiling of chromatin modifications comparing these cell types will be necessary to definitively evaluate this possibility.
Another possibility is that the mesoendodermal lineage is selectively sensitive to entraining maternal cues. Implantation implies more direct exposure to these cues immediately prior to gastrulation, as the uterus communicates extensively with the embryo during and after implantation. The uterus expresses an estrogen-receptive circadian TTFL, and uterine receptivity to implantation is both entrained by systemic circadian cues such as melatonin and impaired by unfavorable photoperiod or jet lag (Mahoney 2010, Nakamura et al. 2008, Zhang et al. 2017). Later in development, the fetus may function in many ways as a peripheral oscillator of the maternal circadian system (Serón-Ferré et al. 2012). Mitotic/metabolic cycling in the mesoendoderm, which unlike ectoderm goes on to form exclusively peripheral oscillators in the adult animal, could be more responsive to maternal entrainment at this early stage.
4.2. Contribution of the Lack of a Circadian Transcriptional-Translational Feedback Loop to the Default Neural Stem Cell Fate
A long-controversial topic in stem cell biology has been whether a neural cell fate is the default specification for pluripotent ESCs, first hypothesized from spontaneous neural differentiation of Xenopus and mammalian cells in culture (Grunz & Tacke 1989, Wiles & Johansson 1999). Various extrinsic cues influence the ectodermal/mesoendodermal specification decision for cells in an intact gastrula. However, both progressive changes in intracellular transduction of those cues and an initial innate bias also seem to play roles, and these cell-autonomous factors do seem to initially favor neural ectodermal cell fate (Smukler et al. 2006, Turner et al. 2014, Zirra et al. 2016).
We propose that the seeming lack of a circadian TTFL in ESCs and iPSCs is one cell-autonomous factor that biases them toward the NSC lineage (Umemura et al. 2017, 2019; Yagita et al. 2010). Notably, NSCs are unusual among adult PSCs in that they lack an autonomous TTFL, a distinction they may only (arguably) share with ISCs out of the adult stem cells we discuss (Malik et al. 2015a,b; Matsu-ura et al. 2016; but see also Karpowicz et al. 2013) (Figure 3). Particularly marked is NSCs’ uniqueness within their lineage in lacking a TTFL, as all other stem cell types we review from the ectodermal (HFSC and EKSC) and related neural crest (MSC and MnSC) lineages have detectable circadian TTFLs (Buhr et al. 2019; Janich et al. 2011, 2013; Wu et al. 2008) (Figure 3). A consequence of this is that NSCs are also unusual in that the initial neural/glial specification decision for their progeny seems to be most directly influenced by the clock-independent functions of Bmal1. Aside from the pro-adipogenic influences of Bmal1 in MSC specification, most other cell fate decisions that Bmal1 contributes to in ectodermal/neural crest lineage adult stem cells appear to occur through Bmal1’s cell-autonomous TTFL function (see Section 3).
4.3. Probable Circadian Transcriptional-Translational Feedback Loop Involvement in Later Lineage Specification
Retinal progenitor stem cells (RSCs) give rise to differentiated cell types in a well-defined order across retinal development (reviewed in Cepko 2014). Chx10Cre>Bmal1 knockout in retinal progenitors early in retinogenesis profoundly reshapes the pool of final cells, depleting early-born populations, such as acetylcholinergic starburst amacrine cells and Brn3b+ retinal ganglion cells, and disrupting the proper distribution of opsin subtypes in the early-born cone population (Sawant et al. 2017, 2019). Moreover, late-born populations such as recoverin-positive bipolar cells and Müller glia are overproduced, and the lamination and morphology of the structurally critical Müller glia population are badly disrupted (Sawant et al. 2019). More Chx10Cre>Bmal1-depleted RSCs appear to undergo symmetrical, self-renewing division at E15 when asymmetrical divisions responsible for producing early-born retinal subtypes should be occurring (Sawant et al. 2019). This suggests that, like neuron/glia balance in the hippocampal niche, clock effects on RSC daughter fates may be secondary to the clock regulation of mitosis. This view is supported by the effects of cyclin D1 loss, which similarly prompt RSCs to overproduce late-born bipolar cell and Müller glia populations (Das et al. 2012). The developmental defects, especially defective Müller glial architecture, described by Sawant et al. (2019) likely contribute to the morphological and functional defects in the eyes of mice with Bmal1-deficient retinas, including thinned retinas and axial elongation of the eye that produces a myopic refractive index, poor visual acuity, and impaired b-wave electroretinogram activity (Baba et al. 2018, Stone et al. 2019).
Tellingly, both systemic Per1/2−/− and CrxCre>Per2–C knockout retinas have complementary S-cone phenotypes to the Bmal1 studies, suggesting that retinogenesis specification events driven by these genes reflect bona fide circadian TTFL activity (Ait-Hmyed et al. 2013, Sawant et al. 2017). This is consistent with the timing of retinogenesis, which begins in mid-gestation and ends in early postnatal life (reviewed in Cepko 2014). While we are unaware of any study examining clock ontogeny specifically within the retina, developmentally related tissues of the SCN—the master light-entrained circadian oscillator of the brain—exhibit circadian TTFL ontogeny at gestation points similar to the onset of retinogenesis (reviewed in Bedont & Blackshaw 2015; see also Carmona-Alcocer et al. 2018, Landgraf et al. 2015, Wreschnig et al. 2014). Indeed, the SCN itself may also feature circadian TTFL involvement in later lineage specification and/or differentiation, as the numbers of certain neuropeptidergic neuron lineages within the nucleus are skewed in Clock−/− mutants (Herzog et al. 2000, Silver et al. 1999). The SCN also provides an elegant example of how developmental neurogenic findings can inform the field of adult neurogenesis, as there is considerable evidence that the SCN may itself contain quiescent NSC potential in adulthood (reviewed in Carmona-Alcocer et al. 2020).
This work suggests that the developmental effects of the circadian TTFL on later branch points in lineage specification and differentiation are worth investigating. Elsewhere in the brain, for instance, cortical progenitors have a developmentally stereotyped progression of distinct earlyand late-born daughter cell types, and this program seems to be largely cell autonomous, much like that in RSCs (Duan et al. 2015, Shen et al. 2006). Such developmental programs could easily be guided by circadian rhythms as the cell-autonomous clock comes online in differentiating progenitors. In another recent example from the endodermal lineage, artificial circadian food entrainment imposed on pancreatic stem cells enhanced the final step of their development, which together with transcriptomic profiling supports a role for a food-entrained TTFL in terminal islet differentiation (Alvarez-Dominguez et al. 2020). More generally, as we discuss, most multipotent stem cells in adult niches express intact circadian TTFLs that regulate daughter cell fate. We propose that, similarly, an active TTFL in mid-embryonic to early-postnatal multipotent stem cells plays a role in regulating proliferation and daughter cell fate during development. Research on the role of the circadian TTFL and related factors on finer cell fate decisions within developmental lineages is unfortunately sparse, and we believe this is a major opportunity for future research.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- Aardal NP, Laerum OD. 1983. Circadian variations in mouse bone marrow. Exp. Hematol. 11(9):792–801 [PubMed] [Google Scholar]
- Abrahamsen JF, Smaaland R, Sothern RB, Laerum OD. 1998. Variation in cell yield and proliferative activity of positive selected human CD34 bone marrow cells along the circadian time scale. Eur. J. Haematol. 60(1):7–15. 10.1111/j.1600-0609.1998.tb00990.x [DOI] [PubMed] [Google Scholar]
- Aguilar-ArnalL HakimO, PatelVR BaldiP, HagerGL Sassone-CorsiP. 2013. Cycles in spatial and temporal chromosomal organization driven by the circadian clock. Nat. Struct. Mol. Biol. 20(10):1206–13. 10.1038/nsmb.2667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aires R, Dias A, Mallo M. 2018. Deconstructing the molecular mechanisms shaping the vertebrate body plan. Curr. Opin. Cell Biol. 55:81–86. 10.1016/j.ceb.2018.05.009 [DOI] [PubMed] [Google Scholar]
- Ait-Hmyed O, Felder-Schmittbuhl M-P, Garcia-Garrido M, Beck S, Seide C, et al. 2013. Mice lacking Period 1 and Period 2 circadian clock genes exhibit blue cone photoreceptor defects.Eur.J.Neurosci.37(7):1048–60. 10.1111/ejn.12103 [DOI] [PubMed] [Google Scholar]
- Akle V, Stankiewicz AJ, Kharchenko V, Yu L, Kharchenko PV, Zhdanova IV. 2017. Circadian kinetics of cell cycle progression in adult neurogenic niches of a diurnal vertebrate. J. Neurosci. 37(7):1900–9. 10.1523/jneurosci.3222-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albaugh BN, Arnold KM, Denu JM. 2011. KAT(ching) metabolism by the tail: insight into the links between lysine acetyltransferases and metabolism. ChemBioChem (12(2):290–98. 10.1002/cbic.201000438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali AAH, Schwartz-Herzke B, Mir S, Sahlender B, Victor M, et al. 2019. Deficiency of the clock gene Bmal1 affects neural progenitor cell migration. Brain Struct. Funct. 224(1):373–86. 10.1007/s00429-018-1775-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali AAH, Schwarz-Herzke B, Stahr A, Prozorovski T, Aktas O, von Gall C. 2015. Premature aging of the hippocampal neurogenic niche in adult Bmal1-deficient mice. Aging 7(6):435–49. 10.18632/aging.100764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida S, Chaves M, Delaunay F. 2020. Control of synchronization ratios in clock/cell cycle coupling by growth factors and glucocorticoids.R.Soc.Open Sci.7(2):192054. 10.1098/rsos.192054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman BJ, Hsieh AL, Sengupta A, Krishnanaiah SY, Stine ZE, et al. 2015. MYC disrupts the circadian clock and metabolism in cancer cells. Cell Metab. 22(6):1009–19. 10.1016/j.cmet.2015.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez JD, Chen D, Storer E, Sehgal A. 2003. Non-cyclic and developmental stage-specific expression of circadian clock proteins during murine spermatogenesis. Biol. Reprod. 69(1):81–91. 10.1095/biolreprod.102.011833 [DOI] [PubMed] [Google Scholar]
- Alvarez JD, Hansen A, Ord T, Bebas P, Chappell PE, et al. 2008. The circadian clock protein BMAL1 is necessary for fertility and proper testosterone production in mice. J. Biol. Rhythms 23(1):26–36. 10.1177/0748730407311254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Dominguez JR, Donaghey J, Rasouli N, Kenty JHR, Helman A, et al. 2020. Circadian entrainment triggers maturation of human in vitro islets. Cell Stem Cell 26(1):108–22.e10. 10.1016/j.stem.2019.11.011 [DOI] [PubMed] [Google Scholar]
- Amano T, Matsushita A, Hatanaka Y, Watanabe T, Oishi K, et al. 2009. Expression and functional analyses of circadian genes in mouse oocytes and preimplantation embryos: Cry1 is involved in the meiotic process independently of circadian clock regulation. Biol. Reprod. 80(3):473–83. 10.1095/biolreprod.108.069542 [DOI] [PubMed] [Google Scholar]
- Ameneiro C, Moreira T, Fuentes-Iglesias A, Coego A, Garcia-Outeiral V, et al. 2020. BMAL1 coordinates energy metabolism and differentiation of pluripotent stem cells. Life Sci. Alliance 3(5):e201900534. 10.26508/lsa.201900534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, et al. 2008. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 134(2):317–28. 10.1016/j.cell.2008.06.050 [DOI] [PubMed] [Google Scholar]
- Baba K, Piano I, Lyuboslavsky P, Chrenek MA, Sellers JT, et al. 2018. Removal of clock gene Bmal1 from the retina affects retinal development and accelerates cone photoreceptor degeneration during aging. PNAS 115(51):13099–104. 10.1073/pnas.1808137115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan A, Stearns AT, Ashley SW, Tavakkolizadeh A, Rhoads DB. 2010. Restricted feeding phase shifts clock gene and sodium glucose cotransporter 1 (SGLT1) expression in rats. J. Nutr. 140(5):908–14. 10.3945/jn.109.116749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsalobre A, Damiola F, Schibler U. 1998. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell 93(6):929–37. 10.1016/s0092-8674(00)81199-x [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. 2011. Regulation of chromatin by histone modifications. Cell Res. 21:381–95. 10.1038/cr.2011.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebas P, Goodall CP, Majewska M, Neumann A, Giebultowicz JM, Chappell PE. 2009. Circadian clock and output genes are rhythmically expressed in extratesticular ducts and accessory organs of mice. FASEB J. 23(2):523–33. 10.1096/fj.08-113191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedont JL, Blackshaw S. 2015. Constructing the suprachiasmatic nucleus: a watchmaker’s perspective on the central clockworks. Front. Syst. Neurosci. 9:e74. 10.3389/fnsys.2015.00074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedont JL, Newman EA, Blackshaw S. 2015. Patterning, specification, and differentiation in the developing hypothalamus. Dev. Biol. 4(5):445–68. 10.1002/wdev.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellet MM, Nakahata Y, Boudjelal M, Watts E, Mossakowska DE, et al. 2013. Pharmacological modulation of circadian rhythms by synthetic activators of the deacetylase SIRT1. PNAS 110(9):3333–38. 10.1073/pnas.1214266110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardos RL, Barthel LK, Meyers JR, Raymond PA. 2007. Late-stage neuronal progenitors in the retina are radial Müller glia that function as retinal stem cells. J. Neurosci. 27(26):7028–40. 10.1523/jneurosci.1624-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatwadekar AD, Beli E, Diao Y, Chen J, Luo Q, et al. 2017. Conditional deletion of Bmal1 accentuates microvascular and macrovascular injury. Am. J. Pathol. 187(6):1426–35. 10.1016/j.ajpath.2017.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatwadekar AD, Yan Y, Qi X, Thinschmidt JS, Neu MB, et al. 2013. Per2 mutation recapitulates the vascular phenotype of diabetes in the retina and bone marrow. Diabetes 62(1):273–82. 10.2337/db12-0172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian S-S, Zheng X-L, Sun H-Q, Chen J-H, Lu Y-L, et al. 2017. Clock1a affects mesoderm development and primitive hematopoiesis by regulating Nodal-Smad3 signaling in the zebrafish embryo. J. Biol. Chem. 292(34):14165–75. 10.1074/jbc.m117.794289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieler J, Cannavo R, Gustafson K, Gobet C, Gatfield D, Naef F. 2014. Robust synchronization of coupled circadian and cell cycle oscillators in single mammalian cells. Mol. Syst. Biol. 10(7):739. 10.15252/msb.20145218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgs L, Beukelaers P, Vandenbosch R, Nguyen L, Moonen G, et al. 2009. Period 2 regulates neural stem/progenitor cell proliferation in the adult hippocampus. BMC Neurosci. 10(1):30. 10.1186/1471-2202-10-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroughs LK, Deberardinis RJ. 2015. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 17(4):351–59. 10.1038/ncb3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard-Cannon P, Mendoza-Viveros L, Yuen A, Kaern M, Cheng H-YM. 2013. The circadian molecular clock regulates adult hippocampal neurogenesis by controlling the timing of cell-cycle entry and exit. Cell Rep. 5(4):961–73. 10.1016/j.celrep.2013.10.037 [DOI] [PubMed] [Google Scholar]
- Boucher H, Vanneaux V, Domet T, Parouchev A, Larghero J. 2016. Circadian clock genes modulate human bone marrow mesenchymal stem cell differentiation, migration and cell cycle. PLOS ONE 11(1):e0146674. 10.1371/journal.pone.0146674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourin P, Ledain AF, Beau J, Mille D, Lévi F. 2002. In-vitro circadian rhythm of murine bone marrow progenitor production. Chronobiol. Int. 19(1):57–67. 10.1081/cbi-120002677 [DOI] [PubMed] [Google Scholar]
- Buhr ED, Vemaraju S, Diaz N, Lang RA, Van Gelder RN. 2019. Neuropsin (OPN5) mediates local light-dependent induction of circadian clock genes and circadian photoentrainment in exposed murine skin. Curr. Biol. 29(20):3478–87.e4. 10.1016/j.cub.2019.08.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byerly MS, Blackshaw S. 2009. Vertebrate retina and hypothalamus development. Syst. Biol. Med. 1(3):380–89. 10.1002/wsbm.22 [DOI] [PubMed] [Google Scholar]
- Calvanese V, Lara E, Suárez-Álvarez B, Abu Dawud R, Vázquez-Chantada M, et al. 2010. Sirtuin 1 regulation of developmental genes during differentiation of stem cells. PNAS 107(31):13736–41. 10.1073/pnas.1001399107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Q, Zhao X, Bai J, Gery S, Sun H, et al. 2017. Circadian clock cryptochrome proteins regulate autoimmunity. PNAS 114(47):12548–53. 10.1073/pnas.1619119114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BW, Finley LWS, Cross JR, Allis CD, Thompson CB. 2015. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518(7539):413–16. 10.1038/nature13981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmel R, Jacobsen DW. 2011. Homocysteine in Health and Disease. New York: Cambridge Univ. Press [Google Scholar]
- Carmona-Alcocer V, Abel JH, Sun TC, Petzold LR, Doyle FJ III, et al. 2018. Ontogeny of circadian rhythms and synchrony in the suprachiasmatic nucleus. J. Neurosci. 38(6):1326–34. 10.1523/jneurosci.2006-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona-Alcocer V, Rohr KE, Joye DAM, Evans JA. 2020. Circuit development in the master clock network of mammals. Eur. J. Neurosci. 51(1):82–108. 10.1111/ejn.14259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright P, McLean C, Sheppard A, Rivett D, Jones K, Dalton S. 2005. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development 132(5):885–96. 10.1242/dev.01670 [DOI] [PubMed] [Google Scholar]
- Casey T, Crodian J, Suárez-Trujillo A, Erickson E, Weldon B, et al. 2016. CLOCK regulates mammary epithelial cell growth and differentiation. Regul. Integr. Comp. Physiol. 311(6):R1125–34. 10.1152/ajpregu.00032.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cela O,Scrima R, Pazienza V, Merla G, Benegiamo G, et al. 2016. Clock genes-dependent acetylation of Complex I sets rhythmic activity of mitochondrial OxPhos. Biochim. Biophys. Acta Mol. Cell Res. 1863(4):596–606. 10.1016/j.bbamcr.2015.12.018 [DOI] [PubMed] [Google Scholar]
- Cepko C 2014. Intrinsically different retinal progenitor cells produce specific types of progeny. Nat. Rev. Neurosci. 15(9):615–27. 10.1038/nrn3767 [DOI] [PubMed] [Google Scholar]
- Cha Y, Han M-J, Cha H-J, Zoldan J, Burkart A, et al. 2017. Metabolic control of primed human pluripotent stem cell fate and function by the MiR-200c-SIRT2 axis. Nat. Cell Biol. 19(5):445–56. 10.1038/ncb3517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaker Z, George C, Petrovska M, Caron J-B, Lacube P, et al. 2016. Hypothalamic neurogenesis persists in the aging brain and is controlled by energy-sensing IGF-I pathway. Neurobiol. Aging 41:64–72. 10.1016/j.neurobiolaging.2016.02.008 [DOI] [PubMed] [Google Scholar]
- Chen Y, Xu X, Tan Z, Ye C, Zhao Q, Chen Y. 2012. Age-related BMAL1 change affects mouse bone marrow stromal cell proliferation and osteo-differentiation potential.Arch.Med.Sci.8(1):30–38. 10.5114/aoms.2012.27277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou Y-Y, Yang Y, Rashid N, Ye R, Selby CP, Sancar A. 2016. Mammalian period represses and de-represses transcription by displacing CLOCK-BMAL1 from promoters in a Cryptochrome-dependent manner. PNAS 113(41):E6072–79. 10.1073/pnas.1612917113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RH, Korst DR. 1969. Circadian periodicity of bone marrow mitotic activity and reticulocyte counts in rats and mice. Science 166(3902):236–37. 10.1126/science.166.3902.236 [DOI] [PubMed] [Google Scholar]
- Clevers H, Loh KM, Nusse R. 2014. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 346(6205):1248012. 10.1126/science.1248012 [DOI] [PubMed] [Google Scholar]
- Costa MJ, So AY-L, Kaasik K, Krueger KC, Pillsbury ML, et al. 2011. Circadian rhythm gene Period 3 is an inhibitor of the adipocyte cell fate. J. Biol. Chem. 286(11):9063–70. 10.1074/jbc.m110.164558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis AM, Seo S, Westgate EJ, Rudic RD, Smyth EM, et al. 2004. Histone acetyltransferase-dependent chromatin remodeling and the vascular clock. J. Biol. Chem. 279(8):7091–97. 10.1074/jbc.m311973200 [DOI] [PubMed] [Google Scholar]
- Das AV, Mallya KB, Zhao X, Ahmad F, Bhattacharya S, et al. 2006. Neural stem cell properties of Müller glia in the mammalian retina: regulation by Notch and Wnt signaling. Dev. Biol. 299(1):283–302. 10.1016/j.ydbio.2006.07.029 [DOI] [PubMed] [Google Scholar]
- Das G, Clark AM, Levine EM. 2012. Cyclin D1 inactivation extends proliferation and alters histogenesis in the postnatal mouse retina. Dev. Dyn. 241(5):941–52. 10.1002/dvdy.23782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBruyne JP, Baggs JE, Sato TK, Hogenesch JB. 2015. Ubiquitin ligase Siah2 regulates RevErbα degradation and the mammalian circadian clock. PNAS 112(40):12420–25. 10.1073/pnas.1501204112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diano S, Horvath TL. 2012. Mitochondrial uncoupling protein 2 (UCP2) in glucose and lipid metabolism. Trends Mol. Med. 18(1):52–58. 10.1016/j.molmed.2011.08.003 [DOI] [PubMed] [Google Scholar]
- Ding L, Morrison SJ. 2013. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495(7440):231–35. 10.1038/nature11885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiTacchio L, Le HD, Vollmers C, Hatori M, Witcher M, et al. 2011. Histone lysine demethylase JARID1a activates CLOCK-BMAL1 and influences the circadian clock. Science 333(6051):1881–85. 10.1126/science.1206022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolatshad H, Cary AJ, Davis FC. 2010. Differential expression of the circadian clock in maternal and embryonic tissues of mice. PLOS ONE 5(3):e9855. 10.1371/journal.pone.0009855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draijer S, Chaves I, Hoekman MFM. 2019. The circadian clock in adult neural stem cell maintenance. Prog. Neurobiol. 173:41–53. 10.1016/j.pneurobio.2018.05.007 [DOI] [PubMed] [Google Scholar]
- Duan L, Peng C-Y, Pan L, Kessler JA. 2015. Human pluripotent stem cell-derived radial glia recapitulate developmental events and provide real-time access to cortical neurons and astrocytes.Transl.Med.4(5):437–47. 10.5966/sctm.2014-0137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong HA, Robles MS, Knutti D, Weitz CJ. 2011. A molecular mechanism for circadian clock negative feedback. Science 332(6036):1436–39. 10.1126/science.1196766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Cheikh R, Bernard S, El Khatib N. 2014. Modeling circadian clock–cell cycle interaction effects on cell population growth rates. J. Theor. Biol. 363:318–31. 10.1016/j.jtbi.2014.08.008 [DOI] [PubMed] [Google Scholar]
- Encinas JM, Michurina TV, Peunova N, Park J-H, Tordo J, et al. 2011. Division-coupled astrocytic differentiation and age-related depletion of neural stem cells in the adult hippocampus. Cell Stem Cell 8(5):566–79. 10.1016/j.stem.2011.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelen E, Janssens RC, Yagita K, Smits VAJ, van der Horst GTJ, Tamanini F. 2013. Mammalian TIMELESS is involved in period determination and DNA damage-dependent phase advancing of the circadian clock. PLOS ONE 8(2):e56623. 10.1371/journal.pone.0056623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etchegaray J-P, Lee C, Wade PA, Reppert SM. 2003. Rhythmic histone acetylation underlies transcription in the mammalian circadian clock. Nature 421(6919):177–82. 10.1038/nature01314 [DOI] [PubMed] [Google Scholar]
- Etchegaray J-P, Yang X, DeBruyne JP, Peters AHFM, Weaver DR, et al. 2006. The polycomb group protein EZH2 is required for mammalian circadian clock function. J. Biol. Chem.281(30):21209–15. 10.1074/jbc.m603722200 [DOI] [PubMed] [Google Scholar]
- Evans MJ, Kaufman MH. 1981. Establishment in culture of pluripotential cells from mouse embryos. Nature 292(5819):154–56. 10.1038/292154a0 [DOI] [PubMed] [Google Scholar]
- Fahrenkrug J, Georg B, Hannibal J, Hindersson P, Gräs S. 2006. Diurnal rhythmicity of the clock genes Per1 and Per2 in the rat ovary. Endocrinology 147(8):3769–76. 10.1210/en.2006-0305 [DOI] [PubMed] [Google Scholar]
- Fan SM-Y, Chang Y-T, Chen C-L, Wang W-H, Pan M-K, et al. 2018. External light activates hair follicle stem cells through eyes via an IpRGC–SCN–sympathetic neural pathway. PNAS 115(29):E6880–89. 10.1073/pnas.1719548115. Corrigendum. 2018. PNAS 115(51):E12121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farshadi E, Yan J, Leclere P, Goldbeter A, Chaves I, van der Horst GTJ. 2019. The positive circadian regulators CLOCK and BMAL1 control G2/M cell cycle transition through Cyclin B1. Cell Cycle 18(1):16–33. 10.1080/15384101.2018.1558638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feillet C, Krusche P, Tamanini F, Janssens RC, Downey MJ, et al. 2014. Phase locking and multiple oscillating attractors for the coupled mammalian clock and cell cycle. PNAS 111(27):9828–33. 10.1073/pnas.1320474111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira MS, Alves PC, Callahan CM, Giska I, Farelo L, et al. 2020. Transcriptomic regulation of seasonal coat color change in hares. Ecol. Evol. 10(3):1180–92. 10.1002/ece3.5956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer B, Bavister BD. 1993. Oxygen tension in the oviduct and uterus of rhesus monkeys, hamsters and rabbits. Reproduction 99(2):673–79. 10.1530/jrf.0.0990673 [DOI] [PubMed] [Google Scholar]
- Fu L, Kettner NM. 2013. The circadian clock in cancer development and therapy. In Progress in Molecular Biology and Translational Science, Vol. 119: Chronobiology: Biological Timing in Health and Disease, ed. Gillette MU, pp. 221–82. Oxford, UK: Acad. Press. 10.1016/b978-0-12-396971-2.00009-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Patel MS, Bradley A, Wagner EF, Karsenty G. 2005. The molecular clock mediates leptin-regulated bone formation. Cell 122(5):803–15. 10.1016/j.cell.2005.06.028 [DOI] [PubMed] [Google Scholar]
- Fu L, Pelicano H, Liu J, Huang P, Lee CC. 2002. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 111(1):41–50. 10.1016/s0092-8674(02)00961-3 [DOI] [PubMed] [Google Scholar]
- Fustin J-M, Doi M, Yamaguchi Y, Hida H, Nishimura S, et al. 2013. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 155(4):793–806. 10.1016/j.cell.2013.10.026 [DOI] [PubMed] [Google Scholar]
- Gallardo A, Molina A, Asenjo HG, Martorell-Marugán J, Montes R, et al. 2020. The molecular clock protein Bmal1 regulates cell differentiation in mouse embryonic stem cells. Life Sci. Alliance 3(5):e201900535. 10.26508/lsa.201900535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-García A, Korn C, García-Fernández M, Domingues O, Villadiego J, et al. 2019. Dual cholinergic signals regulate daily migration of hematopoietic stem cells and leukocytes. Blood 133(3):224–36. 10.1182/blood-2018-08-867648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett RW, Gasiewicz TA. 2006. The aryl hydrocarbon receptor agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin alters the circadian rhythms, quiescence, and expression of clock genes in murine hematopoietic stem and progenitor cells. Mol. Pharmacol. 69(6):2076–83. 10.1124/mol.105.021006 [DOI] [PubMed] [Google Scholar]
- Gérard C, Goldbeter A. 2012. Entrainment of the mammalian cell cycle by the circadian clock: modeling two coupled cellular rhythms. PLOS Comput. Biol. 8(5):e1002516. 10.1371/journal.pcbi.1002516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyfman M, Kumar V, Lui Q, Ruiz R, Gordon W, et al.2012.Brain and muscle Arnt-like protein-1 (BMAL1) controls circadian cell proliferation and susceptibility to UVB-induced DNA damage in the epidermis. PNAS 109(29):11758–63. 10.1073/pnas.1209592109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilhooley MJ, Pinnock SB, Herbert J. 2011. Rhythmic expression of per1 in the dentate gyrus is suppressed by corticosterone: implications for neurogenesis. Neurosci. Lett. 489(3):177–81. 10.1016/j.neulet.2010.12.011 [DOI] [PubMed] [Google Scholar]
- Golan K, Kumari A, Kollet O, Khatib-Massalha E, Subramanian MD, et al. 2018. Daily onset of light and darkness differentially controls hematopoietic stem cell differentiation and maintenance. Cell Stem Cell 23(4):572–85.e7. 10.1016/j.stem.2018.08.002 [DOI] [PubMed] [Google Scholar]
- Gréchez-Cassiau A, Rayet B, Guillaumond F, Teboul M, Delaunay F. 2008. The circadian clock component BMAL1 is a critical regulator of p21WAF1/CIP1 expression and hepatocyte proliferation. J. Biol. Chem. 283(8):4535–42. 10.1074/jbc.m705576200 [DOI] [PubMed] [Google Scholar]
- Grunz H, Tacke L.1989.Neural differentiation of Xenopus laevis ectoderm takes place after disaggregation and delayed reaggregation without inducer. Cell Differ. Dev. 28(3):211–17. 10.1016/0922-3371(89)90006-3 [DOI] [PubMed] [Google Scholar]
- Gu W, Gaeta X, Sahakyan A, Chan AB, Hong CS, et al. 2016. Glycolytic metabolism plays a functional role in regulating human pluripotent stem cell state. Cell Stem Cell 19(4):476–90. 10.1016/j.stem.2016.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilding C, Hughes ATL, Brown TM, Namvar S, Piggins HD. 2009. A riot of rhythms: neuronal and glial circadian oscillators in the mediobasal hypothalamus. Mol. Brain 2(1):28. 10.1186/1756-6606-2-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, Chatterjee S, Li L, Kim JM, Lee J, et al. 2012. The clock gene, brain and muscle Arnt-like 1, regulates adipogenesis via Wnt signaling pathway. FASEB J. 26(8):3453–63. 10.1096/fj.12-205781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Hepp B, Khammash M. 2016. Noise induces the population-level entrainment of incoherent, uncoupled intracellular oscillators. Cell Syst. 3(6):521–31.e13. 10.1016/j.cels.2016.10.006 [DOI] [PubMed] [Google Scholar]
- Haan N, Goodman T, Najdi-Samiei A, Stratford CM, Rice R, et al. 2013. Fgf10-expressing tanycytes add new neurons to the appetite/energy-balance regulating centers of the postnatal and adult hypothalamus. J. Neurosci. 33(14):6170–80. 10.1523/jneurosci.2437-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallows WC, Lee S, Denu JM. 2006. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. PNAS 103(27):10230–35. 10.1073/pnas.0604392103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Ishibashi S, Iglesias-Gonzalez J, Chen Y, Love NR, Amaya E.2018.Ca2+-induced mitochondrial ROS regulate the early embryonic cell cycle. Cell Rep. 22(1):218–31. 10.1016/j.celrep.2017.12.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardman JA, Tobin DJ, Haslam IS, Farjo N, Farjo B, et al. 2015. The peripheral clock regulates human pigmentation. J. Investig. Dermatol. 135(4):1053–64. 10.1038/jid.2014.442 [DOI] [PubMed] [Google Scholar]
- Hawkins KE, Joy S,Delhove JMKM, Kotiadis VN, Fernandez E, et al.2016. NRF2 orchestrates the metabolic shift during induced pluripotent stem cell reprogramming. Cell Rep. 14(8):1883–91. 10.1016/j.celrep.2016.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He P-J, Hirata M, Yamauchi N, Hashimoto S, Hattori M. 2007. Gonadotropic regulation of circadian clockwork in rat granulosa cells. Mol. Cell. Biochem. 302:111–18. 10.1007/s11010-007-9432-7 [DOI] [PubMed] [Google Scholar]
- He Y, Chen Y, Zhao Q, Tan Z. 2013. Roles of brain and muscle ARNT-like 1 and Wnt antagonist Dkk1 during osteogenesis of bone marrow stromal cells. Cell Prolif. 46(6):644–53. 10.1111/cpr.12075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Lin F, Chen Y, Tan Z, Bai D, Zhao Q. 2015. Overexpression of the circadian clock gene Rev-erbα; affects murine bone mesenchymal stem cell proliferation and osteogenesis. Stem Cells Dev. 24(10):1194–204. 10.1089/scd.2014.0437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog ED, Grace MS, Harrer C, Williamson J, Shinohara K, Block GD. 2000. The role of Clock in the developmental expression of neuropeptides in the suprachiasmatic nucleus. J. Comp. Neurol. 424(1):86–98. [DOI] [PubMed] [Google Scholar]
- Hirayama J, Sahar S, Grimaldi B, Tamaru T, Takamatsu K, et al. 2007. CLOCK-mediated acetylation of BMAL1 controls circadian function. Nature 450(7172):1086–90. 10.1038/nature06394 [DOI] [PubMed] [Google Scholar]
- Houghton FD, Thompson JG, Kennedy CJ, Leese HJ. 1996. Oxygen consumption and energy metabolism of the early mouse embryo. Mol. Reprod. Dev. 44(4):476–85. [DOI] [PubMed] [Google Scholar]
- Hsu Y-C, Li L, Fuchs E. 2014. Emerging interactions between skin stem cells and their niches. Nat. Med. 20(8):847–56. 10.1038/nm.3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T-S, Grodeland G, Sleire L, Wang MY, Kvalheim G, Laerum OD. 2009. Induction of circadian rhythm in cultured human mesenchymal stem cells by serum shock and cAMP analogs in vitro. Chronobiol. Int. 26(2):242–57. 10.1080/07420520902766025 [DOI] [PubMed] [Google Scholar]
- Huang Z, Wei H, Wang X, Xiao J, Li Z, et al. 2020. Icariin promotes osteogenic differentiation of BMSCs by upregulating BMAL1 expression via BMP signaling. Mol. Med. Rep. 21(3): 1590–96. 10.3892/mmr.2020.10954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieyasu A, Tajima Y, Shimba S, Nakauchi H, Yamazaki S. 2014. Clock gene Bmal1 is dispensable for intrinsic properties of murine hematopoietic stem cells. J. Negat. Results BioMed. 13:4. 10.1186/1477-5751-13-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada Y, Uchida H, Umemura Y, Nakamura W, Sakai T, et al.2014. Cell and tissue-autonomous development of the circadian clock in mouse embryos. FEBS Lett. 588(3):459–65. 10.1016/j.febslet.2013.12.007 [DOI] [PubMed] [Google Scholar]
- Isagawa T, Nagae G, Shiraki N, Fujita T, Sato N, et al. 2011. DNA methylation profiling of embryonic stem cell differentiation into the three germ layers. PLOS ONE 6(10):e26052. 10.1371/journal.pone.0026052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isern J, García-García A, Martín AM, Arranz L, Martín-Pérez D, et al. 2014. The neural crest is a source of mesenchymal stem cells with specialized hematopoietic stem cell niche function. eLife 3:e03696. 10.7554/elife.03696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M, Cotsarelis G, Kizawa K, Hamada K. 2004. Hair follicle stem cells in the lower bulge form the secondary germ, a biochemically distinct but functionally equivalent progenitor cell population, at the termination of catagen. Differentiation 72(9–10):548–57. 10.1111/j.1432-0436.2004.07209008.x [DOI] [PubMed] [Google Scholar]
- Ito M, Liu Y, Yang Z, Nguyen J, Liang F, et al. 2005. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat. Med. 11(12):1351–54. 10.1038/nm1328 [DOI] [PubMed] [Google Scholar]
- Janich P, Pascual G, Merlos-Suárez A, Batlle E, Ripperger J, et al. 2011. The circadian molecular clock creates epidermal stem cell heterogeneity. Nature 480(7376):209–14. 10.1038/nature10649 [DOI] [PubMed] [Google Scholar]
- Janich P, Toufighi K, Solanas G, Luis NM, Minkwitz S, et al. 2013. Human epidermal stem cell function is regulated by circadian oscillations. Cell Stem Cell 13(6):745–53. 10.1016/j.stem.2013.09.004 [DOI] [PubMed] [Google Scholar]
- Ji A-R, Ku S-Y, Cho MS, Kim YY, Kim YJ, et al. 2010. Reactive oxygen species enhance differentiation of human embryonic stem cells into mesendodermal lineage. Exp. Mol. Med. 42(3):175–86. 10.3858/emm.2010.42.3.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaasik K, Kivimäe S, Allen JJ, Chalkley RJ, Huang Y, et al. 2013. Glucose sensor O-GlcNAcylation coordinates with phosphorylation to regulate circadian clock. Cell Metab. 17(2):291–302. 10.1016/j.cmet.2012.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang B, Li Y-Y, Chang X, Liu L, Li Y-X. 2008. Modeling the effects of cell cycle M-phase transcriptional inhibition on circadian oscillation. PLOS Comput. Biol. 4(3). 10.1371/journal.pcbi.1000019. Corrigendum. 2008. PLOS Comput. Biol. 4(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimian A, Ahmadi Y, Yousefi B. 2016. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 42:63–71. 10.1016/j.dnarep.2016.04.008 [DOI] [PubMed] [Google Scholar]
- Karman BN, Tischkau SA. 2006. Circadian clock gene expression in the ovary: effects of luteinizing hormone. Biol. Reprod. 75(4):624–32. 10.1095/biolreprod.106.050732 [DOI] [PubMed] [Google Scholar]
- Karpowicz P, Zhang Y, Hogenesch JB, Emery P, Perrimon N. 2013. The circadian clock gates the intestinal stem cell regenerative state. Cell Rep. 3(4):996–1004. 10.1016/j.celrep.2013.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katada S, Sassone-Corsi P. 2010. The histone methyltransferase MLL1 permits the oscillation of circadian gene expression. Nat. Struct. Mol. Biol. 17(12):1414–21. 10.1038/nsmb.1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimiwada T, Sakurai M, Ohashi H, Aoki S, Tominaga T, Wada K. 2009. Clock genes regulate neurogenic transcription factors, including NeuroD1, and the neuronal differentiation of adult neural stem/progenitor cells. Neurochem. Int. 54(5–6):277–85. 10.1016/j.neuint.2008.12.005 [DOI] [PubMed] [Google Scholar]
- Kleitman N, Engelmann TG. 1953. Sleep characteristics of infants. J. Appl. Physiol. 6(5):269–82. 10.1152/jappl.1953.6.5.269 [DOI] [PubMed] [Google Scholar]
- Ko MS, Kitchen JR, Wang X, Threat TA, Wang X, et al. 2000. Large-scale cDNA analysis reveals phased gene expression patterns during pre-implantation mouse development. Development 127:1737–49 [DOI] [PubMed] [Google Scholar]
- Kollet O, Vagima Y, D’Uva G, Golan K, Canaani J, et al. 2013. Physiologic corticosterone oscillations regulate murine hematopoietic stem/progenitor cell proliferation and CXCL12 expression by bone marrow stromal progenitors. Leukemia 27(10):2006–15. 10.1038/leu.2013.154 [DOI] [PubMed] [Google Scholar]
- Krishnaiah SY, Wu G, Altman BJ, Growe J, Rhoades SD, et al. 2017. Clock regulation of metabolites reveals coupling between transcription and metabolism. Cell Metab. 25(4):961–74.e4. 10.1016/j.cmet.2017.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, D’Souza SS, Thakur AS. 2019. Understanding the journey of human hematopoietic stem cell development. Stem Cells Int. 2019:2141475. 10.1155/2019/2141475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahne M, Nagashima M, Hyde DR, Hitchcock PF. 2020. Reprogramming Müller glia to regenerate retinal neurons. Annu. Rev. Vis. Sci. 6:171–93. 10.1146/annurev-vision-121219-081808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf D, Achten C, Dallman F, Oster H. 2015. Embryonic development and maternal regulation of murine circadian clock function. Chronobiol. Int. 32(3):416–27. 10.3109/07420528.2014.986576 [DOI] [PubMed] [Google Scholar]
- Lee DA, Bedont JL, Pak T, Wang H, Song J, et al. 2012. Tanycytes of the hypothalamic median eminence form a diet-responsive neurogenic niche. Nat. Neurosci. 15(5):700–2. 10.1038/nn.3079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Lahens NF, Zhang S, Bedont J, Field JF, Sehgal A. 2019. G1/S cell cycle regulators mediate effects of circadian dysregulation on tumor growth and provide targets for timed anticancer treatment. PLOS Biol. 17(4). 10.1371/journal.pbio.3000228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Lee J, Kwon I, Nakajima Y, Ohmiya Y, et al. 2010. Coactivation of the CLOCK-BMAL1 complex by CBP mediates resetting of the circadian clock. J. Cell Sci. 123(20):3547–57. 10.1242/jcs.070300 [DOI] [PubMed] [Google Scholar]
- Lee YL, Peng Q, Fong SW, Chen ACH, Lee KF, et al. 2012. Sirtuin 1 facilitates generation of induced pluripotent stem cells from mouse embryonic fibroblasts through the MiR-34a and p53 pathways. PLOS ONE 7(9):e45633. 10.1371/journal.pone.0045633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees JG, Gardner DK, Harvey AJ. 2020. Nicotinamide adenine dinucleotide induces a bivalent metabolism and maintains pluripotency in human embryonic stem cells. Stem Cells 38(5):625–38. 10.1002/stem.3152 [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, et al. 2007. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445(7124):168–76. 10.1038/nature05453 [DOI] [PubMed] [Google Scholar]
- Li M-D, Ruan H-B, Hughes ME, Lee J-S, Singh JP, et al. 2013. O-GlcNAc signaling entrains the circadian clock by inhibiting BMAL1/CLOCK ubiquitination. Cell Metab. 17(2):303–10. 10.1016/j.cmet.2012.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Liu F, Zhang C, Zhang Z, Guo J, et al. 2009. Pluripotent hair follicle neural crest stem-cell-derived neurons and Schwann cells functionally repair sciatic nerves in rats.Mol.Neurobiol.40(3):216–23. 10.1007/s12035-009-8082-z [DOI] [PubMed] [Google Scholar]
- Lin S, Guo J, Chen S. 2019. Transcriptome and DNA methylome signatures associated with retinal Müller glia development, injury response, and aging. Invest. Opthalmol. Visual Sci. 60(13):4436. 10.1167/iovs.19-27361 [DOI] [PubMed] [Google Scholar]
- Lipton JO, Yuan ED, Boyle LM, Ebrahimi-Fakhari D, Kwiatkowski E, et al. 2015. The circadian protein BMAL1 regulates translation in response to S6K1-mediated phosphorylation. Cell 161(5):1138–51. 10.1016/j.cell.2015.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cai Y, Sothern RB, Guan Y, Chan P. 2007. Chronobiological analysis of circadian patterns in transcription of seven key clock genes in six peripheral tissues in mice. Chronobiol. Int. 24(5):793–820. 10.1080/07420520701672556 [DOI] [PubMed] [Google Scholar]
- Lu C, Yang Y, Zhao R, Hua B, Xu C, et al. 2016. Role of circadian gene clock during differentiation of mouse pluripotent stem cells. Protein Cell 7(11):820–32. 10.1007/s13238-016-0319-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas D, Battista M, Shi PA, Isola L, Frenette PS, et al.2008.Mobilized hematopoietic stem cell yield depends on species-specific circadian timing. Cell Stem Cell 3(4):364–66. 10.1016/j.stem.2008.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney MM. 2010. Shift work, jet lag, and female reproduction. Int. J. Endocrinol. 2010:813764. 10.1155/2010/813764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik A, Jamasbi RJ, Kondratov RV, Geusz ME. 2015b. Development of circadian oscillators in neurosphere cultures during adult neurogenesis. PLOS ONE 10(3):e0122937. 10.1371/journal.pone.0122937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik A, Kondratov RV, Jamasbi RJ, Geusz ME. 2015a. Circadian clock genes are essential for normal adult neurogenesis, differentiation, and fate determination. PLOS ONE 10(10):e0139655. 10.1371/journal.pone.0139655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maryanovich M, Zahalka AH, Pierce H, Pinho S, Nakahara F, et al. 2018. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat. Med. 24(6):782–91. 10.1038/s41591-018-0030-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri S, Patel VR, Eckel-Mahan KL, Peleg S, Forne I, et al. 2013. Circadian acetylome reveals regulation of mitochondrial metabolic pathways. PNAS 110(9):3339–44. 10.1073/pnas.1217632110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri S, Rigor P, Cervantes M, Ceglia N, Sebastian C, et al. 2014. Partitioning circadian transcription by SIRT6 leads to segregated control of cellular metabolism. Cell 158(3):659–72. 10.1016/j.cell.2014.06.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri S, Sassone-Corsi P. 2010. Plasticity and specificity of the circadian epigenome. Nat. Neurosci. 13(11):1324–29. 10.1038/nn.2668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu J, Ruohola-Baker H. 2017. Metabolic remodeling during the loss and acquisition of pluripotency. Development 144(4):541–51. 10.1242/dev.128389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsu-ura T, Dovzhenok A, Aihara E, Rood J, Le H, et al. 2016. Intercellular coupling of the cell cycle and circadian clock in adult stem cell culture. Mol. Cell 64(5):900–12. 10.1016/j.molcel.2016.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo T 2003. Control mechanism of the circadian clock for timing of cell division in vivo. Science 302(5643):255–59. 10.1126/science.1086271 [DOI] [PubMed] [Google Scholar]
- Mauvoisin D, Wang J, Jouffe C, Martin E, Atger F, et al. 2014. Circadian clock-dependent and -independent rhythmic proteomes implement distinct diurnal functions in mouse liver. PNAS 111(1):167–72. 10.1073/pnas.1314066111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQueen CM, Schmitt EE, Sarkar TR, Elswood J, Metz RP, et al. 2018. PER2 regulation of mammary gland development. Development 145(6):dev157966. 10.1242/dev.157966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez-Ferrer S, Lucas D, Battista M, Frenette PS. 2008. Haematopoietic stem cell release is regulated by circadian oscillations. Nature 452(7186):442–47. 10.1038/nature06685 [DOI] [PubMed] [Google Scholar]
- Méndez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, MacArthur BD, et al. 2010. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466(7308):829–34. 10.1038/nature09262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menet JS, Pescatore S, Rosbash M. 2014. CLOCK:BMAL1 is a Pioneer-like transcription factor. Genes Dev. 28(1):8–13. 10.1101/gad.228536.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereness AL, Murphy ZC, Forrestel AC, Butler S, Ko C, et al. 2016. Conditional deletion of Bmal1 in ovarian theca cells disrupts ovulation in female mice. Endocrinology 157(2):913–27. 10.1210/en.2015-1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse D, Cermakian N, Brancorsini S, Parvinen M, Sassone-Corsi P. 2003. No circadian rhythms in testis: Period1 expression is Clock independent and developmentally regulated in the mouse. Mol. Endocrinol. 17(1):141–51. 10.1210/me.2002-0184 [DOI] [PubMed] [Google Scholar]
- Moussaieff A, Rouleau M, Kitsberg D, Cohen M, Levy G, et al. 2015. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 21(3):392–402. 10.1016/j.cmet.2015.02.002 [DOI] [PubMed] [Google Scholar]
- Mulligan P, Yang F, Di Stefano L, Ji J-Y, Ouyang J, et al. 2011. A SIRT1-LSD1 corepressor complex regulates Notch target gene expression and development. Mol. Cell 42(5):689–99. 10.1016/j.molcel.2011.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray IR,West CC, Hardy WR, James AW, Park TS, et al. 2014. Natural history of mesenchymal stem cells, from vessel walls to culture vessels. Cell. Mol. Life Sci. 71(8):1353–74. 10.1007/s00018-013-1462-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najafzadeh N, Esmaeilzade B, Imcheh MD. 2015. Hair follicle stem cells: in vitro and in vivo neural differentiation. World J. Stem Cells 7(5):866. 10.4252/wjsc.v7.i5.866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. 2009. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 324(5927):654–57. 10.1126/science.1170803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura TJ, Sellix MT, Menaker M, Block GD. 2008. Estrogen directly modulates circadian rhythms of PER2 expression in the uterus. Endocrinol. Metab. 295(5):E1025–31. 10.1152/ajpendo.90392.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam HJ, Boo K, Kim D, Han D-H, Choe HK, et al. 2014. Phosphorylation of LSD1 by PKCα is crucial for circadian rhythmicity and phase resetting. Mol. Cell 53(5):791–805. 10.1016/j.molcel.2014.01.028 [DOI] [PubMed] [Google Scholar]
- Naruse Y, Oh-hashi K, Iijima N, Naruse M, Yoshioka H, Tanaka M. 2004. Circadian and light-induced transcription of clock gene Per1 depends on histone acetylation and deacetylation. Mol. Cell. Biol. 24(14):6278–87. 10.1128/mcb.24.14.6278-6287.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neilsen BK, Frodyma DE, McCall JL, Fisher KW, Lewis RE. 2019. ERK-mediated TIMELESS expression suppresses G2/M arrest in colon cancer cells. PLOS ONE 14(1):e0209224. 10.1371/journal.pone.0209224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld-Cohen A, Robles MS, Aviram R, Manella G, Adamovich Y, et al. 2016. Circadian control of oscillations in mitochondrial rate-limiting enzymes and nutrient utilization by PERIOD proteins. PNAS 113(12):E1673–82. 10.1073/pnas.1519650113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Y, Han Y-C, Z Y-R. 2008. CXCR4 is required for the quiescence of primitive hematopoietic cells. J. Exp. Med. 205(4):777–83. 10.1084/jem.20072513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooto S, Akagi T, Kageyama R, Akita J, Mandai M, et al. 2004. Potential for neural regeneration after neurotoxic injury in the adult mammalian retina. PNAS 101(37):13654–59. 10.1073/pnas.0402129101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou J, Li H, Qiu P, Li Q, Chang H-C, Tang Y-C. 2019. CDK9 modulates circadian clock by attenuating REV-ERBα activity. Biochem. Biophys. Res. Commun. 513(4):967–73. 10.1016/j.bbrc.2019.04.043 [DOI] [PubMed] [Google Scholar]
- Pagel R, Bär F, Schröder T, Sünderhauf A, Künstner A, et al. 2017. Circadian rhythm disruption impairs tissue homeostasis and exacerbates chronic inflammation in the intestine. FASEB J. 31(11):4707–19. 10.1096/fj.201700141rr [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paijmans J, Bosman M, ten Wolde PR, Lubensky DK. 2016. Discrete gene replication events drive coupling between the cell cycle and circadian clocks. PNAS 113(15):4063–68. 10.1073/pnas.1507291113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parasram K, Bernardon N, Hammoud M, Chang H, He L, et al. 2018. Intestinal stem cells exhibit conditional circadian clock function. Stem Cell Rep. 11(5):1287–301. 10.1016/j.stemcr.2018.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parasram K, Karpowicz P. 2020. Time after time: circadian clock regulation of intestinal stem cells. Cell. Mol. Life Sci. 77(7):1267–88. 10.1007/s00018-019-03323-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patke A, Young MW, Axelrod S. 2020. Molecular mechanisms and physiological importance of circadian rhythms. Nat. Rev. Mol. Cell Biol. 21(2):67–84. 10.1038/s41580-019-0179-2 [DOI] [PubMed] [Google Scholar]
- Pauklin S, Vallier L. 2013. The cell-cycle state of stem cells determines cell fate propensity.Cell 155(1):135–47. 10.1016/j.cell.2013.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek CB, Affinati AH, Moynihan Ramsey K, Kuo H-Y, Yu W, et al. 2013. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 342(6158):1243417. 10.1126/science.1243417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Martín M, Cifuentes M, Grondona JM, López-Ávalos MD, Gómez-Pinedo U, et al. 2010. IGF-I stimulates neurogenesis in the hypothalamus of adult rats. Eur. J. Neurosci. 31(9):1533–48. 10.1111/j.1460-9568.2010.07220.x [DOI] [PubMed] [Google Scholar]
- Pérez-Roger I, Solomon DLC, Sewing A, Land H. 1997. Myc activation of cyclin E/Cdk2 kinase involves induction of cyclin E gene transcription and inhibition of p27Kip1 binding to newly formed complexes. Oncogene 14(20):2373–81. 10.1038/sj.onc.1201197 [DOI] [PubMed] [Google Scholar]
- Pico MJ, Hashemi S, Xu F, Nguyen KH, Donnelly R, et al. 2016. Glucocorticoid receptor-mediated cis-repression of osteogenic genes requires BRM-SWI/SNF.Bone Rep.5:222–27. 10.1016/j.bonr.2016.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittelli M, Felici R, Pitozzi V, Giovanelli L, Gigagli E, et al.2011. Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol. Pharmacol. 80(6):1136–46. 10.1124/mol.111.073916 [DOI] [PubMed] [Google Scholar]
- Plikus MV, Baker RE, Chen C-C, Fare C, de la Cruz D, et al. 2011. Self-organizing and stochastic behaviors during the regeneration of hair stem cells. Science 332(6029):586–89. 10.1126/science.1201647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plikus MV, Vollmers C, de la Cruz D, Chaix A, Ramos R, et al. 2013. Local circadian clock gates cell cycle progression of transient amplifying cells during regenerative hair cycling. PNAS 110(23):E2106–15. 10.1073/pnas.1215935110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puram RV, Kowalczyk MS, de Boer CG, Schneider RK, Miller PG, et al. 2016. Core circadian clock genes regulate leukemia stem cells in AML. Cell 165(2):303–16. 10.1016/j.cell.2016.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, et al. 2009. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 324(5927):651–54. 10.1126/science.1171641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repouskou A, Prombona A. 2016. c-MYC targets the central oscillator gene Per1 and is regulated by the circadian clock at the post-transcriptional level. Biochim. Biophys. Acta Gene Regul. Mech. 1859(4):541–52. 10.1016/j.bbagrm.2016.02.001 [DOI] [PubMed] [Google Scholar]
- Rey G, Valekunja UK, Feeney KA, Wulund L, Milev NB, et al. 2016. The pentose phosphate pathway regulates the circadian clock. Cell Metab. 24(3):462–73. 10.1016/j.cmet.2016.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rienstein S, Dotan A, Avivi L, Ashkenazi I. 1998. Daily rhythms in male mice meiosis. Chronobiol. Int. 15(1):13–20. 10.3109/07420529808998665 [DOI] [PubMed] [Google Scholar]
- Ripperger JA, Schibler U. 2006. Rhythmic CLOCK-BMAL1 binding to multiple E-box motifs drives circadian Dbp transcription and chromatin transitions. Nat. Genet. 38(3):369–74. 10.1038/ng1738 [DOI] [PubMed] [Google Scholar]
- Rivkees SA. 2003. Developing circadian rhythmicity in infants. Pediatrics 112(2):373–81. 10.1542/peds.112.2.373 [DOI] [PubMed] [Google Scholar]
- Robins SC, Stewart I, McNay DE, Taylor V, Giachino C, et al. 2013. α-Tanycytes of the adult hypothalamic third ventricle include distinct populations of FGF-responsive neural progenitors. Nat. Commun. 4(1):2049. 10.1038/ncomms3049 [DOI] [PubMed] [Google Scholar]
- Rodriguez EM, Blázquez JL, Pastor FE, Peláez B, Peña P, et al. 2005. Hypothalamic tanycytes: a key component of brain-endocrine interaction. Int. Rev. Cytol. 247:89–164. 10.1016/s00747696(05)47003-5 [DOI] [PubMed] [Google Scholar]
- Rompolas P, Mesa KR, Greco V. 2013. Spatial organization within a niche as a determinant of stem-cell fate. Nature 502(7472):513–18. 10.1038/nature12602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Zhang D, Allis CD, Zhao Y. 2017. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 18(2):90–101. 10.1038/nrm.2016.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahar S, Masubuchi S, Eckel-Mahan, Vollmer S, Galla L, et al. 2014. Circadian control of fatty acid elongation by SIRT1 protein-mediated deacetylation of acetyl-coenzyme A synthetase 1. J. Biol. Chem. 289(9):6091–97. 10.1074/jbc.m113.537191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samsa WE, Vasanji A, Midura RJ, Kondratov RV. 2016.Deficiency of circadian clock protein BMAL1 in mice results in a low bone mass phenotype. Bone 84:194–203. 10.1016/j.bone.2016.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassone-Corsi P 2016. The epigenetic and metabolic language of the circadian clock. In A Time for Metabolism and Hormones, ed. Sassone-Corsi P, Christen Y, pp. 1–11. Cham, Switz.: Springer. 10.1007/978-3-319-27069-2_1 [DOI] [PubMed] [Google Scholar]
- Sauer H, Wartenberg M. 2005. Reactive oxygen species as signaling molecules in cardiovascular differentiation of embryonic stem cells and tumor-induced angiogenesis. Antioxid. Redox Signal. 7(11–12):1423–34. 10.1089/ars.2005.7.1423 [DOI] [PubMed] [Google Scholar]
- Sawant OB, Horton AM, Zucaro OF, Chan R, Bonilha VL, et al. 2017. The circadian clock gene Bmal1 controls thyroid hormone-mediated spectral identity and cone photoreceptor function. Cell Rep. 21(3):692–706. 10.1016/j.celrep.2017.09.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawant OB, Jidigam VK, Fuller RD, Zucaro OF, Kpegba C, et al. 2019. The circadian clock gene Bmal1 is required to control the timing of retinal neurogenesis and lamination of Müller glia in the mouse retina. FASEB J. 33(8):8745–58. 10.1096/fj.201801832rr [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheving LE, Burns ER, Pauly JE, Tsai T-H. 1978. Circadian variation in cell division of the mouse alimentary tract, bone marrow and corneal epithelium. Anat. Record 191(4):479–86. 10.1002/ar.1091910407 [DOI] [PubMed] [Google Scholar]
- Schnell A, Chappuis S, Schmutz I, Brai E, Ripperger JA, et al. 2014. The nuclear receptor REV-ERBα regulates Fabp7 and modulates adult hippocampal neurogenesis. PLOS ONE 9(6):e99883. 10.1371/journal.pone.0099883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrima R, Cela O, Merla G, Augello B, Rubino R, et al. 2016. Clock-genes and mitochondrial respiratory activity: evidence of a reciprocal interplay. Biochim. Biophys. Acta Bioenerg. 1857(8):1344–51. 10.1016/j.bbabio.2016.03.035 [DOI] [PubMed] [Google Scholar]
- Sellix MT, Menaker M. 2010. Circadian clocks in the ovary. Trends Endocrinol. Metab. 21(10):628–36. 10.1016/j.tem.2010.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serón-Ferré M, Mendez N, Abarzua-Catalan, Vilches N, Valenzuela FJ, et al. 2012. Circadian rhythms in the fetus. Mol. Cell. Endocrinol. 349(1):68–75. 10.1016/j.mce.2011.07.039 [DOI] [PubMed] [Google Scholar]
- Shen Q, Wang Y, Dimos JT, Fasano CA, Phoenix TN, et al. 2006. The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells. Nat. Neurosci. 9(6):743–51. 10.1038/nn1694 [DOI] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, et al. 2004.Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119(7):941–53. 10.1016/j.cell.2004.12.012 [DOI] [PubMed] [Google Scholar]
- Shimazu T, Hirschey MD, Huang J-Y, Ho LTY, Verdin E, et al. 2010. Acetate metabolism and aging: an emerging connection. Mech. Ageing Dev. 131(7–8):511–16. 10.1016/j.mad.2010.05.001 [DOI] [PubMed] [Google Scholar]
- Shimozaki K 2018. Involvement of nuclear receptor REV-ERBβ in formation of neurites and proliferation of cultured adult neural stem cells. Cell. Mol. Neurobiol. 38(5):1051–65. 10.1007/s10571-018-0576-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraki N, Shiraki Y, Tsuyama T, Obata F, Miura M, et al. 2014. Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab.19(5):780–94. 10.1016/j.cmet.2014.03.017 [DOI] [PubMed] [Google Scholar]
- Shostak A, Ruppert B, Ha N, Bruns P, Toprak UH, et al. 2016. MYC/MIZ1-dependent gene repression inversely coordinates the circadian clock with cell cycle and proliferation. Nat. Commun. 7:11807. 10.1038/ncomms11807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, et al. 2013. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 339(6116):222–26. 10.1126/science.1226603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sifuentes CJ, Kim J-W, Swaroop A, Raymond PA. 2016. Rapid, dynamic activation of Müller glial stem cell responses in zebrafish. Invest. Opthalmol. Visual Sci. 57(13):5148. 10.1167/iovs.16-19973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver R, Sookhoo AI, LeSauter J, Stevens P, Jansen HT, Lehman MN. 1999. Multiple regulatory elements result in regional specificity in circadian rhythms of neuropeptide expression in mouse SCN. NeuroReport 10(15):3165–74. 10.1097/00001756-199910190-00008 [DOI] [PubMed] [Google Scholar]
- Simsek T, Kocabas F, Zheng J, DeBerardinis RJ, Mahmoud AI, et al. 2010. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 7(3):380–90. 10.1016/j.stem.2010.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh KP, Garrett WR, Casado FL, Gasiewicz TA. 2011. Aryl hydrocarbon receptor-null allele mice have hematopoietic stem/progenitor cells with abnormal characteristics and functions. Stem Cells Dev. 20(5):769–84. 10.1089/scd.2010.0333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smaaland R, Laerum OD, Sothern RB, Sletvold O, Bjerknes R, Lote K. 1992. Colony-forming unit-granulocyte-macrophage and DNA synthesis of human bone marrow are circadian stage-dependent and show covariation. Blood 79(9):2281–87. 10.1182/blood.v79.9.2281.2281 [DOI] [PubMed] [Google Scholar]
- Smith E, Shilatifard A.2010.The chromatin signaling pathway: diverse mechanisms of recruitment of histone-modifying enzymes and varied biological outcomes. Mol. Cell 40(5):689–701. 10.1016/j.molcel.2010.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smukler SR, Runciman SB, Xu S, van der Kooy D. 2006. Embryonic stem cells assume a primitive neural stem cell fate in the absence of extrinsic influences. J. Cell Biol. 172(1):79–90. 10.1083/jcb.200508085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperber H, Mathieu J, Wang Y, Ferreccio A, Hesson J, et al. 2015. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat. Cell Biol. 17(12):1523–35. 10.1038/ncb3264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenzinger M, Karpova D, Unterrainer C, Harenkamp S, Wiercinska E, et al. 2019. Hematopoietic-extrinsic cues dictate circadian redistribution of mature and immature hematopoietic cells in blood and spleen. Cells 8(9):1033. 10.3390/cells8091033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes K, Cooke A, Chang H, Weaver DR, Breault DT, Karpowicz P. 2017. The circadian clock gene BMAL1 coordinates intestinal regeneration. Cell. Mol. Gastroenterol. Hepatol. 4(1):95–114. 10.1016/j.jcmgh.2017.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone RA, McGlinn AM, Chakraborty R, Lee DC, Yang V, et al. 2019. Altered ocular parameters from circadian clock gene disruptions. PLOS ONE 14(6):e0217111. 10.1371/journal.pone.0217111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Yang Z, Niu Z, Peng J, Li Q, et al. 2006. MOP3, a component of the molecular clock, regulates the development of B cells. Immunology 119(4):451–60. 10.1111/j.1365-2567.2006.02456.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4):663–76. 10.1016/j.cell.2006.07.024 [DOI] [PubMed] [Google Scholar]
- Takahata S, Ozaki T, Mimura J, Kikuchi Y, Sogawa K, Fujii-Kuriyama Y. 2000. Transactivation mechanisms of mouse clock transcription factors, MClock and mArnt3. Genes Cells 5(9):739–47. 10.1046/j.1365-2443.2000.00363.x [DOI] [PubMed] [Google Scholar]
- Tamai S-I, Sanada K, Fukada Y. 2008. Time-of-day-dependent enhancement of adult neurogenesis in the hippocampus. PLOS ONE 3(12):e3835. 10.1371/journal.pone.0003835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Huang G, Fan W, Chen Y, Ward JM, et al.2014.SIRT1-mediated deacetylation of CRABPII regulates cellular retinoic acid signaling and modulates embryonic stem cell differentiation. Mol. Cell 55(6):843–55. 10.1016/j.molcel.2014.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Xu T, Li Y, Fei W, Yang G, Hong Y. 2020. Inhibition of CRY2 by STAT3/MiRNA-7–5p promotes osteoblast differentiation through upregulation of CLOCK/BMAL1/P300 expression. Mol. Ther. Nucleic Acids 19:865–76. 10.1016/j.omtn.2019.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennen RI, Berber E, Chua KF. 2010. Functional dissection of SIRT6: identification of domains that regulate histone deacetylase activity and chromatin localization. Mech. Ageing Dev. 131(3):185–92. 10.1016/j.mad.2010.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- TeSlaa T, Chaikovsky AC, Lipchina I, Escobar SL, Hochedlinger K, et al. 2016. α-Ketoglutarate accelerates the initial differentiation of primed human pluripotent stem cells. Cell Metab. 24(3):485–93. 10.1016/j.cmet.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thummel R, Kassen SC, Enright JM, Nelson CM, Montgomery JE, et al. 2008. Characterization of Müller glia and neuronal progenitors during adult zebrafish retinal regeneration. Exp. Eye Res. 87(5):433–44. 10.1016/j.exer.2008.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischler J, Gruhn WH, Reid J, Allgeyer E, Buettner F, et al. 2019. Metabolic regulation of pluripotency and germ cell fate through α-ketoglutarate. EMBO J. 38:e99518. 10.15252/embj.201899518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynard P, Feillet C, Soliman S, Delaunay F, Fages F. 2016. Model-based investigation of the circadian clock and cell cycle coupling in mouse embryonic fibroblasts: prediction of RevErb-α up-regulation during mitosis. Biosystems 149:59–69. 10.1016/j.biosystems.2016.07.003 [DOI] [PubMed] [Google Scholar]
- Tsang K, Liu H, Yang Y, Charles JF, Ermann J.2019.Defective circadian control in mesenchymal cells reduces adult bone mass in mice by promoting osteoclast function. Bone 121:172–80. 10.1016/j.bone.2019.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsinkalovsky O, Filipski E, Rosenlund B, Sothern RB, Eiken HG, et al. 2006. Circadian expression of clock genes in purified hematopoietic stem cells is developmentally regulated in mouse bone marrow. Exp. Hematol. 34(9):1248–60. 10.1016/j.exphem.2006.05.008 [DOI] [PubMed] [Google Scholar]
- Tsogtbaatar E, Landin C, Minter-Dykhouse K, Folmes CDL. 2020. Energy metabolism regulates stem cell pluripotency. Front. Cell Dev. Biol. 8:87. 10.3389/fcell.2020.00087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, et al. 2005. Obesity and metabolic syndrome in circadian clock mutant mice. Science 308(5724):1043–45. 10.1126/science.1108750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DA, Trott J, Hayward P, Rué P, Martinez Arias A. 2014. An interplay between extracellular signalling and the dynamics of the exit from pluripotency drives cell fate decisions in mouse ES cells. Biol. Open 3(7):614–26. 10.1242/bio.20148409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda HR, Hayashi S, Chen W, Sano M, Machida M, et al. 2005. System-level identification of transcriptional circuits underlying mammalian circadian clocks. Nat. Genet. 37(2):187–92. 10.1038/ng1504 [DOI] [PubMed] [Google Scholar]
- Umemura Y, Koike N, Ohashi M, Tsuchiya Y, Meng QJ, et al. 2017. Involvement of posttranscriptional regulation of Clock in the emergence of circadian clock oscillation during mouse development. PNAS 114(36):E7479–88. 10.1073/pnas.1703170114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemura Y, Maki I, Tsuchiya Y, Koike N, Yagita K. 2019. Human circadian molecular oscillation development using induced pluripotent stem cells. J. Biol. Rhythms 34(5):525–32. 10.1177/0748730419865436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemura Y, Noike N, Matsumoto T, Yoo S-H, Chen Z, et al. 2014. Transcriptional program of Kpna2/Importin-α2 regulates cellular differentiation-coupled circadian clock development in mammalian cells. PNAS 111(47):E5039–48. 10.1073/pnas.1419272111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemura Y, Yagita K. 2020. Development of the circadian core machinery in mammals. J. Mol. Biol. 432(12):3611–17. 10.1016/j.jmb.2019.11.026 [DOI] [PubMed] [Google Scholar]
- Ünsal-Kaçmaz K, Mullen TE, Kaufmann WK, Sancar A. 2005. Coupling of human circadian and cell cycles by the Timeless protein. Mol. Cell. Biol. 25(8):3109–16. 10.1128/mcb.25.8.3109-3116.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Oudenhove JJ, Grandy RA, Ghule PN, del Rio R, Lian JB, et al. 2016. Lineage-specific early differentiation of human embryonic stem cells requires a G2 cell cycle pause. Stem Cells 34(7):1765–75. 10.1002/stem.2352. Erratum. 2016. Stem Cells 34(10):2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizán P, Beringer M, Ballaré C, Di Croce L. 2015. Role of PRC2-associated factors in stem cells and disease. FEBS J. 282(9):1723–35. 10.1111/febs.13083 [DOI] [PubMed] [Google Scholar]
- Wang J, Lazar MA. 2008. Bifunctional role of Rev-Erbα in adipocyte differentiation. Mol. Cell. Biol. 28(7):2213–20. 10.1128/mcb.01608-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Morita Y, Han B, Niemann S, Löffler B, Rudolph KL. 2016. Per2 induction limits lymphoid-biased haematopoietic stem cells and lymphopoiesis in the context of DNA damage and ageing. Nat. Cell Biol. 18(5):480–90. 10.1038/ncb3342 [DOI] [PubMed] [Google Scholar]
- Wang Z, Fan M, Candas D, Zhang T-Q, Qin L, et al. 2014. Cyclin B1/Cdk1 coordinates mitochondrial respiration for cell-cycle G2/M progression. Dev. Cell 29(2):217–32. 10.1016/j.devcel.2014.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger L, Ayyash M, Novershtern N, Hanna JH. 2016. Dynamic stem cell states: naive to primed pluripotency in rodents and humans. Nat. Rev. Mol. Cell Biol.17(3):155–69. 10.1038/nrm.2015.28 [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. 2009. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324(5930):1076–80. 10.1126/science.1164097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiles MV, Johansson BM. 1999. Embryonic stem cell development in a chemically defined medium. Exp. Cell Res. 247(1):241–48. 10.1006/excr.1998.4353 [DOI] [PubMed] [Google Scholar]
- Wreschnig D, Dolatshad H, Davis FC. 2014. Embryonic development of circadian oscillations in the mouse hypothalamus. J. Biol. Rhythms 29(4):299–310. 10.1177/0748730414545086 [DOI] [PubMed] [Google Scholar]
- Wu X, Yu G, Parks H, Hebert T, Goh BC, et al. 2008. Circadian mechanisms in murine and human bone marrow mesenchymal stem cells following dexamethasone exposure. Bone 42(5):861–70. 10.1016/j.bone.2007.12.226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Ruan G, Dai H, Liu AC, Penn J, McMahon DG. 2016. Mammalian retinal Müller cells have circadian clock function. Mol. Vis. 22:275–83 [PMC free article] [PubMed] [Google Scholar]
- Xu L, Wu T, Li H, Ni Y, Fu Z. 2017. An individual 12-h shift of the light-dark cycle alters the pancreatic and duodenal circadian rhythm and digestive function. Acta Biochim. Biophys. Sin. 49(10):954–61. 10.1093/abbs/gmx084 [DOI] [PubMed] [Google Scholar]
- Xu Z, Robitaille AM, Berndt JD, Davidson KC, Fischer KA, et al. 2016. Wnt/β-catenin signaling promotes self-renewal and inhibits the primed state transition in naïve human embryonic stem cells. PNAS 113(42):E6382–90. 10.1073/pnas.1613849113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagita K, Horie K, Koinuma S, Nakamura W, Yamanaka I, et al. 2010. Development of the circadian oscillator during differentiation of mouse embryonic stem cells in vitro. PNAS 107(8):3846–51. 10.1073/pnas.0913256107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki S, Numano R, Abe M, Hida A, Takahashi R, et al. 2000. Resetting central and peripheral circadian oscillators in transgenic rats. Science 288(5466):682–85. 10.1126/science.288.5466.682 [DOI] [PubMed] [Google Scholar]
- YanJ Goldbeter A. 2019. Robust synchronization of the cell cycle and the circadian clock through bidirectional coupling. J. R. Soc. Interface 16(158):20190376. 10.1098/rsif.2019.0376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Chen L, Grant GR, Paschos G, Song W-L, et al. 2016. Timing of expression of the core clock gene Bmal1 influences its effects on aging and survival. Sci. Transl. Med. 8(324):324ra16. 10.1126/scitranslmed.aad3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Wood PA, Hrushesky WJM. 2010. Mammalian TIMELESS is required for ATM-dependent CHK2 activation and G2/M checkpoint control. J. Biol. Chem. 285(5):3030–34. 10.1074/jbc.m109.050237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuo S, Von Gall C, Weaver DR, Korf H-W. 2008. Rhythmic expression of clock genes in the ependymal cell layer of the third ventricle of rodents is independent of melatonin signaling.Eur.J.Neurosci.28(12):2443–50. 10.1111/j.1460-9568.2008.06541.x [DOI] [PubMed] [Google Scholar]
- Yoo S, Blackshaw S. 2018. Regulation and function of neurogenesis in the adult mammalian hypothalamus. Prog. Neurobiol. 170:53–66. 10.1016/j.pneurobio.2018.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa T, Sellix M, Pezuk P, Menaker M. 2009. Timing of the ovarian circadian clock is regulated by gonadotropins. Endocrinology 150(9):4338–47. 10.1210/en.2008-1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JM, Wu X, Gimble JM, Guan X, Freitas MA, Bunnell BA. 2011. Age-related changes in mesenchymal stem cells derived from rhesus macaque bone marrow. Aging Cell 10(1):66–79. 10.1111/j.1474-9726.2010.00646.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Ma S, Rachmin I, He M, Baral P, et al. 2020. Hyperactivation of sympathetic nerves drives depletion of melanocyte stem cells. Nature 577(7792):676–81. 10.1038/s41586-020-1935-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Khvorostov I, Hong JS, Oktay Y, Vergnes L, et al. 2011. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 30(24):4860–73. 10.1038/emboj.2011.401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhang Z, Wang F, Tian X, Ji P, Liu G. 2017. Effects of melatonin administration on embryo implantation and offspring growth in mice under different schedules of photoperiodic exposure. Reprod. Biol. Endocrinol. 15(1):78. 10.1186/s12958-017-0297-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Choi M, Margineantu D, Margaretha L, Hesson J, et al. 2012. HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/HESC transition. EMBO J. 31(9):2103–16. 10.1038/emboj.2012.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo H, Wang Y, Zhao Q. 2018. The interaction between Bmal1 and Per2 in mouse BMSC osteogenic differentiation. Stem Cells Int. 2018:3407821. 10.1155/2018/3407821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirra A, Wiethoff S, Patani R. 2016. Neural conversion and patterning of human pluripotent stem cells: a developmental perspective. Stem Cells Int. 2016:8291260. 10.1155/2016/8291260 [DOI] [PMC free article] [PubMed] [Google Scholar]