

Summary
Alzheimer’s disease (AD) is associated with both extracellular amyloid-β (Aβ) plaques and intracellular tau-containing neurofibrillary tangles (NFT). We characterized the behavioral, metabolic and lipidomic phenotype of the 5xFADxTg30 mouse model which contains overexpression of both Aβ and tau. Our results independently reproduce several phenotypic traits described previously for this model, while providing additional characterization. This model develops many aspects associated with AD including frailty, decreased survival, initiation of aspects of cognitive decline and alterations to specific lipid classes and molecular lipid species in the plasma and brain. Notably, some sex-specific differences exist in this model and motor impairment with aging in this model does compromise the utility of the model for some movement-based behavioral assessments of cognitive function. These findings provide a reference for individuals interested in using this model to understand the pathology associated with elevated Aβ and tau or for testing potential therapeutics for the treatment of AD.
Subject areas: Animal physiology, Neuroscience, Behavioral neuroscience, Lipidomics
Graphical abstract
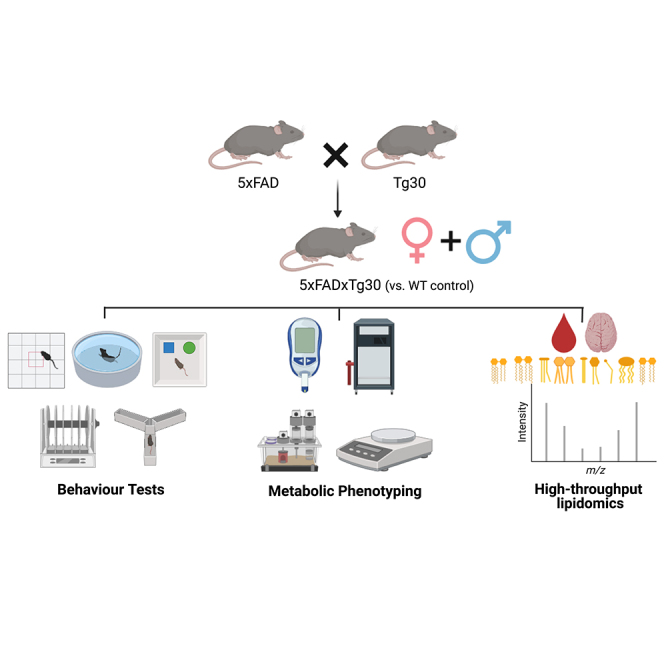
Highlights
-
•
The behavioral, metabolic, and lipidomic profile of the 5xFADxTg30 model was assessed
-
•
Altered body composition, survival, motor ability, and anxiety-like traits were noted
-
•
5xFADxTg30 mice have a distinct plasma lipidome with minor changes observed in the brain
-
•
Sex-specific differences do exist in this model
Animal physiology; Neuroscience; Behavioral neuroscience; Lipidomics
Introduction
Alzheimer’s disease (AD) is a growing global health concern, necessitating appropriate preclinical models to determine underlying pathological mechanisms and to test new therapeutic interventions. While the precise cause of AD is likely multifactorial and dependent on many environmental, lifestyle, genetic, and epigenetic interactions, one prevailing theory has been the amyloid hypothesis (also known as the amyloid cascade hypothesis).1 According to this hypothesis amyloid β (Aβ) peptides accumulate in the brain, causing extracellular senile plaques to form, which in turn causes neurotoxicity and the development of intraneuronal neurofibrillary tangles (NFTs) that contain filaments of the microtubule-associated protein tau. Induction of this tau pathology leads to cell death of neurons and neurodegeneration. Other factors associated with the development and progression of AD such as neuroinflammation2,3 and predisposition through the ε4 allele variant of apolipoprotein E (APOE)4,5 are also linked with Aβ pathology and NFT development. While there is much debate around the main driver(s) in these processes and the precise mechanisms by which this occurs, it is generally agreed that Aβ and tau pathology occur prior to clinical onset and are often hallmarks of AD.6
Studies also suggest that there is interplay between Aβ and tau. For instance, administration of Aβ into the brains of P301L mutant tau transgenic mice caused a five-fold increase in the numbers of NFTs observed,7 while Aβ peptide treatment increased the phosphorylation of tau in cultured neurons.8 Conversely, human tau has the capability to increase Aβ levels9 while intravascular injection of tau proteins increased Aβ burden.10 Together, this suggests a dynamic connection between the two, rather than a simple upstream/downstream relationship. This interplay between Aβ and tau provides a rationale to utilize preclinical models with both pathologies as opposed to studying each model in isolation. Several mouse models have been developed to study concurrent Aβ and tau pathology including the Tg2576/JNPL3 cross,11 the 3 x Tg model containing APPSWE, PS1M146V and tauP301L transgenes,12 hAPPNLI/Tau(P301L),13 the 5xFADxTg30 model,14 the 6 x FAD model,15 the APP-V717I/TauP301L model16 and the dual Tg (hAPP/htau) model.17 Each model displayed aspects of brain pathology and/or behavioral deficits, however, the manner and timing in which the various phenotypes have presented has differed.
The 5xFADxTg30 model14 is an amyloid precursor protein (APP)/Presenilin-1 (PS1)/tau mouse model generated by crossing the single 5xFAD and Tg30 models. To date, only the original description of this model has been published in the literature.14 In this previous paper, the authors extensively characterized the brain pathology, with some minor assessment of whole-body phenotypic traits. Pathological findings from the brain demonstrated that compared with the respective single transgenic models, 5xFADxTg30 mice had elevated NFTs, neuronal loss, and changes to insoluble tau that were more consistent with AD pathology.14 These findings suggested that the combined elevation of tau and Aβ drive pathology in this model, which displayed characteristic brain pathologies that would be anticipated from an experimental model of AD. The study also demonstrated decreased survival and motor function in the 5xFADxTg30 model, and although the data was not included in the manuscript, the authors stated they noted a reduction in body weight after age 6 months, with no difference in spatial memory at age 3 months.
Given the strength of this model in relation to the brain pathology, we set out to independently test whether we could recapitulate the survival, motor impairment and body weight findings in this model and to further characterize the phenotype, including assessing whether any sexual dimorphism existed. We specifically concentrated on a series of behavioral (given the brain pathology) and metabolic (given the body weight finding) tests. Further, given the increasing appreciation of alterations to lipid homeostasis in AD pathology, we conducted in-depth plasma and brain lipidomic analysis to add to our molecular understanding of the impact of APP and tau overexpression on in vivo lipid biology.
Results
We tracked the body weight of mice in Cohort 1 from age 2 months across the study period. From the initial 2-month measurement, the 5xFADxTg30 mice had a lower body weight than WT mice, a finding that was consistent in both male (Figure 1A) and female (Figure 1B) mice. While the WT mice increased in weight over time, the 5xFADxTg30 mice did not, appearing to have reached their maximum weight by age 2 months with a slight dip in body weight after age 7 months. Given this finding, at endpoint we measured tibia length as an indicator of overall mouse size. Tibia length was shorter in 5xFADxTg30 mice, a finding that was more overt in the female mice than the males (Figures 1C and 1D). To determine whether lean mass (the main contributor to body weight) or organ size were altered, we measured the weight of a representative skeletal muscle (tibialis anterior) and the liver. Both the skeletal muscle (Figures 1E and 1F) and the liver (Figures 1G and 1H) of transgenic mice in both sexes weighed significantly less. Brain weight also trended to be lower in the transgenic mice but was not statistically significant (Figures 1I and 1J).
Figure 1.

5xFADxTg30 mice have a decreased body size, muscle size, liver weight and bone length along with decreased lifespan and motor ability
(A) Male and (B) female body weight across the experiment from cohort 1, males n = 6–8 per group, females n = 6–7 per group.
(C and D) Tibia length, males = 7 per group, females n = 5–10 per group.
(E and F) Tibialis anterior weight, males n = 5–8 per group, females n = 3–7 per group.
(G and H) Liver weight, males n = 6–8 per group, females n = 3–7 per group.
(I and J) Brain weight, males n = 4–8 per group, female n = 4–6 per group.
(K) Male mice and (L) female mice Kaplan–Meier percentage survival curve, evaluating mice from cohort 1, by a Gehan-Breslow-Wilcoxon test n = 7–12 males n = 7–10 females.
(M) Male mice and (N) female mice latency to fall off rotarod, displayed between ages 3 and 8 months. Male 5xFADxTg30 displayed significant motor phenotype from 7 months onwards compared to WT. Average of three trials per month. Data assessed by 2-way repeated measures ANOVA or mixed effects analysis or via t-test where appropriate ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 for genotype effect. Data represents mean ± SEM.
A number of 5xFADxTg30 mice were found deceased in their cage or humanely euthanized due to welfare concerns throughout the study period. Survival curve analysis revealed both sexes contributed to this mortality, but to a much greater extent in male mice (Figures 1K and 1L). From all mice, 7/19 5xFADxTg30 mice (37%), died compared to 0/17 WT’s, (p = <0.01). To assess if any motor impairment was associated with the 5xFADxTg30 phenotype, mice underwent rotarod testing. Motor impairment was not observed until after age 6 months, with a steady decline in motor capability from then until endpoint. Males displayed a significant motor phenotype from age 7 months (Figure 1M), and females a trend across age 6–7 months (Figure 1N) before a significant difference at 8 months.
Next, anxiety-like behavior was assessed over time, using the large Open Field maze. At age 3 months, both male and female 5xFADxTg30 mice spent significantly more time in the center of the maze and less time in the corner areas of the maze compared to WT control (Figures 2A and 2B) Female, but not male, 5xFADxTg30 mice also spent greater time in the center at ages 6 and 8 months (Figures 2C–2H). Memory function was assessed via Y-Maze performance conducted monthly to determine whether there was any decline over time. Assessment of memory function at 3, 4, and 5 months (before the appearance of motor deficits as indicated by the rotarod data), or at 6, 7, and 8 months (once motor deficits arose), did not demonstrate any significant differences between genotypes for either sex (Figures 2I–2T). To assess cognitive decline regarding object recognition, a novel object maze was performed at ages 3, 6, 7, and 8 months. At all timepoints, there was no difference between the number of investigations of the novel object between the WT and transgenic mice (Figures 3A–3H).
Figure 2.

5xFADxTg30 model has alterations in fear/anxiety behavior
(A and B) Duration of time spent in the center of a large open field at ages 3 months, (C and D) 6 months, (E and F) 7 months, (G and H) and 8 months in male and female mice. Animals had 10 min in the arena lit with bright lights, corners and center of arena tracked. Male mice, n = 7–10 per group, female mice, n = 6–10 per group.
(I and J) Duration of time spent in the novel arm of a Y-maze at ages 3 months, (K and L) 4 months, (M and N) 5 months, (O and P) 6 months, (Q and R) 7 months and (S and T) 8 months in male and female mice. Animals had 10 min familiarization, before a 5-min trial, 2 h later. No significant differences between genotypes. Male mice, n = 7–9 per group, Female mice, n = 6–10 per group. T-test analysis, ∗p < 0.05 for genotype effect. Data represents mean ± SEM.
Figure 3.

5xFADxTg30 mice do not have a difference in investigative behavior but do have a reduced Morris Water Maze performance that was attributable to an impaired swim velocity at age 8 months
(A and B) Number of bouts of investigative behavior of the novel object at ages 3 months, (C and D) 6 months, (E and F) 7 months, and (G and H) 8 months in male and female mice. Animals had 10 min familiarization with Object 1 and Object 2. Object 1 was then replaced with Novel Object and animals and a 10-min test was recorded. Male mice, n = 7–10 per group, female mice, n = 6–10 per group. Data assessed by Student’s t test ∗p < 0.05 genotype effect. Graph represents Mean ± SEM.
(I and J) Morris water maze learning curve, latency to platform over 7 days at age 3 months in male and female mice. Graph represents Mean of 4 trials per day ±SEM.
(K and L) Probe trial. Animals were given a maximum of 120 s to find the platform and time spent in platform quadrant recorded. Platform was removed on Day 8 and animals swam for 60 s.
(M and N) Learning curve at age 8 months, latency to platform over 7 days with corresponding area under the curve (inset) in male and female mice.
(O and P) Probe trial at age 8 months.
(Q and R) Swim velocity. Graph represents average velocity over 4 trials on day 1 of MWM testing. Male mice, n = 7–8 per group female mice, n = 6–10 per group. Graph indicates mean ± SEM. Data assessed by t-test ∗p < 0.05 ∗∗p < 0.01 genotype effect.
Morris water maze testing was performed at ages 3 and 8 months to assess learning and spatial memory abilities. There were no genotype differences over 7 days of training at 3 months (Figures 3I–3L). At 8 months, there was a difference in latency to platform in the 5xFADxTg30 mice but only reached significance in the female mice (AUC indicated in figure insert) (Figures 3M and 3N) Despite this, time spent in the quadrant area once the platform was removed for the probe trial showed no genotype differences (Figures 3O and 3P). While there was a difference in the latency to platform, improvements in time (i.e., learning curve) over the 7 days were similar between genotypes (Figures 3M and 3N). While this can indicate a decrease in spatial awareness, due to the findings from the rotarod data, it could also indicate a motor phenotype causing a slower swim speed to the platform. Consequently, swim velocity was measured and was indeed significantly reduced in 5xFADxTg30 mice but again only reached statistical difference in the female mice (Figures 3Q and 3R). These results present the possibility that the motor phenotype of the model may compound the effects on the latency data, rather than there being a true spatial cognitive deficit.
Given the body and organ weight findings (Figure 1), and due to AD being associated with metabolic disturbances such as obesity and type 2 diabetes, we bred a second cohort to assess the metabolic phenotype of the model. The differences in body weight were replicated in this cohort (Figures 4A and 4B), despite the mice being housed in a different animal facility. EchoMRI analysis of body composition revealed a reduction in both lean (Figures 4C and 4D) and fat mass (Figures 4E and 4F) in 5xFADxTg30 mice with a trend for lean mass to decrease in the 5xFADxTg30 group as they aged.
Figure 4.

Altered body composition and glucose metabolism in 5xFADxTg30 mice
(A and B) Body weight, (C and D) lean mass and (E and F) fat mass over time in male and female mice. Male animals n = 6–8 per group, female animals, n = 6–7 per group. Data assessed by 2-way repeated measures ANOVA or mixed effects analysis where appropriate ∗p < 0.05 ∗∗p < 0.01 ∗∗∗p < 0.001. Blood glucose levels before and after an oral gavage glucose tolerance test administered at (2gm/kg LBM) at 5 and (50mg glucose) age 9 months with corresponding area under the curve.
(G and H) Fasting blood glucose, (I and J) glucose excursion and (K and L) area under the curve at age 5 months in male and female mice.
(M and N) Fasting blood glucose, (O and P) glucose excursion and (Q and R) area under the curve at age 9 months in male and female mice. Male mice n = 6–8 per group, female mice, n = 6–7 per group. Fasting blood glucose was taken after a 6 h fast and is the ‘0’ time value represented in the OGTT curve. ∗p = <0.05 ∗∗p = <0.01 represent genotype effect between WT and 5xFAD∗Tg30 mice. Fasting blood glucose and AUC assessed by T-Test and glucose curves via repeated measure ANOVA. Graphs indicate mean ± SEM.
To assess glucose metabolism, we measured fasting glucose and conducted two oral glucose tolerance tests (oGTT) (at ages 5 and 9 months). At 5 months, (Figures 4G and 4H), fasting blood glucose was not different between genotypes in either sex. For the oGTT, we dosed the mice with glucose per their amount of lean mass to account for the differences in their lean mass given skeletal muscle is a large driver of glucose disposal.18 There were no differences between genotypes in the glucose curve or calculated AUC in male mice (Figures 4I and 4K), however, in the female mice there was a trend for an improvement (Figures 4J and 4L; p = 0.05 for glucose curve, p = 0.06 for AUC). An oGTT was performed again at age 9 months, this time using a set dose of glucose administered to ensure we did not bias findings at 5 months due to dosing to lean mass in the first analysis (i.e., the female 5xFADxTg30 glucose levels were lower because we gave them less glucose). Interestingly, at this timepoint, there was a trend for reduced fasting glucose in the male 5xFADxTg30 mice (Figure 4M) and a significant decrease in the female 5xFADxTg30 mice (Figure 4N). Over the 120-min test, 5xFADxTg30 mice continued to have a lower blood glucose concentration than WT mice, again, primarily seen in the female animals (Figures 4O–4R). As mice each received the same quantity of glucose, but the 5xFADxTg30 mice had markedly less lean mass (Figures 4C and 4D), the lower glucose excursions have occurred despite receiving more glucose per unit of lean mass.
Assessment of metabolic parameters via the use of a Comprehensive Laboratory Animal Monitoring System (CLAMS) was conducted and assessed during both the light and dark cycle. The respiratory exchange ratio (RER) which assesses the contribution of carbohydrate and lipid utilization was not different between genotypes for either sex (Figures 5A–5D) nor was overall physical activity levels in either the light or dark cycles (Figures 5E–5H). Raw oxygen consumption and energy expenditure was lower in the 5xFADxTg30 male mice during the light phase (Figures 5I and 5Q) but not the dark phase (Figures 5J and 5R), while there was no genotype difference in the females (Figures 5K, 5L, 5S, and 5T). Due to the large differences in body composition, we normalized the oxygen consumption and energy expenditure data per lean mass. This normalization resulted in a significant increase in oxygen consumption and energy expenditure in the male 5xFADxTg30 mice in both the light and dark cycles (Figures 5M, 5N, 5U, and 5V) while still there was no genotype effect in the females (Figures 5O, 5P, 5W, and 5X).
Figure 5.

Metabolic phenotyping of 5xFADxTg30 mice
Aspects of whole-body energy metabolism measured in a CLAMs system for WT control and 5xFADxTg30 mice.
(A–D) Respiratory exchange ratio (RER) in male and female mice during the light and dark cycles.
(E–H) Total movement/activity as measured via beam breaks (x + Y ambulatory plus z breaks) in male and female mice during the light and dark cycles.
(I–L) Raw oxygen consumption in male and female mice during the light and dark cycles.
(M–P) Oxygen consumption normalised to lean tissue mass in male and female mice during the light and dark cycles.
(Q–T) Energy expenditure (Heat), in male and female mice during the light and dark cycles.
(U–X) Energy expenditure (Heat) adjusted to lean mass in male and female mice during the light and dark cycles. Genotype effects in oxygen consumption rate, as well as energy expenditure in males. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p = <0.001 data assessed by a Student’s t test n = 5–8 per group. Graphs indicate mean ± SEM.
An area of AD research receiving attention is the identification of lipid signatures linked to the disease.19 To determine if there was a lipidomic signature associated with this model, we performed plasma and brain lipidomic analysis. Numerous total lipid classes were significantly difference between WT and 5xFADxTg30 mice plasma (Figure 6A). While 15 lipid class totals were different in female mice, none of these remained significantly different following correction for false discovery rate. On the other hand, in male mice, 20 lipid class totals were different, 15 of which remained so after correction, with all 15 classes being decreased in the 5xFADxTg30 mice. While the diacylglycerol class had the greatest decline in the 5xFADxTg30 mice in terms of fold change, statistically, the most significant classes that showed a decline was in the abundance of phospholipid pools, particularly the classes Phosphatidylcholine (PC), Alkylphosphatidylethanolamine (PE(O)), Alkenylphosphatidylcholine (PC(P)), and Lysoalkenylphosphatidylcholine (LPC(P)). Free cholesterol (COH) was also altered to the same degree as the phospholipids (all p = <0.01, Figure 6A; Table S1). In contrast, fewer differences were observed for total lipid classes in the brain lipidome (Figure 6B). 5xFADxTg30 female mice had 7 lipid class totals significantly different but only one, GM3 ganglioside, survived false discovery correction and was higher in the transgenic mice (p = <0.05). Likewise in males, 7 lipid class totals were different but only the diacylglycerol class remained significantly higher after correction (Figure 6B; Table S2), which was despite the diacylglycerol pool being decreased in the plasma of male mice (Figure 6B).
Figure 6.

5xFADxTg30 mice have altered lipidome in both periphery and brain
Linear regression examining the fold difference in lipid class totals between WT and 5xFADxTg30 mice in (A) plasma and (B) brain. Fold differences are presented on a log2 scale such that ‘2’ means 2 times higher in 5xFADxTg30 relative to WT and the equivalent for the inverse (where 5xFADxTg30 is half of WT) is 0.5. Non-filled diamond, uncorrected p value >0.05, Colored diamonds, nominal p value <0.05, black diamond with colored outline, FDR corrected p < 0.05. All results in Tables S1 and S2.
Next, we explored differences in individual lipid species differences between WT and 5xFADxTg30 mice. Of the 790 lipid species measured in the plasma of male mice, 293 were significantly different between WT and 5xFADxTg30, with the vast majority of these being decreased in 5xFADxTg30 mice (Figure 7A). Notably, we observed that numerous related lipid species were altered. Amongst the most significantly decreased lipid species were phospholipids (PLs) containing highly unsaturated 20:4 fatty acyl chains (Figures 7B–7E). Another group of significantly altered lipids were specific sphingolipid (SL) species, in particular ceramide (Figures 7F–7M) and sphingomyelin (SM) (Figures 7N–7Q) species with very long (>20 carbon atoms) N-linked fatty acyl chains; these effects were not observed for ceramides with 16–18 carbon long N-acyl chains, highlighting the specific nature of the changes. Notably, changes in lipid species from the plasma of WT and 5xFADxTg30 female mice (112 lipid species were significantly different out of the 790 measured lipids) were distinct from those seen in the male mice. Once again, the majority of the significantly altered lipid species were decreased in 5xFADxTg30 mice compared with WT mice (Figure 7R). Most prominent among these changes were PC(P) and PE(P) lipid species (Figures 7S–7X), which accounted for 12 of the top 20 most significantly altered lipid species in female plasma.
Figure 7.

Sex and genotype differences in plasma and brain lipid species between WT and 5xFADxTg30 mice
(A) Volcano plot showing changes in plasma lipids in male WT and 5xFADxTg30 mice.
(B–Q) Indicated lipid species in male WT and 5xFADxTg30 mice.
(R) Volcano plot showing changes in plasma lipids in female WT and 5xFADxTg30 mice.
(S–X) Indicated lipid species in female WT and 5xFADxTg30 mice.
(Y and Z) Volcano plot showing changes in brain lipids in male Y) and female Z) WT and 5xFADxTg30 mice. Data shown in B-Q and S-X was analyzed by un-paired t-test. Statistical significance (p value) is shown on each figure. Data in B-Q and S-X are shown as the mean ± SEM. Individual symbols represent biological replicates.
With regards to lipid changes in the brain, a far more modest effect of 5xFADxTg30 was observed. Indeed, after correcting for false discovery, no lipid species were significantly different between the brains of either male or female WT and 5xFADxTg30 mice (Figures 7Y and 7Z). However, in considering the non-FDR corrected data, it was observed 74 lipid species were significantly different (p < 0.05) in brains from male mice. Notably, the most significantly altered lipid species were largely either DG, lyso-PL species (PL containing a single acyl chain), or SM (Figure 7Y). Within female brains, 42 lipid species were significantly different (p < 0.05), although no clear enrichment of specific lipid species was apparent (Figure 7Z).
Finally, to provide additional assessment of the amyloid and tau phenotype in the model, we measured key protein markers in hippocampal tissue. Total tau protein was elevated in both male and female mice (Figure 8A). Given NFTs are composed of hyperphosphorylated tau, we assessed the key tau phosphorylation site Ser202/Thr205 (AT8). Analysis revealed large scale hyperphosphorylation at this site in both male and female mice (Figure 8B). Sadleir et al.20 have previously published findings that suggests a differential transgenic expression level of APP in females compared to males in the 5xFAD model, likely due to an estrogen response element in the Thy1-promoter. This could lead to differences in pathology and or phenotype between sexes. Analysis of APP protein levels in our 5xFADxTg30 tissue demonstrated a genotype but not a sex difference in expression (Figure 8C), while the overexpression of APP resulted in a ∼40-fold increase in Aβ levels in both sexes (Figure 8D). The neuroaxonal protein, NFL, is a blood-based biomarker of neurodegeneration.21 Increased NFL levels in plasma have been shown in several neurodegenerative diseases, including AD, and is indicative of neuroaxonal damage.22,23 We therefore examined plasma NFL levels in mice at endpoint as a measure of neuroaxonal degeneration. Plasma NFL levels were significantly increased in 5xFADxTg30 plasma compared to WT mice in both male and female mice (Figure 8E), indicating substantial neuroaxonal degeneration at endpoint. These neuropathological markers provide confirmation of the expected manifestation of the disease to correlate with the phenotyping analysis.
Figure 8.

Characterization of brain and biomarker pathology associated with APP and tau
(A) Total tau western images and densitometry, (B) phosphorylated tau (Ser202/Thr205) western images and densitometry, (C) APP protein levels and densitometry, and (D) Amyloid β expression levels and densitometry in male and female mice, n = 5–7. Brain tissue obtained from the hippocampus, “MW” indicates molecular weight marker.
(E) Plasma neurofilament light (NFL) levels, n = 4–5. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p = <0.001 data assessed by a Student’s t test or two-way ANOVA. Graphs indicate mean ± SEM.
Discussion
Genetic mouse models allow scientists to study key pathological and physical hallmarks of disease and provide a tool to trial therapeutic interventions in a pre-clinical setting. However, all fields utilizing genetic models, including neuroscience, have been hampered by a lack of reproducible results due to a multitude of factors including different environmental conditions, lack of experimental details, lack of appropriate controls, underpowered studies and inexperienced personnel conducting the experiments.24 This highlights the importance of independent verification of phenotypes. Herein, we were able to replicate numerous findings reported in the original description of the 5xFADxTg30 mouse model14 and further our knowledge of this model by investigating sex-specific differences, the long-term behavioral phenotype, metabolic characteristics and the plasma/brain lipidomic profile of this model.
When comparing our findings to the original description of this model we were able to replicate the observation of lower body weight in 5xFADxTg30 mice. While the previous report14 indicated a reduction in body weight from age 6 months, both of our cohorts exhibited body weight differences from age 2 months. To comprehensively characterize the body weight findings, we conducted longitudinal EchoMRI studies that demonstrated the weight difference was due to reductions in both whole-body lean and fat mass. The loss of lean mass can likely be attributed to the tau mutation, which is known to cause progressive motor impairment with neurogenic muscle atrophy.25 To provide an indication as to whether the overall size of the mice was smaller, we measured tibia length which was significantly shorter in the transgenic mice. Together, overall body size, lean mass development and fat accumulation were all compromised in this combined APP/PS1/tau model.
Another key finding from Heraud et al. was the large reduction in survival in the 5xFADxTg30 mice. Extrapolating from their published figures, the 5xFADxTg30 mice exhibited a death rate approaching ∼40% greater than WT mice.14 In our hands, we observed a 37% decrease in survival out to 10 months in the 5xFADxTg30 mice. Although more prominent in males, premature death was not limited to any one sex, with both male and female 5xFADxTg30 mice succumbing before the endpoint. Rotarod analysis also reproduced the previous findings, with both datasets demonstrating no motor impairment at age 3 months but a significant decline in motor ability by age 6 monthswhich worsened further at age 8 months in the 5xFADxTg30 mice. Interestingly, this 6-month timepoint corresponded with the dip in lean mass of the transgenic mice (Figures 5D–5F) which could indicate a functional decline due to loss of muscle tissue. Finally, in terms of comparisons between studies, both assessed memory and exploratory behavior via the Y-maze test at age 3 months with both studies observed no differences between genotypes, a finding we confirmed as the mice aged throughout our study and this was supported by no significant differences in novel object recognition testing.
We furthered knowledge of this model by conducting extended behavioral analysis. When testing anxiety-like behavior, using the large Open Field maze we observed that at 3, 6, and 8 months, female 5xFADxTg30 mice spent significantly more time in the center of the maze suggesting that they had a decrease in their aversion to the bright light. This loss of fear associated with vulnerability in the open environment is likely a result of changes in the brain as it was observed before any motor impairment was obvious (not observed until 6 months) on the rotarod. The deficit in identifying a potentially hazardous situation (exposure in a brightly lit arena) could be suggestive of misprocessing in the areas of thinking and reasoning. The same cannot be definitively stated about the Morris water maze testing. While at age 8 months the female 5xFADxTg30 mice took longer to reach the platform (suggestive of a decrease in spatial reference memory), we did observe a slower swimming speed in these animals. The decrease in muscle size and motor ability likely decreased the swimming capability of the transgenic mice and accounted for some if not all of the difference between the groups.
During the behavioral characterization of the 5xFADxTg30 model, it became apparent that there were complications assessing markers of cognition due to the confounding nature of their motor phenotype. In humans, the frailty phenotype defines frailty as a syndrome meeting at least three of five phenotypic criteria: weakness, slowness, low level of physical activity, self-reported exhaustion, and unintentional weight loss,26 As there was evidence of weakness (swim test, rotarod), slowness (swim test), and unintentional weight loss (muscle size, lean mass composition), along with coordination issues evidence by the rotarod data, the model demonstrates characteristics of frailty. Frailty is a common symptom in AD patients and data have suggested an association between these two conditions. A scoping review that analyzed eight studies that included measures of both frailty and an AD biomarker such as Aβ or tau fluid, PET imaging or structural MRI showing atrophy, demonstrated that all reported positive relationships between biomarkers of AD and frailty.27 In some of these cases, frailty was related to a more rapid cognitive decline. This relationship suggests that AD and frailty share some type of common biological mechanism or etiology. Furthermore, in a study of older individuals, baseline frailty levels and the rate of increasing frailty were associated with an increase in the risk of the development of AD and of cognitive decline.28 Complementing this finding is a study of 165 older persons which identified that physical frailty proximate to death was related to AD pathology postmortem, but was not related to other common age-related pathologies such as the presence of cerebral infarcts or Lewy body disease.29 These findings raise the possibility that AD pathology may be a contributing factor to frailty or that frailty and AD pathology share a common etiopathogenesis. Therefore, a common physiological mechanism that could be targeted for both conditions concurrently would be optimal. While the frailty phenotype in the transgenic mice provides some difficulties for movement-based behavioral testing, having a degree of frailty in a pre-clinical mouse model of AD is likely a clinically relevant trait to have present. Furthermore, frailty is associated with increasing age and age-specific phenotypes were observed in the current study. For instance, rotarod differences in the 5xFADxTg30 mice were not observed until after age 6 months; glucose homeostasis differences were more obvious at the later time point measured and data obtained from the Morris water maze was more exaggerated between genotypes with age.
Despite those with diabetes being more susceptible to AD,30 we observed an improvement in glucose homeostasis in the 5xFADxTg30 mice with aging, an effect that was more prominent in female mice. Fasting blood glucose levels were significantly lower in the 5xFADxTg30 mice, and glucose levels continued to trend lower after glucose administration. This finding was despite the fact that at age 9 months we dosed the mice with a set dose of glucose, much like an OGTT conducted in humans, meaning that the 5xFADxTg30 mice received more glucose per unit of their lean mass than the WT mice. As skeletal muscle is the main component of lean mass and the major tissue for insulin-stimulated glucose disposal, it would be expected that blood glucose would be higher in the transgenic mice. While deciphering the exact mechanism of action behind this finding would involve comprehensive radioactive tracer experiments such as the hyperinsulinemia-euglycemic clamp technique and is outside the scope of the current study, it is possible that the atrophic nature of the skeletal muscle results in an increase in glucose disposal into the muscle to try to provide sufficient nutrients for maintenance of muscle function. Alternatively, various physiological stressors at the muscle site are known to induce induction of mitokines such as Fibroblast growth factor 21 (FGF21) and growth differentiation factor 15 (GDF15) which are known to play a role in systemic glucose homeostasis.31,32,33,34 Whilst whole-body glucose metabolism is not commonly assessed in AD models, it would be of interest to do so in other models, particularly those containing both amyloid and tau mutations.
Plasma and brain lipidomic analysis was performed to investigate lipid abundance in the 5xFADxTg30 model. Significant evidence has accumulated in the literature on lipid changes both in the periphery and centrally in the context of AD.19,35,36,37,38,39,40 Indeed, the major genetic risk factor determined for late-onset AD is possession of the ε4 allele of APOE.41,42 The primary role of the apoE protein in the brain and peripherally is to coordinate the transport and metabolism of cholesterol and other lipids with alterations to apoE4 known to modify lipid metabolism.43,44,45,46 Little is known about how alterations to APP and tau in pre-clinical models alter lipid handling, particularly at the molecular lipid species level. Interestingly in males, but not females, there was a decrease in the total free cholesterol pool in the plasma but not brain of the 5xFADxTg30 mice. Also, in male 5xFADxTg30 mice, there were significant declines in the phosphoethanolamine ether lipid classes, PE(O) and PE(P). PE(O) and PE(P) species have been reported to be negatively associated with both prevalent and incident AD in plasma of several human clinical cohorts of AD.19,35,40 In the brain, total GM3 ganglioside was elevated in the 5xFADxTg30 female mice. The involvement of gangliosides with AD19 and brain amyloid pathology47,48 is well documented. One recent study demonstrated alleviation of neuropathology in transgenic mice when treated with inhibitors within the GM3 pathway.49 In the brain of male mice, total diacylglycerol pool was elevated, in contrast to the plasma where it was reduced. Diacylglycerol has been shown to be elevated in the frontal cortex of patients with AD,50,51 however, why there is a sex difference and opposing results between plasma and brain in our model is unknown.
In analyzing individual lipid species, we observed that the profile of changes was distinct in male and female mice. Furthermore, we identified specific clusters of related lipids that were altered in 5xFADxTg30 mice. One of the most prominent differences we observed in plasma from male 5xFADxTg30 mice was a decrease in the abundance of 20:4 poly-unsaturated fatty acids across multiple PL classes. Additionally, we found that multiple ceramide and SM species containing very long chain fatty acids (>20 carbons long) were reduced in the plasma of male 5xFADxTg30 mice. These findings were particularly interesting as they parallel previous work from our group showing that decreases in SM and ceramides with 22:0 and 24:0 N-acyl chains are negatively associated with prevalent AD.19 Notably, the most significantly altered plasma lipid species in female mice were distinct from those in male mice, primarily being PC and PE plasmalogen species. Interestingly, the specific lipid species changes we observed in the plasma lipidome were not seen within the brain lipidome. As the diets of all mice used within this study were standardized, alterations in lipids from the diet were not the source of these differences. It is possible that changes in the regulation of one or more of the elongases that produce these very long chain fatty acids contributes to these lipid changes.
In contrast to the plasma, the differences in the brain lipid composition of the 5xFADxTg30 mice were much smaller. Indeed, when controlling for false discoveries, no individual lipid species was significantly altered. Nonetheless, in considering the data without FDR correction, a number of consistent changes in specific lipid species was observed, specifically DG, lyso-PLs and some SM were increased in the brain of male 5xFADxTg30 mice.
Here, we have reproduced the findings of the initial description of this model in relation to their observation of early death, decreased motor control, and reduced body weight. Moreover, we have expanded on these findings to demonstrate that the body weight phenotype is due to a decrease in both lean and fat mass, that in females the model exhibits alterations in fear/anxiety and have improved glucose control with aging and that there are distinct genotype and sex lipidomes. There are likely complications of the motor defect on the outcome of behavioral tests that involve movement in determining cognition status in this model, and further cognitive analysis with tests designed to require less movement would be beneficial. Finally, identifying the precise cause of early death in this model would be of interest in future studies.
Limitations of the study
Limitations of the study include a lack of specific skeletal muscle strength measures and the fact that due to the welfare of the mice we needed to cease the studies at age 10 months and could not age them out any further. This meant that at endpoint our mice would be considered “mature adult” as opposed to “old”. More advanced age may have revealed further age-specific phenotypes. Finally, as with all animal modeling of human disease, translating behavioral change is animal models to cognitive or behavioral changes in humans is challenging and such preclinical tools need to be used as the basis for further investigation to answer specific targeted questions.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GAPDH (14C10) Rabbit mAb | Cell Signaling Technology | Cat#2118; RRID:AB_561053 |
| APP | Invitrogen | Cat#36-6900 |
| Polyclonal Rabbit Anti-Tau | Dako | Cat#A0024; RRID:AB_10013724 |
| Phospho-Tau Monoclonal Antibody (AT8) Ser202/Thr205 | Invitrogen | Cat#MN1020; RRID:AB_223647 |
| Beta Amyloid Antibody (MOAB-2) | Novus Biologicals | Cat#NBP2-13075 |
| Anti-rabbit IgG Antibody | Cell Signaling Technology | Cat#7074; RRID:AB_2099233 |
| Anti-Mouse IgG, HRP-linked | Cell Signaling Technology | Cat#7076; RRID:AB_330924 |
| Biological samples | ||
| Tissues from WT and 5xFADxTg30 mice | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| SuperSignal West Femto | Thermo Scientific | Cat#34094 |
| Trans-Blot Turbo RTA Mini 0.2 μm Nitrocellulose Transfer Kit | BIO-RAD | Cat#1704270 |
| Laemmli Buffer | BIO-RAD | Cat#1610747 |
| RIPA Buffer | Thermo Scientific | Cat#89900 |
| Protease and Phosphatase Inhibitor | Thermo Scientific | Cat#A32959 |
| Ponceau S Staining Solution | Thermo Scientific | Cat#A40000279 |
| Critical commercial assays | ||
| Simoa® NF-Light™ Advantage (SR-X) kit | Quanterix™ | Cat#103400 |
| Pierce BCA Protein Assay Kit | Thermo Scientific | Cat#23225 |
| Experimental models: Organisms/strains | ||
| Mus musculus 5xFAD mice: B6.Cg-Tg(APPSwFlLon, PSEN1∗M146L∗L286V)6799Vas/Mmjax | Mutant Mouse Resource Center (MMRCRC) | RRID:MMRRC_034848-JAX |
| Mus musculus Tg30 mice: Tg(Thy1-MAPT∗)30Schd | Gift from Luc Buee | N/A -gift |
| Software and algorithms | ||
| GraphPad Prism v8.3.1 | GraphPad software | https://graphpad.com |
| ImajeJ-win64 | NIH, USA | https://imagej.nih.gov/ij |
| CleverSys TopScan Tracking Software | Cleversys, VA, USA | N/A |
| Oxymax for Windows 0233-127M | Columbus Instruments | https://www.colinst.com/downloads/files/oxymax_software.pdf |
| Mass Hunter B.07.00 | Agilent Technologies | https://www.agilent.com/en/product/software-informatics/mass-spectrometry-software |
| Other | ||
| CLAMS Comprehensive Laboratory Animal Monitoring System | Columbus Instruments | https://www.colinst.com/default.aspx |
| EchoMRI 4 in1 system | EchoMRI | www.echomri.com/Body_Composition_4_in_1.aspx |
| Rotarod | Ugo Basile | https://ugobasile.com |
| AccuCheck Glucometer | Roche, Diabetes Care | https://www.accu-chek.com.au/about-roche-diabetes-care |
| Laboratory Scales | Mettler Toledo | https://www.mt.com/au/en/home.html |
| 6490 mass spectrometer with Agilent 1290 series HPLC system and a ZORBAX eclipse plus C18 column | Agilent Technologies | https://www.agilent.com/en/product/liquid-chromatography-mass-spectrometry-lc-ms |
| Mini-PROTEAN Tetra Cell | BIO-RAD | Cat#1658000 |
| Trans-Blot Turbo transfer system | BIO-RAD | Cat#1704150 |
| Normal chow diet (NC) | Specialty Feeds | https://www.specialtyfeeds.com/products/standard-diet/ |
| 4-15% Mini-PROTEAN gels | BIO-RAD | Cat#4561086 |
| Fujifilm LAS-3000 Intelligent Dark Box | N/A | N/A |
| TECAN Infinite M200Pro | N/A | https://www.tecan.com |
| SR-X™ Biomarker Detection System | Quanterix™ | https://www.quanterix.com/simoa-technology/ |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Darren Henstridge (darren.henstridge@utas.edu.au).
Materials availability
This study did not generate any new materials.
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Ethics and animals
All activities involving the use of animals for research were approved by the Alfred Medical Research Education Precinct Animal Ethics Committee (AMREP AEC) and conducted according to the guidelines of the National Health and Medical Research Council of Australia for animal experimentation. Hemizygous single transgenic 5xFAD (strain name: B6.Cg-Tg(APPSwFlLon, PSEN1∗M146L∗L286V)6799Vas/Mmjax originally described in52) sourced from the Mutant Mouse Resource Center (MMRCRC) and Tg30 mice (strain name Tg(Thy1-MAPT∗)30Schd originally described in25,53) kindly supplied by Prof Luc Buée were crossed to produce the double transgenic, 5xFADxTg30 model. Mice were genotyped and confirmed from ear clip samples performed by Transnetyx (TN, USA) using real-time PCR. WT animals were non-transgenic littermate controls from the heterozygous breeding. All transgenes are under the control of a Thy-1 brain specific promoter and both lines on a C57BL/6J genetic background. Mice were fed a normal chow diet (NC) (14.0 MJ/kg, 75.2% kJ from carbohydrate, 4.8% from fat, 20% from protein; Specialty Feeds, Glen Forrest, Western Australia, Australia). During the experiment, mice had free access to food and water (except for in fasting periods before a glucose tolerance test) and were housed at 22 ± 1°C on a 12 h light/dark cycle. A cohort of animals (Cohort 1) was relocated to the Florey Institute of Neuroscience and Mental Health (Melbourne, Australia) at two months of age for behavioral tests, with a one-month familiarisation period, while a separate cohort (Cohort 2) remained at Baker Heart and Diabetes Institute for metabolic analyses. The investigators conducting experiments were blinded to the genotype until following the conclusion of the study. See Figures S1 and S2 for animal flow charts detailing the animal numbers utilised and the break down by sex. Both male and female mice were utilised in the studies to determine any sex-specific phenotypic traits. This is important as the Thy1-promoter (which drives the transgene in our model) has an estrogen response element and therefore this could influence sex-specific differences in phenotypes. Indeed it has been previously shown that female 5xFAD mice have elevated amyloid deposition due to higher levels of APP transgene expression.20 Age at which each procedure was tested is provided in the figures and figure legends. All animals were euthanised at 10 months age, via injection of Lethabarb.
Method details
Behavioral tests
A battery of behavioral tests were performed to track cognitive performance over time. To assess short term memory and exploratory behavior, time spent in a novel arm was analyzed in mice using a Y-Maze.54 Briefly, animals were placed in a two arm Y-maze and left to explore for 10 min. After 2 h, animals were replaced into the Y-Maze with access to 3 arms and the percentage of time spent in the novel arm was recorded. Arms were marked by a unique cue to the end to differentiate it from others. The cues were 2-dimensional pictures of black and white symbols including a circle, stripes and triangle. Motor function and coordination was assessed via the Rotarod.55 Briefly, mice were initially subjected to a training session that consisted of 3, 5-min trials on a spinning rod with a constant speed of 4rpm. The test consisted of 3 trials with the rotating rod progressively accelerating from 4 to 40rpm over 300s. Latency to fall off was recorded-any animals still on the rod at the end of the test were removed and a latency of 300s was recorded. To assess novel object recognition, animals were placed in a field, and given a 10min habituation period. One object (a 50mL falcon tube wrapped in foil) was replaced with a novel object (an old soda can) and the mouse was again allowed to explore for 10 min. All objects were cleaned between each trial. Time spent exploring the novel object was recorded. Anxiety-like behavior was assessed using the large open field.54 Animals were allowed to explore the large open field for 10 min and only the first 5 min were analyzed. Anxiety-like behavior was measured by the time spent in the center of the field (perceived as more threatening). Finally, to assess learning, memory and spatial awareness, the Morris Water Maze was conducted over a 7-day training period.54 Briefly, animals were provided 60 s to find the platform and if it did not do so in this time was gently guided onto the platform. The mouse remained on the platform for 15 s before being removed to its cage. Over 6 days, the animals were given 4 trials per day, each from one of four starting positions and allowed 120 s to find the submerged platform. On day 7, a probe trial was conducted to assess reference memory where the platform was removed. Age at which each of these tests were conducted is provided on the figures and in the figure legends. When multiple tests were conducted within the same month, the tests were conducted in the following order: Rotarod, Y-maze, large open field test, novel object recognition, morris water maze.
Metabolic tests
Body composition (Fat mass and lean mass) were measured using a 4-in-1 EchoMRI (EchoMRI, Houston, TX, USA) and standard laboratory scales were used for body mass (Mettler Toledo, Greifensee, Switzerland).56 Oral glucose tolerance tests (oGTT) were performed on fasted (6h) mice at 5 months and 9 months. At 5 months, mice received an oral gavage of 2g glucose/kg lean body mass (25% w/v glucose solution), and blood glucose levels were measured via a glucometer (AccuCheck, Roche Diabetes Care, NSW, Australia) at the indicated times on the blood that was collected from the tail. At 9 months, mice received a set dose of glucose at 50mg/mouse in an attempt to elucidate potential biases due to the large discrepancies in lean mass between the genotypes. A Comprehensive Laboratory Animal Monitoring System (CLAMS, Columbus Instruments, Columbus, OH, USA) was utilized to measure various metabolic parameters.56 Mice were placed into individually housed chambers and oxygen consumption (VO2), respiratory exchange ratio (RER), Energy expenditure (heat) and total movement (beam breaks) were recorded over 48h. The first 24h served as an acclimatisation period and the 24-48h period served as the period that was analyzed.
Laboratory analysis
Lipidomic profiling
Lipidomics was performed on plasma samples obtained at endpoint of the studies.57 Lipids were extracted from the plasma with butanol/methanol solution containing ammonium formate and internal standards.58 Analysis of extracts was performed on an Agilent 6490 mass spectrometer with an Agilent 1290 series HPLC system and a ZORBAX eclipse plus C18 column set at 45°C. An additional passivation step was performed prior to the run, where 0.5% phosphoric acid in 9:1 acetonitrile:water was run through the HPLC system for an hour and subsequently washed with 1:9 acetonitrile:water overnight. This was done to improve chromatographic peak shape for certain anionic lipid classes.
Mass spectrometry analysis was performed in positive ion mode with dynamic scheduled multiple reaction monitoring. Lipids run on the Agilent 6490 were measured with the following conditions: isolation widths for Q1 and Q3 were set to “unit” resolution (0.7 amu), gas temperature 150°C, nebulizer 20 psi, sheath gas temperature 200°C, gas flow rate 17 L/min, capillary voltage 3500 V and sheath gas flow 10 L/min. Chromatographic peaks were integrated using the Mass Hunter (B.07.00, Agilent Technologies) software and assigned to a specific lipid species based on MRM (precursor/product) ion pairs and retention time. Relative quantification was achieved by using the ratio of each analyte peak with the corresponding internal standard. Plasma lipids were standardised to volume extracted (pmol per mL of plasma, or nM) and brain lipids were normalised to total brain lipids and expressed as pmol of lipid per μmol of total lipid.
Plasma NFL biomarker assay
To measure plasma neurofilament light (NFL) levels in the plasma, the NF-Light (SR-X Version) single molecule array (Simoa) assay from Quanterix was used). Mouse plasma samples and assay calibrators and controls were thawed at room temperature for 1 h, vortexed for 10 s. Plasma was then centrifuged at 10,000 x g for 5 min prior to loading on the assay plate. Calibration samples were run neat, in duplicate, control samples were run at a 1/4 dilution in duplicate and mouse plasma samples were run in single at a dilution of 1/8. The assay was run as per manufacturer’s protocol.
Western blotting
Protein was extracted from the frozen brain tissue of WT and 5xFADxTg30 mice using RIPA buffer (Thermo Scientific, Rockford, IL, USA) with protease and phosphatase inhibitors (Thermo Scientific, Rockford, IL, USA). A BCA protein assay (Thermo Scientific, Rockford, IL, USA) performed to determine protein concentration on a multimode plate reader (Tecan, Mannedorf, Switzerland). The lysates were heated in Laemmli sample buffer (Bio-Rad, Gladesville, NSW, Australia) and mercaptoethanol for 5 min at 95°C. Gel electrophoresis was performed in a Mini-PROTEAN Tetra Cell (Bio-Rad, Gladesville, NSW, Australia) using 4–15% gradient polyacrylamide gels (Bio-Rad, Gladesville, NSW, Australia). Transfer was performed in the Trans-Blot Turbo Semi-Dry Transfer System (Bio-Rad, Gladesville, NSW, Australia) according to manufacturer instructions (except when probing for MOAB-2 which was conducted via wet transfer). Transfer quality was visually inspected via Ponceau staining (Sigma-Aldrich, Macquarie Park, NSW, Australia). Antibodies were sourced from the following companies. GAPDH, Anti-mouse IgG HRP-linked and anti-rabbit HRP-linked from Cell Signaling Technology (CST, Danvers, MA, USA) IL, USA. Phospho-Tau Monoclonal Antibody (AT8) Ser202/Thr205 and APP from Invitrogen (Waltham, Massachusetts, USA). Polyclonal Rabbit Anti-Tau (Dako/Agilent, Santa Clara, CA, USA), Beta-Amyloid Antibody (MOAB-2) (Novus Biologicals, Centennial, CO, USA). All primary antibodies were diluted 1:2000 before use except for GAPDH and the secondary antibody (#7074/#7076), which were diluted 1:5000. Bands were visualized electrochemically using the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific, Rockford, IL, USA) on a Fujifilm LAS-3000 Imager (FUJI PHOTO FILM CO LTD, Japan).
Quantification and statistical analysis
Band intensities from western blots were quantified and normalised in ImageJ software (National Institutes of Health, Bethesda, MD, USA) based on pixel brightness/gray value and count and normalised to an internal loading control. Data was assessed via unpaired T-Test when comparing two groups. When comparisons were made over time, a 2-way repeated measures ANOVA or mixed effects model was used where appropriate (such as when data points were missing due to the early death of mice in the 5xFADxTg30 model). Kaplan-Meier analysis was conducted using GraphPad PRISM v8.3.1 for survival analysis. Lipidomic data was analyzed using R version 4.1.3 and was log10 transformed prior to statistical analysis. Linear regression was used to identify class level differences between WT and 5xFADxTg30 mice and reported as fold-difference. Correction for multiple correction/false discovery rate was done using the approach by Benjamini and Hochberg separately for lipid class totals and species. Statistics are indicated in the figure legends. p < 0.05 was considered significant.
Acknowledgments
With thanks to: Dr Luc Buée for providing the Tg30 mouse line. We would like to acknowledge funding support provided from The Yulgibar Foundation, The Dementia Australia Research Foundation and the Shine on Foundation. ACC was funded by a National Heart Foundation of Australia Fellowship (105631). We acknowledge the assistance of the Animal Services staff from at the Baker Heart and Diabetes Institute and The Florey Institute for their care and maintenance of the mice. Graphical abstract created with Biorender.com.
Author contributions
Conceptualization, M.A.F, P.A.A., and D.C.H.; Methodology, K.H., T.F., P.J.M., P.A.A., and D.C.H.; Formal Analysis, J.P.S.M., K.H., G.I.L., A.L., and D.C.H.; Investigation, J.P.S., K.H., G.I.L., J.N., J.M.C., G.P., A.L., T.F., N.A.M., and D.C.H.; Resources, B.G.D., A.C.C., A.E.K., P.J.M., M.A.F., P.A.A., and D.C.H.; Writing – Original Draft, J.P.S.M., K.H., P.A.A., and D.C.H.; Writing – Review and Editing G.I.L., B.G.D., and M.A.F.; Visualization, J.P.S.M., K.H., G.I.L., and D.C.H.; Supervision, G.I.L., B.G.D., A.C.C., A.E.K., P.J.M., M.A.F., P.A.A., and D.C.H.; Funding Acquisition, J.P.S.M., M.A.F., and D.C.H.
Declaration of interests
The authors declare no competing interests.
Published: January 4, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.108800.
Supplemental information
References
- 1.Hardy J.A., Higgins G.A. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 2.Kinney J.W., Bemiller S.M., Murtishaw A.S., Leisgang A.M., Salazar A.M., Lamb B.T. Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement. 2018;4:575–590. doi: 10.1016/j.trci.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ising C., Venegas C., Zhang S., Scheiblich H., Schmidt S.V., Vieira-Saecker A., Schwartz S., Albasset S., McManus R.M., Tejera D., et al. NLRP3 inflammasome activation drives tau pathology. Nature. 2019;575:669–673. doi: 10.1038/s41586-019-1769-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu C.C., Zhao N., Fu Y., Wang N., Linares C., Tsai C.W., Bu G. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron. 2017;96:1024–1032.e3. doi: 10.1016/j.neuron.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shi Y., Yamada K., Liddelow S.A., Smith S.T., Zhao L., Luo W., Tsai R.M., Spina S., Grinberg L.T., Rojas J.C., et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549:523–527. doi: 10.1038/nature24016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy M.P., LeVine H. Alzheimer's Disease and the Amyloid-beta Peptide. J. Alzheimers Dis. 2010;19:311–323. doi: 10.3233/JAD-2010-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Götz J., Chen F., van Dorpe J., Nitsch R.M. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 8.Zheng W.H., Bastianetto S., Mennicken F., Ma W., Kar S. Amyloid beta peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience. 2002;115:201–211. doi: 10.1016/s0306-4522(02)00404-9. [DOI] [PubMed] [Google Scholar]
- 9.Bright J., Hussain S., Dang V., Wright S., Cooper B., Byun T., Ramos C., Singh A., Parry G., Stagliano N., Griswold-Prenner I. Human secreted tau increases amyloid-beta production. Neurobiol. Aging. 2015;36:693–709. doi: 10.1016/j.neurobiolaging.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 10.Houben S., de Fisenne M.A., Ando K., Vanden Dries V., Poncelet L., Yilmaz Z., Mansour S., De Decker R., Brion J.P., Leroy K. Intravenous Injection of PHF-Tau Proteins From Alzheimer Brain Exacerbates Neuroinflammation, Amyloid Beta, and Tau Pathologies in 5XFAD Transgenic Mice. Front. Mol. Neurosci. 2020;13:106. doi: 10.3389/fnmol.2020.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewis J., Dickson D.W., Lin W.L., Chisholm L., Corral A., Jones G., Yen S.H., Sahara N., Skipper L., Yager D., et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 12.Oddo S., Caccamo A., Shepherd J.D., Murphy M.P., Golde T.E., Kayed R., Metherate R., Mattson M.P., Akbari Y., LaFerla F.M. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 13.Paulson J.B., Ramsden M., Forster C., Sherman M.A., McGowan E., Ashe K.H. Amyloid plaque and neurofibrillary tangle pathology in a regulatable mouse model of Alzheimer's disease. Am. J. Pathol. 2008;173:762–772. doi: 10.2353/ajpath.2008.080175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Héraud C., Goufak D., Ando K., Leroy K., Suain V., Yilmaz Z., De Decker R., Authelet M., Laporte V., Octave J.N., Brion J.P. Increased misfolding and truncation of tau in APP/PS1/tau transgenic mice compared to mutant tau mice. Neurobiol. Dis. 2014;62:100–112. doi: 10.1016/j.nbd.2013.09.010. [DOI] [PubMed] [Google Scholar]
- 15.Kang S., Kim J., Chang K.A. Spatial memory deficiency early in 6xTg Alzheimer's disease mouse model. Sci. Rep. 2021;11:1334. doi: 10.1038/s41598-020-79344-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Terwel D., Muyllaert D., Dewachter I., Borghgraef P., Croes S., Devijver H., Van Leuven F. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am. J. Pathol. 2008;172:786–798. doi: 10.2353/ajpath.2008.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lippi S.L.P., Smith M.L., Flinn J.M. A Novel hAPP/htau Mouse Model of Alzheimer's Disease: Inclusion of APP With Tau Exacerbates Behavioral Deficits and Zinc Administration Heightens Tangle Pathology. Front. Aging Neurosci. 2018;10:382. doi: 10.3389/fnagi.2018.00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sylow L., Tokarz V.L., Richter E.A., Klip A. The many actions of insulin in skeletal muscle, the paramount tissue determining glycemia. Cell Metab. 2021;33:758–780. doi: 10.1016/j.cmet.2021.03.020. [DOI] [PubMed] [Google Scholar]
- 19.Huynh K., Lim W.L.F., Giles C., Jayawardana K.S., Salim A., Mellett N.A., Smith A.A.T., Olshansky G., Drew B.G., Chatterjee P., et al. Concordant peripheral lipidome signatures in two large clinical studies of Alzheimer's disease. Nat. Commun. 2020;11:5698. doi: 10.1038/s41467-020-19473-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sadleir K.R., Eimer W.A., Cole S.L., Vassar R. Abeta reduction in BACE1 heterozygous null 5XFAD mice is associated with transgenic APP level. Mol. Neurodegener. 2015;10:1. doi: 10.1186/1750-1326-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barro C., Chitnis T., Weiner H.L. Blood neurofilament light: a critical review of its application to neurologic disease. Ann. Clin. Transl. Neurol. 2020;7:2508–2523. doi: 10.1002/acn3.51234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewczuk P., Ermann N., Andreasson U., Schultheis C., Podhorna J., Spitzer P., Maler J.M., Kornhuber J., Blennow K., Zetterberg H. Plasma neurofilament light as a potential biomarker of neurodegeneration in Alzheimer's disease. Alzheimer's Res. Ther. 2018;10:71. doi: 10.1186/s13195-018-0404-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forgrave L.M., Ma M., Best J.R., DeMarco M.L. The diagnostic performance of neurofilament light chain in CSF and blood for Alzheimer's disease, frontotemporal dementia, and amyotrophic lateral sclerosis: A systematic review and meta-analysis. Alzheimers Dement. 2019;11:730–743. doi: 10.1016/j.dadm.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Onos K.D., Sukoff Rizzo S.J., Howell G.R., Sasner M. Toward more predictive genetic mouse models of Alzheimer's disease. Brain Res. Bull. 2016;122:1–11. doi: 10.1016/j.brainresbull.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leroy K., Bretteville A., Schindowski K., Gilissen E., Authelet M., De Decker R., Yilmaz Z., Buée L., Brion J.P. Early axonopathy preceding neurofibrillary tangles in mutant tau transgenic mice. Am. J. Pathol. 2007;171:976–992. doi: 10.2353/ajpath.2007.070345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X., Mao G., Leng S.X. Frailty syndrome: an overview. Clin. Interv. Aging. 2014;9:433–441. doi: 10.2147/CIA.S45300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wallace L., Theou O., Rockwood K., Andrew M.K. Relationship between frailty and Alzheimer's disease biomarkers: A scoping review. Alzheimers Dement. 2018;10:394–401. doi: 10.1016/j.dadm.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buchman A.S., Boyle P.A., Wilson R.S., Tang Y., Bennett D.A. Frailty is associated with incident Alzheimer's disease and cognitive decline in the elderly. Psychosom. Med. 2007;69:483–489. doi: 10.1097/psy.0b013e318068de1d. [DOI] [PubMed] [Google Scholar]
- 29.Buchman A.S., Schneider J.A., Leurgans S., Bennett D.A. Physical frailty in older persons is associated with Alzheimer disease pathology. Neurology. 2008;71:499–504. doi: 10.1212/01.wnl.0000324864.81179.6a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chatterjee S., Mudher A. Alzheimer's Disease and Type 2 Diabetes: A Critical Assessment of the Shared Pathological Traits. Front. Neurosci. 2018;12:383. doi: 10.3389/fnins.2018.00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johann K., Kleinert M., Klaus S. The Role of GDF15 as a Myomitokine. Cells. 2021;10 doi: 10.3390/cells10112990. ARTN 2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Macia L., Tsai V.W.W., Nguyen A.D., Johnen H., Kuffner T., Shi Y.C., Lin S., Herzog H., Brown D.A., Breit S.N., Sainsbury A. Macrophage Inhibitory Cytokine 1 (MIC-1/GDF15) Decreases Food Intake, Body Weight and Improves Glucose Tolerance in Mice on Normal & Obesogenic Diets. PLoS One. 2012;7:e34868. doi: 10.1371/journal.pone.0034868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu J., Stanislaus S., Chinookoswong N., Lau Y.Y., Hager T., Patel J., Ge H., Weiszmann J., Lu S.C., Graham M., et al. Acute glucose-lowering and insulin-sensitizing action of FGF21 in insulin-resistant mouse models--association with liver and adipose tissue effects. Am. J. Physiol. Endocrinol. Metab. 2009;297:E1105–E1114. doi: 10.1152/ajpendo.00348.2009. [DOI] [PubMed] [Google Scholar]
- 34.Oost L.J., Kustermann M., Armani A., Blaauw B., Romanello V. Fibroblast growth factor 21 controls mitophagy and muscle mass. J. Cachexia Sarcopenia Muscle. 2019;10:630–642. doi: 10.1002/jcsm.12409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goodenowe D.B., Cook L.L., Liu J., Lu Y., Jayasinghe D.A., Ahiahonu P.W.K., Heath D., Yamazaki Y., Flax J., Krenitsky K.F., et al. Peripheral ethanolamine plasmalogen deficiency: a logical causative factor in Alzheimer's disease and dementia. J. Lipid Res. 2007;48:2485–2498. doi: 10.1194/jlr.P700023-JLR200. [DOI] [PubMed] [Google Scholar]
- 36.Han X., Holtzman D.M., McKeel D.W., Jr. Plasmalogen deficiency in early Alzheimer's disease subjects and in animal models: molecular characterization using electrospray ionization mass spectrometry. J. Neurochem. 2001;77:1168–1180. doi: 10.1046/j.1471-4159.2001.00332.x. [DOI] [PubMed] [Google Scholar]
- 37.Igarashi M., Ma K., Gao F., Kim H.W., Rapoport S.I., Rao J.S. Disturbed choline plasmalogen and phospholipid fatty acid concentrations in Alzheimer's disease prefrontal cortex. J. Alzheimers Dis. 2011;24:507–517. doi: 10.3233/JAD-2011-101608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mapstone M., Cheema A.K., Fiandaca M.S., Zhong X., Mhyre T.R., MacArthur L.H., Hall W.J., Fisher S.G., Peterson D.R., Haley J.M., et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 2014;20:415–418. doi: 10.1038/nm.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamoto N., Igbabvoa U., Shimada Y., Ohno-Iwashita Y., Kobayashi M., Wood W.G., Fujita S.C., Yanagisawa K. Accelerated Abeta aggregation in the presence of GM1-ganglioside-accumulated synaptosomes of aged apoE4-knock-in mouse brain. FEBS Lett. 2004;569:135–139. doi: 10.1016/j.febslet.2004.05.037. [DOI] [PubMed] [Google Scholar]
- 40.Yamashita S., Kiko T., Fujiwara H., Hashimoto M., Nakagawa K., Kinoshita M., Furukawa K., Arai H., Miyazawa T. Alterations in the Levels of Amyloid-beta, Phospholipid Hydroperoxide, and Plasmalogen in the Blood of Patients with Alzheimer's Disease: Possible Interactions between Amyloid-beta and These Lipids. J. Alzheimers Dis. 2016;50:527–537. doi: 10.3233/JAD-150640. [DOI] [PubMed] [Google Scholar]
- 41.Holtzman D.M., Herz J., Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012;2:a006312. doi: 10.1101/cshperspect.a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loy C.T., Schofield P.R., Turner A.M., Kwok J.B.J. Genetics of dementia. Lancet. 2014;383:828–840. doi: 10.1016/S0140-6736(13)60630-3. [DOI] [PubMed] [Google Scholar]
- 43.Hamanaka H., Katoh-Fukui Y., Suzuki K., Kobayashi M., Suzuki R., Motegi Y., Nakahara Y., Takeshita A., Kawai M., Ishiguro K., et al. Altered cholesterol metabolism in human apolipoprotein E4 knock-in mice. Hum. Mol. Genet. 2000;9:353–361. doi: 10.1093/hmg/9.3.353. [DOI] [PubMed] [Google Scholar]
- 44.Jeong W., Lee H., Cho S., Seo J. ApoE4-Induced Cholesterol Dysregulation and Its Brain Cell Type-Specific Implications in the Pathogenesis of Alzheimer's Disease. Mol. Cells. 2019;42:739–746. doi: 10.14348/molcells.2019.0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dupuy A.M., Mas E., Ritchie K., Descomps B., Badiou S., Cristol J.P., Touchon J. The relationship between apolipoprotein E4 and lipid metabolism is impaired in Alzheimer's disease. Gerontology. 2001;47:213–218. doi: 10.1159/000052801. [DOI] [PubMed] [Google Scholar]
- 46.Yang L.G., March Z.M., Stephenson R.A., Narayan P.S. Apolipoprotein E in lipid metabolism and neurodegenerative disease. Trends Endocrinol. Metab. 2023;34:430–445. doi: 10.1016/j.tem.2023.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oikawa N., Yamaguchi H., Ogino K., Taki T., Yuyama K., Yamamoto N., Shin R.W., Furukawa K., Yanagisawa K. Gangliosides determine the amyloid pathology of Alzheimer's disease. Neuroreport. 2009;20:1043–1046. doi: 10.1097/WNR.0b013e32832e4b9d. [DOI] [PubMed] [Google Scholar]
- 48.McLaurin J., Franklin T., Fraser P.E., Chakrabartty A. Structural transitions associated with the interaction of Alzheimer beta-amyloid peptides with gangliosides. J. Biol. Chem. 1998;273:4506–4515. doi: 10.1074/jbc.273.8.4506. [DOI] [PubMed] [Google Scholar]
- 49.Dodge J.C., Tamsett T.J., Treleaven C.M., Taksir T.V., Piepenhagen P., Sardi S.P., Cheng S.H., Shihabuddin L.S. Glucosylceramide synthase inhibition reduces ganglioside GM3 accumulation, alleviates amyloid neuropathology, and stabilizes remote contextual memory in a mouse model of Alzheimer's disease. Alzheimer's Res. Ther. 2022;14:19. doi: 10.1186/s13195-022-00966-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chan R.B., Oliveira T.G., Cortes E.P., Honig L.S., Duff K.E., Small S.A., Wenk M.R., Shui G., Di Paolo G. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J. Biol. Chem. 2012;287:2678–2688. doi: 10.1074/jbc.M111.274142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wood P.L., Barnette B.L., Kaye J.A., Quinn J.F., Woltjer R.L. Non-targeted lipidomics of CSF and frontal cortex grey and white matter in control, mild cognitive impairment, and Alzheimer's disease subjects. Acta Neuropsychiatr. 2015;27:270–278. doi: 10.1017/neu.2015.18. [DOI] [PubMed] [Google Scholar]
- 52.Oakley H., Cole S.L., Logan S., Maus E., Shao P., Craft J., Guillozet-Bongaarts A., Ohno M., Disterhoft J., Van Eldik L., et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schindowski K., Bretteville A., Leroy K., Bégard S., Brion J.P., Hamdane M., Buée L. Alzheimer's disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am. J. Pathol. 2006;169:599–616. doi: 10.2353/ajpath.2006.060002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Daglas M., Truong P.H., Miles L.Q., Juan S.M.A., Rao S.S., Adlard P.A. Deferiprone attenuates neuropathology and improves outcome following traumatic brain injury. Br. J. Pharmacol. 2023;180:214–234. doi: 10.1111/bph.15950. [DOI] [PubMed] [Google Scholar]
- 55.Beauchamp L.C., Liu X.M., Vella L.J., Adlard P.A., Bush A.I., Finkelstein D.I., Barnham K.J. ATH434 Rescues Pre-motor Hyposmia in a Mouse Model of Parkinsonism. Neurotherapeutics. 2022;19:1966–1975. doi: 10.1007/s13311-022-01300-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lancaster G.I., Henstridge D.C. Body Composition and Metabolic Caging Analysis in High Fat Fed Mice. J. Vis. Exp. 2018:57280. doi: 10.3791/57280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huynh K., Barlow C.K., Jayawardana K.S., Weir J.M., Mellett N.A., Cinel M., Magliano D.J., Shaw J.E., Drew B.G., Meikle P.J. High-Throughput Plasma Lipidomics: Detailed Mapping of the Associations with Cardiometabolic Risk Factors. Cell Chem. Biol. 2019;26:71–84.e4. doi: 10.1016/j.chembiol.2018.10.008. [DOI] [PubMed] [Google Scholar]
- 58.Alshehry Z.H., Barlow C.K., Weir J.M., Zhou Y., McConville M.J., Meikle P.J. An Efficient Single Phase Method for the Extraction of Plasma Lipids. Metabolites. 2015;5:389–403. doi: 10.3390/metabo5020389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.