

Summary
Ribosome profiling is a sequencing technique that provides a global picture of translation across a genome. Here, we present iRibo, a software program for integrating any number of ribosome profiling samples to obtain sensitive inference of annotated or unannotated translated open reading frames. We describe the process of using iRibo to generate a species’ translatome from a set of ribosome profiling samples using S. cerevisiae as an example.
For complete details on the use and execution of this protocol, please refer to Wacholder et al. (2023).1
Subject areas: Bioinformatics, Sequence analysis, Genomics, Model Organisms
Graphical abstract
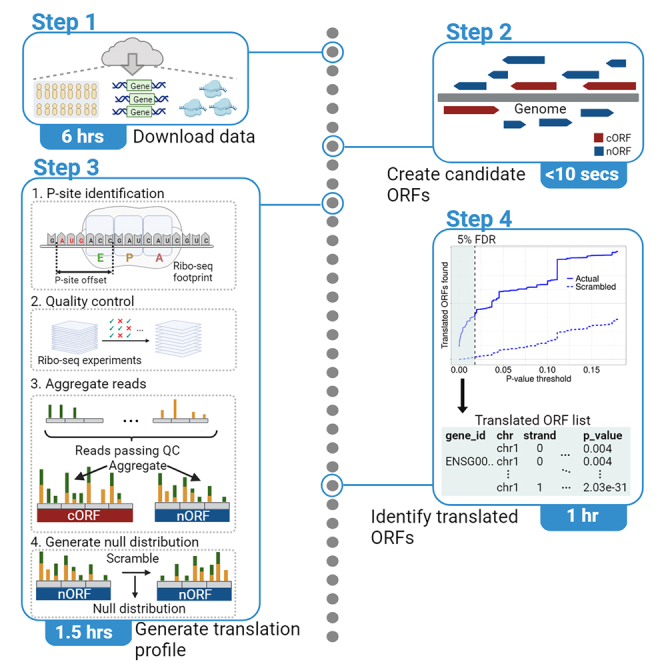
Highlights
-
•
Steps for inferring genome-wide translation using ribosome profiling data
-
•
Identify candidate open reading frames and assess them for translation
-
•
Sensitive detection through aggregation of reads across many experiments
-
•
Detect unannotated coding sequences with desired false discovery rate
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Ribosome profiling is a sequencing technique that provides a global picture of translation across a genome. Here, we present iRibo, a software program for integrating any number of ribosome profiling samples to obtain sensitive inference of annotated or unannotated translated open reading frames. We describe the process of using iRibo to generate a species’ translatome from a set of ribosome profiling samples using S. cerevisiae as an example.
Before you begin
Timing: 6–16 h
This protocol provides a guide for using iRibo to identify a set of translated open reading frames (ORFs) in a species of interest using a collection of ribosome profiling (ribo-seq) experiments. iRibo can infer translation of both canonical open reading frames (cORFs, i.e., annotated coding sequences) and noncanonical open reading frames (nORFs, i.e., unannotated coding sequences). We demonstrate the process by identifying the translatome of S. cerevisiae. This section describes the hardware requirements and input files needed to run iRibo.
Hardware
A UNIX computing environment that possesses at minimum a CPU with 64 GB of memory and 100 GB of free storage.
Downloading software
This step will set up the necessary computing environment to run iRibo.
-
1.Installing iRibo.
-
a.iRibo can be downloaded from https://github.com/CarvunisLab/iRibo. Click the ‘Code’ tab in the top right and download the zip folder.
-
b.After downloading the zip, extract the files:>unzip iRibo-main.zip
-
c.Move into the directory containing the iRibo files and compile iRibo with the following commands:>cd iRibo-main>make
-
a.
-
2.Installing R.
-
a.Install R (version 4.2.2+). Download and documentation is available at https://www.r-project.org/.
-
b.Create a conda environment and download packages.
-
a.
> conda create -n iRibo_r_env r-base r-scales r-ggplot2 r-future.apply r-argparse
-
3.Installing Samtools.
-
a.Install Samtools 1.10+. Download and documentation is available at https://www.htslib.org.
-
a.
Data collection and preprocessing
In this step, the genome sequence, genome annotation and ribosome profiling sequencing data are obtained and processed. This data will be used in future steps to detect translated ORFs.
-
4.Download annotations and genome.
-
a.Download the genome sequence (FASTA format) and annotation (GFF3 or GTF format) for your organism of interest. These can usually be found in databases such as REFSEQ, GENCODE, Ensembl, or species-specific databases.
-
i.For following the example in this protocol, we have provided S. cerevisiae genome and annotation files as part of the iRibo-main.zip package downloaded earlier, so no additional download is needed.
-
i.
-
a.
-
5.Obtain transcriptome (optional).
-
a.Download or assemble a transcriptome (GFF3 or GTF formation) for your organism of interest.
-
a.
CRITICAL: iRibo can identify translated ORFs in either a transcriptome or directly from a genome. As multi-exon ORFs are relatively rare in S. cerevisiae and the genome is small, most potentially translated ORFs can be inferred directly from the genome sequence and so defining the transcriptome is not needed. For many eukaryotes, a transcriptome will be necessary to obtain comprehensive results. If a transcriptome is not used, all ORFs will be inferred from the genome sequence alone and no multi-exonic ORFs will be identified. If a transcriptome is used, it is recommended to use as comprehensive a transcriptome as possible in order to obtain the largest coverage of the translatome; for example, MiTranscriptome2 in human contains many transcripts that are not present in other annotations. If no appropriate transcriptome is available, transcriptomes may also be assembled from RNA-seq reads (popular tools include Trinity3 and StringTie4), or created by merging an existing set of transcriptomes using a tool such as Cufflinks5 or StringTie.4
-
6.Produce a set of SAM/BAM files containing alignments of ribo-seq reads to the genome/transcriptome.
-
a.For conducting new analyses:
-
i.If not using one’s own experimental data, ribo-seq experiments can be found by searching published papers, or repositories such as Sequence Read Archive (SRA), Gene Expression Omnibus (GEO), or European Nucleotide Archive (ENA). Sequencing files for the ribo-seq experiments (FASTQ format) can be downloaded from SRA using fastq-dump.
- ii.
- iii.
-
i.
-
a.
Note: To follow this protocol, 412 SAM files that we constructed can be downloaded from https://zenodo.org/record/8187381 and https://zenodo.org/record/8187637. Each SAM file was produced by aligning a FASTQ file containing ribosome profiling reads to the S. cerevisiae genome as described in Wacholder et al. 2023.1 These can be downloaded with the commands:
> wgethttps://zenodo.org/record/8187381/files/iRibo_S288C_SAM_Files.zip.
> wgethttps://zenodo.org/record/8187637/files/iRibo_S288C_SAM_Files_2.zip.
> unzip iRibo_S288C_SAM_Files.zip
> unzip iRibo_S288C_SAM_Files_2.zip
> gunzip iRibo_S288C_SAM_Files/∗.gz
> gunzip iRibo_S288C_SAM_Files_2/∗.gz
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Aligned ribosome profiling reads | This paper |
https://doi.org/10.5281/zenodo.8187637 https://doi.org/10.5281/zenodo.8187381 |
| Saccharomyces cerevisiae S288C reference genome | Saccharomyces Genome Database | S288C reference sequence R64.2.1 |
| Saccharomyces cerevisiae S288C reference annotation | Saccharomyces Genome Database | R64.2.1 |
| Software and algorithms | ||
| iRibo | This paper | https://github.com/CarvunisLab/iRibo |
| R v4.2.2+ | The R Foundation | https://www.r-project.org |
| Samtools v1.10+ | Genome Research | https://www.htslib.org |
| Other | ||
| High performance computing cluster environment with Intel Xeon E5-2650 CPU, 64 GB memory | Intel | intel.com |
Step-by-step method details
Here we describe a step-by-step methodology for detecting translation across a genome with iRibo.
Create candidate ORFs
In this step, iRibo scans the genome or transcriptome to identify candidate ORFs. Each candidate ORF will be assessed for translation in later steps. Candidates are identified as canonical if present as protein-coding genes in the genome annotations, noncanonical otherwise. The list of candidate ORFs will be output in a file called “candidate_orfs.”
-
1.
Generate candidate ORFs from the genomic sequence.
> ./iRibo --RunMode=GetCandidateORFs --Genome=S288C_example/S288C_sequence_iRibo.fsa --Annotations=S288C_example/saccharomyces_cerevisiae_iRibo.gff --Output=iRibo_yeast --Threads=8
Note: iRibo generates candidate ORFs by first identifying all sequences starting with ATG and ending with an in-frame stop codon. If a canonical and noncanonical ORF overlap by at least one nucleotide in the same frame, only the canonical ORF is kept. If two noncanonical or two canonical ORFs overlap by at least one nucleotide in the same frame, only the longest is kept. If the user wants to assess another set of candidate ORFs for translation, the “candidate_orfs” output file generated in this step can be replaced with a custom list of ORFs, with specified coordinates, in the same format.
Note: To generate ORFs on a transcriptome rather than genome, the path to the transcriptome will need to be given as indicated below. This enables iRibo to detect multi-exon ORFs. This is the only change needed; all other steps will be the same whether using ORFs derived from a transcriptome or directly from a genome.
> ./iRibo --RunMode=GetCandidateORFs --Genome=path/to/genome.fa --Annotations=path/to/annotations.gtf --Transcriptome=path/to/transcriptome.gtf
Generate translation profile
In this step, iRibo uses ribo-seq read alignment files (SAM or BAM format) to construct translation profiles consisting of the counts of inferred ribosome P-sites at each position for each ORF (Figure 1A).1 For quality control, each read length for each file is tested to ensure it exhibits three nucleotide periodicity (read counts following a high-low-low pattern) among annotated ORFs. Three nucleotide periodicity is a strong signature of translation and the signal iRibo uses to distinguish genuine translation from other processes that could generate ribo-seq reads. Reads from all files and read lengths that show periodicity are aggregated and associated with each candidate ORF.
-
2.Create a list of paths to each SAM/BAM file, all in a single input file called sam_list.txt, with each path on its own line.> ls -d iRibo_S288C_SAM_Files/∗.sam > sam_list.txt> ls -d iRibo_S288C_SAM_Files_2/∗.sam >> sam_list.txt
-
a.The first few lines in the file should look like this:iRibo_S288C_SAM_Files/ERR3218434_iribo.sam iRibo_S288C_SAM_Files/ERR3218435_iribo.sam iRibo_S288C_SAM_Files/ERR3218438_iribo.sam iRibo_S288C_SAM_Files/ERR3218439_iribo.sam iRibo_S288C_SAM_Files/SRR1002819_iribo.sam
-
a.
-
3.
Run the command below to generate a translation profile:
> ./iRibo --RunMode=GenerateTranslationProfile --Genome=S288C_example/S288C_sequence_iRibo.fsa --CandidateORFs=iRibo_yeast/candidate_orfs --Riboseq=sam_list.txt --Output=iRibo_yeast --Threads=8 --QC_Positions=true --Min_Length=25 --Max_Length=35 --P_Site_Distance=20 --QC_Count=10000 --QC_Periodicity=2.0
Note: Min_Length and Max_Length specify the range of ribo-seq read lengths to consider in nucleotides; reads that show three nucleotide periodicity are generally between 25 and 35 nt long. P_Site_Distance sets the maximum distance from the mapped position of aligned reads to search for the ribosome P-site. QC_Count sets the minimum number of total reads required to include a read length/file combination in the analysis. For a full list of possible parameters, please see the iRibo manual on the GitHub page.
Note: Running this step will generate an output file with statistics about the ribo-seq reads mapping to each candidate ORF (translation_calls). Additionally, it will generate two .wig files containing tracks of all the reads (riboseq_reads_plus.wig, riboseq_reads_minus.wig). The wig and gff3 files can be input into a program like Integrative Genomics Viewer10 to visualize ribo-seq reads across the genome, including reads that do and do not map to candidate ORFs. See the iRibo manual located on the iRibo GitHub page for a full description of output files.
Figure 1.

Thousands of noncanonical S. cerevisiae ORFs identified by iRibo
(A) The process by which iRibo generates a translation profile using ribo-seq data. Mapped ribo-seq reads from each experiment are grouped by read length, and for each length the P-site is inferred. Reads are then shifted to the P-site. For quality control, three-nucleotide periodicity is then checked among canonical ORFs. For each read length in which a P-site can be inferred, and which shows periodicity among canonical ORFs, all reads are then aggregated in the genome to allow determination of which ORFs are translated.
(B) Translated nORFs found by iRibo at a range of p-value thresholds. A 5% false discovery rate was identified at a p-value threshold of 0.037 for nORFs, indicating 19269 translated nORFs. The dashed blue line represents the average number of nORFs discovered performing the same test on 100 scrambled distributions of reads.
(C) Translated cORFs found by iRibo at a range of p-value thresholds. At the p-value threshold of .037, set to obtain a 5% FDR among nORFs, 5519 cORFs are detected. The dashed red line represents the average number of cORFs discovered performing the same test on 100 scrambled distributions of reads.
Generate translatome
This step uses the pattern of ribo-seq reads across each candidate ORF to infer which are translated. Every ORF will be assessed for three-nucleotide periodicity using a binomial test. The p-values for the binomial tests will then be used as a confidence score to construct a list of translated ORFs at a desired false discovery rate (FDR).
-
4.
Run the command below:
> conda run -n iRibo_r_env Rscript GenerateTranslatome.R --FDR=0.05 --CandidateORFs=iRibo_yeast/candidate_orfs --TranslationCalls=iRibo_yeast/translation_calls --NullDistribution=iRibo_yeast/null_distribution --ExcludeCHR=chrM --ExcludeOverlapGene=True --Output=iRibo_yeast
Note: FDR specifies the desired false discovery rate, in this case 5%. The FDR only considers noncanonical translated ORFs; i.e., if a 5% FDR is set it is expected that 5% of the noncanonical ORFs called translated will be false positives, though translation calls will also be made for canonical ORFs at the same threshold. ExcludeCHR is a list of chromosomes or contigs to exclude from the translatome, separated by commas. ExcludeOverlapGene excludes noncanonical ORFs that overlap canonical ORFs on the same strand. This may be desired because overlapping ORFs obscure signals of translation.
Expected outcomes
This protocol will provide a high confidence list of translated canonical and noncanonical ORFs and their expression levels for any organism given a set of ribosome profiling samples (Table 1). A simple binomial test for three-nucleotide periodicity and empirical false discovery rate makes the results both robust and interpretable.
Table 1.
Most highly translated noncanonical intergenic ORFs in S. cerevisiae
| Coordinates | Ribo-seq reads per base | p value |
|---|---|---|
| chrVI: 1312102–1312494+ | 78.12027 | 1.83E-40 |
| chrIV: 1312102-1312494- | 1.651399 | 6.08E-37 |
| chrXIII: 397355-397690- | 1.458333 | 7.81E-27 |
| chrXIII: 619099-619371- | 4.62271 | 3.60E-26 |
| chrXV: 853623-853826- | 1.921568627 | 1.17E-20 |
The list of inferred translated ORFs will be output to a file called “translated_orfs.csv”, and plots showing the number of translated ORFs found at different p-value thresholds for noncanonical and canonical candidate ORFs will also be output in the files “nORF Discovery.png” and “cORF_discovery.png”, respectively (Figures 1B and 1C). See the iRibo manual located on the iRibo GitHub page for full description of the output files. Integrating the 412 ribo-seq samples we obtained for Saccharomyces cerevisiae reveals a noncanonical translatome consisting of nearly 20,000 noncanonical ORFs.
Limitations
This protocol uses three nucleotide periodicity in ribo-seq reads to distinguish translation from other biological processes and ensure robustness. Therefore, it will likely not work well with ribo-seq that does not have nucleotide-level resolution. Overlapping translated ORFs are challenging to detect even with nucleotide-level resolution. In the candidate ORF creation step, iRibo considers only the longest possible ORF among ORFs that overlap in the same frame, and only ORFs with ATG start codons. However, users may also supply their own lists of ORFs that do not follow these rules. ORFs can be detected in nucleotide sequence using tools such as ORF Finder.11
Troubleshooting
Problem 1
The GetCandidateORFs step job gets killed (related to step 1 in step-by-step method details).
Potential solution
This could be because the organism has a very large transcriptome. Try using a computer with more memory. For reference, the mouse transcriptome takes around 30 GB of memory to process. Additionally, try using less threads, as this will reduce memory usage.
Problem 2
GenerateTranslationProfile.log shows the P-sites are all unidentifiable for a sample (related to step 3 in step-by-step method details).
Potential solution
-
•
First, this could be caused by a low quality ribo-seq sample. If other P-sites can be identified in other samples, this is most likely the problem.
-
•
Second, the problem could be caused by improperly trimmed adapters, which will generally lead to read lengths above the 25–35 nt typical of ribo-seq. If few reads are in the expected length range, ensure that adapters are correctly trimmed.
-
•
Third, make sure there are canonical genes present in the candidate_orfs file, indicated by a gene identifier in the gene_id column rather than an X. If not, make sure the annotations file is in proper gtf or gff3 format and has annotated coding sequences, indicated by ‘CDS’ in the third column. Then, rerun GetCandidateORFs.
-
•
Fourth, make sure the chromosome or contig names match up between the candidate_orfs file and the SAM files. See the example scripts under Data Collection and Preprocessing for more information.
Problem 3
GenerateTranslatome.R fails and generates an error message “No reads detected. Make sure any read lengths passed quality control” (related to step 4 in step-by-step method details).
Potential solution
Make sure that there were read lengths that passed quality control in the dataset. To check this, go to the GenerateTranslationProfile output folder, check GenerateTranslationProfile.log, and check if any read lengths in any samples detected a P-site and passed quality control. If no read lengths passed, higher quality data will be needed to identify the translatome.
Problem 4
GenerateTranslatome.R is taking a long time to run (related to step 4 in step-by-step method details).
Potential solution
If it needs to be run faster and many samples passed quality control, you can lower the number of random scrambles to consider with the option --Scrambles = 10. This is acceptable because the sample size (candidate ORFs with reads) will be high enough to stay robust.
Problem 5
GenerateTranslatomeProfile and GetCandidateORFs takes a long time to run (related to steps 1 and 3 in step-by-step method details).
Potential solution
Increasing the thread count decreases the runtime near-linearly up to a point. 8 threads or above should be optimal for performance.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Aaron Wacholder (acw87@pitt.edu).
Techinical contact
Technical questions on executing this protocol should be directed to and will be answered by the technical contacts Alistair Turcan (alt245@pitt.edu) and Aaron Wacholder (acw87@pitt.edu).
Materials availability
This study did not generate unique reagents.
Data and code availability
iRibo is available at https://github.com/CarvunisLab/iRibo. The SAM files used were deposited at https://zenodo.org/record/8187381 and https://zenodo.org/record/8187637.
Acknowledgments
This work was supported by funds provided by Alfred P. Sloan Foundation, Sloan Research Fellowship number FG-2021-15678 awarded to A.-R.C.; the National Science Foundation grant MCB-2144349 awarded to A.-R.C.; the National Institute of General Medical Sciences of the National Institutes of Health grant DP2GM137422 awarded to A.-R.C.; and the National Center for Complementary and Integrative Health of the National Institutes of Health grant R01AT012826 awarded to A.-R.C. The graphical abstract and figure were created with BioRender.com.
Author contributions
A.T. wrote, tested, and analyzed the code and drafted the manuscript. A.W. conceptualized the idea, wrote the initial code, and edited the manuscript. J.L. tested the code, edited the manuscript, and created figures including the graphical abstract. A.-R.C. conceptualized the idea, edited the manuscript, and supervised the project.
Declaration of interests
A.-R.C. is a member of the Scientific Advisory Board for ProFound Therapeutics.
Contributor Information
Alistair Turcan, Email: alt245@pitt.edu.
Aaron Wacholder, Email: acw87@pitt.edu.
Anne-Ruxandra Carvunis, Email: anc201@pitt.edu.
References
- 1.Wacholder A., Parikh S.B., Coelho N.C., Acar O., Houghton C., Chou L., Carvunis A.-R. A vast evolutionarily transient translatome contributes to phenotype and fitness. Cell Syst. 2023;14:363–381.e8. doi: 10.1016/j.cels.2023.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iyer M.K., Niknafs Y.S., Malik R., Singhal U., Sahu A., Hosono Y., Barrette T.R., Prensner J.R., Evans J.R., Zhao S., et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015;47:199–208. doi: 10.1038/ng.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grabherr M.G., Haas B.J., Yassour M., Levin J.Z., Thompson D.A., Amit I., Adiconis X., Fan L., Raychowdhury R., Zeng Q., et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shumate A., Wong B., Pertea G., Pertea M. Improved transcriptome assembly using a hybrid of long and short reads with StringTie. PLoS Comput. Biol. 2022;18 doi: 10.1371/journal.pcbi.1009730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10. [Google Scholar]
- 7.Babraham Bioinformatics . Babraham Institute; 2019. Trim Galore. [Google Scholar]
- 8.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wheeler D.L., Church D.M., Federhen S., Lash A.E., Madden T.L., Pontius J.U., Schuler G.D., Schriml L.M., Sequeira E., Tatusova T.A., Wagner L. Database resources of the National Center for Biotechnology. Nucleic Acids Res. 2003;31:28–33. doi: 10.1093/nar/gkg033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
iRibo is available at https://github.com/CarvunisLab/iRibo. The SAM files used were deposited at https://zenodo.org/record/8187381 and https://zenodo.org/record/8187637.