

Summary
The native, perennial shrub American hazelnut (Corylus americana) is cultivated in the Midwestern United States for its significant ecological benefits, as well as its high‐value nut crop. Implementation of modern breeding methods and quantitative genetic analyses of C. americana requires high‐quality reference genomes, a resource that is currently lacking. We therefore developed the first chromosome‐scale assemblies for this species using the accessions ‘Rush’ and ‘Winkler’. Genomes were assembled using HiFi PacBio reads and Arima Hi‐C data, and Oxford Nanopore reads and a high‐density genetic map were used to perform error correction. N50 scores are 31.9 Mb and 35.3 Mb, with 90.2% and 97.1% of the total genome assembled into the 11 pseudomolecules, for ‘Rush’ and ‘Winkler’, respectively. Gene prediction was performed using custom RNAseq libraries and protein homology data. ‘Rush’ has a BUSCO score of 99.0 for its assembly and 99.0 for its annotation, while ‘Winkler’ had corresponding scores of 96.9 and 96.5, indicating high‐quality assemblies. These two independent assemblies enable unbiased assessment of structural variation within C. americana, as well as patterns of syntenic relationships across the Corylus genus. Furthermore, we identified high‐density SNP marker sets from genotyping‐by‐sequencing data using 1343 C. americana, C. avellana and C. americana × C. avellana hybrids, in order to assess population structure in natural and breeding populations. Finally, the transcriptomes of these assemblies, as well as several other recently published Corylus genomes, were utilized to perform phylogenetic analysis of sporophytic self‐incompatibility (SSI) in hazelnut, providing evidence of unique molecular pathways governing self‐incompatibility in Corylus.
Keywords: Corylus americana, hazelnut, de novo genome assembly, comparative genomics, population genetics, sporophytic self‐incompatibility
Introduction
Species of hazelnut (Corylus spp.) are cultivated globally, and generate an annual production of 1.1 million tons of in‐shell nuts, produced across 34 countries (FAOSTAT, 2023). This production is driven by a wide range of market uses across diverse industries, including food products, pharmaceuticals and dietary supplements (Ceylan et al., 2022). Currently, commercial cultivation relies overwhelmingly on cultivars of European hazelnut (C. avellana) adapted to Mediterranean climates (Di Lena et al., 2022) whose moderate winters satisfy their chilling requirements (Mehlenbacher, 1991). The distribution of hazelnut production reflects these climatic requirements, with major centres of production in Turkey, Italy and Spain, with Turkey alone producing over 65% of the global supply (Semih Uzundumlu et al., 2022). In the United States, this narrow range of adaptation currently restricts production to relatively small regions such as the Willamette Valley in Oregon (Revord et al., 2020).
As a woody perennial crop, hazelnuts have substantial potential to provide ecosystem services such as reduced soil erosion and nutrient runoff (Demchik et al., 2014), while also sequestering carbon in above‐ and below‐ground biomass (Granata et al., 2020). Expanding the climatic conditions under which commercially viable hazelnut varieties can be grown would not only make production more resilient in the face of changing climates, but provide high‐value alternative crops to producers across wider geographies. One possible method for the development of commercially viable hazelnut varieties adapted to novel growing regions is the improvement of the native North American shrub C. americana, which possesses disease resistance and cold hardiness traits lacking in most cultivated forms of European hazelnut (Molnar et al., 2018). Genomics‐assisted breeding of C. americana oriented towards such improvement efforts, utilizing methods such as marker‐assisted selection and genomic prediction, would be greatly aided by a high‐quality reference genome assembly for the species. Such a resource would, for example, facilitate the rapid identification and alignment of genetic polymorphisms across experimental and breeding populations (Kang et al., 2016). In addition, well‐annotated assemblies provide the opportunity to investigate the genetic features underlying specific biomolecular pathways involved in abiotic and biotic stress resistance, enabling methods ranging from fine mapping of quantitative trait loci (QTL) to gene editing (Bolger et al., 2017; Dmitriev et al., 2022).
To facilitate such applied and experimental approaches, this study reports the first chromosome‐scale reference assemblies for C. americana, built from two accessions – ‘Rush’ and ‘Winkler’ – which have historically been widely used in hazelnut breeding in the Eastern United States, largely as sources of resistance to the endemic fungal pathogen Anisogramma anomala (Eastern Filbert Blight; EFB) (Bhattarai et al., 2017). Constructed using a combination of long‐read sequencing, chromosome conformation capture and genetic mapping, these chromosome‐scale reference assemblies are of high quality and can immediately be deployed to advance breeding and genetic research objectives. The potential of these pseudohaploid assemblies is illustrated in this study through the identification of high‐density single nucleotide polymorphism (SNP) markers for over 1343 hazelnut plants, drawn from Midwestern natural and breeding populations. These markers provide a detailed assessment of population structure in C. americana and interspecific hybrids between C. americana and C. avellana. 20th‐century hazelnut breeding in the Midwestern United States involved interspecific hybridization with C. avellana (Rutter, 1987; Weschcke, 1954), but generations of open pollination and lack of detailed pedigree records obfuscate the genetic background of current Midwestern varieties. An analysis of population structure helps to resolve this question. Understanding the proportional representation of C. americana and C. avellana genetic backgrounds in varieties which have been successfully selected under Midwestern conditions will help determine breeding strategies for key traits.
In addition, the last 2 years have seen the release of chromosome‐scale genome assemblies for several other Corylus species: C. avellana (cultivars ‘Tombul’ (Lucas et al., 2021) and ‘Tonda Gentile della Langhe’ (‘TGdL’) (Pavese et al., 2021)), C. heterophylla (accessions from Siping City, Jilin (Liu et al., 2021) and Yanqing, Beijing (Zhao et al., 2021)) and a wild specimen of C. mandshurica (Li et al., 2021). Together with the two C. americana genomes reported here, this study presents the first comparative genomic analyses across nearly half of the Corylus genus.
Comparative genetic analysis also is useful for analysing specific traits, such as self‐incompatibility, an understanding of which is critical to both hazelnut breeding and production. Plants have evolved numerous strategies to increase the frequency of outcrossing, ranging from variation in the timing of floral development, to genetically regulated mechanisms. With respect to the latter, two primary classes of genetic self‐incompatibility have been identified: gametophytic self‐incompatibility (GSI) and sporophytic self‐incompatibility (SSI) (Silva and Goring, 2001). The former is typified by Rosaceae and Solanaceae species, wherein the haploid genotype of the male gamete at an ‘S‐locus’ (often encoding an F‐box (SLF/SFB) protein) is detected by the female parent, which blocks the growth of the germinating pollen tube (Sassa, 2016; Sijacic et al., 2004). The latter is exemplified by Brassicaceae species, where the diploid genotype of the male parent at the S‐locus (often encoding a cysteine‐rich protein (SCR/SP11)) mediates detection by the female parent and a suppression of pollen germination (Hiscock and McInnis, 2003). Self‐incompatibility in Corylus represents a form of SSI, regulated by a single S‐locus (Mehlenbacher and Thompson, 1988). Fluorescent microscopic observation of pollen tube growth has identified 33 alleles with 8 levels of linear dominance at this S‐locus (Mehlenbacher, 1997, 2014). We apply a phylogenetic approach to the analysis of SSI using the transcriptomes reported here, and this analysis sheds additional light on the evolution of self‐incompatibility in Corylus.
Results
Sequencing and assembly
Table 1 summarizes the raw sequence data used in assembling the two genomes. For ‘Rush’, 14.97 Gb of circular consensus sequencing (CCS) reads (44.32× coverage) and 13.60 Gb of ONT sequence (40.29× coverage) were generated. For ‘Winkler’, 19.60 Gb of CCS reads (58.02×) and 12.69 Gb of ONT sequence (37.57× coverage) was generated. Short‐read sequencing of Arima Hi‐C libraries generated 78.7× and 114× coverage for ‘Rush’ and ‘Winkler’, respectively. While ‘Winkler’ libraries, therefore, generated substantially more data across all sequencing platforms, in the case of both accessions, coverage was judged to be more than sufficient for the assembly of a diploid organism, assumed to have a relatively small genome size of ~350–370 Mb, based on comparisons with previous Corylus assemblies (Li et al., 2021; Liu et al., 2021; Lucas et al., 2021; Pavese et al., 2021; Zhao et al., 2021). The contact map for ‘Winkler’, showing contigs scaffolded into chromosome‐scale pseudomolecules, is shown in Figure 1a (visualized using Juicebox (https://github.com/aidenlab/Juicebox)).
Table 1.
Summary of sequence data generated on PacBio, Oxford Nanopore and Illumina sequencing platforms
| Sample | Sequence type | Reads | Avg. length (bp) | Coverage |
|---|---|---|---|---|
| ‘Rush’ | PacBio CCS | 1 026 165 | 14 584 | 44.3× |
| ‘Winkler’ | PacBio CCS | 1 264 282 | 15 496 | 58.0× |
| ‘Rush’ | ONT | 581 538 | 22 053 | 40.3× |
| ‘Winkler’ | ONT | 616 903 | 21 814 | 37.6× |
| ‘Rush’ | Arima Hi‐C | 88 601 323 | 2 × 150 | 78.7× |
| ‘Winkler’ | Arima Hi‐C | 129 091 816 | 2 × 150 | 114.7× |
Figure 1.
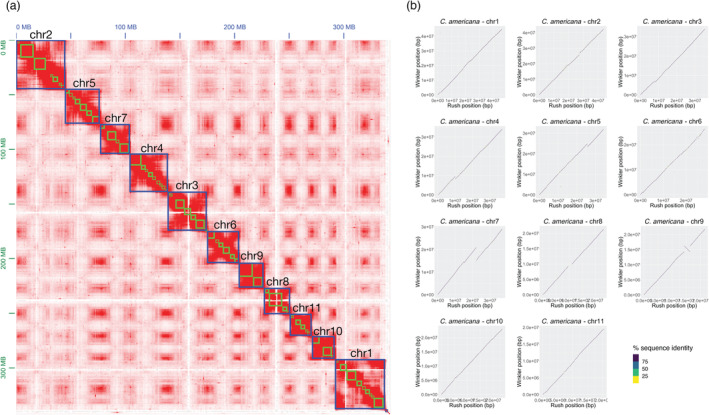
(a) Contact map showing aligned and sorted Arima Hi‐C data for ‘Winkler’ as visualized in Juicebox; green squares indicate ordered and oriented contigs, and blue squares indicate chromosomes. (b) Whole genome comparison of the ‘Rush’ and ‘Winkler’ chromosome‐scale assemblies. Dotplots of raw windowed alignment were generated by GENESPACE. While small indels are evident, largely consistent synteny is observed across all 11 chromosomes. A single sizeable inversion on chromosome 9 is highlighted in red. Potential inclusion of both haplotypes is only evident in ‘Rush’.
Table 2 presents statistics summarizing the quality and contiguity of the ‘Rush’ and ‘Winkler’ assemblies. While both genomes exhibit high quality and contiguity, with N50 scores >30 Mb, and over 90% of the genome assembled into 11 pseudomolecules, for both accessions there are clear differences which reflect the divergence in assembly methods. Both assemblies are close in length to previous reports of the size of Corylus species' genomes; however, the ‘Rush’ assembly is over 50 Mb larger than the ‘Winkler’ assembly. This is likely due to unresolved duplication, apparent also in Figure 1b, due to the fact that Hi‐C sequence data was not be included in the assembly, which is also reflected in the many more contigs present in ‘Rush’ compared to ‘Winkler’. While including Hi‐C data for ‘Winkler’ led to a reduction in total contigs from 398 (following hifiasm and haplotig_purge) to the 264 reported in Table 1, including Hi‐C data for ‘Rush’ led to a fragmentation of the assembly, increasing the number of contigs to 1845. Benchmarking Universal Single Copy Orthologs (BUSCOs) were calculated for ‘Rush’ and ‘Winkler’: while both assemblies had nearly identical complete and single‐copy BUSCOs represented (95.1% and 94.6%, respectively) ‘Rush’ contained more duplicates (3.8% vs. 1.4%).
Table 2.
Summary of statistics related to genome assemblies. BUSCO score codes: C: complete; S: complete and single copy; D: complete and duplicated; F: fragmented; M: missing
| Statistic | ‘Winkler’ | ‘Rush’ |
|---|---|---|
| Assembly size (bp) | 337 645 099 | 388 211 906 |
| Number of contigs | 264 | 1118 |
| GC content (%) | 36.05 | 36.68 |
| Assembly L50 (#) | 5 | 5 |
| Contig N50 (bp) | 31 913 876 | 35 381 848 |
| Maximum contig size (bp) | 44 790 255 | 46 319 783 |
| Genome in chromosomes (%) | 97.1 | 90.2 |
| BUSCO (C [S, D]; F; M) (%) | 96 [94.6, 1.4]; 0.9; 3.1 | 98.9 [95.1, 3.8]; 0.2; 0.9 |
This finding and the slightly larger maximum contig size also suggest unresolved artificial duplication. The duplicated regions visible in Figure 1B appear to be flanked by repetitive content, suggesting both haplotypes have been included in this draft assembly. While not conclusive, these dotplots also highlight the fact that Winkler has no such identifiable duplications.
Both assemblies were annotated for several genomic features: gene models were predicted using a combination of RNAseq data and protein homology data; transposable elements were identified using RepeatModeler; tandem repeats were identified using TandemRepeatFinder; and tRNAs were predicted using tRNAscan‐SE. These features are presented in a Circos‐style plot in Supp. Figure S2. Specific statistics related to the gene model predictions are summarized in Table 3.
Table 3.
Summary of statistics for the annotation of ‘Winkler’ and ‘Rush’
| Statistic | ‘Winkler’ | ‘Rush’ |
|---|---|---|
| Primary transcripts | 23 331 | 24 562 |
| Alternative transcripts | 5701 | 6174 |
| Median exon length (bp) | 159 | 159 |
| Median intron length (bp) | 245 | 248 |
| Average number of exons per gene | 5.7 | 5.7 |
These annotations appear extremely similar in terms of the structure of the gene models that were predicted. Slightly fewer genes were predicted in ‘Winkler’, which in this case is likely not the consequence of greater unresolved duplication in ‘Rush’, as the genomes were soft masked for repetitive elements. Finally, additional statistics related to the genome annotation are provided in Table 4, which compares the ‘Rush’ and ‘Winkler’ assemblies to the five currently published chromosome‐scale Corylus assemblies.
Table 4.
Comparison with annotations of previously published Corylus species. BUSCO scores refer to all complete representatives, both single and duplicated
| Species and cultivar name or accession | Genome size (11 chromosomes) | Repeat content (%) | Predicted genes | Genome BUSCO | Annotation BUSCO |
|---|---|---|---|---|---|
| C. americana (‘Winkler’) | 327.7 | 48.4 | 23 331 | 96.9 | 96.5 |
| C. americana (‘Rush’) | 350.2 | 49.0 | 24 562 | 99.0 | 99.0 |
| C. mandshurica | 367.7 | 68.7 | 25 923 | 97.1 | 92.2 |
| C. heterophylla (Beijing) | 361.9 | 56.7 | 27 591 | 93.5 | 92.0 |
| C. heterophylla (Jilin) | 342.9 | 58.0 | 22 319 | 94.7 | 92.7 |
| C. avellana (‘TGdL’) | 373.1 | 41.5 | 27 791 | 96.2 | 92.0 |
| C. avellana (‘Tombul’) | 369.8 | 38.1 | 27 270 | 97.0 | 76.8 |
The total number of predicted genes is within the range previously reported in Corylus. In addition, the BUSCO scores for ‘Rush’ and ‘Winkler’ are substantially higher than the scores reported for any previous Corylus genome annotation and are very close to the assembly BUSCO scores. This suggests not only that these annotations are highly complete, but also provides support for the accuracy of the predictive procedure described above.
Comparison across the Corylus genus
Comparisons across multiple species can be made simultaneously or in a pairwise manner. Using the five previously published Corylus assemblies, which contain comparable annotations to the genomes presented here, together with the outgroups silver birch (Betula pendula) and apple (Malus domestica), the GENESPACE package (Lovell et al., 2022) was utilized to visualize syntenic relationships in both manners, as well as construct a pangenome assembly anchored to C. americana ‘Winkler’. Figure 2 illustrates the general result that across these Corylus genomes, there is extremely broad synteny between all defined blocks, illustrating the high degree of relatedness between each pair of species. Differences in pseudomolecule numbering in C. avellana ‘Tonda Gentile della Langhe’, C. heterophylla ‘Jilin’ and C. mandshurica is an artefact of chromosome numbering not being standardized across species. Instead, numbering is currently made according to descending physical contig size; while Corylus chromosome sizes are generally very similar, assemblies had slight variations in contig lengths. Figure S3 shows dot plots representing each pairwise comparisons between syntenic blocks for each of the seven Corylus genomes. Despite the high degree of synteny across all Corylus species on a chromosome‐scale, it is clear that C. americana exhibits nearly perfect synteny with the two C. avellana assemblies. This decreases marginally when compared against the two C. heterophylla assemblies, with small inversions evident on a number of chromosomes. C. mandshurica very clearly exhibits the least amount of synteny, with large inversions and rearrangements of syntenic blocks across most chromosomes.
Figure 2.
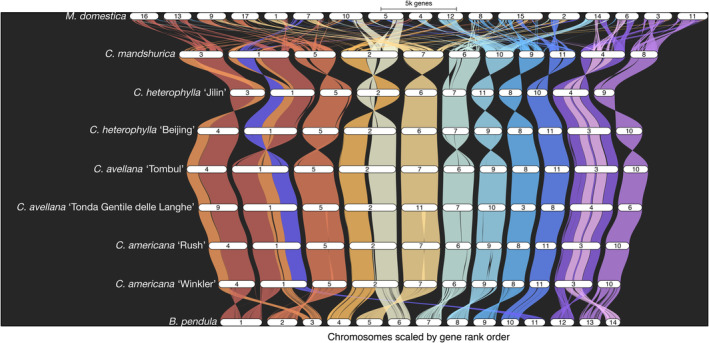
Riparian plot generated by GENESPACE. Single‐copy orthologues were used to generate and visually compare synteny between ‘Rush’ and ‘Winkler’, the five other published Corylus genomes, and the outgroups Betula pendula and Malus domestica. In addition to illustrating consistent genome‐wide inter‐species synteny across the 11 chromosomes of Corylus, this plot clearly illustrates the large‐scale relationships between these 11 chromosomes and the 14 chromosomes of B. pendula.
With respect to B. pendula, this figure makes clear the specific large‐scale rearrangements which relate the 14 chromosomes of B. pendula to the 11 of Corylus spp. Specifically, sections of chromosomes 1 and 3 in B. pendula are rearranged in chromosomes 1 and 4 in Corylus spp., with B. pendula chromosome 11 also being fused with the end of chromosome 1 in Corylus. In addition, chromosomes 4 and 6 in B. pendula together constitute chromosome 2 in Corylus, while chromosomes 12 and 14 constitute chromosome 3 in Corylus.
Single‐copy orthologues unique to C. americana
It is also possible to use the orthogroups generated by OrthoFinder to determine, instead of synteny, specifically those putative genes in C. americana which possess no orthologue in any currently annotated Corylus species, nor the other two outgroups included in this analysis: B. pendula and M. domestica. This subset of the transcriptome consists of 66 predicted single‐copy orthologues present in both ‘Rush’ and ‘Winkler’, but absent in all other analysed genomes (File S1). By filtering for only those single‐copy orthologues present in the predicted gene sets for ‘Rush’ and ‘Winkler’ limits the possibility that these unique genes are an artefact of spurious assembly errors in either of the independent ‘Rush’ or ‘Winkler’ assemblies. Similarly, including a diversity of assemblies of other Corylus species, as well as the Betula and Malus outgroups, limits the potential that these genes have been identified as unique simply as a result of more successful gene prediction for ‘Rush’ and ‘Winkler’, which due to the high BUSCO scores for the annotations reported here, would otherwise be a concern.
Many of these genes logically did not return hits when the Viridiplantae database was queried using NCBI‐BLAST. Several, however, are predicted to be involved in defence response pathways. Of particular note is the gene CamerWinkler.08G009300 (homologous to CamerRush.08G012000.1 in ‘Rush’), which is characterized as involved in ‘defense response to fungus’. Given C. americana's high level of resistance to the endemic fungal pathogen EFB, this gene would be a valuable target for future functional characterization.
Contributions of C. americana and C. avellana to current Midwest germplasm
A total of 1343 individual plants were sequenced using GBS, and SNP markers were called using the TASSEL GBSv2 pipeline (Bradbury, 2007). Sampled plants represented wild C. americana, cultivars from breeding programmes in Oregon, New Jersey and Minnesota, and F1 populations comprising controlled crosses between these cultivars. A biplot for the first two principal components of the distance matrix constructed using these markers is shown in Figure 3. This visualization makes evident the wide genetic diversity represented in current Midwestern hazelnut varieties and lends support to the significant C. avellana contribution to specific selections, such as Rose9‐2. Many currently named Midwestern varieties, on the other hand, cluster extremely closely with ‘Rush’, ‘Winkler’ and wild C. americana from the DNR, suggesting that while an interspecific cross may have occurred in the past, subsequent selection of progeny greatly favoured C. americana genetic contributions.
Figure 3.

PCA biplot of SNPs identified in C. americana, C. avellana and interspecific hybrids. On the left, wild C. americana sourced from the DNR clusters together with ‘Rush’ and ‘Winkler’, along with a number of Midwestern varieties and an F1 family produced through a cross between two of them (light and dark green dots, respectively). In the middle, an outlier Midwest variety (Rose9‐2) exhibits PC1 scores similar to an F1 between Eric4‐21 (a Midwest variety that appears similar to C. americana) x Jefferson (a C. avellana variety from Oregon State University). On the right are C. avellana varieties from Oregon State University and Rutgers University, along with an interspecific F1 population between a Rutgers cultivar and Rose9‐2. This plot illustrates that while most Midwestern varieties appear to be genetically quite similar to C. americana, there is substantial diversity among them (more so than what is observed between the limited number of C. avellana varieties included in the analysis) with some appearing quite similar to interspecific F1 hybrids – a finding supported by the historical record of interspecific hybridization in Midwestern breeding efforts.
Phylogenetic analysis of sporophytic self‐incompatibility
The locus regulating sporophytic self‐incompatibility (SSI) in hazelnut has been fine‐mapped in C. avellana (Hill et al., 2021) and C. heterophylla x C. avellana interspecific hybrids (Hou et al., 2022). These studies identified three MIK2 homologues believed to be responsible for SSI in these two Corylus species. In order to examine their phylogenetic relationship, multiple sequence alignment was performed between the top BLAST hit for each of these three genes and all currently available Corylus transcriptomes. B. pendula and B. oleracea, which both also exhibit SSI (Hynynen et al., 2010; Kitashiba and Nasrallah, 2014), as well as M. domestica, a more distantly related species that is a well‐studied example of gametophytic self‐incompatibility (Cheng et al., 2006; Minamikawa et al., 2010), were included as outgroups. This phylogenetic tree is shown in Figure 4 and illustrates the genus‐specific evolution of SSI in Corylus, represented by several clades containing orthologous sequences only found in Corylus species.
Figure 4.
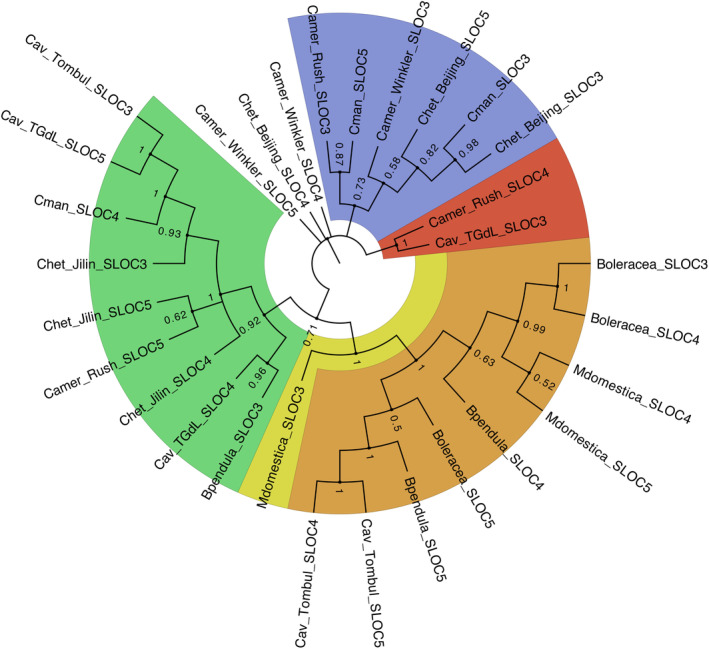
A phylogenetic tree showing the evolution of genes comprising the self‐incompatibility locus in Corylus across the seven available Corylus genomes, with B. pendula, M. domestica, and B. oleracea included as outgroups. Node labels represent bootstrap values based on 100 permutations. Colour overlays illustrate evident clades, and highlight the fact that several lineages of S‐locus genes are entirely specific to Corylus.
Discussion
These assemblies represent a critical step towards developing the use of next‐generation sequencing data in the improvement of C. americana, while also enabling comparisons of structural variation across Corylus species.
Variable contiguity of two de novo assemblies
The fragmentation of the ‘Rush’ assembly by the 3D‐DNA pipeline (using Arima Hi‐C sequence data) was unexpected, given the widespread and successful use of this method in building chromosome‐scale assemblies (Ghurye et al., 2019), and in particular, the substantial improvements in contiguity that was observed when using this data to refine the initial ‘Winkler’ assembly. Two possible explanations for this discrepancy suggest themselves. First, the ‘Rush’ accession generated less absolute sequence compared to the ‘Winkler’ assembly on all sequencing platforms, and as a result, there was less sequence data to include in each bioinformatic step. The raw assemblies generated by hifiasm were as a result also more contiguous for Winkler compared to ‘Rush’.
Nevertheless, with coverage >40× for all sequence data types, the failure of Hi‐C sequence to improve contiguity would not appear to be solely a consequence of a lack of data. Indeed, when reads were randomly removed from the Hi‐C sequence data for ‘Winkler’, to simulate the coverage of Hi‐C reads obtained for ‘Rush’ (78.7×), no decrease in the contiguity of the resulting assembly was observed, relative to the assembly generated using the full set of ‘Winkler’ Hi‐C reads. Artificially reducing coverage of the ‘Winkler’ Hi‐C sequence data to even 30× still resulted in an assembly with a larger N50, and equivalent L50, relative to the assembly prior to running 3D‐DNA. The fragmentation of the ‘Rush’ assembly caused by the 3D‐DNA pipeline, therefore, cannot solely be explained by the number of Hi‐C reads generated. A second likely factor is a difference in the relative heterozygosity of ‘Rush’ and ‘Winkler’. Heterozygous single‐individual assemblies are more challenging to construct, relative to fully homozygous individuals (Garg, 2021). Using the 53 696 GBS‐derived SNPs described above it was possible to directly estimate genome‐wide heterozygosity as a ratio of heterozygous sites to total polymorphic sites. By this metric, 22% heterozygosity was observed for ‘Rush’, while only 17% heterozygosity was observed for ‘Winkler’, suggesting that this could have contributed to the challenges in scaffolding ‘Rush’ using Hi‐C data.
Regardless of the specific underlying factors complicating this stage of genome assembly, it is clear that even in diploid species with relatively small genome sizes, integrating chromosomal conformation capture sequence data into genome assembly pipelines does not always lead to more contiguous assemblies, with the outcome likely dependent on sequence coverage, heterozygosity and the quality of the initial draft assembly. At the same time, the use of high‐coverage long‐read sequence data, together with modern assemblers for scaffolding and polishing chromosome‐scale genomes, can generate high‐quality reference assemblies, even in outcrossing, heterozygous organisms.
PCA‐modelled population structure
This study reports the first combined analysis of relatedness between commercial varieties of hazelnuts from the three main breeding programmes in the United States: Oregon State University, Rutgers University and the Upper Midwest Hazelnut Development Initiative. This evaluation of population structure, by combining a diversity of clonal varieties with F1 full‐sib families produced through multiple pairwise controlled crosses of these clones, help elucidate a long‐outstanding question regarding the degree to which Midwest‐bred hazelnut accessions remain closely related to C. avellana. 20th‐century Midwest breeding populations included interspecific crosses with C. avellana, but the precise frequency of such hybridization events, and the fact that they were often followed by several generations of open pollination within predominantly C. americana breeding orchards, has left this question unresolved. Figure 3 shows clearly that the majority of these Midwest accessions are indeed closely related to wild C. americana sourced from the Department of Natural Resources, as well as ‘Rush’ and ‘Winkler’ themselves. A clear outlier in this respect is the variety Rose9‐2, which very closely resembles other F1 interspecific crosses.
Cross‐genus level analysis
Transcriptomic analysis of the seven Corylus genomes included in this study reveals a consistent distribution of genes across the 11 chromosomes. This analysis considers nearly half of the recognized species in Corylus (Thompson et al., 1996). The observed consistency in gene distribution suggests a relatively recent common ancestor of these hazelnut species. This finding is important for understanding the evolutionary relationships between these species and could have implications for hazelnut breeding and genetic improvement programmes by providing a foundation for identifying useful traits and genes that are present in all Corylus species. These observations should not, however, be considered a substitute for phylogenetic analyses, constructed either with genome‐wide polymorphisms or organellar sequence data. Two such phylogenies have recently been reported (Helmstetter et al., 2019; Zhao et al., 2020), which suggest a closer relationship between C. americana and the Asian taxa, relative to C. avellana, which is a finding similar to that seen in the phylogenetic trees built using SSI genes presented above. These genome‐wide syntenic relationships are not direct evidence against this finding, given that the algorithms for estimating phylogenetic relationships are not based on chromosome‐scale evaluation of syntenic relationships in gene features. In addition, published phylogenies have included a far greater number of samples, relative to the limited number of chromosome‐scale genome assemblies available for the comparative analyses carried out in this study. However, our results do provide important context when interpreting such phylogenies, demonstrating both a substantial level of genome‐wide similarity across numerous clades within Corylus, as well as marginally greater synteny in the nuclear genome between species that are phylogenetically more diverged.
In addition to identifying general synteny on a genome‐wide level, these analyses also made possible the identification of those single‐copy orthologues with no representation in any other Corylus species. In particular, a gene found in both ‘Rush’ (CamerRush.08G012000.1) and ‘Winkler’ (CamerWinkler.08G009300) was ontologically associated with defence responses to fungal infection.
In particular, it appears to be a defensin‐like (DEFL) gene, cysteine‐rich antimicrobial proteins (Tesfaye et al., 2013). This gene (CamerWinkler.08G009300) is, therefore, a promising target for future functional characterization. Quantitative genetic analyses have revealed numerous loci in C. americana that are associated with EFB resistance (Komaei Koma et al., 2021; Lombardoni et al., 2022; Revord et al., 2020), and the gene reported here could be productively included in any future studies aimed at using expression analysis or forward genetic approaches to validating their role in plant defence pathways.
Sporophytic self‐incompatibility
Attempts at genetically characterizing the SSI S‐locus in Corylus have included hybridization‐based staining of genes identified in Brassica oleracea L., which showed irregular hybridization (Hampson et al., 1996). Similarly, gene expression studies identified only 61% homology between B. oleracea alleles and S alleles in Corylus (Torello Marinoni et al., 2009). Finally, transcriptomic analysis of the fine‐mapped S‐locus region in multiple Corylus species suggest Corylus may harbour a novel SSI molecular mechanism that differs from Brassica (Hill et al., 2021; Hou et al., 2022). The analysis presented here suggests independent evolution of at least part of the sporophytic self‐incompatibility mechanism that is unique to Corylus and not shared by other members of the Betulaceae, nor model species of SSI such as Brassica oleracea. Only two implicated SSI genes, both from C. avellana ‘Tombul’, appear within a clade shared by B. oleracea, although occupying a relatively distinct sub‐lineage. This shared ancestry, albeit limited, could suggest that a shared SSI system evolved prior to the species' divergence.
While gametophytic self‐incompatibility (GSI) necessitates codominance between S alleles to prevent self‐fertilization, SSI does not, allowing for the evolution of complex hierarchies of dominance and codominance (Brennan et al., 2011), characteristics observed in both Corylus and Brassica (Mehlenbacher, 2014; Prigoda et al., 2005). Dominance in SSI has been implicated in accelerating rates of evolution within these systems, due to selective pressure for heterozygous genotypes to maximize their compatibility with the greatest number of S‐locus genotypes (Schoen and Busch, 2009). This rapid rate of diversification within SSI systems may explain the inconclusive results observed in the gene tree in Figure 4 and makes the investigation of whether SSI evolved independently in the Brassicales and Fagales challenging. As long‐read sequencing increasingly makes the assembly of high‐quality, annotated genome assemblies more accessible to previously understudied species, future phylogenetic studies will hopefully be able to repeat the analyses performed here with a greater degree of resolution, helping to elucidate the specific point or points at which SSI systems evolved and diverged.
Conclusion
High‐quality, annotated, chromosome‐scale genome assemblies are essential tools for using modern genetic methods in the investigation of the molecular control of important traits, as well as the application of advanced breeding methods. These two assemblies provide an important resource which we hope will enable the application of such methods to the study and improvement of both C. americana, as well as interspecific hybrids which expand the range over which hazelnuts can be cultivated. Our analysis demonstrates highly conserved genome‐wide synteny across Corylus species. As such, genomic tools, analyses and resources developed in one species may be more broadly useful to breeding programmes in other species within the genus.
Experimental procedures
Plant material collection
Tissue was collected from C. americana accessions ‘Rush’ and ‘Winkler’ maintained at the National Clonal Plant Germplasm Repository (NCGR) in Corvallis, Oregon (PI 557022 and PI 557019, respectively). ‘Rush’ is a specimen collected around 1900 by J.F. Jones in Lancaster, Pennsylvania, while ‘Winkler’ was collected in 1910 by Wendell Williams in Danville, Iowa (Molnar, 2011). These two varieties were historically significant in early‐ and mid‐20th century hazelnut breeding programmes and as such represent genetically relevant, pure C. americana selections, which also reflect the wide geographic distribution of C. americana in the Eastern United States. Today, ‘Rush’ remains a widely used source of EFB resistance (Bhattarai et al., 2017). Photos of the clones located at the NCGR are shown in Figure S1. Tissue was collected on April 13th, 2020, from bushes that had been etiolated for 3 days prior to sampling in order to minimize the concentration of volatiles and secondary metabolites in leaf tissue. Samples were immediately flash‐frozen using liquid nitrogen and stored at −80 °C until DNA extraction.
Long‐read sequencing
High‐molecular‐weight DNA was extracted following the protocol described by Vaillancourt and Buell (2019). In brief, leaf tissue was homogenized in liquid nitrogen, lysed with Carlson lysis buffer and purified with chloroform and Qiagen Genomic‐tips (Qiagen N.V., Venlo, The Netherlands). Purity of extracted DNA was assessed spectrophotometrically using a NanoDrop™ One (Thermo Fisher Scientific, Waltham, Massachusetts). DNA was quantified a using Qubit™ dsDNA High Sensitivity kit (Thermo Fisher Scientific, Waltham, Massachusetts), and diluted and assessed for size using an Agilent FemtoPulse System (Santa Clara, California).
Pacific Biosciences HiFi libraries were prepared according to PN 101‐853‐100 Version 03 (Pacific Biosciences, Menlo Park, California). Modifications included shearing with a Covaris g‐TUBE (Covaris, Woburn, Massachusetts) and size selecting with BluePippin (Sage Science, Beverly, MA). Libraries were sequenced on a PacBio Sequel II using the Sequel Polymerase Binding Kit 2.2 at the University of Wisconsin‐Madison Biotechnology Center's DNA Sequencing Facility. Oxford Nanopore libraries were prepared following the Native Barcoding Expansion protocol (Oxford Nanopore Technologies, Oxford, United Kingdom). The library was sequenced on a R9.4.1 flowcell on an Oxford Nanopore PromethION, also at the UW‐Madison Biotechnology Center. Partway through the sequencing run, DNA was flushed with the Oxford Nanopore Technologies Flow Cell Wash Kit (EXP‐WSH004), and the additional library was loaded.
Hi‐C sequencing
Nuclei were extracted from flash‐frozen leaf tissue using a Sigma CelLytic™ PN Plant Nuclei Isolation/Extraction Kit (Sigma‐Aldrich, Burlington, Massachusetts). Crosslinking was performed following low‐input protocols from Arima Genomics (Arima Genomics, Carlsbad, California). Crosslinked nuclei were quantified using a Qubit™ dsDNA High Sensitivity kit, samples were sheared to 600 bp, and library preparation was performed using a KAPA® Hyper Prep kit (Roche, Basel, Switzerland). The final library was assessed for quality using the Agilent TapeStation System with a D1000 kit (Agilent Technologies, Santa Clara, California), and paired‐end, 150 bp paired‐end sequences were generated using an Illumina NovaSeq 6000 (Illumina, San Diego, California).
RNA extraction, library preparation and sequencing
Prior to RNA extraction, Tissuelyser II adapters, safelock tubes and 5 mm stainless steel beads were chilled at −80 °C for 2 h. Working on dry ice, leaf and kernel tissue was added to prechilled tubes and disrupted using the Tissuelyser II (Qiagen N.V., Venlo, The Netherlands) for 2 rounds at 30 Hz for 1 min each round. Samples were then processed using the Qiagen Plant RNeasy (Qiagen N.V., Venlo, The Netherlands) with an on‐column DNA digest. Total RNA was assayed for purity and integrity using a NanoDrop One™ Spectrophotometer and Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, California), respectively. RNA libraries were prepared from samples that met the Illumina TruSeq® Stranded Total RNA Sample Preparation Guide (15 031 048 E) input guidelines using the Illumina TruSeq® Stranded Total (Plant) RNA Sample Preparation kit (Illumina Inc., San Diego, California). For each library preparation, cytoplasmic, mitochondrial and chloroplast ribosomal RNA was removed using biotinylated target‐specific oligos combined with paramagnetic beads tagged with streptavidin. Following purification, the reduced RNA was fragmented using divalent cations under elevated temperature. Fragmented RNA was copied into first‐stranded cDNA using SuperScript II Reverse Transcriptase (Invitrogen, Carlsbad, California) and random primers. Second‐strand cDNA was synthesized using a modified dNTP mix (dTTP replaced with dUTP), DNA Polymerase I and RNase H. Double‐stranded cDNA was cleaned with AMPure XP Beads (1×) (Beckman Coulter, Brea, California). The cDNA products were incubated with Klenow DNA Polymerase to add a single ‘A’ nucleotide to the 3′ end of the blunt DNA fragments. Unique dual indexes (UDI) were ligated to the DNA fragments and cleaned with two rounds of AMPure XP beads (0.8×). Adapter‐ligated DNA was amplified by PCR and cleaned with AMPure XP beads (0.8×). Final libraries were assessed for size and quantity using an Agilent DNA1000 chip and a Qubit® dsDNA HS Assay Kit (Invitrogen, Carlsbad, California), respectively. Libraries were standardized to 2 nm, and 150‐bp paired‐end sequencing was performed on an Illumina NovaSeq 6000.
Genome assembly
An initial assembly was created using PacBio CCS reads with the program hifiasm v0.16.0‐r369 (Cheng et al., 2021) on a Unix server with 160 cores and 3TB of RAM. This draft assembly exhibited higher contiguity than references generated with Canu using ONT reads, HiCanu and Flye using either HiFi or ONT reads. The program purge_haplotigs v1.1.1 (Roach et al., 2018) was then utilized to reduce artificial genome duplication caused by incorporation of non‐collapsed haplotigs in the final assembly. Hi‐C reads were aligned, filtered and binned using the haplotig‐purged assembly using the programme juicer (Durand et al., 2016). Contigs were then scaffolded and ordered using the program 3D‐DNA (Dudchenko et al., 2017).
While integration of Hi‐C data led to improved contiguity and reduced artificial duplication in the ‘Winkler’ assembly, contigs in ‘Rush’ were fragmented following the use of juicer and 3D‐DNA. As a result, this step was omitted for ‘Rush’. The assemblies were next iteratively polished three times with Racon (Vaser et al., 2017), using the original PacBio reads. As hifiasm performs polishing internally, this step was superfluous and to ensure it did not introduce artificial errors by re‐inserting alleles from the alternate haplotype, we used merqury (Rhie et al., 2020) to confirm that the quality and completeness of the assembly were unaffected by Racon. We then used a recently constructed linkage map to detect erroneous inversions in the physical assemblies (Brainard et al., 2023), using visual inspection of heatmaps of recombination frequencies in an F1 population. The program Ragtag v2.1.0 (Alonge et al., 2022), which is used to carry out reference‐guided assembly, was then used to correct observed errors using ONT reads that overlapped any identified inversions. Initial quality assessments were made using BUSCO v5.4.3 (Manni et al., 2021) with the eudicots_odb10 database and quast v5.2.0 (Mikheenko et al., 2018) with default parameters.
Genome annotation
Transcript assemblies for both ‘Rush’ and ‘Winkler’ were made from ~200 M 2 × 150 bp stranded paired‐end Illumina RNAseq reads using the software PERTRAN, which conducts genome‐guided transcriptome short‐read assembly via GSNAP (Wu and Nacu, 2010) and builds splice alignment graphs after alignment validation, realignment and correction. These outputs were subsequently used to construct 37 794 and 36 419 transcript assemblies for ‘Rush’ and ‘Winkler’, respectively, using the program PASA (Haas et al., 2003). Loci were determined by transcript assembly alignments and/or EXONERATE alignments of proteins from Mimulus guttatus, Arabidopsis thaliana, Gossypium raimondii, Betula platyphylla, Carya illinoinensis, Castanea dentata, Quercus rubra, Prunus persica, Fragaria vesca, Glycine max, Medicago truncatula, Vitis vinifera, Liriodendron tulipifera, Juglans microcarpa, Populus trichocarpa, Beta vulgaris, Solanum lycopersicum, Sorghum bicolor, Oryza sativa and Swiss‐Prot (release 2022_04 of eukaryote proteomes), with up to 2 k bp extension on both ends unless extending into another locus on the same strand. Alignments were made to the respective genome assemblies following softmasking for repetitive elements. The repeat library consisted of de novo repeats identified by RepeatModeler2 (Flynn et al., 2020), generated using both the ‘Rush’ and ‘Winkler’ assemblies, as well as C. americana repeats identified in the databases RepBase and Dfam. Gene models were predicted by homology‐based predictors: FGENESH+ (Salamov and Solovyev, 2000), FGENESH_EST (which is similar to FGENESH+, but uses EST to compute splice site and intron input instead of protein/translated ORF), EXONERATE (Slater and Birney, 2005), PASA assembly ORFs (a homology constrained ORF finder) and AUGUSTUS (Stanke et al., 2006) trained by the high confidence PASA assembly ORFs and with intron hints from short read alignments. The best‐scored predictions for each locus were selected using multiple positive factors including EST and protein support and overlap with repeats as a negative factor. The selected gene predictions were improved by PASA. The improvement included adding UTRs, splicing correction and adding alternative transcripts. PASA‐improved gene model proteins were subject to protein homology analysis to the above‐mentioned proteomes to obtain a Cscore and protein coverage. Cscore is a protein BLASTP score ratio to the mutual best hit (MBH) BLASTP score, and protein coverage is the highest percentage of protein aligned to the best homologues. PASA‐improved transcripts were selected based on Cscore, protein coverage, EST coverage and their CDS overlap with repeats. The transcripts were selected if their Cscore and protein coverage were ≥0.5 or if covered by ESTs. For gene models whose CDS were overlapped by repeats by more than 20%, their Cscore was required to be at least 0.9, and homology coverage at least 70%, in order to be selected. The selected gene models were subject to Pfam analysis, and gene models whose proteins were more than 30% overlapped by Pfam TE domains were removed as weak gene models. Incomplete gene models, low homology‐supported gene models without full transcriptome support, short single exon (<300 bp CDS) with neither protein domains nor good expression, and repetitive gene models without strong homology support were manually filtered out.
Functional annotation
Every peptide sequence in the dataset was analysed with a computational pipeline that includes the standard InterProScan (Jones et al., 2014) suite of programs to determine protein domains and other sequence features, E2P2 (Chae et al., 2014; Schläpfer et al., 2017) for enzyme assignments (EC) and PathoLogic (Karp et al., 2021) for metabolic pathway assignments. Additional processing was used to determine Eukaryotic Orthologous Groups (KOG) gene assignment using a modified mutual best hit algorithm. Results of the InterProScan calculations were used to assign standard InterPro protein domain associations and from these, gene ontology (GO) terms. Protein domains inferred from these calculations were used to develop a putative gene functional assignment which includes a count of the multiplicity of the assignment in the proteome set.
Genotyping by sequencing
To better compare genomes of C. avellana and C. americana, we used genotyping‐by‐sequencing (GBS) to genotype a population of interspecific hybrids and a population of C. americana. Tissue from 1343 samples of breeding lines from the Upper Midwest Hazelnut Development Initiative (UMHDI), Oregon State University, in Corvallis, Oregon and Rutgers University, in New Brunswick, New Jersey, along with full‐sib F1 populations derived from controlled crosses between these varieties, and a wild Midwestern population of C. americana sourced from the Wisconsin Department of Natural Resources (DNR) and planted in Barneveld, WI, was sampled following budbreak in May of 2020. Genomic DNA was extracted and libraries for GBS were prepared using a double digestion with the restriction enzymes NsiI and BfaI following the methodology described by Elshire et al. (2011). This specific double digest was selected based on analysis of k‐mer distributions of sequence libraries generated with ApeKI alone, NsiI and MspI, PstI and BfaI, PstI and MspI, ApeKI and BfaI, and ApeKI and MspI. Illumina GBS barcodes and adapters were ligated, and paired‐end reads (2 × 150 bp, 10 million reads/sample) were generated using an Illumina NovaSeq 6000.
Due to the high levels of synteny across the genomes described above, direct use of Illumina sequence data to infer variable degrees of interspecific hybridization, using tools such as sppIDer (Langdon et al., 2018), is limited. Biallelic SNPs were, therefore, identified using the TASSEL GBSv2 pipeline (Bradbury et al., 2007) and filtered for missing data (<10% across all samples), minor allele frequency (>0.05), linkage disequilibrium (r 2 < 0.75) and allele depth (80% of samples with a depth >8) using bcftools (Danecek et al., 2021). Population structure analysis using principal components analysis (Price et al., 2006) has been shown to be a simple and efficient alternative to more complex model‐based approaches such as STRUCTURE (Falush et al., 2007) and ADMIXTURE (Alexander et al., 2009). This filtered VCF file was, therefore, converted to a Euclidean distance matrix using R (R Core Team, 2021), and multi‐dimensional scaling (which permits some missing data) was then performed to obtain measures analogous to scores along the first two principal components of the distance matrix.
Comparative analyses of Corylus
Comparisons across the Corylus genus were made utilizing the five currently available chromosome‐scale genome assemblies, as well as the C. americana assemblies reported here for ‘Rush’ and ‘Winkler’. This included two C. avellana assemblies (cultivars ‘Tombul’ (Lucas et al., 2021) and ‘Tonda Gentile della Langhe’ (Pavese et al., 2021)), two C. heterophylla assemblies (an accession from Siping City, Jilin (Liu et al., 2021) and an accession from Yanqing, Beijing (Zhao et al., 2021)) and one C. mandshurica assembly (a wild specimen from Xinglong (Li et al., 2021)). All of these assemblies were annotated using similar ab initio prediction methods which combined RNAseq data and protein homology data, and thus their transcriptomes were well‐suited to comparative analyses with ‘Rush’ and ‘Winkler’.
GENESPACE v0.94 (Lovell et al., 2022) was used to investigate genome‐wide syntenic relationships, with Betula pendula (silver birch) and Malus domestica (apple) included as outgroups (gene models being obtained from https://genomevolution.org/coge/ and https://www.rosaceae.org, respectively). In brief, GENESPACE utilizes the program OrthoFinder (Emms and Kelly, 2019) to identify orthogroups from predicted gene models and then parses orthologues to define syntenic blocks across species using BLAST and MCScanX (Wang et al., 2012). Genes in C. americana with no orthologous sequences in any other Corylus species were also identified using OrthoFinder.
Phylogenetic analysis of sporophytic self‐incompatibility (SSI)
Sequences for three S‐locus genes (MIK3 homologues) reported by Hou et al. (2022) were obtained from the transcriptome reported in Zhao et al. (2021). NCBI‐BLAST v2.6.0+ (Altschul et al., 1990) was used to identify homologous sequences in all Corylus species, as well as B. pendula, M. domestica and Brassica oleracea. These latter three species were included as variably related outgroups which also exhibit self‐incompatibly: SSI in the case of B. oleracea and B. pendula and GSI in the case of M. domestica. The B. oleracea transcriptome was obtained from http://brassicagenome.net/. Multiple sequence alignments were made using muscle v5.1 (Edgar, 2021). These 84 alignments were then imported into MEGA11 (Tamura et al., 2021) and used to build a phylogenetic tree using maximum likelihood and the Jones‐Taylor‐Thornton matrix‐based model (Jones et al., 1992). A bootstrap consensus tree was inferred from 100 replicates, in which branches present in <50% of these replicates were collapsed. Trees were visualized and clades labelled using the program FigTree v1.4.4.
Funding
Sequencing and bioinformatics was supported by the Jewett Prize of the Arnold Arboretum and USDA‐NIFA SCRI Grant No. H007913501, with matching funds from the Savanna Institute and The Grantham Foundation for the Protection of the Environment. Genome annotation conducted at the U.S. Department of Energy Joint Genome Institute (https://ror.org/04xm1d337) was supported by the Office of Science of the U.S. Department of Energy under Contract No. DE‐AC02‐05CH11231.
Conflict of interest
All authors declare no conflict of interest with the research and findings reported here.
Author contributions
SHB and JCD carried out conceptualization, methodology design and project administration. SHB, DMS and TB performed data curation. SHB, DMS, TB, SS and JCD conducted formal analysis. SHB performed visualization. SHB, JCD and TB wrote the manuscript. SHB and JCD were responsible for funding acquisition.
Supporting information
Figure S1 Clockwise from top left: the typical involucres, growth habit and nuts of C. americana ‘Rush’, held at the NCGR in Corvallis, Oregon.
{kind=link}
Figure S2 Circular plot of genomic features in ‘Winkler’. From the outside in: ideogram of the 11 chromosomes of C. americana; total gene content, as identified by gene prediction and annotation; transposable elements, as identified by RepeatModeler; heatmap of tandem repeats; line graph of tRNA content.
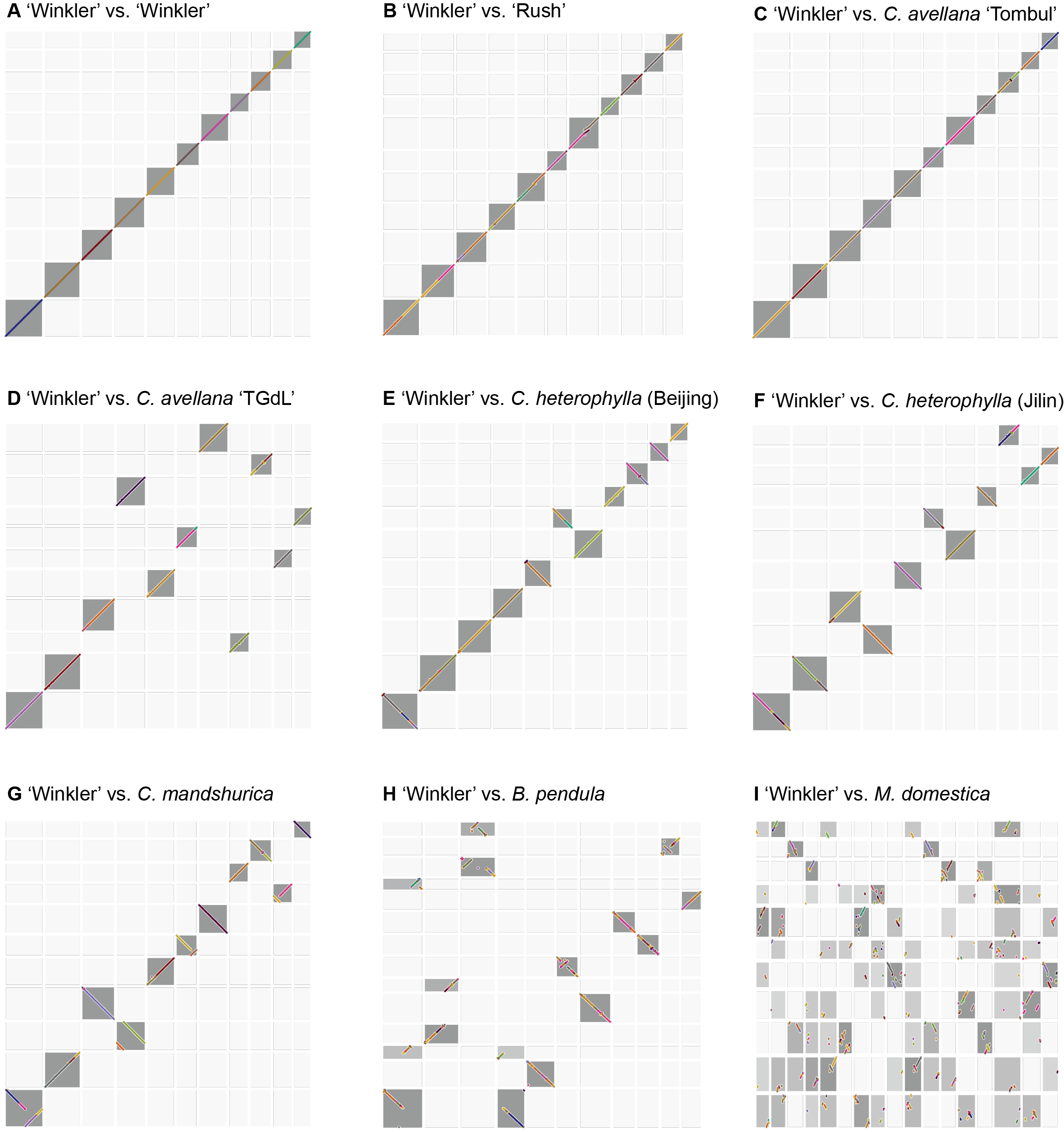
Figure S3 Pairwise dotplots visualizing pairwise BLAST hits between C. americana ‘Winkler’, and each of the other eight genomes included in the GENESPACE analysis.
{kind=link}
File S1 Gene IDs for a subset of the C. americana transcriptome consisting of 66 predicted single copy orthologs present in both ‘Rush’ and ‘Winkler’, but absent in all other analyzed genomes.
Acknowledgements
We thank Al Kovalevski, Samridhi Chaturvedi and Ashely Yow for providing helpful advice in the performance of phylogenetic analysis. In addition, Nahla Bassil, Shawn Mehlenbacher, Tom Molnar, Malachi Persche, Lois Braun and Mark Hamann assisted with the collection of tissue samples used in this study. We thank Thomas Hickey and Marissa Nix as well as the staff of the University of Wisconsin‐Madison Biotechnology Center DNA Sequencing Facility and the USDA National Clonal Germplasm Repository in Corvallis, OR for logistical support of the research. We also thank the staff of the Bioinformatics Resource Center, in particular Derek Pavelec, Olaf Mueller and Mark Berres. We thank Chuck and Gerta Zinda for allowing us to study the C. americana planted on their property in Barneveld, WI for our research.
Data availability statement
Genome assemblies, as well as short‐ and long‐read sequence data (from Arima Hi‐C, Oxford Nanopore, PacBio and RNAseq libraries) are available through the NCBI Genome & Sequence Read Archive, respectively, under BioProject ID: PRJNA939214 BioSample accessions: SAMN33458638 (‘Rush’), SAMN33458639 (‘Winkler’). Genome assemblies, GFF3 models of predicted genes, functionally annotated peptide sequences, anchored pangenome and VCF of GBS‐derived SNPs are also available at https://doi.org/10.5281/zenodo.7439335. Finally, genomes and annotations have been added to Phytozome: https://phytozome.jgi.doe.gov/info/Camericanavar_winkler_v1_1 (for Winkler); and https://phytozome.jgi.doe.gov/info/Camericanavar_rush_v1_1 (for Rush). Bioinformatic pipelines for genome assembly and polymorphism identification are published at: https://github.com/shbrainard/Camericana_pipelines.
References
- Alexander, D.H. , Novembre, J. and Lange, K. (2009) Fast model‐based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonge, M. , Lebeigle, L. , Kirsche, M. , Jenike, K. , Ou, S. , Aganezov, S. , Wang, X. et al. (2022) Automated assembly scaffolding using RagTag elevates a new tomato system for high‐throughput genome editing. Genome Biol. 23, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S.F. , Gish, W. , Miller, W. , Myers, E.W. and Lipman, D.J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Bhattarai, G. , Mehlenbacher, S.A. and Smith, D.C. (2017) Eastern filbert blight disease resistance from Corylus americana ‘Rush’ and selection ‘Yoder #5’ maps to linkage group 7. Tree Genet. Genomes 13, 1–10. [Google Scholar]
- Bolger, M. , Schwacke, R. , Gundlach, H. , Schmutzer, T. , Chen, J. , Arend, D. , Oppermann, M. et al. (2017) From plant genomes to phenotypes. J. Biotechnol. 261, 46–52. [DOI] [PubMed] [Google Scholar]
- Bradbury, P.J. , Zhang, Z. , Kroon, D.E. , Casstevens, T.M. , Ramdoss, Y. and Buckler, E.S. (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. [DOI] [PubMed] [Google Scholar]
- Brainard, S.H. , Dawson, J.C. , Fischbach, J.A. and Braun, L.C. (2023) Improving selection efficiency in C. americana × C. avellana interspecific hybrids through the development of an indel‐based genetic map. Acta Hortic, 1–7. 10.1101/2023.02.05.527175 [DOI] [Google Scholar]
- Brennan, A.C. , Tabah, D.A. , Harris, S.A. and Hiscock, S.J. (2011) Sporophytic self‐incompatibility in Senecio squalidus (Asteraceae): S allele dominance interactions and modifiers of cross‐compatibility and selfing rates. Heredity 106, 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceylan, F.D. , Adrar, N. , Bolling, B.W. and Capanoglu, E. (2022) Valorisation of hazelnut by‐products: current applications and future potential. Biotechnol. Genet. Eng. Rev. 1–36. 10.1080/02648725.2022.2160920 [DOI] [PubMed] [Google Scholar]
- Chae, L. , Kim, T. , Nilo‐Poyanco, R. and Rhee, S.Y. (2014) Genomic signatures of specialized metabolism in plants. Science 344, 510–513. [DOI] [PubMed] [Google Scholar]
- Cheng, J. , Han, Z. , Xu, X. and Li, T. (2006) Isolation and identification of the pollen‐expressed polymorphic F‐box genes linked to the S‐locus in apple (Malus × domestica). Sex. Plant Reprod. 19, 175–183. [Google Scholar]
- Cheng, H. , Concepcion, G.T. , Feng, X. , Zhang, H. and Li, H. (2021) Haplotype‐resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Bonfield, J.K. , Liddle, J. , Marshall, J. , Ohan, V. , Pollard, M.O. , Whitwham, A. et al. (2021) Twelve years of SAMtools and BCFtools. GigaScience 10, giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demchik, M. , Fischbach, J. , Kern, A. , Lane, J. , McCown, B. , Zeldin, E. and Turnquist, K. (2014) Selection of American hazelnut as a potential oilseed crop. Agroforestry Syst. 88, 449–459. [Google Scholar]
- Di Lena, B. , Curci, G. , Vergni, L. and Farinelli, D. (2022) Climatic suitability of different areas in Abruzzo, Central Italy, for the cultivation of hazelnut. Horticulturae 8, 1–16. [Google Scholar]
- Dmitriev, A.A. , Pushkova, E.N. and Melnikova, N.V. (2022) Plant genome sequencing: modern technologies and novel opportunities for breeding. Mol. Biol. 56, 495–507. [DOI] [PubMed] [Google Scholar]
- Dudchenko, O. , Batra, S.S. , Omer, A.D. , Nyquist, S.K. , Hoeger, M. , Durand, N.C. , Shamim, M.S. et al. (2017) De novo assembly of the Aedes aegypti genome using Hi‐C yields chromosome‐length scaffolds. Science 356, 92–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand, N.C. , Shamim, M.S. , Machol, I. , Rao, S.S.P. , Huntley, M.H. , Lander, E.S. and Aiden, E.L. (2016) Juicer provides a one‐click system for analyzing loop‐resolution Hi‐C experiments. Cell Syst. 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R.C. (2021) High‐accuracy alignment ensembles enable unbiased assessments of sequence homology and phylogeny. bioRxiv, 1–30. 10.1101/2021.06.20.449169 [DOI] [PMC free article] [PubMed]
- Elshire, R.J. , Glaubitz, J.C. , Sun, Q. , Poland, J.A. , Kawamoto, K. , Buckler, E.S. and Mitchell, S.E. (2011) A Robust, simple genotyping‐by‐sequencing (GBS) approach for high diversity species. PloS One 6, e19379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms, D.M. and Kelly, S. (2019) OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush, D. , Stephens, M. and Pritchard, J.K. (2007) Inference of population structure using multilocus genotype data: dominant markers and null alleles: technical article. Mol. Ecol. Notes 7, 574–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAOSTAT . (2023). Value of Agricultural Production Database. https://www.fao.org/faostat/en/#data/QV. [Accessed 14th October 2023].
- Flynn, J.M. , Hubley, R. , Goubert, C. , Rosen, J. , Clark, A.G. , Feschotte, C. and Smit, A.F. (2020) RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg, S. (2021) Computational methods for chromosome‐scale haplotype reconstruction. Genome Biol. 22, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghurye, J. , Rhie, A. , Walenz, B.P. , Schmitt, A. , Selvaraj, S. , Pop, M. , Phillippy, A.M. et al. (2019) Integrating Hi‐C links with assembly graphs for chromosome‐scale assembly. PLoS Comput. Biol. 15, e1007273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granata, M.U. , Bracco, F. and Catoni, R. (2020) Carbon dioxide sequestration capability of hazelnut orchards: daily and seasonal trends. Energy, Ecol. Environ. 5, 153–160. [Google Scholar]
- Haas, B.J. , Delcher, A.L. , Mount, S.M. , Wortman, J.R. , Smith, R.K. , Hannick, L.I. , Maiti, R. et al. (2003) Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampson, C.R. , Coleman, G.D. and Azarenko, A.N. (1996) Does the genome of Corylus avellana L. contain sequences homologous to the self‐incompatibility gene of Brassica? Theor. Appl. Genet. 93, 759–764. [DOI] [PubMed] [Google Scholar]
- Helmstetter, A.J. , Buggs, R.J.A. and Lucas, S.J. (2019) Repeated long‐distance dispersal and convergent evolution in hazel. Sci. Rep. 9, 16016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, R.J. , Baldassi, C. , Snelling, J.W. , Vining, K.J. and Mehlenbacher, S.A. (2021) Fine mapping of the locus controlling self‐incompatibility in European hazelnut. Tree Genet. Genomes 17, 6. [Google Scholar]
- Hiscock, S.J. and McInnis, S.M. (2003) Pollen recognition and rejection during the sporophytic self‐incompatibility response: Brassica and beyond. Trends Plant Sci. 8, 606–613. [DOI] [PubMed] [Google Scholar]
- Hou, S. , Zhao, T. , Yang, Z. , Liang, L. , Ma, W. , Wang, G. and Ma, Q. (2022) Stigmatic transcriptome analysis of self‐incompatible and compatible pollination in Corylus heterophylla Fisch. × Corylus avellana L. Front Plant Sci 13, 1‐14. 10.3389/fpls.2022.800768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynynen, J. , Niemistö, P. , Viherä‐Aarnio, A. , Brunner, A. , Hein, S. and Velling, P. (2010) Silviculture of birch (Betula pendula Roth and Betula pubescens Ehrh.) in northern Europe. Forestry: Int. J. Forest Res. 83, 103–119. [Google Scholar]
- Jones, D.T. , Taylor, W.R. and Thornton, J.M. (1992) The rapid generation of mutation data matrices from protein sequences. Bioinformatics 8, 275–282. [DOI] [PubMed] [Google Scholar]
- Jones, P. , Binns, D. , Chang, H.‐Y. , Fraser, M. , Li, W. , McAnulla, C. , McWilliam, H. et al. (2014) InterProScan 5: genome‐scale protein function classification. Bioinformatics 30, 1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, Y.J. , Lee, T. , Lee, J. , Shim, S. , Jeong, H. , Satyawan, D. , Kim, M.Y. et al. (2016) Translational genomics for plant breeding with the genome sequence explosion. Plant Biotechnol. J. 14, 1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp, P.D. , Midford, P.E. , Billington, R. , Kothari, A. , Krummenacker, M. , Latendresse, M. , Ong, W.K. et al. (2021) Pathway Tools version 23.0 update: software for pathway/genome informatics and systems biology. Brief. Bioinform. 22, 109–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitashiba, H. and Nasrallah, J.B. (2014) Self‐incompatibility in Brassicaceae crops: lessons for interspecific incompatibility. Breed Sci. 64, 23–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komaei Koma, G. , Şekerli, M. , Snelling, J.W. and Mehlenbacher, S.A. (2021) New sources of eastern filbert blight resistance and simple sequence repeat markers on linkage group 6 in hazelnut (Corylus avellana L.). Front. Plant Sci 12, 684122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langdon, Q.K. , Peris, D. , Kyle, B. and Hittinger, C.T. (2018) sppIDer: A species identification tool to investigate hybrid genomes with high‐throughput sequencing. Mol. Biol. Evol. 35, 2835–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Sun, P. , Lu, Z. , Chen, J. , Wang, Z. , Du, X. , Zheng, Z. et al. (2021) The Corylus mandshurica genome provides insights into the evolution of Betulaceae genomes and hazelnut breeding. Hortic. Res. 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. , Wei, H. , Zhang, X. , He, H. , Cheng, Y. and Wang, D. (2021) Chromosome‐level genome assembly and HazelOmics database construction provides insights into unsaturated fatty acid synthesis and cold resistance in hazelnut (Corylus heterophylla). Front. Plant Sci. 12, 1–15. 10.3389/fpls.2021.766548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardoni, J.J. , Honig, J.A. , Vaiciunas, J.N. , Revord, R.S. and Molnar, T.J. (2022) Segregation of eastern filbert blight disease response and single nucleotide polymorphism markers in three European–American interspecific hybrid hazelnut populations. J. Am. Soc. Hort. Sci. 147, 196–207. [Google Scholar]
- Lovell, J.T. , Sreedasyam, A. , Schranz, M.E. , Wilson, M. , Carlson, J.W. , Harkess, A. , Emms, D. et al. (2022) GENESPACE tracks regions of interest and gene copy number variation across multiple genomes. eLife 11, e78526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas, S.J. , Kahraman, K. , Avşar, B. , Buggs, R.J.A. and Bilge, I. (2021) A chromosome‐scale genome assembly of European hazel (Corylus avellana L.) reveals targets for crop improvement. Plant J. 105, 1413–1430. [DOI] [PubMed] [Google Scholar]
- Manni, M. , Berkeley, M.R. , Seppey, M. , Simão, F.A. and Zdobnov, E.M. (2021) BUSCO Update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlenbacher, S.A. (1991) Chilling requirements of hazelnut cultivars. Sci. Hortic. 47, 271–282. [Google Scholar]
- Mehlenbacher, S.A. (1997) Revised dominance hierarchy for S‐alleles in Corylus avellana L. Theor. Appl. Genet. 94, 360–366. [DOI] [PubMed] [Google Scholar]
- Mehlenbacher, S.A. (2014) Geographic distribution of incompatibility alleles in cultivars and selections of European hazelnut. J. Am. Soc. Hort. Sci. 139, 191–212. [Google Scholar]
- Mehlenbacher, S.A. and Thompson, M.M. (1988) Dominance relationships among S‐alleles in Corylus avellana L. Theor. Appl. Genet. 76, 669–672. [DOI] [PubMed] [Google Scholar]
- Mikheenko, A. , Prjibelski, A. , Saveliev, V. , Antipov, D. and Gurevich, A. (2018) Versatile genome assembly evaluation with QUAST‐LG. Bioinformatics 34, i142–i150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamikawa, M. , Kakui, H. , Wang, S. , Kotoda, N. , Kikuchi, S. , Koba, T. and Sassa, H. (2010) Apple S locus region represents a large cluster of related, polymorphic and pollen‐specific F‐box genes. Plant Mol. Biol. 74, 143–154. [DOI] [PubMed] [Google Scholar]
- Molnar, T.J. (2011) Corylus. In Wild Crop Relatives: Genomic and Breeding Resources( Kole, C. , ed), pp. 15–48. Berlin Heidelberg: Springer. [Google Scholar]
- Molnar, T.J. , Honig, J.A. , Mayberry, A. , Revord, R.S. , Lovell, S.T. , Mehlenbacher, S.A. and Capik, J.M. (2018) Corylus americana: a valuable genetic resource for developing hazelnuts adapted to the eastern United States. Acta Hortic. 1226, 115–122. [Google Scholar]
- Pavese, V. , Cavalet‐Giorsa, E. , Barchi, L. , Acquadro, A. , Torello Marinoni, D. , Portis, E. , Lucas, S.J. et al. (2021) Whole‐genome assembly of Corylus avellana cv ‘Tonda Gentile delle Langhe’ using linked‐reads (10× Genomics). G3 Genes|Genomes|Genetics 11, jkab152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, A.L. , Patterson, N.J. , Plenge, R.M. , Weinblatt, M.E. , Shadick, N.A. and Reich, D. (2006) Principal components analysis corrects for stratification in genome‐wide association studies. Nat. Genet. 38, 904–909. [DOI] [PubMed] [Google Scholar]
- Prigoda, N.L. , Nassuth, A. and Mable, B.K. (2005) Phenotypic and genotypic expression of self‐incompatibility haplotypes in Arabidopsis lyrata suggests unique origin of alleles in different dominance classes. Mol. Biol. Evol. 22, 1609–1620. [DOI] [PubMed] [Google Scholar]
- R Core Team (2021) R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Revord, R.S. , Lovell, S.T. , Capik, J.M. , Mehlenbacher, S.A. and Molnar, T.J. (2020) Eastern filbert blight resistance in American and interspecific hybrid hazelnuts. J. Am. Soc. Hort. Sci. 145, 162–173. [Google Scholar]
- Rhie, A. , Walenz, B.P. , Koren, S. and Phillippy, A.M. (2020) Merqury: reference‐free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach, M.J. , Schmidt, S.A. and Borneman, A.R. (2018) Purge Haplotigs: allelic contig reassignment for third‐gen diploid genome assemblies. BMC Bioinform. 19, 1–10. 10.1186/s12859-018-2485-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter, P.A. (1987) Badgersett research farm – plantings, projects and goal. Northern Nut. Growers Ann. Reprot. 78, 173–186. [Google Scholar]
- Salamov, A.A. and Solovyev, V.V. (2000) Ab initio gene finding in Drosophila genomic DNA. Genome Res. 10, 516–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassa, H. (2016) Molecular mechanism of the S‐RNase‐based gametophytic self‐incompatibility in fruit trees of Rosaceae . Breed. Sci. 66, 116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schläpfer, P. , Zhang, P. , Wang, C. , Kim, T. , Banf, M. , Chae, L. , Dreher, K. et al. (2017) Genome‐wide prediction of metabolic enzymes, pathways, and gene clusters in plants. Plant Physiol. 173, 2041–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoen, D.J. and Busch, J.W. (2009) The evolution of dominance in sporophytic self‐incompatibility systems. II. Mate availability and recombination. Evolution 63, 2099–2113. [DOI] [PubMed] [Google Scholar]
- Semih Uzundumlu, A. , Kurtoğlu, S. and Şerefoğlu, C. (2022) The role of Turkey in the world hazelnut production and exporting. Emirates J. Food Agric. 34, 117–127. [Google Scholar]
- Sijacic, P. , Wang, X. , Skirpan, A.L. , Wang, Y. , Dowd, P.E. , McCubbin, A.G. , Huang, S. et al. (2004) Identification of the pollen determinant of S‐RNase‐mediated self‐incompatibility. Nature 429, 302–305. [DOI] [PubMed] [Google Scholar]
- Silva, N.F. and Goring, D.R. (2001) Mechanisms of self‐incompatibility in flowering plants. Cell. Mol. Life Sci. 58, 1988–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater, G.S.C. and Birney, E. (2005) Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 6, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M. , Schöffmann, O. , Morgenstern, B. and Waack, S. (2006) Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. , Stecher, G. and Kumar, S. (2021) MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesfaye, M. , Silverstein, K.A. , Nallu, S. , Wang, L. , Botanga, C.J. , Gomez, S.K. , Costa, L.M. et al. (2013) Spatio‐temporal expression patterns of Arabidopsis thaliana and Medicago truncatula defensin‐like genes. PloS One 8, e58992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, M. , Lagerstedt, H. and Mehlenbacher, S. (1996) Hazelnuts. In Fruit Breeding: Nuts( Janick, J. and Moore, J. , eds), pp. 125–184. New York, NY: Wiley. [Google Scholar]
- Torello Marinoni, D. , Beltramo, C. , Akkak, A. , Destefanis, M.L. , Boccacci, P. and Botta, R. (2009) Gene expression and sporophytic self‐incompatibility in hazelnut. Acta Hortic. 845, 227–232. [Google Scholar]
- Vaillancourt, B. and Buell, C.R. (2019) High molecular weight DNA isolation method from diverse plant species for use with Oxford Nanopore sequencing [JOURNAL/ISSUE] 783159. 10.1101/783159 [DOI]
- Vaser, R. , Sović, I. , Nagarajan, N. and Šikić, M. (2017) Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Tang, H. , DeBarry, J.D. , Tan, X. , Li, J. , Wang, X. , Lee, T.‐H. et al. (2012) MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weschcke, C. (1954) Growing Nuts in the North: A Personal Story of the author's Experience of 33 Years with Nut Culture in Minnesota and Wisconsin. St. Paul, Minnesota: Webb Publishing Company. [Google Scholar]
- Wu, T.D. and Nacu, S. (2010) Fast and SNP‐tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, T. , Wang, G. , Ma, Q. , Liang, L. and Yang, Z. (2020) Multilocus data reveal deep phylogenetic relationships and intercontinental biogeography of the Eurasian‐North American genus Corylus (Betulaceae). Mol. Phylogenet. Evol. 142, 106658. [DOI] [PubMed] [Google Scholar]
- Zhao, T. , Ma, W. , Yang, Z. , Liang, L. , Chen, X. , Wang, G. , Ma, Q. et al. (2021) A chromosome‐level reference genome of the hazelnut, Corylus heterophylla Fisch. GigaScience 10, giab027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Clockwise from top left: the typical involucres, growth habit and nuts of C. americana ‘Rush’, held at the NCGR in Corvallis, Oregon.
Figure S2 Circular plot of genomic features in ‘Winkler’. From the outside in: ideogram of the 11 chromosomes of C. americana; total gene content, as identified by gene prediction and annotation; transposable elements, as identified by RepeatModeler; heatmap of tandem repeats; line graph of tRNA content.
Figure S3 Pairwise dotplots visualizing pairwise BLAST hits between C. americana ‘Winkler’, and each of the other eight genomes included in the GENESPACE analysis.
File S1 Gene IDs for a subset of the C. americana transcriptome consisting of 66 predicted single copy orthologs present in both ‘Rush’ and ‘Winkler’, but absent in all other analyzed genomes.
Data Availability Statement
Genome assemblies, as well as short‐ and long‐read sequence data (from Arima Hi‐C, Oxford Nanopore, PacBio and RNAseq libraries) are available through the NCBI Genome & Sequence Read Archive, respectively, under BioProject ID: PRJNA939214 BioSample accessions: SAMN33458638 (‘Rush’), SAMN33458639 (‘Winkler’). Genome assemblies, GFF3 models of predicted genes, functionally annotated peptide sequences, anchored pangenome and VCF of GBS‐derived SNPs are also available at https://doi.org/10.5281/zenodo.7439335. Finally, genomes and annotations have been added to Phytozome: https://phytozome.jgi.doe.gov/info/Camericanavar_winkler_v1_1 (for Winkler); and https://phytozome.jgi.doe.gov/info/Camericanavar_rush_v1_1 (for Rush). Bioinformatic pipelines for genome assembly and polymorphism identification are published at: https://github.com/shbrainard/Camericana_pipelines.