

Summary
Cas9 protein‐mediated gene editing has revolutionized genetic manipulation in most organisms. There are many cases where multiplexed gene editing is needed. Cas9 is capable of multiplex gene editing when expressed with multiple guide RNAs. Conventional cloning methods for multiplexed gene editing vector is not efficient due to repeated use of a single‐guide RNA scaffold and inefficient ligation. In this study, we conducted structure‐guided mutagenesis and random mutagenesis on the original sgRNA scaffold and identified a large number of functional sgRNA scaffold variants. With these scaffold variants and different tRNAs, fusion polymerase chain reaction protocol was developed to rapidly synthesize spacer‐scaffold‐tRNA‐spacer units with up to 9 targets. In conjunction with golden gate cloning, gene editing vectors with up to 24 target sites were efficiently cloned in one‐step cloning. One such gene editing vector targeting 12 genes in tomato were tested in stable transformation and 10 out of the 12 genes were found mutated in a single transgenic line. To facilitate the application of multiplexed gene editing using these scaffold variants and tRNAs from different species, a webserver was created to generate primer sets and provide template sequences for the synthesis of large sgRNA expression units based on the user‐supplied target sequences and species.
Keywords: Cas9, multiplex editing, fusion PCR, scaffold variant, sgRNA
Introduction
Clustered regularly interspaced short palindromic repeat (CRISPR) and CRISPR‐associated (Cas) protein consist of a natural immune system against phage invasion in bacteria (Barrangou et al., 2007; Horvath and Barrangou, 2010). Some of the CRISPR‐Cas systems, such as the Streptococcus pyogenes CRISPR‐Cas9 system, are capable of directing sequences specific double‐stranded DNA break, which can also function in eukaryotic cells and induce short nucleotide insertion or deletion (INDEL) via non‐homologous end joining (NHEJ). Thus, many CRISPR‐Cas systems were developed into targeted gene editing tools, which revolutionized the genetic analysis in life science (Cong et al., 2013; Jinek et al., 2012; Mao et al., 2013). In S. pyogenes, Cas9 is a nuclease with two DNA cleavage centre and associates with a crRNA and a trans‐activating CRISPR (tracrRNA) to form a functional site‐specific dsDNA cleavage enzyme guided by a 20 nucleotide spacer sequences in the 5′ part of the crRNA (Jinek et al., 2012). For the simplicity of application in gene editing, the dual crRNA‐tracrRNA were engineered into single guide RNA (sgRNA), which keeps the spacer sequence and the secondary structure involving crRNA‐tracrRNA‐Cas9 interaction (Jinek et al., 2012). The Cas9‐sgRNA complex scans and invades dsDNA when a spacer and a protospacer adjacent motif (PAM, e.g. NGG) is found, and then double‐stranded break is generated (Jinek et al., 2014; Nishimasu et al., 2014; Sternberg et al., 2014).
CRISPR system has the built‐in capacity to cleave multiple target sites, for example, Cas9 programmed by multiple sgRNAs can mutate multiple genes simultaneously (Cong et al., 2013). In plants, gene editing usually was carried out by stable transformation using Agrobacterium, which delivers T‐DNA encoding Cas9 protein, multiple sgRNA and selection marker. Currently, there are mainly two strategies to express multiple sgRNAs for editing multiple genes simultaneously. The first one is the concatenation of multiple sgRNA expression cassettes, each of which contains a pol III promoter and terminator, by golden gate cloning. In this way, up to 8 sgRNA expression cassettes had been cloned into Cas9‐based gene editing vector and mutated 7 out of the 8 target genes in three T0 transgenic individual lines (Ma et al., 2015). The second one is an expression of multiple sgRNAs separated by cleavable RNAs, such as ribozyme, pre‐tRNA and intron, in one transcription unit. Multiple sgRNAs are released after RNA processing (Cermak et al., 2017; Ding et al., 2018; Xie et al., 2015). Both strategies require golden gate ligation, which efficiency usually decreases as the number of DNA fragments or the length of individual DNA fragments to be ligated increases. By reducing sgRNA promoter length, 12 sgRNA expression units were efficiently cloned into a gene editing vector (Hao et al., 2020).
In Cas9‐mediated gene editing experiments, variation of sgRNA activity across different target sites remains an important limitation. Sequence features in spacer sequences, variable nucleotide in the PAM and target site position in the gene structure all had a significant impact on gene editing activity (Doench et al., 2014; Moreno‐Mateos et al., 2015; Wang et al., 2014). To increase the possibility of obtaining targeted mutation, multiple sgRNAs were usually tested in the protoplast of recipient plants to select sgRNAs of high efficiency before conducting transformation, which costed a lot extra effort. Having a way to clone and express a large number of sgRNA is of significant interest in gene editing research.
Here we developed a novel approach to improve the efficiency of cloning large sgRNA clusters. Structure‐guided site mutagenesis combined with random library screening was conducted to select over 35 sequence variants of sgRNA scaffold, of which their efficiency in mediating target gene cleavage was validated in transient expression. With these sgRNA scaffold variants and different tRNAs, fusion PCR protocol was made possible to synthesize sgRNA‐tRNA subcluster with eight to nine spacers. Three such subclustered were cloned into a backbone vector to construct a gene editing vector with 24 targets with high cloning efficiency. One such vector targeting 12 genes in tomato was transformed in tomato and 10 out of 12 genes were edited among 10 T0 plants and one transgenic line contained mutations in 9 targeted genes in the T1 generation. Our work provided valuable materials and optimized protocol for the rapid assembly of large sgRNA cluster for efficient gene editing. To further facilitate its application, a webserver, SupClusterWriter, was developed which could help design primers for sgRNA supercluster synthesis and recommend a strategy for cloning. SupClusterWriter is available at http://lifenglab.hzau.edu.cn/Tools/index.php?tid=SupClusterWriter.
Results
Identification of gRNA scaffold variants through structure‐guided mutagenesis
Structural biology studies have obtained detailed mechanisms for the formation of functional gRNA‐Cas9 complex (Nishimasu et al., 2014). During the complex formation, only a few nucleotides (class I) in the gRNA scaffold formed base‐specific interaction with Cas9 protein while the other nucleotides (class II) contributed through the formation of secondary structure or interacting with Cas9 via phosphate and ribose backbone (Figure 1a). We reasoned that permutation of class II nucleotide, which maintains gRNA secondary structures, would generate gRNA sequence variants that still maintain its gene editing function.
Figure 1.
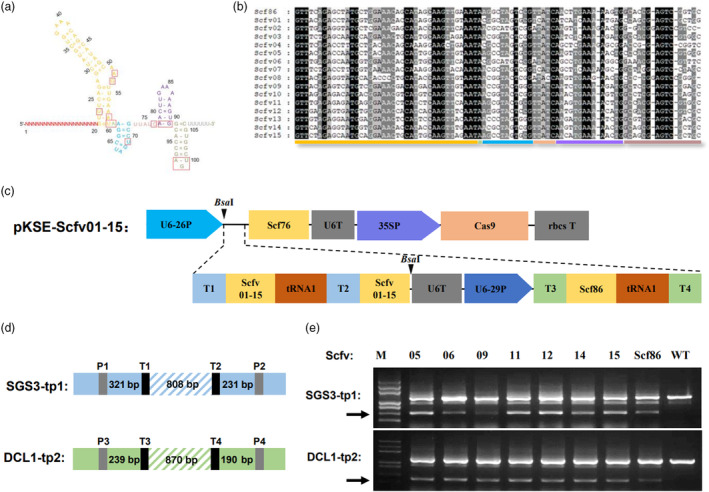
Generation and functional selection of gRNA scaffold variants by structure‐directed mutagenesis. (a) Secondary structure of gRNA. Letters in the red square box indicate nucleotides forming base‐specific interactions with Cas9 protein. (b) Sequence alignment of scaffold variants generated by directed mutagenesis. Nucleotide groups are indicated at the bottom; mutated nucleotides are shaded. (c) Structure of the gene editing construct for testing scaffold function. U6‐26P, U6‐26 promotor from Arabidopsis; Scf76, original 76‐nt scaffold; U6T, U6 terminator from Arabidopsis; 35SP, 35S promoter; Cas9, Cas9 open reading frame; rbcsT, terminator from pea rubisco gene; T1‐4, spacer sequences for target site 1‐4; Scfv01‐15, scaffold variant 01 to 15; tRNA1, tRNA1 sequence; U6‐29P, U6‐29 promotor from Arabidopsis. (d) Diagram showing the relative position of gene editing target sites and PCR primers on SGS3 and DCL1 gene. (e) Gel analysis of SGS3 and DCL1 gene editing results. Arrows point to PCR products amplified from the edited gene sequences resulting from the deletion of sequences between two editing sites. ID of Scaffolds that fused to T1 and T2 is indicated on top of each lane.
To test this hypothesis, we divided the class II nucleotides into several groups, within each group nucleotides formed base‐pairing (Figure 1a). Scaffold variants were generated by mutations in multiple groups which kept the nucleotide base‐pairing within each group (Figure 1b). These scaffold variants were fused with two spacer sequences targeting the validated SGS3‐tp1 sites (See Methods and Figure S1) to form two gRNAs separated by tRNA1 (Figure 1c,d). Then these dual gRNA clusters were fused to the U6 RNA terminator and a control dual‐gRNA cluster targeting the validated DCL1‐tp2 sites and cloned into pKSE401 vector by Bsa I restriction and ligation (Figure 1c,d). The resultant constructs were transformed into agrobacterium and tested in N. benthamiana leaves by agroinfiltration. Gene editing efficacy of these different scaffolds on SGS3‐tp1 target sites was tested by PCR using primers flanking target sites (Figure 1d). As expected, the control gRNAs directed similar levels of editing at DCL1‐tp2 sites resulting in similar intensity of a shorter DNA band detected after PCR (Figure 1e bottom). However, gene editing efficiency at the SGS3‐tp1 sites differed significantly among gRNAs with different scaffold variants and the Scfv05, ‐06, ‐09, ‐11, ‐12, ‐14 and ‐15 showed similar efficiency as the wild type scaffold Scf86 (Figures 1e and S1d).
In summary, through structure‐guided mutagenesis, we can generate sequence variation in the gRNA scaffold that maintained its activity in gene editing.
Efficient assembly of guide RNA cluster with multiple tRNAs and gRNA scaffold variants
The expression of multiple gRNAs interspaced by tRNAs (MGT) to produce multiple individual gRNA for simultaneously editing many targets is widely used in gene editing studies (Xie et al., 2015). Fusion PCR is an efficient method to quickly piece together short DNA fragments (Horton et al., 1989). We first tested whether MGT could be assembled by fusion PCR using a single gRNA scaffold and single tRNA (Figure 2a). Two round PCR reactions were conducted to synthesize 7 units of spacer‐scaffold‐tRNA‐spacer (SSTS) fragments (Figure 2a). In the 1st round PCR, two sub fragments with overlapping sequences at the scaffold‐tRNA junction were synthesized (Figure S2a). Then the two subfragments were used as templates in fusion PCR to produce SSTS unit in the 2nd round PCR (Figure S2d). The first two round of PCRs were successful, but in the 3rd round PCR to generate the 4‐unit and 3‐unit subfragments the product of expected sizes were not seen, and not to our surprise, the 4th round PCR failed to produce the 7‐unit final product (Figure 2b). We reasoned that repeated scaffold and tRNA1 sequences may cause the failure of fusion PCR (Ji et al., 1994; Viguera et al., 2001).
Figure 2.
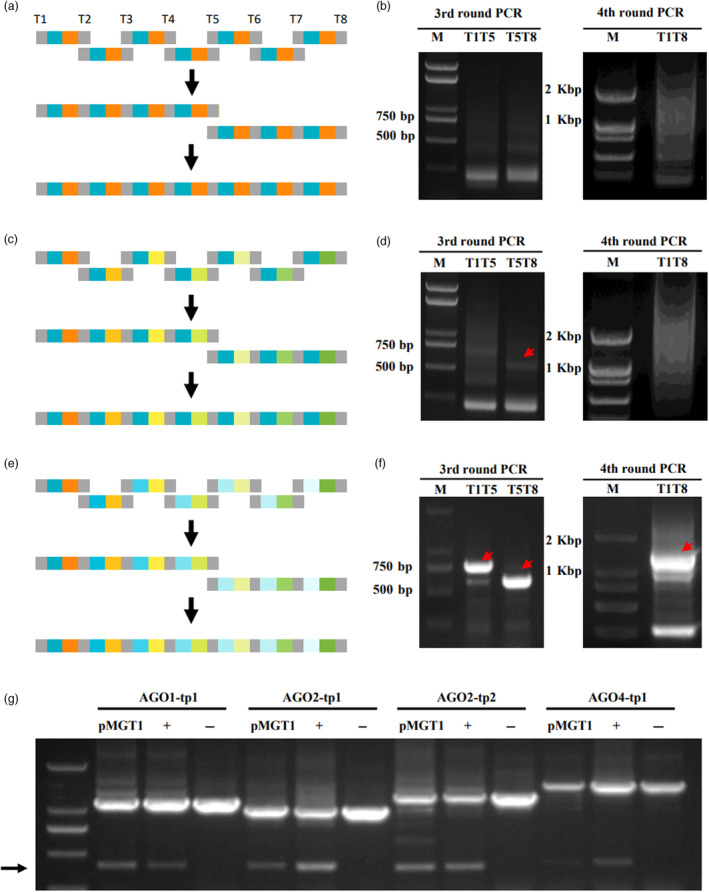
Strategy to make MGT by fusion PCR. (a) Making MGT using wild‐type scaffold Scf86 and tRNA1. Grey boxes represent spacer sequences for different targets indicated on top. Blue and orange boxes represent Scf86 and tRNA1. The top row shows the 2nd round PCR product, SSTS unit. The second row shows the 3rd round PCR products, the 4‐unit and 3‐unit subfragments. The third row shows the 7‐unit final MGT fragment. T1 and T2 are spacers for target pair AGO1‐tp1; T3/T4, T5/T6 and T7/T8 are for AGO2‐tp1, AGO2‐tp2 and AGO4‐tp1, respectively. (b)Gel picture of the 3rd (left) and the 4th (right) round PCR products using one scaffold (Scf86) and one tRNA (tRNA1). (c) Making MGT using wild‐type scaffold Scf86 and tRNA1‐7. Similar to in (a), boxes with brown to green colour series represent tRNA1‐7. (d) Gel picture of the 3rd (left) and the 4th (right) round PCR products using one scaffold (Scf86) and seven tRNAs (tRNA1‐7). Red arrowheads point to 4‐unit, and 3‐unit subfragments (from left to right). (e) Making MGT using Scf86 and variants and tRNA1‐7. Boxes with blue to light blue colour series represent Scf86, Scfv06, Scfv11, Scfv12, Scfv05, Scfv14 and Scfv15 from left to right. Boxes with brown to green colour series represent tRNA1‐7. (f) Gel picture of the 3rd (left) and the 4th (right) round PCR products using different scaffolds and different tRNAs. Red arrowheads point to 4‐unit, 3‐unit subfragments and 7‐unit final MGT (from left to right). (g) Target gene editing results in transient assay. Lanes 2–4, Lanes 5–7, Lanes 8–10 and Lanes 11–13 PCR conducted using primers (see Table S1) analysing AGO1‐tp1, AGO2‐tp1, AGO2‐tp2 and AGO4‐tp1 editing. PCR products in Lanes 2, 5, 8 and 11 were amplified from pMGT1‐expressed samples while those in Lanes 3, 6, 9 and 12 were from leaves expressing positive control pAGO1‐tp1, pAGO2‐tp1, pAGO2‐tp2 and pAGO4‐tp1, respectively. Lane 1, DNA ladder. Lanes 4, 7, 10 and 13 were samples amplified from untreated wild‐type N. benthamiana DNA template as negative control. The black arrow points to the diagnostic bands amplified from gRNA pair‐directed cleavage and relegation.
To solve the problem, a series of tRNAs were first tested in their ability to release individual gRNAs targeting validated SGS3‐tp1 (Figure S1e,f). Transient expression analysis showed that except constructs with tRNA11, all other constructs generated diagnostic SGS3 editing band like that with tRNA1 while control gRNAs targeting DCL1‐tp2 sites in all constructs generated similar DCL1 diagnostic bands (Figure S1g), suggesting tRNA2‐10 and tRNA12‐16 were functional in splitting multiple gRNAs. Next, the wild‐type scaffold was combined with tRNA1‐7 to generate the 7 SSTS units through two rounds of PCR (Figures 2c and S2b,e). In the 3rd round PCR, a faint band corresponding to the 3‐unit subfragment was produced but the majority of the products were not of the right sizes (Figure 2d left panel). The final product was also not successfully generated in the 4th round of PCR using these two products as templates (Figure 2d right panel). These data suggested that increasing heterogeneity by using different tRNA sequences can slightly increase the efficiency of fusion PCR but not enough to produce a major specific band of expected size.
To further increase the heterogeneity, the wild‐type scaffold Scf86 and functional variants Scfv05, ‐06, ‐11, ‐12, ‐14 and ‐15 were combined with tRNA1‐7 to generate the 7 SSTS units through two rounds of PCR (Figures 2e and S2c,f). Interestingly, major bands of 4‐unit and 3‐unit subfragments were detected in the 3rd round PCR products and a major band of 7‐unit final gRNA fragments was also detected in the 4th round PCR using the 3rd round PCR products as a template (Figure 2f), indicating that the strategy to increase heterogeneity using different scaffold variants and different tRNAs successfully enabled MGT assembly by fusion PCR.
To validate the function of the assembled MGT, the major band of the 4th round PCR products was gel purified and cloned into a gene editing vector to produce pMGT1, which targets four pairs of validated sites on different NbeAGO genes, AGO1‐tp1, AGO2‐tp1, AGO2‐tp2 and AGO4‐tp1 (Figure S1c). The pMGT1 vector was tested in N. benthamiana by agrobacterium‐mediated transient expression and previously validated vectors targeting one pair of the AGO sites were used as positive control. DNA samples from the pMGT1 and control vector expressed leaves were analysed by PCR using validated primer pairs. The results showed that diagnostic smaller bands were detected for each pair of target sites of similar efficiency between the control editing vector and the pMGT1 vector expressed samples (Figure 2g). To further validate this strategy, we shuffled the scaffold sequences in the final gRNA fragments and repeated the fusion PCR and cloning process to produce pMGT2, which has the same order of spacers but a different order of scaffold sequences as pMGT1 (Figure S2g–j). Transient assay in N. benthamiana also validated the editing efficiency of pMGT2 (Figure S2k).
The above results showed that functional MGT can be efficiently assembled by fusion PCR with functional scaffold variants and different tRNAs due to the heterogeneity they brought to the final MGT sequences.
Identification of gRNA scaffold variants through random mutagenesis
With structure‐guided point mutation, scaffold variants were identified, which enabled assembly of MGT by fusion PCR. While some scaffold variants, such as Scfv06 and Scfv09, slightly reduced their efficiency in direct target cleavage (Figure 1e). Limited number and efficiency of scaffold variants would restrict the application of assembly of MGT by fusion PCR. To increase the number and efficiency of scaffold variants, we set to perform a functional screen from a library of scaffold variants made by error‐prone PCR (Figure 3a, see Method section).
Figure 3.
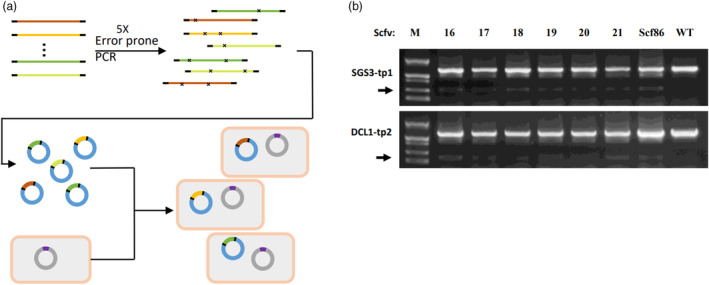
Identification of functional scaffold variants through library screening. (a) Pipeline of library screen. Lines with different colours represent different parent scaffold variants. Black lines flanking the coloured lines represent common primer binding sites. ‘x’ on each line represents point mutation generated through error‐prone PCR. Blue circles represent pWT021Scfv vectors with different scaffold variants. Grey square with brown line represents E. coli cell and the grey circle inside the cells represents the plasmid pTarget. (b) Gel analysis of SGS3‐tp1 and DCL1‐tp2 cleavage products directed by editing vector containing Scfv16‐21 as indicated on top of each lane.
For the functional screen, a previously reported base‐editing system in E. coli was employed, in which the pWT021a vector was capable of C‐to‐T editing (Tang and Liu, 2018). This vector was modified into pWT021TS1.0 and the scaffold variant library was cloned into this vector to replace its own scaffold to produce pWT021Scfv library (see Method section, Figure S3a). An editing target vector, pTarget was made and co‐transformed with pWT021Scfv library into E. coli (Figure 3a). Eight hundred and ten single colonies were picked up and target sequences were amplified and analysed by high‐throughput sequencing. This analysis yields 40 colonies with target sequences edited with higher efficiency than wild‐type scaffold. Scaffold variants were then amplified from these colonies and sequenced by Sanger sequencing and eventually 18 new scaffold variants were identified that capable of C‐to‐T editing in E. coli (Table S3).
Next, the 18 new scaffold variants were cloned into the pKSE vector and tested their function in directing SGS3 cleavage as the previous section. Interestingly, these 18 scaffolds showed significant variation in directing SGS3 cleavage in plants (Figure S3b) though they were all functional in directing C‐to‐T base editing in E. coli. Six new scaffold variants with stable performance in plants were selected and named Scfv16‐21 (Figure 3b and Data S1). Further analysis showed that all these six variants were derived from Scfv05 and Scfv12 and only a single base changed from parent sequences (Figure S3c,e). As these single‐base point mutation did not affect scaffold function, we tried to make combinations of these mutations to further increase the sequence diversity of functional scaffold variants. Thirty‐two combinations (Scfv22‐53) were generated from the six‐point mutations identified in the screen and incorporated into the parent scaffold (Scfv05 and Scfv12) (Data S1) and cloned into pKSE vector to test their function in directing SGS3 cleavage. The results showed that all these new variants directed cleavage with efficiency comparable to that of Scf86 (Figure S3d,f). The editing efficiency of different scaffold variants was further evaluated using high‐throughput sequencing (see Method section). The results showed that about 22% and 20% of sequencing reads from Scf86‐treated sample bearing InDels in the predicted editing window at the target1 and target2 sites of SGS3‐tp1, respectively (Figure S4). Most variants showed wild‐type level editing efficiency at the target1 site and 80%–100% of the wild‐type editing efficiency at the target2 site (Figure S4), which was consistent with their functionality in generating deletion mutation detected by PCR analysis.
In summary, library screening in conjunction with a combination of point mutations generated a large number of new scaffold variants that function in gene editing in plants. These new variants provided rich resources for producing MGT using fusion PCR.
Efficient assembly of 24‐spacer gRNA cluster using PCR and golden gate cloning
To further demonstrate the potential of scaffold variants and different tRNAs in facilitating MGT cloning, we set to clone a 24‐spacer gRNA cluster targeting 12 tomato genes, with two sites on each gene using two strategies (Figure 4a). In strategy I, 23 designed SSTS units were amplified from scaffold‐tRNA chimera in pMGT1 (Figures 4a and S5a) in the first‐round reaction. Six subfragments were then generated by fusion PCR in the second round, five with 4‐SSTS units and one with 3‐SSTS units (Figures 4a and S5b). In the third‐round reaction, two 8‐unit (9 spacers) and one 7‐unit (8 spacers) subfragments were produced by fusion PCR (Figure S5c). And then cloned the three fragments were cloned into pTX vector (Shang et al., 2021) by golden gate cloning; in strategy II, 23 SSTS units were directly cloned via golden gate cloning (Figures 4a and S5d). As controls, a set of fragments with 3‐, 6‐, 9‐ and 12‐SSTS units were also cloned by strategy II. The ligation mix was transformed into E. coli competent cells and colony PCR was performed to select positive clones. PCR products of expected size were detected in 31 colonies out of 60 colonies picked. Seven positive clones were selected for sequencing analysis and four of them contained vectors with gRNA clusters identical to designed sequences, resulting in successful cloning of pMGT3. However, out of 48 colonies picked, no clone produced a PCR product of expected size in the colony PCR test. As expected, in the control experiments, the cloning efficiency of strategy II increased as the unit number decreased (Figure 4b). The efficiency of strategy I to clone 23‐SSTS units gRNA cluster (with 24 spacers) is comparable to golden gate cloning a 6‐SSTS unit gRNA cluster (with 7 spacers).
Figure 4.
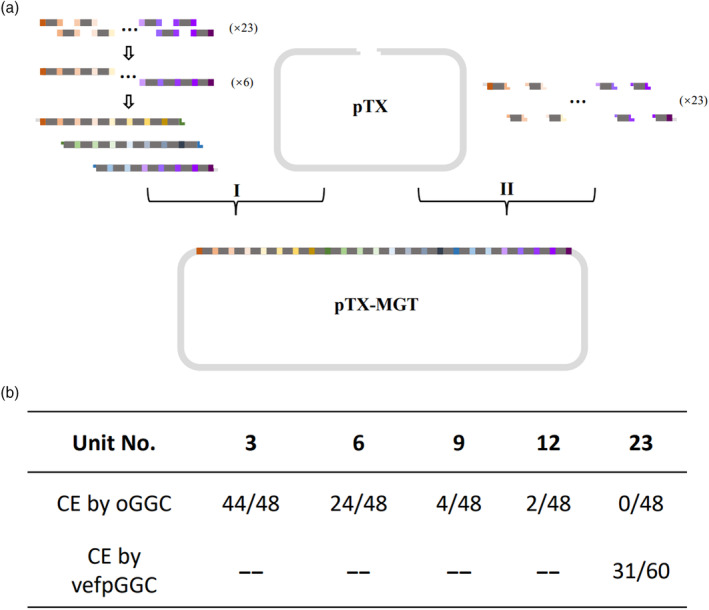
Efficient cloning of 23‐unit gRNA cluster using fusion PCR combined with golden gate cloning. (a) Two strategies to clone 23‐unit gRNA cluster. In strategy I, three subfragments with 9, 9 and 8 spacers, respectively, were synthesized by fusion PCR and then cloned into a pTX vector by golden gate cloning. The 9th (dark green boxes) and 17th (dark blue boxes) spacers are attached to BsaI site to make compatible sticky ends. In strategy II, 23 SSTS units were directly cloned into pTX vector by golden gate cloning. Each spacer is attached to BsaI site to make compatible sticky ends between adjacent SSTS units. The grey bars represent scaffold‐tRNA fusion consisting of different scaffold variants and tRNAs. (b) Cloning efficiency (CE) comparison between strategy I and II.
These results showed that the new scaffold‐enabled fusion PCR method had great potential to increase the efficiency of cloning multiple gRNA clusters in conjunction with golden gate cloning.
Editing multiple tomato genes with large gRNA cluster built from gRNA scaffold variants
In order to test whether the scaffold variant can direct mutagenesis in stable transformation, the pMGT3 vector was used in tomato transformation. Twenty‐six T0 lines with T‐DNA insertion were identified. The pMGT3 vector targeted 12 tomato genes, of which CRISPR mutants had been reported, including SlySE3.1 (Shang et al., 2021), SlySdeA1 (Lopez et al., 2021), SlyHD8 (Hua et al., 2021), SlyNOR (Gao et al., 2019), SlyTAGL1 (Liu et al., 2020), SlyLOL1 (Borovsky et al., 2019), SlyRIN (Li et al., 2020), SlyMLO1 (Nekrasov et al., 2017), SlyCPK28 (Hu et al., 2021), SlySp5G (Soyk et al., 2017), SlyRCM1 (Liu et al., 2021) and SlyPIF4 (Pan et al., 2021). Target site sequences were amplified from 10 T‐DNA positive lines and analysed by Sanger sequencing. The results showed that nine lines had a mutation in at least one targeted gene (Data S3). Among these lines, Line25 and Line32 contained 8 and 7 edited genes, respectively and together these two lines contained 10 edited genes out of the 12 targets. The SlyHD8 and SlyLOL1 were not mutated in any analysed lines. The Line32 T0 plant flowered but no fruit set, thus not studied further. The T0 plant of Line25 set fruits, which showed light green to milk white before the mature green stage and orange to light red at the ripen stage, showing clear mutant phenotype in fruit maturation (Figure 5a). There are three fruit maturation‐related genes, RCM1, TAGL1 and NOR that were at homozygous mutation status (Figure 5b). However, individual CRISPR mutations of these genes showed different maturation phenotypes (Gao et al., 2019; Liu et al., 2020, 2021), suggesting the mutant phenotype was due combinatory effect of multiple mutations.
Figure 5.
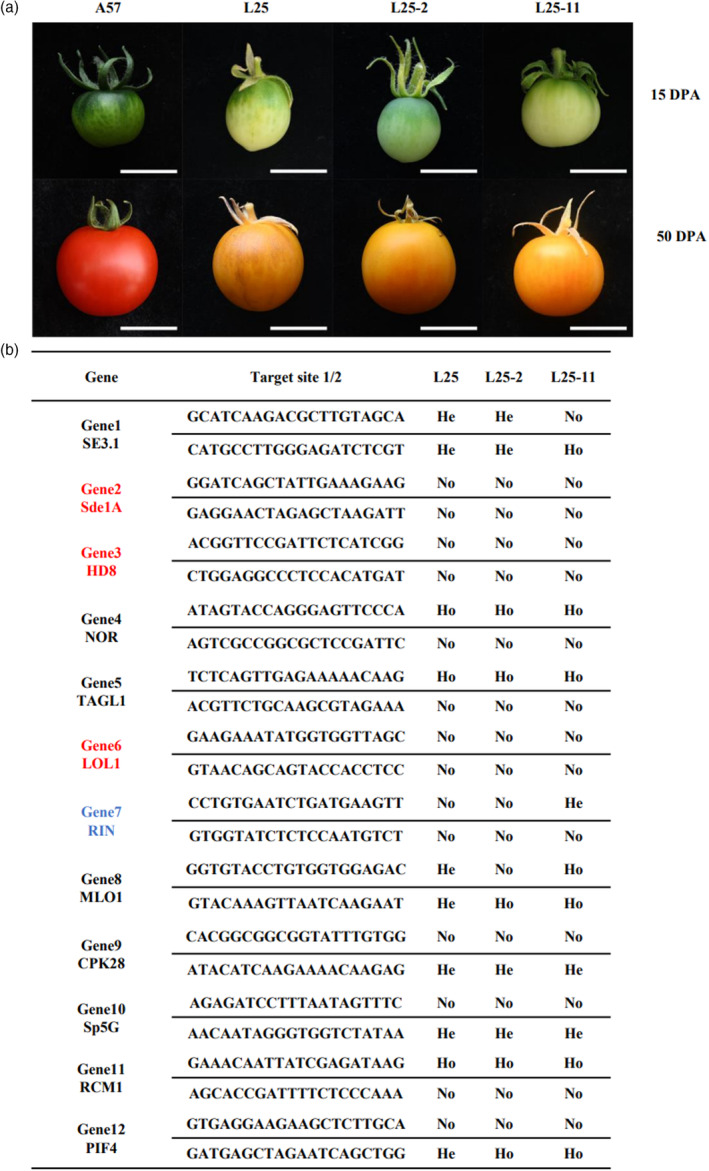
Multiple gene editing in tomato directed by pTX‐MGT made of different scaffold variants. (a) Fruit phenotype of gene‐edited tomato in T0 and T1 generation. A57 indicates fruit from the wild‐type control. L25, Line25 of T0 generation; L25‐2 and L25‐11 of T1 generation. Top panel, fruits photographed at 15 days post anthesis (DPA); bottom panel, fruits at 50 DPA. Scale bar is 1 cm. (b) Summary of sequencing analysis of each editing site in T0 and T1 plants. He, heterozygous mutation; Ho, homozygous mutation; No, no editing. Genes in red, targets edited in neither T0 nor T1 plants. Gene in blue, target edited in T1 but not T0 plants.
T1 generation from Line25 was further analysed to study the inheritance of the induced mutations. Eleven T1 plants were tested PCR using Cas9 primers and 7 plants produced DNA band of expected size (Figure S6a). Fruit phenotypes in T1 plants with or without Cas9 were the same as in T0 plants (Figures 5a and S6b), confirmed the heritability and homozygous status of mutations in three ripening genes. Target sequences from Line25‐2 (L25‐2, without Cas9) and Line25‐11 (L25‐11, with Cas9) were analysed by Sanger sequencing. The results showed that 8 out of 12 genes were edited in one or both target sites in T0 Line 25 (L25) plants (Figure 5b, Data S3). In the L25‐2 plant, the edited mutations were all inherited and no new mutations occurred while in the L25‐11 plant new mutation in one target site of RIN gene was observed, which was not present in the T0 plant (Figure 5b, Data S3), presumably due to continuous activity of Cas9. These results showed that the scaffold variants were functional in directing site‐specific and heritable mutations in plants.
SupClusterWriter, a webserver for gRNA cluster design using scaffold variants
In order to provide a simpler and better option for the synthesis of large gRNA cluster, 24 combinations of scaffold‐tRNA were cloned into pEASY cloning vectors. We further optimized the PCR protocol, and the SSTS unit was directly amplified from the cloned scaffold‐tRNA templates instead of by a two‐step fusion PCR (Figure S7a–c vs Figure S2a–f). These pEASY‐ST vectors were divided into three template sets, each was tested in three‐step PCR to produce an 8‐unit SSTS gRNA cluster (Figure S7d,e). To facilitate the design of gRNA cluster using the scaffold variants, a webserver, SupClusterWrite, was set up which can design gRNA cluster containing up to 25 spacers. On the homepage, there are four user input fields (Figure 6a left panel). The first one is a pull‐down menu for the user to select a template set to produce eight individual SSTS units, which contain tRNAs suitable for different groups of plants, for example, set A contains conserved tRNA for tobacco and tomato, set B contains tRNAs from Arabidopsis and set C contains tRNAs conserved for rice and corn. The second and the third ones are text fields for the user to fill in a 5′ and 3′ sticky ends which are compatible with the gene editing vector digested with BsaI. The last one is the text filed for inputting spacer sequences for selected targets. A maximum of 25 spacers can be pasted. After submission, the server generates a table listing the paired forward and reverse primers and corresponding pEASY‐ST template ID, and the complete gRNA cluster sequence plus flanking adapter sequences is also listed in the end (Figure 6a right panel). A fusion PCR pipeline for piecing together 4 to 8 SSTS units (5 to 9 targets) was provided (Figure 6b). For gRNA cluster with 9 to 16 SSTS units (10 to 17 targets), the first 8 SSTS unit and the remaining SSTS units are fused together individually into two fragments; the 9th spacer is split in half and BsaI adapter sequences are added to each half so that after golden gate cloning the 9th spacer is reconstituted. For gRNA cluster with 17 to 24 SSTS units, the first 16 SSTS units are fused together individually into two fragments and the remaining units are fused into a third fragment; the 9th and the 17th spacers are split in half and BsaI adapter sequences are added to each half for reconstitution of these two spacers in golden gate cloning (Figure 6c). This web application greatly simplified the supper gRNA cluster and primer design process.
Figure 6.
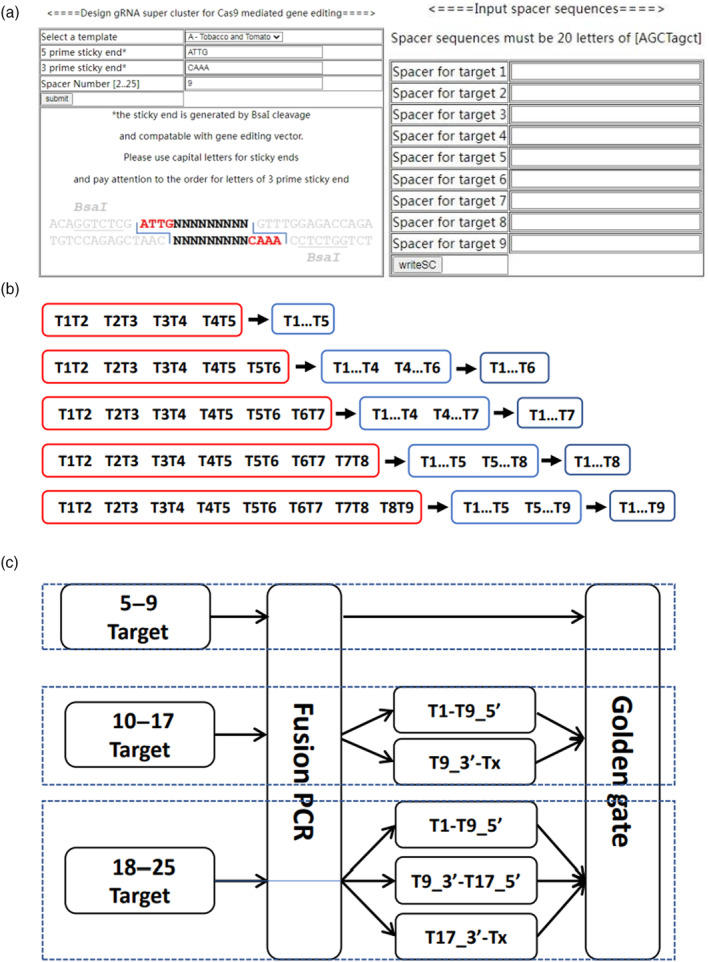
Web application for large gRNA cluster design and synthesis. (a) Interface of SupClusterWriter. (b) Suggested fusion PCR strategies for gRNA clusters with 5 to 9 spacers. Red box, first‐round PCR for SSTS units; blue box, second‐round PCR for subcluster(s); black box, third‐round PCR for end products. (c) Webserver recommended strategies for cloning gRNA clusters with different number of targets.
Discussion
Even though the development of Cas9‐based gene editing technology revolutionized life science in the past decay. Versatile research needs promoted continuous improvements and even revolution of this technology itself. In basic and applied research, there are many scenarios where multiple sites in the genome need to be edited simultaneously. Elucidation of redundant gene function for a gene family requires higher order multiple mutants (Bouche et al., 2006), which took a lot of effort to obtain through conventional cross and not possible for asexually propagated organisms. In crop improvements, large number of promoter variation of the same tomato gene were generated using gene editing vector with 8 sgRNA targeting various positions in the promoter region, resulting in rich variation in tomato fruit morphology, inflorescence and plant architectures (Rodriguez‐Leal et al., 2017). As sgRNA activity is affected by many factors, it is always good to use multiple sgRNAs to target a single gene to ensure the mutation of the target gene in gene editing experiments. Thus, an easy way to clone a large number of sgRNA expression clusters is of significant value in general.
Current methods for cloning multiple sgRNA clusters usually rely on the concatenation of a single sgRNA unit by golden gate cloning. In this study, we developed fusion PCR‐based golden gate cloning, in which fusion PCR was conducted to quickly assemble sgRNA subcluster with up to eight sgRNA units and then ligated into editing vector by golden gate cloning, enabling efficient cloning of editing vector targeting 24 sites at once. Fusion PCR assembly of sgRNA subcluster was not possible with a single sgRNA scaffold or tRNAs, due to recombination occurred during PCR and multiple identical repeats in the template (Meyerhans et al., 1990). To circumvent this problem, we conducted structure base site mutagenesis in conjunction with a random library screen to identify sgRNA scaffold variants that contained multiple sequence variations from wild‐type scaffold and retained similar cleavage function in plants (Figures 1, 3 and S3). Different tRNAs were also tested for splitting sgRNAs for multiplex editing (Figure S1g). Only when both scaffold variants and multiple tRNAs were used in the sgRNA subcluster, fusion PCR was able to successfully assemble specific sgRNA subcluster DNA fragments (Figure 2), which highlighted the importance of scaffold variants in this technology. Transformation experiments in tomato with a 24‐spacer editing vector targeting 12 genes obtained one T1 plant with nine genes mutated out of ten T0 lines (Figure 5), validated the function of the scaffold variant in inducing stable mutations.
Further analysis showed that the majority of the newly identified scaffolds were functional with high editing efficiency like the wild‐type scaffold in the tomato transgenic plant, except Scfv09 (Figure S6c). However, it was functional in the transient assay in N. benthamiana with slightly reduced activity (Figure 1e). Further examination revealed that this spacer was preceded with tRNA05 from N. benthamiana which had 4 mismatches to its tomato homologue (Figure S6d,e, Data S4). The sequence divergence in tRNA05 may affect the processing of spacer‐Scfv09, thus abolishing the activity of this sgRNA in conjunction with slightly reduced scaffold activity. These results suggested that further optimization in different organisms with highly conserved or native tRNA may further improve the editing efficiency of large sgRNA clusters made from these scaffold variants. The other possibility is that Scfv09 may not function well at the selected target site due to site‐specific effects and slightly reduced editing efficiency of Scfv09 as revealed by high‐throughput sequencing analysis (Figure S4 and Table S4). To simplify the vector design and synthesis for different plant species, we developed a webserver, SupClusterWriter, which can design sgRNA clusters and primers based on input spacers and selection of species. As tRNAs may impact sgRNA processing, different sets of tRNAs were provided for application in different species.
In this study, we found that some sgRNAs were capable of guiding base‐editing in E. coli but not able to guide cleavage in N. benthamiana (Figure S3b), which suggested that target binding and target cleavage may be uncoupled by these scaffold variants. Thus, these scaffold variants identified in this study not only enabled fusion PCR‐based assembly of large sgRNA clusters, they were also valuable materials to study the mechanism of sgRNA‐Cas9‐DNA triple‐interactions.
Online materials and methods
Plant materials and growth conditions
Tomato (Solanum lycopersicum) cultivar A57 and wild‐type tobacco (N. Benthamiana) were used in this study. A57 and N. benthamiana seedlings were grown in a greenhouse under long‐day conditions (light/dark: 16 h/8 h) at a temperature of 25 °C.
Selection of gene editing target sites in N. benthamiana genome
Target gene screening vectors were made based on pKSE401 vector, which was kindly provided by Professor Chen Qijun (Xing et al., 2014). To select target sites, the gDNA sequences of NbAGO, NbDCL and NbSGS3 were downloaded from the NCBI (www.ncbi.nlm.nih.gov/) website and analysed using the CRISPR2.0 (crispr.hzau.edu.cn/CRISPR2/) website (Bombarely et al., 2012; Liu et al., 2017; Table S2). A total of 17 groups of target sites were selected for screening, and each group of target sites contained two target sites T1 and T2. First, T1 was attached to the 5′ end of Scf86 and T2 was attached to the 3′ end of tRNA1 by primers. At the same time, two BsaI cleavage sites in opposite directions were introduced at the 5′ end of T1 and the 3′ end of T2. Then, the first 20 bp of tRNA1 was used as the overlapping region, and the two fragments were connected by fusion PCR to generate a BsaI‐T1‐Scf86‐tRNA1‐T2‐BsaI fragment (Figure S1a). Finally, the fragment was ligated into the pKSE401 vector using the Golden gate cloning method to generate the pKSE‐AGOX‐tpY, pKSE‐DCLX‐tpY and pKSE‐SGS3‐tp1 series of vectors (Table S2). These constructs were then transformed into agrobacterium and their gene editing efficiency was tested by agroinfiltration in 5‐week‐old N. benthamiana leaves. DNA samples were extracted from infiltrated leaves 3 day post infiltration and primers flanking the cleavage sites were used in PCR tests (Figure S1b). The results showed that clear diagnostic bands were detected for AGO1‐tp1, AGO1‐tp2, AGO2‐tp1, AGO2‐tp2, AGO4‐tp1, AGO4‐tp2, AGO7‐tp1, AGO16‐tp1, DCL1‐tp1, DCL1‐tp2, DCL2‐tp1, DCL4‐tp1 and SGS3‐tp1, which sizes were consistent with the size of edited gene with deletion of sequences between two target sites (Figure S1c).
Construction of tRNA screening vectors
A control module consisting U6‐26T‐U6‐29P‐TDCL1‐tp2‐1‐Scf86‐tRNA1‐TDCL1‐tp2‐2 fragment was synthesized by fusion PCR and then the fragment was inserted into the pKSE401 vector using the Golden gate ligation method to generate the intermediate vector pKSE401‐Interim, which contains two BsaI restriction sites were used for subsequent fragment insertion. Sequences of sixteen N. benthamiana tRNAs were downloaded from the tRNA database (Lowe and Eddy, 1997) and named tRNA1‐tRNA16, respectively. Sixteen different BsaI‐TSGS3‐tp1‐1‐Scf86‐(tRNA1‐tRNA16)‐TSGS3‐tp1‐2‐Scf86‐BsaI fragments were synthesized and inserted into the pKSE401‐Interim vector by the Golden gate ligation method to construct the pKSE‐tRNAx vector series (Figure S1e).
Construction of scaffold variants screening vectors
Based on Scf86, a series of scaffold variants were obtained by site‐directed mutagenesis and random mutation, and different BsaI‐TSGS3‐tp1‐1‐Scfv (1–53)‐tRNA1‐TSGS3‐tp1‐2‐Scfv (1–53) ‐BsaI fragment were synthesized by fusion PCR, which was inserted into the pKSE401‐interim to make the scaffold variants pKSE‐Scfvx vector series.
Construction of bacterial gene editing vector pWT021TS1.0 and pTarget
For screen functional gRNA scaffold variant in bacterium, the pWT021a vector, which was functional in C‐to‐T editing in E. coli (Tang and Liu, 2018), was purchased from Addgene and modified: the original tet promoter for CBE was replaced with the T7 promoter; a Pac I site was replaced with a Stu I site for cloning purpose; the original Scf76 scaffold was replaced with Scf86 to generate pWT021TS1.0. The target vector for base editing in bacteria was constructed by inserting the complementary sequence of spacer sequences in pWT021TS1.0 into the pEASY‐T1 vector (at 1158 bp). First, a linear vector fragment was amplified by a pair of primers bearing the target site using pEASY‐T1 as the template, and then the amplified fragment was subjected to T4 PNK treatment and ligated using T4 DNA Ligase to generate pTarget vector. The editing function of pWT021TS1.0 was verified by co‐transformation with pTarget and sequencing analysis (Figure 3a).
Screen error‐prone PCR library of scaffold variants
A scaffold mutant library was established by five rounds of error‐prone PCR using Scfv05, 06, 11, 12, 14 and 15 mixtures as templates. The error‐prone PCR was performed as described earlier (Cirino et al., 2003). Subsequently, the library was cloned into pWT021TS1.0 by Homologous recombination ClonExpress II One Step Cloning Kit (Vazyme, China) to generate a series of pWT021Scfv vectors. The pWT021Scfv vectors (cloning reaction) were transformed into DH5α containing pTarget vector. Transformants were grown overnight on plates containing spectinomycin (100 μg/mL) and ampicillin (50 μg/mL) in a 37 °C incubator. The next day, single clones were picked and inoculated into 2 mL of liquid LB medium containing spectinomycin and ampicillin, and incubated overnight in a 37 °C shaker. On the third day, 40 μL bacterial culture was inoculated with 2 mL fresh liquid LB medium with antibiotics, incubated in a 37 °C shaker until the OD600 value reached 0.6–0.8, and then 2 μL of 1 M IPTG inducer was added and placed in a 28 °C shaker for 16 h. After three rounds of IPTG induction and expression, a pair of primers was used to perform PCR amplification on the target site. The amplified products were subjected to high‐throughput sequencing to calculate the base editing efficiency of different Scfvs in bacteria using a set of Perl scripts. The clones with editing efficiency higher than Scf86 were selected. Scfv in these clones were amplified and sequenced by Sanger sequencing for subsequent studies.
Agrobacterium‐mediated transient expression in N. benthamiana
Agrobacterium strain GV3101 was transformed by heat shock protocol. Fully expanded leaves of N. benthamiana of 5 weeks old were infiltrated with these strains at an OD600 value of 1.0 in Agro‐infiltration buffer (10 mm MgCl2, pH 5.7 and 150–200 μm acetosyringone). Leaf samples were harvested for DNA assays 3 days after infiltration.
Tomato transformation and characterization
The pMGT3 vector was introduced into Agrobacterium strain GV3101 and used for Agrobacterium‐mediated transformation. Tomato cultivar A57 was transformed as described before (Ouyang et al., 2005). Transgenic plants were tested for the presence of the transgene using Cas9 primers (Table S1). The target sequences of T‐DNA positive lines were amplified using the primers flanking the two target sites in each gene (Table S3) and sequenced by Sanger sequencing. The results of the Sanger sequencing were uploaded to DSDecode website (Liu et al., 2015) for target site mutation analysis.
Evaluation of gene editing efficiency of different scaffold variants by high‐throughput sequencing
DNA sample was prepared from N. benthamiana leaves infiltrated with agrobacteria harbouring various pKSE‐Scfv gene editing vectors. A pair of primers were designed to amplify about 200 bp DNA fragment with the predicted cleavage site in the middle. The amplified PCR products were mixed in equal mass amount and ligated with sequencing adapters using T4 DNA ligase at 16 °C for 12 h. After ligation, 1 μL reaction was used as a template for PCR amplification using Index primers and gel purified. The resulting libraries were sent for high‐throughput sequencing using the Illumina platform. The clean read files were analysed using in‐house Perl scripts to calculate the mismatch and InDel rates for each target site.
Funding
This work was supported by grants from the National Natural Science Foundation of China (32272491, 91940301) and the National Key Research and Development Program of China (2018YFD1000800).
Supporting information
Table S1 Primers used in this study.
Table S2 Target sequences on N. benthamiana and S. lycopersicum genes.
Table S3 C‐to‐T editing efficiency in E. coli of scaffold variants newly selected from error‐prone PCR library.
Table S4 Statistical results of editing efficiency of SGS3 by different Scfvs.
Data S1 Alignment of all functional scaffold variants identified in this study.
Data S2 Sequences of gene editing vectors cloned in this study.
Data S3 Sequencing analysis of gene editing in T0 and T1 tomato plants.
Data S4 Sequence alignments of tRNAs in different plant species.
Figure S1 Screen for functional scaffold variation and different tRNAs for making gene editing construct.
Figure S2 Producing MGT by fusion PCR.
Figure S3 Functional analysis of new scaffold variants from library screen.
Figure S4 Indels rates for different Scfvs targeting the SGS3 gene, measured by deep sequencing (see method section).
Figure S5 Analysis of PCR products for synthesis of 24‐spacer gRNA cluster.
Figure S6 Gene editing in T0 and T1 tomato plants.
Figure S7 Assembly of 8‐unit SSTS fragments with different template sets.
Acknowledgements
We thank the current lab member Mr. Zheng Kuijun for growth room maintenance and all other members for their contribution to essential reagents for this work.
References
- Barrangou, R. , Fremaux, C. , Deveau, H. , Richards, M. , Boyaval, P. , Moineau, S. , Romero, D.A. et al. (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. [DOI] [PubMed] [Google Scholar]
- Bombarely, A. , Rosli, H.G. , Vrebalov, J. , Moffett, P. , Mueller, L.A. and Martin, G.B. (2012) A draft genome sequence of Nicotiana benthamiana to enhance molecular plant‐microbe biology research. Mol. Plant Microbe Interact. 25, 1523–1530. [DOI] [PubMed] [Google Scholar]
- Borovsky, Y. , Monsonego, N. , Mohan, V. , Shabtai, S. , Kamara, I. , Faigenboim, A. , Hill, T. et al. (2019) The zinc‐finger transcription factor CcLOL1 controls chloroplast development and immature pepper fruit color in Capsicum chinense and its function is conserved in tomato. Plant J. 99, 41–55. [DOI] [PubMed] [Google Scholar]
- Bouche, N. , Lauressergues, D. , Gasciolli, V. and Vaucheret, H. (2006) An antagonistic function for Arabidopsis DCL2 in development and a new function for DCL4 in generating viral siRNAs. EMBO J. 25, 3347–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermak, T. , Curtin, S.J. , Gil‐Humanes, J. , Cegan, R. , Kono, T.J.Y. , Konecna, E. , Belanto, J.J. et al. (2017) A multipurpose toolkit to enable advanced genome engineering in plants. Plant Cell 29, 1196–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirino, P.C. , Mayer, K.M. and Umeno, D. (2003) Generating mutant libraries using error‐prone PCR. Methods Mol. Biol. 231, 3–9. [DOI] [PubMed] [Google Scholar]
- Cong, L. , Ran, F.A. , Cox, D. , Lin, S.L. , Barretto, R. , Habib, N. , Hsu, P.D. et al. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, D. , Chen, K.Y. , Chen, Y.D. , Li, H. and Xie, K.B. (2018) Engineering introns to express RNA guides for Cas9‐and Cpf1‐mediated multiplex genome editing. Mol. Plant 11, 542–552. [DOI] [PubMed] [Google Scholar]
- Doench, J.G. , Hartenian, E. , Graham, D.B. , Tothova, Z. , Hegde, M. , Smith, I. , Sullender, M. et al. (2014) Rational design of highly active sgRNAs for CRISPR‐Cas9‐mediated gene inactivation. Nat. Biotechnol. 32, 1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, Y. , Zhu, N. , Zhu, X.F. , Wu, M. , Jiang, C.Z. , Grierson, D. , Luo, Y.B. et al. (2019) Diversity and redundancy of the ripening regulatory networks revealed by the fruitENCODE and the new CRISPR/Cas9 CNR and NOR mutants. Hortic. Res.‐England 6, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao, Y. , Zong, W.B. , Zeng, D.C. , Han, J.L. , Chen, S.F. , Tang, J.N. , Zhao, Z. et al. (2020) Shortened snRNA promoters for efficient CRISPR/Cas‐based multiplex genome editing in monocot plants. Sci. China‐Life Sci. 63, 933–935. [DOI] [PubMed] [Google Scholar]
- Horton, R.M. , Hunt, H.D. , Ho, S.N. , Pullen, J.K. and Pease, L.R. (1989) Engineering hybrid genes without the use of restriction enzymes‐gene‐splicing by overlap extension. Gene 77, 61–68. [DOI] [PubMed] [Google Scholar]
- Horvath, P. and Barrangou, R. (2010) CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–170. [DOI] [PubMed] [Google Scholar]
- Hu, Z.J. , Li, J.X. , Ding, S.T. , Cheng, F. , Li, X. , Jiang, Y.P. , Yu, J.Q. et al. (2021) The protein kinase CPK28 phosphorylates ascorbate peroxidase and enhances thermotolerance in tomato. Plant Physiol. 186, 1302–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua, B. , Chang, J. , Xu, Z.J. , Han, X.Q. , Xu, M.Y. , Yang, M.N. , Yang, C.X. et al. (2021) HOMEODOMAIN PROTEIN8 mediates jasmonate‐triggered trichome elongation in tomato. New Phytol. 230, 1063–1077. [DOI] [PubMed] [Google Scholar]
- Ji, W. , Zhang, X.Y. , Warshamana, G.S. , Qu, G.Z. and Ehrlich, M. (1994) Effect of internal direct and inverted Alu repeat sequences on PCR. PCR Methods Appl. 4, 109–116. [DOI] [PubMed] [Google Scholar]
- Jinek, M. , Chylinski, K. , Fonfara, I. , Hauer, M. , Doudna, J.A. and Charpentier, E. (2012) A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek, M. , Jiang, F. , Taylor, D.W. , Sternberg, S.H. , Kaya, E. , Ma, E. , Anders, C. et al. (2014) Structures of Cas9 endonucleases reveal RNA‐mediated conformational activation. Science 343, 1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S. , Zhu, B.Z. , Pirrello, J. , Xu, C.J. , Zhang, B. , Bouzayen, M. , Chen, K.S. et al. (2020) Roles of RIN and ethylene in tomato fruit ripening and ripening‐associated traits. New Phytol. 226, 460–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, W.Z. , Xie, X.R. , Ma, X.L. , Li, J. , Chen, J.H. and Liu, Y.G. (2015) DSDecode: a web‐based tool for decoding of sequencing chromatograms for genotyping of targeted mutations. Mol. Plant 8, 1431–1433. [DOI] [PubMed] [Google Scholar]
- Liu, H. , Ding, Y. , Zhou, Y. , Jin, W. , Xie, K. and Chen, L.L. (2017) CRISPR‐P 2.0: an improved CRISPR‐Cas9 tool for genome editing in plants. Mol. Plant 10, 530–532. [DOI] [PubMed] [Google Scholar]
- Liu, G.Z. , Li, C.X. , Yu, H.Y. , Tao, P.W. , Yuan, L. , Ye, J. , Chen, W.F. et al. (2020) GREEN STRIPE, encoding methylated TOMATO AGAMOUS‐LIKE 1, regulates chloroplast development and Chl synthesis in fruit. New Phytol. 228, 302–317. [DOI] [PubMed] [Google Scholar]
- Liu, G.Z. , Yu, H.Y. , Yuan, L. , Li, C.X. , Ye, J. , Chen, W.F. , Wang, Y. et al. (2021) SlRCM1, which encodes tomato Lutescent1, is required for chlorophyll synthesis and chloroplast development in fruits. Hortic. Res.‐England 8, 128–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez, H. , Schmitz, G. , Thoma, R. and Theres, K. (2021) Super determinant1A, a RAWULdomain‐containing protein, modulates axillary meristem formation and compound leaf development in tomato. Plant Cell 33, 2412–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe, T.M. and Eddy, S.R. (1997) tRNAscan‐SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, X.L. , Zhang, Q.Y. , Zhu, Q.L. , Liu, W. , Chen, Y. , Qiu, R. , Wang, B. et al. (2015) A robust CRISPR/Cas9 system for convenient, high‐efficiency multiplex genome editing in monocot and dicot plants. Mol. Plant 8, 1274–1284. [DOI] [PubMed] [Google Scholar]
- Mao, Y. , Zhang, H. , Xu, N. , Zhang, B. , Gou, F. and Zhu, J.K. (2013) Application of the CRISPR‐Cas system for efficient genome engineering in plants. Mol. Plant 6, 2008–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerhans, A. , Vartanian, J.P. and Wain‐Hobson, S. (1990) DNA recombination during PCR. Nucleic Acids Res. 18, 1687–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno‐Mateos, M.A. , Vejnar, C.E. , Beaudoin, J.D. , Fernandez, J.P. , Mis, E.K. , Khokha, M.K. and Giraldez, A.J. (2015) CRISPRscan: designing highly efficient sgRNAs for CRISPR‐Cas9 targeting in vivo. Nat. Methods 12, 982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrasov, V. , Wang, C.M. , Win, J. , Lanz, C. , Weigel, D. and Kamoun, S. (2017) Rapid generation of a transgene‐free powdery mildew resistant tomato by genome deletion. Sci. Rep. 7, 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu, H. , Ran, F.A. , Hsu, P.D. , Konermann, S. , Shehata, S.I. , Dohmae, N. , Ishitani, R. et al. (2014) Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang, B. , Chen, Y.H. , Li, H.X. , Qian, C.J. , Huang, S.L. and Ye, Z.B. (2005) Transformation of tomatoes with osmotin and chitinase genes and their resistance to Fusarium wilt. J. Hortic. Sci Biotechnol. 80, 517–522. [Google Scholar]
- Pan, C.T. , Yang, D.D. , Zhao, X.L. , Liu, Y. , Li, M.Z. , Ye, L. , Ali, M. et al. (2021) PIF4 negatively modulates cold tolerance in tomato anthers via temperature‐dependent regulation of tapetal cell death. Plant Cell 33, 2320–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Leal, D. , Lemmon, Z.H. , Man, J. , Bartlett, M.E. and Lippman, Z.B. (2017) Engineering quantitative trait variation for crop improvement by genome editing. Cell 171, 470–480. [DOI] [PubMed] [Google Scholar]
- Shang, L.L. , Song, J.W. , Yu, H.Y. , Wang, X. , Yu, C.Y. , Wang, Y. , Li, F.M. et al. (2021) A mutation in a C2H2‐type zinc finger transcription factor contributed to the transition toward self‐pollination in cultivated tomato. Plant Cell 33, 3293–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soyk, S. , Muller, N.A. , Park, S.J. , Schmalenbach, I. , Jiang, K. , Hayama, R. , Zhang, L. et al. (2017) Variation in the flowering gene SELF PRUNING 5G promotes day‐neutrality and early yield in tomato. Nat. Genet. 49, 162–168. [DOI] [PubMed] [Google Scholar]
- Sternberg, S.H. , Redding, S. , Jinek, M. , Greene, E.C. and Doudna, J.A. (2014) DNA interrogation by the CRISPR RNA‐guided endonuclease Cas9. Nature 507, 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, W.X. and Liu, D.R. (2018) Rewritable multi‐event analog recording in bacterial and mammalian cells. Science 360, eaap8992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viguera, E. , Canceill, D. and Ehrlich, S.D. (2001) Replication slippage involves DNA polymerase pausing and dissociation. EMBO J. 20, 2587–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, T. , Wei, J.J. , Sabatini, D.M. and Lander, E.S. (2014) Genetic screens in human cells using the CRISPR‐Cas9 system. Science 343, 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, K.B. , Minkenberg, B. and Yang, Y.N. (2015) Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA‐processing system. Proc. Natl. Acad. Sci. USA 112, 3570–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing, H.L. , Dong, L. , Wang, Z.P. , Zhang, H.Y. , Han, C.Y. , Liu, B. , Wang, X.C. et al. (2014) A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol. 14, 327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Primers used in this study.
Table S2 Target sequences on N. benthamiana and S. lycopersicum genes.
Table S3 C‐to‐T editing efficiency in E. coli of scaffold variants newly selected from error‐prone PCR library.
Table S4 Statistical results of editing efficiency of SGS3 by different Scfvs.
Data S1 Alignment of all functional scaffold variants identified in this study.
Data S2 Sequences of gene editing vectors cloned in this study.
Data S3 Sequencing analysis of gene editing in T0 and T1 tomato plants.
Data S4 Sequence alignments of tRNAs in different plant species.
Figure S1 Screen for functional scaffold variation and different tRNAs for making gene editing construct.
Figure S2 Producing MGT by fusion PCR.
Figure S3 Functional analysis of new scaffold variants from library screen.
Figure S4 Indels rates for different Scfvs targeting the SGS3 gene, measured by deep sequencing (see method section).
Figure S5 Analysis of PCR products for synthesis of 24‐spacer gRNA cluster.
Figure S6 Gene editing in T0 and T1 tomato plants.
Figure S7 Assembly of 8‐unit SSTS fragments with different template sets.