

Abstract
The selective removal of dysfunctional mitochondria, a process termed mitophagy, is critical for cellular health and impairments have been linked to aging, Parkinson disease, and other neurodegenerative conditions. A central mitophagy pathway is orchestrated by the ubiquitin (Ub) kinase PINK1 together with the E3 Ub ligase PRKN/Parkin. The decoration of damaged mitochondrial domains with phosphorylated Ub (p-S65-Ub) mediates their elimination though the autophagy system. As such p-S65-Ub has emerged as a highly specific and quantitative marker of mitochondrial damage with significant disease relevance. Existing p-S65-Ub antibodies have been successfully employed as research tools in a range of applications including western blot, immunocytochemistry, immunohistochemistry, and ELISA. However, physiological levels of p-S65-Ub in the absence of exogenous stress are very low, therefore difficult to detect and require reliable and ultrasensitive methods. Here we generated and characterized a collection of novel recombinant, rabbit monoclonal p-S65-Ub antibodies with high specificity and affinity in certain applications that allow the field to better understand the molecular mechanisms and disease relevance of PINK1-PRKN signaling. These antibodies may also serve as novel diagnostic or prognostic tools to monitor mitochondrial damage in various clinical and pathological specimens.
Keywords: autophagy, mitochondria, mitophagy, Parkin, Parkinson disease, phospho-ubiquitin, PINK1, PRKN, recombinant antibody, ubiquitin
INTRODUCTION
Mitophagy is a cytoprotective mechanism for the selective and timely removal of dysfunctional or superfluous mitochondria through autophagy. One central mitophagy mechanism that provides specificity for damaged mitochondria is the phospho-ubiquitylation of mitochondrial proteins, which fosters the recruitment of autophagy receptors only to those organelles that need to be degraded [1]. PTEN-induced kinase 1 (PINK1) and the E3 ubiquitin (Ub) ligase Parkin (PRKN) are the key players of this Ub-tagging process and complete loss of either enzyme leads to early-onset Parkinson disease (PD). Upon mitochondrial damage, PINK1 locally stabilizes and phosphorylates serine 65 on Ub (p-S65-Ub) that is either free or already attached to outer mitochondrial proteins as monomer or different types of poly-Ub chains [2,3]. PINK1 also phosphorylates the E3 Ub ligase PRKN at a conserved serine at position 65 within its Ub-like domain [4,5]. Both PRKN phosphorylation and allosteric p-S65-Ub binding fully activates its E3 Ub ligase activity [6–8]. Activated PRKN attaches additional Ub moieties to mitochondrial proteins, which can be further phosphorylated by PINK1 and serve as additional docking stations to recruit even more cytosolic PRKN to mitochondria. This positive feedback loop between PINK1 and PRKN amplifies the extent of p-S65-Ub coating of damaged mitochondria, which drives their elimination via mitophagy.
Given the causal link between the loss of function in PINK1 or PRKN and early-onset PD, mitophagy dysfunction has been long considered to play an important role in disease pathogenesis [9]. Mitochondrial impairments are commonly found in aging and age-related diseases including neurodegenerative disorders, and mitophagy defects are likely also widespread [10]. As the joint product of PINK1 and PRKN enzymatic activity, p-S65-Ub can be used as a quantitative measure of mitochondrial health and mitophagy alteration. Absent or reduced p-S65-Ub levels result from failure to properly activate mitophagy in model organisms with PINK1 or PRKN knockout or mutations [11–13]. In contrast, elevated p-S65-Ub levels can be detected with age and in many diseases and may originate from increased mitochondrial damage or rather may accumulate due to a downstream block of the autophagic-lysosomal degradation system [11,13–15]. p-S65-Ub therefore represents a promising and potent diagnostic marker for damaged mitochondria in cell and animal models as well as in human biofluids and postmortem brain [11,13]. Indeed, p-S65-Ub is currently being tested in several preclinical studies as an early biomarker in different neurodegenerative diseases. Existing p-S65-Ub antibodies show encouraging results in ELISA-based applications. However, the overall p-S65-Ub signal in human biofluids at baseline and under pathological conditions is very low and therefore difficult to detect in a reliable manner. Similarly, sensitive histopathological assessment of damaged mitochondria undergoing mitophagy in human brain is currently very limited and would greatly benefit from more sensitive and reliable tools to further validate injury-prone brain regions and subcellular locations that are positive for the p-S65-Ub signal. In general, the detection of endogenous p-S65-Ub in vivo at baseline or under low (chronic) stress is key and requires more sensitive techniques and antibodies.
To expand the existing repertoire of tools, we here generated and thoroughly characterized a large series of recombinant, rabbit monoclonal p-S65-Ub antibodies (Fig. 1). The top performing p-S65-Ub antibody clones for each category were affinity-purified and further interrogated in cells and brain tissue of mouse and human origin and across a range of different applications. The top antibody candidates target p-S65-Ub with high specificity and affinity in different applications and are available as a novel resource for the community to study PINK1-PRKN signaling under normal, stress, or disease conditions.
Figure 1.

Schematic overview of recombinant p-S65-Ub antibody clone generation, selection, and validation. The illustration summarizes the entire process of recombinant, rabbit monoclonal antibody production starting from testing of the initial polyclonal bleeds to the isolation and screening of B cells, and the subsequent cloning of variable heavy and light chain antibody regions into an IgG vector frame. Recombinant antibodies were then sequenced, expressed in HEK293 cells and supernatants were screened by western blot and immunocytochemistry. The five most promising antibody clones were affinity purified and re-validated by a series of biochemical and imaging analyses using recombinant proteins as well as cells and brain tissues from mouse and human origin. PBMC - peripheral blood mononuclear cell, Lc - light chain, Hc - heavy chain, ICC - immunocytochemistry, IHC – immunohistochemistry. Created with BioRender.com.
RESULTS
Generation and screening of polyclonal rabbit antibodies
Four rabbits were immunized with a combination of N- or C-terminally conjugated p-S65-Ub peptides. Initial bleeds from these animals were tested for immunoreactivity in dot blots towards the respective phosphorylated- and non-phosphorylated Ub and PRKN peptides (Fig. S1A). Based on their stronger immunoreactivity, rabbits (#15021 and #15022) were further selected to receive additional p-S65-Ub peptide booster injections. The resulting affinity-purified polyclonal antibodies were tested in western blots using wild-type (WT) and negative control PINK1 knockout (KO) human embryonic kidney (HEK293E) cells that were each treated with the mitochondrial stressor carbonyl cyanide 3-chlorophenylhydrazone (CCCP) (Fig. S1B) [13]. Affinity purified bleeds from rabbit #15022 but not rabbit #15021 demonstrated strong p-S65-Ub immunoreactivity in CCCP-treated WT cells, which was comparable to the commercially available reference antibody. Additional immuno-depletion further reduced the non-specific signal in PINK1 KO cells. Given the superior signal-to-noise ratio, rabbit #15022 was selected for isolation of peripheral blood mononuclear cells (PBMCs).
Recombinant monoclonal antibody generation and clonal selection
Collected PBMCs were enriched for antigen-specific B cells, and subsequently diluted for single B cell cultivation. Cell supernatants from a total of 215 single B cell cultures were tested by direct ELISA for immunoreactivity towards phosphorylated (serine 65) and non-phosphorylated full-length, monomeric Ub protein as well as non-/phosphorylated Ub and PRKN peptides surrounding the respective serine 65 residues (Table S1). We identified 48 candidates that consisted of 36 B cell cultures producing antibodies that strongly targeted both p-S65-Ub protein and peptide, four with similar specificity but weaker immunoreactivity, four that detected p-S65-Ub protein only but not peptide, and another four that also recognized the corresponding p-S65-PRKN peptide (Table S1). Variable regions of both heavy and kappa chains were subsequently cloned, sequenced, and co-expressed in HEK293 cells to give rise to 47 recombinant IgG antibody clones. A phylogenetic tree was created to determine their clonal relationship (Fig. S2).
We next screened all 47 recombinant clonal supernatants in western blots for affinity and selectivity (Fig. S3). Ten supernatants showed substantially stronger signal with lysates from CCCP-treated WT HEK293E compared to PINK1 KO cells and were selected for additional validation by immunocytochemical analysis. Primary dermal fibroblasts from a PD patient with homozygous PINK1 Q456X loss-of-function mutation and matching WT cells were left untreated or were challenged with the mitochondrial depolarizer valinomycin and then stained with the top ten clonal supernatants (Fig. S4A). Most supernatants showed only minimal staining in untreated WT cells but detected a robust signal in stressed WT compared to PINK1 mutant cells. Clones 11H2K3, 29H1K3, 29H2K2, and 30H3K1 had the most intense staining, while two clones showed no real signal increase above background levels (Fig. S4B).
Taken together and considering different biochemical and immunocytochemical validation criteria, cell supernatants from five recombinant p-S65-Ub clones (11H2K3, 19H1K1, 29H2K2, 30H3K1, and 41H1K3) were selected for subsequent affinity purification and further detailed characterization.
Western blot assessment of the top affinity purified recombinant p-S65-Ub antibodies
Supernatant from the five top p-S65-Ub clones were next affinity-purified and first re-validated to confirm the previous outcome before expanding to additional analyses. Briefly, WT and PINK1 KO HEK293E cells (Fig. 2A) as well as WT and pink1 KO mouse embryonic fibroblasts (MEFs) (Fig. 2B) were treated with mitochondrial stressors and their lysates were analyzed by western blot. All recombinant affinity-purified p-S65-Ub antibodies were used in identical concentration, except the reference antibody, which was used five times diluted. All antibodies gave increased signal in WT but not the corresponding PINK1/pink1 KO cells of human and mouse origin (Fig. 2A, B). In WT HEK293E cells, recombinant p-S65-Ub clones 29H2K2 and 30H3K1 were able to detect p-S65-Ub with highest intensity, closely followed by clone 11H2K3 and subsequently clones 19H1K1 and 41H1K3. Different from HEK293E cells, all clones except 41H1K3 performed comparably in treated WT MEFs. Overall, all five affinity-purified clones showed very little background signal in western blots of human and mouse PINK1/pink1 KO cells treated with mitochondrial stressors, suggesting high specificity.
Figure 2.

Western blot assessment of the top p-S65-Ub antibodies in cells. (A) HEK293E cells and (B) mouse embryonic fibroblasts with (WT) or without PINK1 expression (PINK1 KO or pink1 KO) were treated with mitochondrial depolarizer and analyzed by western blot. Representative western blot images are shown for the performance of the top five p-S65-Ub antibody clones in each cell type. Note that concentrations of the tested p-S65-Ub antibodies are five times higher compared to the reference antibody. GAPDH was used as loading control. A densitometric quantification of p-S65-Ub relative to GAPDH is shown alongside after normalization by the reference antibody set to 1. Ref. Ab - reference antibody, KO - knockout, WT - wild-type.
Testing antibodies with recombinant p-S65-Ub chains using sandwich ELISAs
We next tested the purified recombinant p-S65-Ub antibodies in a sandwich ELISA format on a Meso Scale Discovery (MSD) platform. Rabbit p-S65-Ub antibodies were used as capturing agents and combined with a mouse total Ub antibody (P4D1) as detecting reagent as published previously [13]. We first tested recombinant p-S65-Ub tetramers with K48 or K63 chain linkage, the two most abundant Ub chain types in cells, and used their non-phosphorylated counterparts as negative controls and determined the Limit of Blank (LoB), the Limit of Detection (LoD), and the Limit of Quantification (LoQ) according to Armbruster and Pry (2008) [16] (Table 1; Fig. 3A). The LoB represents the relative noise or background, and was relatively high for clones 41H1K3, 11H2K3, and 29H2K2. The LoD was similar compared to the corresponding LoB and consequently also relatively high for the same clones mentioned (41H1K3, 11H2K3, and 29H2K2). Importantly, the LoQ for K48-linked p-S65-Ub tetramers was lowest for clone 19H1K1 at 100 fg/ml, which was even lower compared to the reference antibody.
Table 1.
LoB, LoD, and LoQ of p-S65-Ub MSD ELISA with recombinant protein
| ref. Ab | 11H2K3 | 19H1K1 | 29H2K2 | 30H3K1 | 41H1K3 | |
|---|---|---|---|---|---|---|
|
| ||||||
| LoB (MSD-ECL [AU]) | 469 | 1162 | 522 | 774 | 578 | 2340 |
| LoD for K48 p-S65-Ub4 (MSD-ECL [AU]) | 472 | 1268 | 538 | 860 | 590 | 2454 |
| LoD for K63 p-S65-Ub4 (MSD-ECL [AU]) | 473 | 1178 | 522 | 804 | 912 | 2349 |
| LoQ for K48 p-S65-Ub4 (fg/ml) | 1x104 | 1x106 | 100 | 1x107 | 1x105 | 1x106 |
| LoQ for K63 p-S65-Ub4 (fg/ml) | 1x106 | 1x107 | 1x106 | 1x107 | 1x107 | 1x105 |
| Chain preference | K48 >> K63 | K48 >> K63 | K48 >> K63 | K48 = K63 | K48 >> K63 | K48 << K63 |
ref.Ab - reference antibody, LoB - Limit of Blank, LoD - Limit of Detection, LoQ - Limit of Quantitation, Ub - ubiquitin tetramer, K48 - polyubiquitin chain linked by lysine residue 48, K63 - polyubiquitin chain linked by lysine residue 63.
Figure 3.

Assessing the top p-S65-Ub antibodies in ELISA using recombinant protein. (A) Sandwich ELISA results for the detection of recombinant K48 (solid line) and K63 (dash line) p-S65-Ub tetramers (Ub4) that were serially diluted to the range of 1–1,000,000 pg/ml. (B) Direct ELISA results for the detection of p-S65-PRKN and nonphosphorylated PRKN recombinant proteins that were serially diluted to the range of 1–1,000,000 pg/ml. N = 2. Data are shown as mean with SD.
Of note, the LoQs for K63-linked p-S65-Ub tetramers were in most cases substantially higher compared to those for K48-linked p-S65-Ub tetramers, which is in line with previously published results [13]. Two exceptions were clones 41H1K3 and 29H2K2. Remarkably, clone 41H1K3 had a stronger preference towards Ub tetramers with K63-linkage relative to those with K48-linkage, which is reflected in a 10-fold lower LoQ for K63-linkage compared to Ub chains of the K48-linkage type. In contrast, the LoQ for both K48 and K63-linked p-S65-Ub were both similarly high (10 ng/ml each) for clone 29H2K2 suggesting that this antibody may not be the best option for ELISA-based detection (Table 1; Fig. 3A). The range of linearity in this assay differed between p-S65-Ub species used and was between 10 ng/ml and 1 μg/ml for K63 p-S65-Ub tetramers and between 1 ng/ml and 1 μg/ml for K48 p-S65-Ub tetramers. The coefficient of variation was excellent for all clones and matrices analyzed and was always well below 10% (data not shown).
Given the sequence similarity of phosphorylated epitopes around serine 65 in both Ub versus PRKN, we tested all purified recombinant antibodies towards phosphorylated and non-phosphorylated PRKN protein in a direct ELISA on an MSD platform (Fig. 3B). It is noteworthy that all antibodies including the reference antibody showed no cross-reactivity towards p-S65-PRKN or total PRKN except for the highest concentration tested (1 μg/ml). At this concentration, equimolar amounts of p-S65-Ub monomer resulted in almost 600-fold fold higher MSD-ECL signal for the reference antibody and between 2600 to 6600-fold higher MSD-ECL signal for all other recombinant p-S65-Ub clones. Lack of significant cross-reactivity with recombinant p-S65-PRKN protein was further confirmed by western blots (data not shown).
ELISA-based p-S65-Ub detection in cells and in mouse and human brain lysates
We next tested the purified p-S65-Ub antibodies for ELISA using cell culture lysates (Fig. 4A). We used WT and PINK1 KO HEK293E cells treated with CCCP for 24 h and compared to vehicle (DMSO)-treated WT cells. It is of note that all antibodies except clone 29H2K2 were able to show significant differences in p-S65-Ub levels at baseline condition between DMSO-treated WT and CCCP-treated PINK1 KO cells. In line with previous results using recombinant p-S65-Ub chains, clone 19H1K1 and the reference p-S65-Ub antibody showed most sensitive p-S65-Ub detection and comparable results for both baseline (1.8- vs. 2.1-fold increases compared DMSO-treated WT to CCCP-treated PINK1 KO cells) and upon stress (91- vs. 144-fold increases compared CCCP-treated WT over CCCP-treated PINK1 KO cells) (Fig. 4A; Table 2). Interestingly, clone 41H1K3 that showed a higher sensitivity for recombinant K63- over K48-linked p-S65-Ub chains showed the lowest fold changes for p-S65-Ub detection in the cell samples, both at baseline and under mitochondrial stress (Table 2).
Figure 4.

Assessing the top p-S65-Ub antibodies by sandwich ELISA using lysates from cells, mouse brain, and human brain. (A) ELISA detection of p-S65-Ub levels in cell lysates from CCCP or DMSO treated WT and PINK1 KO HEK293E cells. One-way ANOVA. N = 2. Data are shown as mean with SEM. (B) ELISA detection of p-S65-Ub levels in hemibrain lysates from unstimulated WT and pink1 KO mice. Unpaired t-test. N = 6 per group. Data are shown as mean with SEM. (C) ELISA detection of p-S65-Ub levels in human brain lysates from neurological normal controls and AD cases. Mann-Whitney U test. N = 10 per group. Data are shown as median with interquartile range. * p<0.05, ** p<0.005, *** p<0.0001. Asterisks (*) indicate the comparison to the untreated WT or the comparison between two groups under the brackets. Ref. Ab - reference antibody, WT - wild-type, KO - knockout, AD - Alzheimer disease, n.s. - non significant.
Table 2.
Comparison of MSD ELISA detection of p-S65-Ub with different testing materials
| Fold change in MSD ELISA detection | ref. Ab | 11H2K3 | 19H1K1 | 29H2K2 | 30H3K1 | 41H1K3 |
|---|---|---|---|---|---|---|
|
| ||||||
| HEK293E cells baseline detection (WT vs. PINK1 KO) | 2.1 | 1.3 | 1.8 | 1.1 | 1.4 | 1.1 |
| HEK293E cells stressed condition (treated WT vs. PINK1 KO) | 143.5 | 52 | 91.2 | 5.6 | 80.2 | 4.8 |
| Mouse brain baseline detection (WT vs. pinkl KO) | 1.5 | 1.1 | 1.6 | 1 | 1.2 | 1 |
| Human brain disease condition (control vs. AD) | 9.3 | 10.8 | 2.6 | 4.9 | 3.7 | 2 |
WT - wild-type, KO - knockout, AD - Alzheimer disease.
We then tested the performance of recombinant p-S65-Ub antibodies in mouse hemibrain lysates from unstimulated WT and pink1 KO mice (Fig. 4B). As seen with recombinant Ub chains and in cells, clone 19H1K1 and the reference p-S65-Ub antibody again showed comparable results in mouse brains. Both antibodies were able to distinguish between the two genotypes with similar fold changes (1.6 vs. 1.5) (Table 2). A weaker detection of p-S65-Ub in unstimulated WT mice was found when using clone 30H3K1, while none of the remaining antibodies showed differences in p-S65-Ub levels between WT and pink1−/− mice (Fig. 4B; Table 2).
Finally, we tested the performance of purified p-S65-Ub clones in human autopsy brain lysates from the frontal cortex of Alzheimer disease (AD) cases compared to sex- and age-matched neurologically normal controls (Fig. 4C). Different from the results with mouse brain lysates, all p-S65-Ub clones detected significant increases of p-S65-Ub levels in AD cases compared to controls. The highest fold change between the two groups were obtained with clone 11H2K3 (10.8-fold) and the reference antibody (9.3-fold), followed by clones 29H2K2 and 30H3K1 and finally clones 19H1K1 and 41H1K3 (Fig. 4C; Table 2). It should be noted that the reference antibody, clones 19H1K1 and 30H3K1 showed the largest differences between control tissue and the buffer blank (data not shown), suggesting a greater assay window with potentially better detectability of low p-S65-Ub amounts.
Characterization of p-S65-Ub clones via immunocytochemistry in PINK1 patient fibroblasts
To further confirm the specificity and sensitivity of the top five p-S65-Ub clones in other applications, we next validated them by immunocytochemistry in WT and homozygous PINK1 loss-of-function mutation (PINK1Q456X) skin fibroblasts. For this, both WT and PINK1 mutant fibroblasts were left untreated or were treated with the mitochondrial stressor valinomycin, stained with each of the antibodies using the same concentration and measured by high content imaging (Fig. 5). Both unstressed WT cells and stressed PINK1 mutant cells were expected to show minimal p-S65-Ub signal and served as negative controls. Valinomycin-induced mitophagy led to a significant elevation of p-S65-Ub signal in WT fibroblasts as confirmed by the previously validated reference antibody [13] (Fig. 5A). All five clones also showed robust immunostaining of treated WT cells but only minimal immunopositive signals in untreated cells or treated PINK1 mutant cells (Fig. 5A), indicating excellent specificity towards p-S65-Ub detection. In the treated WT cells, the highest signal was observed with antibody clones 30H3K1, 11H2K3, and 29H2K2. Clone 19H1K1 showed a moderate intensity, and 41H1K3 resulted in the lowest signal (Fig. 5B). The performance of the top three clones was comparable to the reference antibody. The specificity of the antibodies was further confirmed by the strong co-localization of p-S65-Ub positive signals with the mitochondrial marker HSP60 upon mitochondrial stress in WT cells (Fig. 5C).
Figure 5.

Comparison of the top p-S65-Ub antibodies using immunocytochemistry staining of human dermal fibroblasts. All five p-S65-Ub recombinant antibodies and the reference antibody were evaluated by immunocytochemistry in human primary dermal fibroblasts treated with 2 μM valinomycin for 0 or 24 hours. All antibodies were used at 1 μg/ml. (A) Representative images of p-S65-Ub immunoreactive signals (green) in fibroblasts carrying WT or homozygous PINK1Q456X mutation. (B) Fluorescence intensities of each antibody was quantified by high content imaging and compared signals from treated WT to the signals from the corresponding PINK1 mutant fibroblasts. Fold changes are labeled at top of each bar. Samples stained without primary antibody was used a negative control (no Ab). N = 2. Data are shown as mean with SEM. (C) Representative zoom-in images of p-S65-Ub and HSP60 immunoreactive signals in treated WT fibroblasts. 24 hours of valinomycin treatment induced mitochondrial aggregation (HSP60, red) and the accumulation of p-S65-Ub (green). Nuclei were stained with Hoechst (blue). Scale bar: 50 μm. Ref. Ab - reference antibody, WT - wild-type.
Immunohistological assessment of p-S65-Ub clones in transgenic mouse and human AD brain
In addition to immunocytochemical staining in cultured fibroblasts, we next assessed p-S65-Ub labeling in the hippocampus of transgenic mice overexpressing human tau protein (rTg4510 mice). We have shown previously that p-S65-Ub is robustly induced in this tauopathy model and can reliably be detected by both immunohistochemical methods and MSD-ELISA [12,13,15]. Given that the reference antibody performs poorly in immunohistochemistry [13], the five p-S65-Ub recombinant antibodies were compared with a previously validated in-house p-S65-Ub antibody [11,14] (Fig. 6). Non-transgenic (nonTg) littermates were used as controls. Typical, punctate p-S65-Ub immunopositive structures (dark brown) were detected by all p-S65-Ub clones at identical concentrations with high specificity in sections from 9–10 months old transgenic but not nonTg mice. However, intensity levels were quite different for different recombinant clones and p-S65-Ub positive structures were most prominently detected with clone 41H1K3 followed by our in-house antibody [11,14,15], and minimal signal was found with the reference antibody (Fig. 6).
Figure 6.
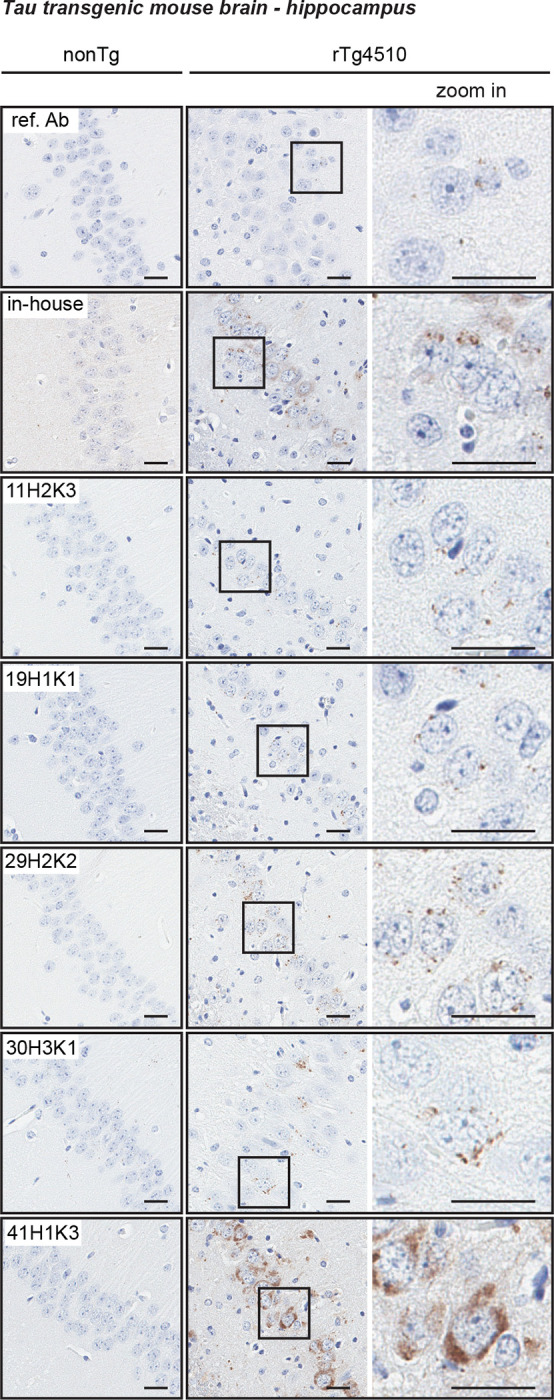
Characterization of the top p-S65-Ub antibodies by immunohistochemical staining of mouse brain. All five p-S65-Ub recombinant antibodies together with our in-house p-S65-Ub antibody and the reference antibody were evaluated by immunohistochemistry in brain sections from 9- to 10-month-old nonTg and rTg4510 mice. Same concentration (1.56 μg/ml) was used for all antibodies. Zoom-in images of the highlighted regions are shown to the right. Typical punctate p-S65-Ub immunopositive structures (dark brown) were detected by all clones but with different intensity. No signal was observed in nonTg mice. Scale bar: 50 μm. Ref. Ab - reference antibody, nonTg – non-transgenic.
Next, we tested the antibody clones on hippocampal regions of human AD brain (Fig. 7) [15]. Again, we used the same concentration (0.78 μg/ml) for all tested antibodies to directly compare the sensitivity. As previously reported, the in-house antibody detects small granular, cytoplasmic p-S65-Ub immunopositive structures in the AD hippocampus [15]. Similar p-S65-Ub structures were also observed when stained with clones 19H1K1, 29H2K2, and 41H1K3 with lower, similar, or higher signal intensity compared to the in-house antibody, respectively. Because of the sensitivity of 41H1K3 we diluted this antibody further and were still able to clearly label p-S65-Ub positive granules at a much lower concentration of 0.125 μg/ml (Fig. 7). In contrast to the abovementioned three clones, clone 11H2K3 showed undefined and rather diffuse cytoplasmic staining that has not been previously observed and only very minimal immunoreactive signal was found in clone 30H3K1-stained section.
Figure 7.

Characterization of the top p-S65-Ub antibodies by immunohistochemical staining of Alzheimer disease brain. All five p-S65-Ub recombinant antibodies together with our in-house p-S65-Ub antibody and the reference antibody were evaluated by immunohistochemistry of hippocampal sections from Alzheimer disease brain. Same concentration (0.78 μg/ml) was used for all antibodies. Zoom-in images of the highlighted regions are shown to the right. Typical punctate p-S65-Ub immunopositive structures (dark brown) were detected by most of the antibodies at different sensitivity. The superior sensitivity of 41H1K3 allowed detection of p-S65-Ub at a much lower concentration (0.125 μg/ml). Scale bar: 50 μm. Ref. Ab - reference antibody.
Taken together, selected clones showed great sensitivity for p-S65-Ub detection in immunostaining in fibroblasts (clones 11H2K3, 29H2K2, and 30H3K1) and brain tissue (clone 41H1K3). While similar characteristic morphologies were detected upon mitochondrial stress in cells, in tauopathy mouse brain, and in human disease autopsy brain, the clones that worked the best for immunostaining in cells and tissue did not appear to overlap.
DISCUSSION
We here developed and thoroughly characterized a new series of recombinant, rabbit monoclonal antibodies targeting the mitophagy tag p-S65-Ub. Starting from an immunization of four animals, we screened over 200 B cell lines by direct ELISA and cloned and sequenced 47 recombinant antibodies that were further evaluated. The top five clones were affinity purified and their sensitivity and specificity were validated using a range of biochemical and imaging techniques with recombinant Ub chains, gene-edited cells, and patient fibroblasts as well as mouse and human brain tissue. We compared their respective signal-to-noise ratios in samples with diverse complexity and levels of the mitophagy tag at baseline and upon acute, mitochondrial stress or in chronic, diseased conditions, and across applications. The top performing antibodies were selected for each category and are now available to the community, expanding the toolkit to monitor PINK1-PRKN signaling in different sample types and with different methods (see Table 3 for a summary).
Table 3.
Top recombinant p-S65-Ub antibodies for different applications
| Matrix | Cells | Brain tissue | |||
|---|---|---|---|---|---|
|
| |||||
| WB | ELISA | ICC | ELISA | IHC | |
| 30H3K1* | 30H3K1* | 30H3K1* | 30H3K1* | 41H1K3* | |
| human | 19H1K1 | 19H1K1 | 19H1K1 | ||
| 41H1K3 | |||||
| mouse | 30H3K1* 19H1K1 |
ND | ND | 19H1K1* 30H3K1 |
41H1K3* |
= denotes best clone for the application in that sample type. WB - western blot, ICC - immunocytochemistry, IHC - immunohistochemistry, ND - not defined.
Since its discovery [17–19], p-S65-Ub has been recognized as a highly specific and quantitative mitochondrial damage marker of disease relevance [14,20,21]. By now p-S65-Ub has been employed for larger-scale, functional and genetic screening of mitophagy modifiers in cell culture [22,23] and in human autopsy brain [24]. p-S65-Ub can also be detected in clinical samples and may be useful as a biomarker of mitochondrial health and function [13,25]. Yet, physiological levels of p-S65-Ub can vary between different cells and tissues and the normal range of the p-S65-Ub signal needs to be established for each case and sample type. While p-S65-Ub is strongly elevated with stress, age, and various disease conditions in cell and animal models and in humans [11–13,15,26], the underlying causes may differ. It is of note that while p-S65-Ub is a direct result of PINK1 and PRKN enzymatic activities, this mitophagy label is only transitory and highly dynamic in nature as damaged labeled mitochondria are rapidly degraded under healthy, physiological conditions. Greater levels of p-S65-Ub can result from primarily increased mitochondrial stress and damage or from an impaired turnover and reduced flux through the autophagy-lysosome system. Monitoring p-S65-Ub levels over time may allow to better determine mitophagic flux and could help discriminate between both scenarios. Therefore, it may be useful not only as a diagnostic and prognostic tool, but also as pharmacodynamic and therapeutic biomarker.
Although mitochondrial proteins are the primary targets of p-S65-Ub, the exact nature and composition of the phosphorylated Ub chains is not well understood but could have important implications for outcomes and detection of the signal. A preference for phosphorylation of multi-mono-Ub moieties or short poly-Ub chains has been suggested in cell lines [27], but an increase particularly in K63-linked poly-Ub chains has been noted on mitochondria and appears to be the preferred mitophagy signal in neurons [28,29]. The temporal order of events as well as a certain stoichiometry of the p-S65-Ub signal could also be important for proper induction and flux of mitophagy. As such p-S65-Ub specific antibodies that are also able to better discriminate between different Ub linkages might be particularly useful to further resolve the mitophagy signal in vivo. In that regard it is worth noting that some of the clones developed herein seemed to have somewhat distinct chain linkage preferences. While most antibodies to date including clones 11H2K3, 19H1K1, and 30H3K1, seem to have a higher affinity towards recombinant K48-linked p-S65-Ub chains, clone 41H1K3 preferred phosphorylated K63-linked chains. Clone 29H2K2 seemed to recognize both types equally well. Certainly, much more work is needed to fully characterize the binding profiles and determine selectivity and affinity of the antibody clones, but a toolkit consisting of clones with differential binding abilities combined with sophisticated mass spectrometry approaches [30] may provide a unique opportunity to decode the complexity of the p-S65-Ub signal in health and disease.
We interrogated the top five antibodies across a range of methods using cell and brain tissue samples of mouse and human origin alongside with appropriate controls for each setting. It is noteworthy that none of the top five clones performed best in all tested applications or with all sample types. Instead, 30H3K1, 19H1K1, and 41H1K3 were selected as the best clones for different applications and are currently in production for public access (see details in Materials and Methods). Clone 30H3K1 convinced particularly through its performance in western blots and immunocytochemistry, clone 41H1K3 showed the highest sensitivity for immunohistochemistry in mouse and human brain tissue, and clone 19H1K1 worked best in sandwich ELISAs using cell culture and (mouse) brain lysates. Although other clones such as 11H2K3 also performed quite well in MSD-ELISA and perhaps captured even better the peak intensities under more severe conditions (e.g., human AD brain tissue), clone 19H1K1 had a bigger assay window towards the lower end of the signal. It appeared most useful to also detect the basal, physiological p-S65-Ub levels and subtle changes within cells, normal mouse brain, and human control brain. Whether differences in the sample matrix, changes in the Ub chain composition, or linkage preferences influenced the clones’ overall performance in different applications or sample types remains to be determined. However, testing of the clones should be expanded to clinical biofluids in order to determine their full potential for also ultrasensitive detection of p-S65-Ub.
Here we capitalized on recent technological developments in the field and generated first recombinant, rabbit monoclonal antibodies against the mitophagy tag p-S65-Ub. Compared to conventional monoclonal antibodies derived from hybridoma cells lines, recombinant antibodies are not susceptible to genetic drift, gene loss, or mutations. As such recombinant antibodies are more stable and better scalable with minimal variability in specificity and excellent batch-to-batch reproducibility. Antibodies may also be recloned into backbones from a different species or conjugated with other epitope tags or direct site-specific labeling. Overall, this could generate more flexibility and enhance their versatility across applications where the host species may be limiting their utility [31]. Given the known sequences of the heavy and light chain variable regions, recombinant antibodies can be further engineered. For instance, the variable regions could be modified to further modulate the affinity to different Ub linkages, making them more selective for a certain Ub chain type. It was of interest that clones 11H2K3 and 30H3K1 were located on one end, while clone 41H1K3 was located on the opposite end on the phylogenetic tree. It is however not clear whether there is a correlation between the distance and their binding preferences with regards to Ub chain linkages.
In summary, we here introduce and characterize novel recombinant p-S65-Ub antibodies that can now be further exploited by the field to better understand mechanisms and nature of the PINK1-PRKN mediated p-S65-Ub signaling in a range of applications as well as its underlying implication in various disease-associated conditions. Our study also highlights the biomarker potential of the mitophagy tag in various clinical and pathological specimens as a diagnostic and prognostic tool or for therapeutic assessment.
MATERIAL AND METHODS
Peptide synthesis, rabbit immunization, and polyclonal antibody production
Synthetic peptides and the polyclonal antibodies were manufactured by 21st Century Biochemicals, Marlborough, MA. Briefly, peptides were manufactured by Fmoc-based solid phase peptide synthesis, HPLC purified to >90%, and conjugated via the cysteine thiol to a mixture of carrier proteins. Each peptide was covalently attached to Optimized Affinity Resin (21st Century Biochemicals) containing an extended spacer arm and a thiol-reactive moiety. All rabbit animal work was conducted at Pacific Immunology (San Diego, CA; NIH and USDA compliant). After an initial rabbit immunization and multiple boosts, the serum was collected at specific intervals and analyzed by dot blots at 21st Century Biochemicals after which appropriate production bleeds were pooled. The serum then underwent two purification processes. Antibodies were first recovered by affinity purification using p-S65-Ub peptides covalently attached to Optimized Affinity Resin through a cysteine residue added to either the N- or C-terminus. A portion of the resulting antibodies were further subjected to an additional immunodepletion step with both the nonphosphorylated Ub peptides (Ub Ser-OH N-/C-terminus) and PRKN peptides (p-S65-PRKN control and Ser-OH65-PRKN control). Peptide affinity columns were incubated with serum, washed multiple times including a 0.5 M NaCl wash in phosphate buffer, and the antibodies eluted under acidic conditions. After dialysis against PBS, the antibodies were stored in PBS with 50% glycerol.
PBMC isolation, B cell selection, screening, and recombinant antibody generation
Following the final boost, 30 ml of rabbit peripheral blood was collected for PBMC isolation and sent to ImmunoPrecise Antibodies Ltd. In short, the whole blood from each animal was diluted one to one in serum free medium and subjected to a density separation medium, Lympholyte-Mammal (Cedarlane Laboratories, L5110). Following the manufacturer’s protocol, the enriched PBMCs at the middle layer were carefully removed and washed twice in 1% BSA (VWR, 9048–45-8) in DMEM (Corning, 15–018-CV). The isolated PBMCs were then subjected to an antigen specific enrichment process as part of the ImmunoPrecise Antibodies Ltd. Proprietary B Cell Select platform and were diluted in growth medium for single B cell cultivation. On day 7, the supernatant of each single B cell well was screened for antigen specificity through direct ELISA for targeting full-length monomeric p-S65-Ub protein (Boston Biochem, U-102), free p-S65-Ub peptides 1 and 2 and BSA-conjugated p-S65-Ub peptides 1 and 2 (provided by 21st Century Biochemicals). Non-phosphorylated Ub protein (Boston Biochem, U-100H), free non-phosphorylated, hydroxylated Ub peptides 1 and 2, free p-S65-PRKN peptide, and free hydroxylated p-S65-PRKN peptide (provided by 21st Century Biochemicals) were used as negative controls.
The top 48 performing B cells from the screening were assessed and selected for recombinant expression. Briefly, mRNA was isolated (Dynabeads, Invitrogen, 61006) from lysed B cell cultures and RT-PCR was performed for the conversion to cDNA, followed by amplification of targeted heavy and light chains using specific primer sets. Variable regions of G1 and kappa chains were separately ligated into expression vectors containing respective constant regions. Plasmids encoding both the antibody heavy chain and light chain, housed within an expression vector driven by the CMV promoter were co-transfected into HEK293 cells, which had been adapted for suspension culture in a serum-free medium. Following transfection using the polyethylenimine-mediated gene delivery method, the cells were allowed to incubate for a further six days, resulting in 47 single clones that were producing recombinant antibodies. Variable region sequences of antigen specific antibodies were determined by analysis of samples submitted to Psomagen, MD, USA (Table S1; Fig. S2).
Sequences for the top five recombinant antibodies were sent to Abcam for production of purified recombinant antibodies and commercial distribution. The five top performing antibodies produced in the supernatant were captured by Protein A ligands, specifically binding to the constant region (Fc) of the IgG antibodies (CH2-CH3 region) as the final purification step. The three best clones 30H3K1 (Abcam, ab309155, 30H3/30K1), 19H1K1 (Abcam, ab314901, 19H1/19K1), and 41H1K3 (Abcam, ab314903, 41H1/41K3) for different applications as recommended in Table 3 are publicly available (https://www.michaeljfox.org/research-tools-catalog).
Dot blots using peptides
In peptide dot blots, phosphorylated and non-phosphorylated Ub 13-mers with central Ser65 respective PRKN 12mers (Ser65 in 6th position) were used in decrements of 5 (left to right) starting with 25 μM, 5 μM, 1 μM and 0.2 μM concentrations on 0.2 μm Nitrocellulose membrane (BioRad, 10484059). Ub peptides 1 and 2 had identical peptide sequences but different N- and C-terminal linker residues e.g., 6-Aminohexanoic Acid. The membrane was placed between 2 sheets of filter paper (Whatman, 1001917) and dried overnight. The next morning the membrane was rinsed in Tris-buffered saline (TBS; 50 mM Tris [Millipore, G48311], 150 mM NaCl [FisherScientific, BP358], pH 7.4) for ~10 seconds, blocked in 5% skim milk (Sysco, 5398953) in TBS with 0.1% Tween (TBS-T) for 30 minutes at room temperature. Primary antibodies were then added in a concentration of 1 μg/ml in 5% BSA (Boston BioProducts, P-753) in TBS-T and incubated for 2 hours at room temperature. Membranes were subsequently washed four times for 5 minutes in TBS-T before secondary antibody (Jackson ImmunoResearch, 711–035-152, 1:15,000) was added for 1 hour at room temperature in 5% skim milk. Membranes were washed five times for 10 minutes each in TBS-T and detected with Immobilon Western Chemiluminescent HRP Substrate (Millipore Sigma, WBKLS0500) on Pro Signal Blotting film (Genesee Scientific, 30–810L).
Generation of MEFs
The pink1 KO mice was obtained from Jackson Laboratories (RRID:IMSR_JAX:017946). Experiments were conducted in accordance with the Animal Scientific Procedures Act (1986) and with the Directive 2010/63/EU of the European Parliament and of the Council on the protection of animals used for scientific purposes (2010, no. 63). MEFs were generated using E13.5 embryos. Embryos were separated from placenta and individually placed into a single dish with cold DPBS, no calcium, no magnesium (Gibco, 14190094). After dissection of the head and all internal organs, embryo bodies were chopped with a spring scissor to mince the tissue of 1–2 mm in Petri dish. Small piece of tail was collected for genotyping. Tissues were digested for 15 min at 37 °C with digestion medium [0.025% Trypsin-EDTA (Gibco, 25300054); 0.125mg/mL DNase I (Merck, 11284932001) in HBSS (Gibco, 14025050)]. After enzymatic digestion, tissue dissociation was completed by pipette trituration and trypsin neutralised with 5 ml of culturing media [DMEM (Gibco, 11960–085); 20% heat-inactivated Fetal Bovine Serum (Gibco, 10500064); 1% Penicillin-Streptomycin (Gibco, 15140122), 1% L-Glutamine (Gibco, 25030024); 1X Non-essential Amino acid (Gibco, 11140–035), 1X Sodium pyruvate (Gibco, 11360–039)]. Then, dissociated tissues were centrifuged at 1200 rpm for 5 min and resuspend in 5 ml culturing media. Cell suspension was filtered using a 70 μm cell strainer in order to remove any undigested tissue pieces or aggregates from the cells. Cells were seeded in 10 cm dishes, containing 10 ml of pre-warmed culturing media, and cultured at 37°C in a humidified incubator with 5% CO2. Media was changed every 5 days and cells split at 90% confluence. A detailed protocol describing the preparation of mouse embryonic fibroblasts has been reported [32].
Cell Culture and treatment
WT and PINK1 KO HEK293E cells were previously described [13] and were cultured in Dulbecco’s modified Eagle medium (Thermo Fisher, 11965118) supplemented with 10% fetal bovine serum (Neuromics, FBS001800112). Primary human dermal fibroblasts were collected from a homozygous PINK1Q456X carrier and a healthy relative to serve as negative control [33]. All fibroblasts were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum, 1% PenStrep (Thermo Fisher, 15140122), and 1% non-essential amino acids (Thermo Fisher, 11140050). WT and pink1 KO MEF cells have been described previously [28]. MEFs were grown in Dulbecco’s modified Eagle medium supplemented with 20% fetal bovine serum, sodium pyruvate (Fisher Scientific, 11360–070), non-essential amino acids, and PenStrep. All cells were grown at 37°C, 5% CO2:air in humidified atmosphere. To induce mitophagy, HEK293E cells, MEFs, and human dermal fibroblasts were treated with 20 μM CCCP (Sigma-Aldrich, C2759), 5 μM valinomycin (Cayman Chemicals, 10009152), or 2 μM valinomycin, respectively, for 24 hours. DMSO (Sigma-Aldrich, D4540) was used as vehicle treatment.
Western blotting
HEK293E cells and MEFs were lysed in RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% NP-40 [United States Biological, 19628], 0.5% deoxycholate [Sigma-Aldrich, D6750], 0.1% SDS [Bio-Rad, 1610302]) containing protease inhibitor cocktail and phosphatase inhibitors (Sigma-Aldrich, 11697498001 and 04906837001). Cell lysates were then cleared for 15 minutes, 4°C at 20817 × g and protein concentrations were determined by BCA assay (Thermo Fisher, 23225). Cell lysates (10 μg for HEK293E cells or 15 μg for MEFs) were diluted in Laemmli buffer (62.5 mM Tris, pH 6.8, 1.5% SDS, 8.33% glycerol [Fisher Scientific, BP2291], 1.5% β-mercaptoethanol [Sigma-Aldrich, M3148], 0.005% bromophenol blue [Sigma-Aldrich, B5525]) and subjected to SDS-PAGE using 8–16% Tris-glycine gels (Invitrogen, EC60485BOX). Proteins were transferred onto polyvinylidene fluoride membranes (Millipore, Immobilon PSQ, ISEQ00010 or PVH00010) and the membranes were then blocked for 1 hour in 5% non-fat milk in TBS-T and incubated in primary antibodies overnight in 5% BSA in TBS-T at 4°C. 1 μg/ml of recombinant antibodies and 0.2 μg/ml of the reference antibody (Cell Signaling Technology, 62802) were used. GAPDH (Meridian Life Science, H86504M, 0.025 μg/ml) and vinculin/VCL (Sigma, V9131, 0.016 μg/ml) were used as loading controls. The following day, membranes were subjected to 1 hour incubation with secondary antibodies (Jackson ImmunoResearch, 711–035-152 and 715–035-150) at room temperature. Proteins were visualized using Immobilon Western Chemiluminescent HRP Substrate (Millipore Sigma, WBKLS0500) on Pro Signal Blotting film (Genesee Scientific, 30–810L) or on a Chemidoc MP imaging system (Bio-Rad, Hercules, CA, USA). Quantification of western blots was performed using Image Studio Lite software (Li-COR Biosciences, Version 5.2). Intensity levels per area were background subtracted and normalized to the loading controls.
Tissue collection and preparation of mouse and human brain lysates
A total of 12 WT and pink1 KO mice (N = 3 per sex/genotype) at age of three months were anesthetized and intracardially perfused with cold PBS (Invitrogen, 14190–235). Brains were removed and immediately flash-frozen in liquid nitrogen. All animal procedures were in accordance with the ethical standards established by Mayo Clinic and approved by the Institutional Animal Care and Use Committee at Mayo Clinic. Additionally, frozen human frontal cortex from ten AD and ten age- and sex-matched neurologically normal control cases were obtained from the Mayo Clinic Florida Brain Bank. All samples are from autopsies performed after approval by the legal next-of-kin. Research on de-identified postmortem brain tissue is considered exempt from human subjects’ regulations by the Mayo Clinic Institutional Review Board.
For protein extraction, frozen brain tissues were homogenized in 5x volumes of ice-cold RIPA buffer supplemented with protease and phosphatase inhibitors using glass Dounce tissue grinders (Fisher Scientific, K885300–0002). After 30 minutes incubation on ice, brain lysates were cleared by two times centrifugation at 20,000 × g, 4°C for 15 minutes. Resulting supernatant was collected as final lysates and protein concentrations were determined by BCA assay.
Sandwich MSD ELISA using recombinant protein, cell lysates, and tissue lysates
Similar to the p-S65-Ub sandwich MSD ELISA that has been described previously [13], the top five recombinant p-S65-Ub antibodies or the reference antibody (Cell Signaling Technology, 62802, E2J6T) were used as capturing agents. Detection was performed with a mouse Ub antibody (ThermoFisher, 14–6078-37, P4D1) followed by a Sulfo-tag labeled anti-mouse antibody (MSD, R32AC-1). All capturing antibodies were used at a concentration of 1 μg/ml in 200 mM sodium carbonate buffer pH 9.7 and coated overnight at 4°C with 30 μl per well in 96-well MSD plate (MSD, L15XA-3). The next morning MSD plates were washed two times with TBS-T and incubated with 300 μl filtered blocking buffer (150 mM Tris, pH 7.4, 150 mM NaCl, 0.1% Tween-20, 1% BSA) for 1 hour at 22°C. Antigen incubation was performed for 2 hours at room temperature at 600 rpm on a microplate mixer (USA Scientific, 8182–2019). After washing, 30 μl of detecting mouse Ub antibody (ThermoFisher, 14–6078-37, P4D1, 5 μg/ml) were added to each well and incubated again for 2 hours. 50 μl Sulfo-tag labeled goat anti-mouse antibody (MSD, R32AC-1, 1:500) was next added and incubated for 1 hour. The plates were then read on a MESO QuickPlex SQ 120 reader (Meso Scale Diagnostics, Rockville, MD, USA) after addition of 150 μl MSD GOLD Read Buffer (MSD, R92TG-2).
For detection of recombinant proteins, K48 (Boston Biochem, UC-250) and K63 (Boston Biochem, UC-350) p-S65-Ub tetramers were serially diluted to a final concentration of 10 fg/ml to 1 μg/ml. Their nonphosphorylated counterparts (Boston Biochem, UC-210b and UC-310b) were used as negative controls. For detection in HEK293E cell lysates, 10 μg of protein was used with CCCP-treated PINK1 KO cells serving as negative control and CCCP-treated WT cells serving as positive control. For detection in mouse and human brain lysates, 30 μg of protein was used. All samples were run in duplicates and diluted in blocking buffer.
The LoB, LoD, and LoQ were calculated using MSD-ECL values from recombinant p-S65-Ub chains and according to Armbruster and Pry [16]. In brief, the LoBs were determined using the formula LoB = mean blank + 1.645(SD blank) where blocking buffer served as blank. The LoD was determined using serial dilutions of p-S65-Ub analytes and calculated with the following formula: LoD = LoB + 1.645(SD low concentration sample). The LoQ was determined as the lowest concentration of analyte that could be measured with a predefined goal for imprecision (here under 10%) or is clearly distinguishable from the LoB with more than 3 times the standard deviation.
Direct MSD ELISA using recombinant protein
Recombinant p-S65-PRKN (Boston Biochem, E3–166), non-phosphorylated PRKN (Boston Biochem, E3–160) and p-S65-Ub monomer (Boston Biochem, U102) were initially dissolved in RIPA buffer (stock concentration of 1 μg/μl), aliquoted in 1 μl aliquots, flash-frozen in liquid nitrogen and stored at −80°C. Recombinant PRKN and p-S65-PRKN aliquots were serially (10-fold) diluted in 200 mM sodium carbonate buffer pH 9.7 with a starting concentration of 1 μg/ml towards 10 fg/ml and used to coat the MSD plates overnight at 4°C without shaking with 50 μl/well. Recombinant p-S65-Ub monomer (positive control) was used in equimolar concentration relative to the highest p-S65-PRKN concentration of 1 μg/ml. On the next morning, plates were washed twice with TBS-T and incubated for 1 hour with blocking buffer without shaking at 22°C. Recombinant p-S65-Ub antibodies and the reference antibody in blocking buffer were subsequently incubated at a concentration of 1 μg/ml with 30 μl/well for 2 hours at room temperature at 600 rpm on a microplate mixer. 50 μl Sulfo-tag labeled goat anti-rabbit Ab (MSD, R32AB-1, 1:500) was next added and incubated for 1 hour. The plates were then read on a MESO QuickPlex SQ 120 reader after addition of 150 μl MSD GOLD Read Buffer.
Immunofluorescence staining of fibroblasts followed by high content imaging
To evaluate top clones for immunocytochemistry and quantify p-S65-Ub levels using automated high-content imaging, WT and homozygous PINK1Q456X mutant fibroblasts were seeded in 96-well imaging plates (Fisher Scientific, 08772225) and allowed to attach overnight. Cells were treated for 2 μM valinomycin (Enzo Life Science, BML-KC140–0025) or vehicle DMSO (Sigma-Aldrich, D4540) for 24 hours and then fixed in 4% paraformaldehyde (Sigma-Aldrich, 441244) after one wash with PBS. Fibroblasts were immunostained with all clones or reference antibody against p-S65-Ub (Cell Signaling Technology, 62802, E2J6T) at 1 μg/ml and with HSP60 (arigo Biolaboratories, ARG10757, 1:2,000) followed by incubation with secondary antibody (Invitrogen, A-11034 and A-11041; 1:1,000) and Hoechst 33342 (Invitrogen, H21492; 1:5,000). Plates were imaged using an Operetta CLS High Content imaging system (Perkin Elmer, Waltham, MA, USA) with a 20x water immersion objective using a 2×2 montage (no gaps) per well. Raw images were processed using the built-in Harmony analysis software to calculate the average cytoplasmic p-S65-Ub intensity per cell in each well.
Processing of fixed mouse and human tissue and immunohistochemistry
Without doxycycline administration, rTg4510 mice used here constitutively express high levels of human P301L tau in the hippocampus. Brain tissue from one nonTg and one rTg4510 transgenic mice at 9 months of age were used for immunohistochemistry. All procedures were in accordance with the ethical standards established by Mayo Clinic and approved by the Institutional Animal Care and Use Committee at Mayo Clinic. Additionally, autopsy brain from a single AD case was retrieved for immunohistochemistry from the Mayo Clinic Florida Brain Bank.
Paraffin embedded hippocampal tissue was cut into 5 μm consecutive sections and allowed to dry overnight at 60°C. Slides were deparaffinized and rehydrated, followed by antigen retrieval in steaming deionized water for 30 minutes. After blocking with 0.03% hydrogen peroxide (Sigma, 216763) and 5% normal goat serum (Invitrogen, 16210072), sections were incubated with all five clones, reference p-S65-Ub antibody (Cell Signaling Technology, 62802, E2J6T), or in-house p-S65-Ub antibody at 1.56 μg/ml (in mouse brain), 0.78 μg/ml (in human brain), or 0.125 μg/ml (only for clone 41H1K3 in human brain), followed by rabbit-labeled polymer HRP (Agilent, K4011) at room temperature. Peroxidase labeling was visualized with the chromogen solution 3,3’-diaminobenzidine (Agilent, K4011). The sections were then counterstained with Lerner 1 hematoxylin (Fisher Scientific, CS400–1D) and coverslipped with Cytoseal mounting medium (Thermo Scientific, 8310). After immunohistochemical staining, all sections were imaged with an Aperio AT2 digital pathology scanner at 40x magnification (Leica Biosystems, Wetzlar, Germany).
Statistical analysis
Data analysis was performed in GraphPad Prism version 9 software. One-way ANOVA followed by Tukey’s multiple comparison test was used for the HEK293E cell ELISA analysis. Unpaired t-test and Mann-Whitney U test were used for mouse and human brain ELISA analysis, respectively.
Supplementary Material
ACKNOWLEDGEMENTS
We thank The Michael J. Fox Foundation for Parkinson’s Research for their guidance and leadership in this collaborative effort. We are also grateful to the patients and their families who made this study possible. We thank the Center for Regenerative Medicine and the Neuroregeneration Laboratory for biobanking of patient cells at Mayo Clinic Florida.
FUNDING
This study was supported through grants from the Michael J. Fox Foundation for Parkinson’s Research (017399 to W.S., 017367 to J.B.F., 017367.01 to K.P., and 022155 to R.K.). Work in the authors labs is further funded by the National Institute of Neurological Disorders and Stroke (NINDS: RF1 NS085070, R01 NS110085, and U54 NS110435 to W.S.), the National Institute on Aging (NIA: R56 AG062556 to W.S.), the Department of Defense Congressionally Directed Medical Research Programs (CDMRP) (W81XWH-17–1-0248 to W.S.), the Florida Department of Health - Ed and Ethel Moore Alzheimer’s Disease Research Program (9AZ10 to W.S. and 22A07 to F.C.F.), the Ted Nash Long Life Foundation (W.S.) and The Michael J. Fox Foundation for Parkinson’s Research (019046, 019258, and 021186 to W.S. and 021182 to F.C.F.). W.S. is also supported by Mayo Clinic Foundation, Mayo Clinic Center for Biomedical Discovery (CBD), and Mayo Clinic Robert and Arlene Kogod Center on Aging. F.C.F. is further supported by Mayo Clinic Center for Biomedical Discovery (CBD), a Gerstner Family Career Development Award from the Mayo Clinic Center for Individualized Medicine (CIM), and the Mayo Clinic Younkin Scholar Program. X.H. is supported by Mayo Clinic Alzheimer Disease Research Center (ADRC, P30 AG062677) pilot and developmental project grants and research fellowships from the American Parkinson Disease Association (APDA) and the Alzheimer’s Association (AARF-22–973152). Mayo Clinic Florida is an American Parkinson Disease Association (APDA) Center for Advanced Research (W.S., F.C.F., Z.K.W., and D.W.D.). Z.K.W. is partially supported by the National Institute of Aging (U19 AG063911), Mayo Clinic Center for Regenerative Medicine (CRM), gifts from the Donald G. and Jodi P. Heeringa Family, the Haworth Family Professorship in Neurodegenerative Diseases fund, the Albertson Parkinson’s Research Foundation, and the PPND Family Foundation.
ABBREVIATIONS
- AD
Alzheimer disease
- CCCP
carbonyl cyanide 3-chlorophenylhydrazone
- ELISA
enzyme-linked immunosorbent assay
- HEK293E
cell human embryonic kidney E cell
- KO
knockout
- LoB
Limit of Blank
- LoD
Limit of Detection
- LoQ
Limit of Quantification
- MEF
mouse embryonic fibroblast
- nonTg
non-transgenic
- PBMC
peripheral blood mononuclear cell
- PD
Parkinson disease
- p-S65-PRKN
phosphorylated PRKN at serine 65
- p-S65-Ub
phosphorylated Ub at serine 65
- Ub
Ubiquitin
- VCL
vinculin
- WT
wild-type
Footnotes
DISCLOSURE STATEMENT
Mayo Clinic, F.C.F., and W.S. have filed a patent related to PRKN activators. Additional funding sources to disclose but not pertinent to the current study include a grant from Amazentis SA (to W.S.). Z.K.W. serves as PI or Co-PI on projects/grants from Biohaven Pharmaceuticals, Inc. and Vigil Neuroscience, Inc., and is an external advisory board member for Vigil Neuroscience, Inc., and a consultant on neurodegenerative medical research for Eli Lilly & Company. J.B.F. was CEO at 21st Century Biochemicals Inc., K.P. is an employee of ImmunoPrecise Antibodies Ltd., and R.K. is an employee of Abcam Plc.. All other authors declare they have no competing interests. This research was conducted in compliance with Mayo Clinic conflict of interest policies.
REFERENCES
- 1.Tanaka K. The PINK1-Parkin axis: An Overview. Neurosci Res. 2020. Oct;159:9–15. [DOI] [PubMed] [Google Scholar]
- 2.Hasson SA, Kane LA, Yamano K, et al. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature. 2013. Dec 12;504(7479):291–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lazarou M, Jin SM, Kane LA, et al. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012. Feb 14;22(2):320–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kondapalli C, Kazlauskaite A, Zhang N, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012. May;2(5):120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shiba-Fukushima K, Imai Y, Yoshida S, et al. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep. 2012;2:1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ordureau A, Sarraf SA, Duda DM, et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell. 2014. Nov 6;56(3):360–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wauer T, Simicek M, Schubert A, et al. Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature. 2015. Aug 20;524(7565):370–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng X, Hunter T. Pink1, the first ubiquitin kinase. EMBO J. 2014. Aug 1;33(15):1621–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012. Jul;393(7):547–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amorim JA, Coppotelli G, Rolo AP, et al. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat Rev Endocrinol. 2022. Apr;18(4):243–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hou X, Fiesel FC, Truban D, et al. Age- and disease-dependent increase of the mitophagy marker phospho-ubiquitin in normal aging and Lewy body disease. Autophagy. 2018;14(8):1404–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watzlawik JO, Fiesel FC, Fiorino G, et al. Basal activity of PINK1 and PRKN in cell models and rodent brain. Autophagy. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watzlawik JO, Hou X, Fricova D, et al. Sensitive ELISA-based detection method for the mitophagy marker p-S65-Ub in human cells, autopsy brain, and blood samples. Autophagy. 2021. Sep;17(9):2613–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fiesel FC, Ando M, Hudec R, et al. (Patho-)physiological relevance of PINK1-dependent ubiquitin phosphorylation. EMBO Rep. 2015. Sep;16(9):1114–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hou X, Watzlawik JO, Cook C, et al. Mitophagy alterations in Alzheimer’s disease are associated with granulovacuolar degeneration and early tau pathology. Alzheimers Dement. 2020. Oct 8;17(3):417–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Armbruster DA, Pry T. Limit of blank, limit of detection and limit of quantitation. Clin Biochem Rev. 2008. Aug;29 Suppl 1(Suppl 1):S49–52. [PMC free article] [PubMed] [Google Scholar]
- 17.Kane LA, Lazarou M, Fogel AI, et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014. Apr 28;205(2):143–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kazlauskaite A, Kondapalli C, Gourlay R, et al. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J. 2014. May 15;460(1):127–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koyano F, Okatsu K, Kosako H, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014. Jun 5;510(7503):162–6. [DOI] [PubMed] [Google Scholar]
- 20.Fiesel FC, Springer W. Disease relevance of phosphorylated ubiquitin (p-S65-Ub). Autophagy. 2015. Nov 2;11(11):2125–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shiba-Fukushima K, Ishikawa KI, Inoshita T, et al. Evidence that phosphorylated ubiquitin signaling is involved in the etiology of Parkinson’s disease. Hum Mol Genet. 2017. Aug 15;26(16):3172–3185. [DOI] [PubMed] [Google Scholar]
- 22.Potting C, Crochemore C, Moretti F, et al. Genome-wide CRISPR screen for PARKIN regulators reveals transcriptional repression as a determinant of mitophagy. Proc Natl Acad Sci U S A. 2018. Jan 9;115(2):E180–E189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soutar MPM, Melandri D, O’Callaghan B, et al. Regulation of mitophagy by the NSL complex underlies genetic risk for Parkinson’s disease at 16q11.2 and MAPT H1 loci. Brain. 2022. Dec 19;145(12):4349–4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hou X, Heckman MG, Fiesel FC, et al. Genome-wide association study identifies APOE and ZMIZ1 variants as mitophagy modifiers in Lewy body disease. medRxiv. 2023. Oct 16. [Google Scholar]
- 25.Chin RM, Rakhit R, Ditsworth D, et al. Pharmacological PINK1 activation ameliorates Pathology in Parkinson’s Disease models. bioRxiv. 2023. Feb 15. [Google Scholar]
- 26.Hou X, Chen TH, Koga S, et al. Alpha-synuclein-associated changes in PINK1-PRKN-mediated mitophagy are disease context dependent. Brain Pathol. 2023. Sep;33(5):e13175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swatek KN, Usher JL, Kueck AF, et al. Insights into ubiquitin chain architecture using Ub-clipping. Nature. 2019. Aug;572(7770):533–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antico O, Ordureau A, Stevens M, et al. Global ubiquitylation analysis of mitochondria in primary neurons identifies endogenous Parkin targets following activation of PINK1. Sci Adv. 2021. Nov 12;7(46):eabj0722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ordureau A, Paulo JA, Zhang W, et al. Dynamics of PARKIN-Dependent Mitochondrial Ubiquitylation in Induced Neurons and Model Systems Revealed by Digital Snapshot Proteomics. Mol Cell. 2018. Apr 19;70(2):211–227 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deol KK, Strieter ER. The ubiquitin proteoform problem. Curr Opin Chem Biol. 2021. Aug;63:95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trimmer JS. Recombinant Antibodies in Basic Neuroscience Research. Curr Protoc Neurosci. 2020. Dec;94(1):e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Antico O., Ordureau A., Stevens M., et al. Reconstitution of Parkin ubiquitin ligase activity using mouse and human mitochondria. protocols io. 2021. 10.17504/protocols.io.bxmypk7w [DOI] [Google Scholar]
- 33.Siuda J, Jasinska-Myga B, Boczarska-Jedynak M, et al. Early-onset Parkinson’s disease due to PINK1 p.Q456X mutation--clinical and functional study. Parkinsonism Relat Disord. 2014. Nov;20(11):1274–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.