

Abstract
Previously we demonstrated frequent homologous crossovers among molecules of the RNA3 segment in the tripartite brome mosaic bromovirus (BMV) RNA genome (A. Bruyere, M. Wantroba, S. Flasinski, A. Dzianott, and J. J. Bujarski, J. Virol. 74:4214-4219, 2000). To further our knowledge about mechanisms of viral RNA genome variability, in this paper we have studied homologous recombination in BMV RNA1 and RNA2 components during infection. We have found that basal RNA-RNA crossovers could occur within coding regions of both RNAs, although recombination frequencies slightly varied at different RNA sections. In all cases, the frequencies were much lower than the rate observed for the intercistronic recombination hot spot in BMV RNA3. Probability calculations accounted for at least one homologous crossover per RNA molecule per replication cycle. In addition, we have demonstrated an efficient repair of mutations within the conserved 3′ and 5′ noncoding regions, most likely due to error-prone BMV RNA replication. Overall, our data verify that homologous crossovers are common events a during virus life cycle, and we discuss their importance for viral RNA genetics.
Enormous genetic variability is one of the unusual features of RNA viruses. Numerous experiments reveal two main sources of genetic polymorphism that contribute to the rapid evolution of RNA viruses: error-prone replication and RNA recombination (25). The former introduces into the viral RNA genome a wide spectrum of point mutations at the rate of 10−4 to 10−5 per nucleotide per replication cycle (33, 34). The latter is a widespread phenomenon described in many groups of RNA viruses, including picornaviruses (3, 16, 20), coronaviruses (5, 41), and alphaviruses (15) and in plant viruses: plum pox potyvirus (9), bromoviruses (4, 7, 8, 13), alfalfa mosaic virus (36), cucumber mosaic virus (CMV) (1, 10), tobacco mosaic virus (2), turnip crinkle virus (29, 30), and tomato bushy stunt tombusvirus (38). RNA recombination is also seen in bacteriophage Qβ (31), in negative RNA viruses (11), in double-stranded RNA viruses (35), and in retroviruses (17, 24) as well as during the formation of defective interfering RNAs (38).
In spite of intensive studies, the mechanism of RNA recombination is not well understood. The copy choice mechanism, which is the most widely accepted (21), assumes that RNA recombinants result from template switching by viral RNA polymerase (RdRp) during RNA replication. Depending on the primary structure of the recombining molecules and on the location of junction sites, two types of recombination events have been recognized: homologous and nonhomologous (21), with the former being 10-fold higher than the latter in the case of brome mosaic bromovirus (BMV) (26).
There is little information about homologous recombination in natural virus populations, because recombination products do not differ from parental RNAs. The crossovers in poliovirus RNA tended to occur within potential inter- and intramolecular heteroduplex regions (3, 20), whereas in mouse hepatitis coronavirus the crossovers were found at apparent hot spots. However, the hot spots in mouse hepatitis virus appeared to result from selection pressure rather than from molecular constraints (21, 37, 42).
The relationship between RNA recombination and RNA replication has been studied extensively in the case of BMV (27, 28). The BMV RNA genome consists of three messenger sense RNA segments, RNA1, RNA2 and RNA3, each carrying the conservative 200-nucleotide (nt) 3′ region. RNA1 codes for 1a protein which carries both the helicase and methyltransferase domains, RNA2 codes for 2a protein, the RNA polymerase component, and RNA3 codes for two proteins: 3a (movement protein) and coat protein. The two genes in RNA3 are separated by an intercistronic region that carries the subgenomic RNA4 promoter (sgp). It has been demonstrated that the frequency of homologous crossovers within the intercistronic region of BMV RNA3 reaches 71% (6, 39) and that they map to the polyU tract of the sgp on minus strands (40). It has been speculated that the polyU tract promotes replicase detachment (28), while the adjacent sgp core acts as the reinitiation site of RNA synthesis (40).
To expand our knowledge on recombination events over all three segments of the BMV RNA genome, in this work we have studied homologous crossovers among RNA1 or RNA2 molecules. We have observed the accumulation of RNA1-RNA1 and RNA2-RNA2 recombinants. The crossovers occurred at a basal frequency that was much lower than the rate within the sgp sequence in RNA3. In addition, we have demonstrated an efficient repair of mutations within the conserved 3′ and 5′ regions in BMV RNA1 and RNA2 molecules which most likely reflects the error-prone RNA replication process. We have concluded that homologous crossovers are common events during the BMV life cycle and endow the virus with means of both variability and stability.
MATERIALS AND METHODS
Materials.
Plasmids pB1TP3, pB2TP5, and pB3TP7 (19) were used as templates to synthesize in vitro the infectious capped full-length transcripts of wild-type (wt) BMV RNA1, RNA2, and RNA3, respectively, using the MEGAscript T7 kit (Ambion, Austin, Tex.). Plasmids PM1-RNA2, pM2-RNA2, pM34-RNA2, pM5-RNA1, pM6-RNA2, pM1345-RNA2, pO1-RNA1, pO2-RNA1, pO3-RNA1, pO4-RNA1, pO5-RNA1, pO6-RNA1, pO7-RNA1, and pO8-RNA1 (see below) were used to synthesize mutant RNA1 and RNA2 transcripts. The Moloney murine leukemia virus reverse transcriptase and Taq DNA polymerase were purchased from MBI Fermentas. Restriction enzymes, Pfu DNA polymerase, and dNTPs were purchased from Promega Corporation, whereas the QIAGEN PCR cloning kit was from QIAGEN GmbH.
Generation of M-RNA2-BMV and O-RNA1-BMV mutants.
To generate the series of M-RNA2-BMV and O-RNA1-BMV viral mutants, plasmids pB2TP5 and pB1TP3, respectively, were subjected to site-specific mutagenesis by means of replacing unmutated portions of the DNA sequences of interest with the altered fragments that had been generated by PCR using the mutating primers (Table 1). In the case of the pM-RNA2 constructs, PCRs were with Taq DNA polymerase, and the DNA fragment was ligated into the pB2TP5 plasmid. For pO-RNA1 constructs, PCR amplifications were done on pB1TP3 by using Pfu DNA polymerase and the mutagenizing primers (Table 1). The introduced marker restriction sites did not change the amino acid coding sequences.
TABLE 1.
Sequences of the primers used
| Primer or primer pair | Sequence from 5′ to 3′ enda |
|---|---|
| R2A | CATGCCTGCAGGTCGAC |
| R2B | TTTTCGAAGACATCTTGGGGATCCTAGAAAG |
| R2G | GGAGGCCTAACGTCAGTTGATGC |
| R2H | CACCCGCATTCTAAGACTTGCTATGCAAGCCCATGCAGGATCCCCTAAGG |
| AN1a | GGTTCAATCCCTTTTCGAACACGGTTCTGCTAC |
| AN1b | GTAGCAGAACCGTGTTCGAAAAGGGATTGAACC |
| AN7 | GGAGAGCCCTGTTCGAAGTAGGAACGTTGTGG |
| AN8 | CCACAACGTTCCTACTTCGAACAGGGCTCTCC |
| 139 | GTAAACCACGGAACGAGGTTC |
| R2B2 | GAACCATTTGTTGGACGGTGTCGCAAATGGATCC |
| R2C | TTCCATGGACTTTGATAGGATCCTAGAAAG |
| R2D | CTGGTACCGACATAAATCAGTC |
| R2E | TGACGCGTTTACATATTTCGGTAATACTCTGGTCACCATGGC |
| R2F | GGGAGCTCCTTAGTACTACAG |
| R2G2 | GGAGGCCTAACGTCAGTTGATGCATTG |
| 1st | CAGTGAATTCTGGTCTTTTTAGAGATTTACAG |
| ANB/ANC | GAAAATCAACGTTCGAAATAAGCTCTCTATTG CAATAGAGAGCTTATTTCGAACGTTGATTTTC |
| R1A/R1B | GTGCCGCATGCGAAAGATCTTGCAAGAAAGCGATG CATCGCTTTCTTGCAAGATCTTTCGCATGCGGCAC |
| AN3 | CACTGTGTATGGTTCGAAGACATATCTAAG |
| AN4 | CTTAGATATGTCTTCGAACCATACACAGTG |
| AN5 | CCGTATGGGAGAAGATCTAATTGTGACGGCG |
| AN6 | CGCCGTCACAATTAGATCTTCTCCCATACGG |
| R1E | GTTTCTGACTCGTCTTTCGAACGTTGAAGAATTTG |
| R1F | CAAATTCTTCAACGTTCGAAAGACGAGTCAGAAAC |
| R1G | CGGCGAGCTCGTCGGAGATCTGATCTTTAATTGTG |
| R1H | CACAATTAAAGATCAGATCTCCAGCGAGCTCGCCG |
| 119 | CGGTCACTATTGAAGAGC |
| AN1.3 | GTAGTACGCGTACTGCATGC |
| AN4.1 | CAAACGTGATATCGGTACAG |
| AN5.1 | CTGTACCGATATCACGTTTG |
| seq3.2 | GGACTCAGGGCTCAACTC |
Marker restriction sites are underlined and nucleotide substitutions are in bold type. Pairs of primers used in PCR with Pfu DNA polymerase have both sequences on one line.
Each of the two of BMV RNA1 and RNA2 mutants carried the marker restriction sites within either 5′ or 3′ noncoding regions. pM1-RNA2 had a BamHI site at position 93 (5′ noncoding region), created by three substitutions (93 C→G, 94 T→G, and 98 A→C). This was accomplished by amplification of the pB2TP5 fragment between nucleotides 6014 (i.e., the position within the vector part, just upstream of the 5′ end of the RNA2 insert) and 116, using the pair of primers R2A and R2B (Table 1). Cloning was between unique BstBI and PstI restriction sites. pM5-RNA2 carried the BamHI marker restriction site at position 2780 (3′ noncoding region) created by nucleotide substitutions at positions 2782 (T→A) and 2785 (G→C). Here, the PCR product was amplified between RNA2 nucleotides 2498 and 2822 using primers R2G and R2H, and the cloning was between unique StuI and BsaMI restriction sites. pO1-RNA1 carried the BstBI marker site at position 30 (5′ noncoding region) due to a double substitution at nucleotides 30 (G→T) and 35 (C→A) via PCR with primers AN1a and AN1b (complementary to nucleotide positions 17 to 49 in pB1TP3). pO7-RNA1 carried a BstBI marker site at nt 3004 (3′ noncoding region) due to double substitutions at positions 3007 (C→G) and 3009 (G→A). They were introduced by PCR amplification with primers AN7 and AN8, complementary to nt 2991 to 3024 in pB1TP3.
Ten mutant constructs carried the marker sites within coding regions. pM2-RNA2, which carried a BamHI marker site at position 326 of BMV RNA2, was created by two substitutions (328 C→A, 331 T→C) by PCR from pB2TP5 with primers R2A and R2B2, and the fragment was subcloned between nucleotides 6014 (PstI restriction site) and 360 (PflMI site). pM34-RNA2, carrying a BamHI marker at position 897 and a BstEII marker at position 1708 (coding region of RNA2), was generated as follows. To generate a BamHI site, the T at position 902 (902-T) was mutated into C by amplification between nucleotides 880 and 1255 with primers R2C and R2D and ligation between NcoI and KpnI restriction sites. To generate BstEII, the substitutions (1708 T→G, 1714 T→C) were generated by PCR between nucleotides 1678 and 2379 with primers R2E and R2F followed by subcloning into pB2TP5 between the MluI and SacI restriction sites. The third RNA2 mutant, pM6-RNA2, carried the NsiI marker at position 2517 that was created due to a single substitution (2521 T→A) in a PCR product amplified between nucleotides 2498 and 2865 with primers R2G2 and 1st and religation between the StuI and BsaMI sites. Finally, the quadruple mutant pM1345-RNA2 was constructed similarly as mutants pM1-, pM34-, and pM5-RNA2.
Among six RNA1 mutants, the BstB1 marker site in pO2-RNA1 was introduced due to a 221-C→A substitution with primers ANB and ANC. pO3-RNA1 carried a BglII marker that was generated by 512-A→G and 514-T→C substitutions with primers R1A and R1B covering nucleotides 498 to 531 on pB1TP3. pO4-RNA1 was created with primers AN3 and AN4 (positions 978 to 1007 on pB1TP3), which substituted 992-T→C, giving a BstBI site. pO5-RNA1 carried a BglII site that was created by double substitution (2183-G→A and 2185-C→T) with primers AN5 and AN6 that hybridized between nucleotides 2171 to 2201 on pB1TP3. pO6-RNA1 carried a double substitution (1472-C→T and 1475-A→G) that generated a BstBI site with primers R1E and R1F that hybridized between nucleotides 1457 to 1491 on pB1TP3. Finally, a pO8-RNA1 BglII site was created by double substitution (2933-T→A and 2937-T→C) with primers R1G and R1H that hybridized between nucleotides 2918 to 2952 on pB1TP3.
The resulting mutant plasmids were propagated in DH5α Escherichia coli cells, and the presence of the marker sites was confirmed by sequencing and restriction digestion.
Determination of infectivity and stability.
Viral RNAs were synthesized by in vitro transcription with T7 RNA polymerase from linearized mutant plasmids, and 1 μg per leaf of each of the BMV RNA transcripts was coinoculated on Chenopodium quinoa or on barley (Hordeum sativum) leaves as described previously (19). After 10 days postinoculation, the local lesions that developed on C. quinoa leaves were counted to confirm the infectivity of the viral mutants, and the total RNA was extracted from the infected tissue (7). The BMV RNA was converted into the single-stranded DNA counterparts by using the Moloney murine leukemia virus reverse transcriptase and primer 1st. The resulting single-stranded DNA product was then amplified by PCR using Taq DNA polymerase and different pairs of primers, depending on the position of the particular mutation (Table 1). Specifically, for M1- and M2-RNA2, primers 139 and R2D were used. For M34-RNA2, primers 139 and R2F were used, while for M5- and M6-RNA2, primers 1st and 119 (complementary to nucleotides 1404 to 1387) were used. For O1- and O3-RNA1, primers R2A and AN1.3 (complementary to nucleotides 659 to 679) were used. For O4-RNA1, primers R2A and AN4.1 were used. For O7- and O8-RNA1, primers AN5.1 and 1st were used. For O6-RNA1, primers AN3 and sek3.2 (complementary to nucleotides 1679 to 1661) were used.
The resulting cDNA products were cloned into the pDRIVE vector (QIAGEN PCR cloning kit) as specified by the manufacturer, subjected to restriction analysis, and sequenced to confirm the presence of marker mutations.
In vivo recombination assays.
Barley and C. quinoa plants were inoculated with equal amounts (5 μg) of pairs of previously prepared M-RNA2-BMV or O-RNA1-BMV mutants (22). After 10 days postinoculation, leaf tissues were collected and the progeny BMV RNAs were extracted. The following mutant pairs were used for inoculations on C. quinoa or barley hosts: M2-/M34-RNA2-BMV, M34-/M6-RNA2-BMV, O3-/O6-RNA1-BMV, and O6-/O8-RNA1-BMV. The inoculated plants were maintained in a greenhouse for 10 days, and then the total RNA was extracted. In the case of C. quinoa, total RNA extracts were obtained either from separate local lesions or from whole leaves.
Cloning and analysis of recombinants.
The BMV RNA2- or RNA1-specific sequences were amplified by reverse transcription-PCR (RT-PCR) as described above, and the products were cloned and sequenced in order to determine the recombination frequency. The recombination frequency was defined as the fraction of recombined clones in a total number of cDNA clones. For M2-/M34-RNA2-BMV, the region of RNA2 between marker mutations was amplified with primers 139 and R2F, while for the pair M34-/M6-RNA2-BMV, with primers 119 and 1st. For the pair O3-/O6-RNA1-BMV, the region of RNA1 between marker mutations was amplified with primers ANB and sek3.2, while for the pair O6-/O8-RNA1-BMV, with primers AN3 and 1st. The PCR products were ligated into the pDRIVE vector, cloned, and analyzed as described above.
The control RT-PCR was performed on separately inoculated barley plants with either M34-RNA2-BMV or M6-RNA2-BMV. After 10 days postinoculation, total RNA was extracted separately, the resulting RNA preparations were mixed together, and the RT-PCR amplifications were performed with primers 119 and 1st. The resulting cDNA products were cloned and analyzed as described above.
RESULTS
Generation and infectivity of M-RNA2-BMV and O-RNA1-BMV mutants.
Marker mutations were introduced at both coding and noncoding regions of BMV RNAs 2 and 1. Figure 1 and Table 2 show the locations of sequence modifications present in six variants of RNA2 (M1-, M2-, M34-, M5-, M6-, and M1345-RNA2) and eight variants of RNA1 (O1-, O2-, O3-, O4-, O5-, O6-, O7-, and O8-RNA1). The mutations in the coding regions were created without changing the encoded amino acid sequences. The infectivity of the modified BMV RNAs was determined on C. quinoa (a local lesion host) and barley (systemic host) seedlings. All mutants except for O2-RNA1-BMV and O5-RNA1-BMV were infectious to both hosts, and the times necessary for symptom development were similar among mutants and for the wt virus (Table 2). Thus, the introduced mutations did not noticeably change the in vivo characteristics of the generated mutants.
FIG. 1.

Location of marker mutations in the M-RNA2- (A) and O-RNA1-BMV (B) mutants. White boxes represent noncoding regions and grey boxes represent open reading frames for 2a or 1a proteins. The sites of the newly created restriction markers are marked by arrows, and the detailed nucleotide substitutions (highlighted with bold capital letters) and their nucleotide positions (counting from the 5′ end of wt RNA3) are shown on the right hand side.
TABLE 2.
Characterization of the infectivity and stability of M- RNA2-BMV and O- RNA1-BMV mutants, as determined for the C. quinoa hosta
| BMV mutant | Location of mutation | Marker(s)c | No. of local lesions | Days until symptom developmentd | Stability or infectivity of mutationse |
|---|---|---|---|---|---|
| wt | n/ab | n/a | 36 | 9 | n/a |
| M1- RNA2-BMV | RNA2 5′ UTR | M1-93 BamHI | 33 | 9 | Unstable |
| M2- RNA2-BMV | RNA2 ORF | M2-326 BamHI | 27 | 10 | Stable |
| M34- RNA2-BMV | RNA2 ORF | M3-897 BamHI | |||
| M4-1708 BstEII | 31 | 9 | Stable | ||
| M5- RNA2-BMV | RNA2 3′ UTR | M5-2780 BamHI | 37 | 9 | Unstable |
| M6- RNA2-BMV | RNA2 ORF | M6-2517 NsiI | 22 | 11 | Stable |
| M1345-RNA2-BMV | RNA2 5′ UTR, ORF, 3′ UTR | M1-93 BamHI | M1-unstable | ||
| M3-897 BamHI | 29 | 10 | M3-stable | ||
| M4-1708 BstEII | M4-stable | ||||
| M5-2780 BamHI | M5-unstable | ||||
| O1- RNA1-BMV | RNA1 5′ UTR | O1-30 BstBI | 35 | 9 | Unstable |
| O2- RNA1-BMV | RNA1 ORF | O2-217 BstBI | 0 | 10 | Not infectious |
| O3- RNA1-BMV | RNA1 ORF | O3-511 BglII | 28 | 10 | Stable |
| O4- RNA1-BMV | RNA1 ORF | O4-990 BstBI | 25 | 9 | Stable |
| O5- RNA1-BMV | RNA1 ORF | O5-2183 BglII | 0 | 10 | Not infectious |
| O6- RNA1-BMV | RNA1 ORF | O6-1472 BstBI | 27 | 10 | Stable |
| O7- RNA1-BMV | RNA1 3′ UTR | O7-3004 BstBI | 38 | 9 | Unstable |
| O8- RNA1-BMV | RNA1 ORF | O8-2933 BglII | 24 | 10 | Stable |
Since BMV carries the tripartite RNA genome, the host plants were coinoculated with equal amounts of the mutant RNA segment plus the remaining two wt BMV RNAs, all synthesized by in vitro transcription. See Materials and Methods for further details.
n/a, not applicable.
Newly created restriction site.
The numerals show the number of days postinoculation necessary for symptom appearance.
The stability of marker mutations was determined by restriction analysis of 24 cDNA clones and sequencing of 3 to 5 cDNA clones obtained from total RNAs combined from 3 to 4 C. quinoa leaves per mutation.
Stability of M-RNA2-BMV and O-RNA1-BMV mutants.
In order to analyze the stability of the introduced mutations, progeny BMV RNA was extracted from infected C. quinoa or barley leaves, and the RNA2 and RNA1 sequences were amplified by RT-PCR and cloned. A set of approximately 24 clones for every mutant was analyzed by restriction digestion at the marker restriction site, and a few clones were sequenced. As summarized in Table 2, marker mutations in three BMV RNA2 variants (M2-, M34-, and M6-RNA2-BMV) were maintained during infection.
In contrast, the sequences of M1- and M5-RNA2-BMV, carrying marker mutations within both 5′ and 3′ noncoding regions, reverted to wt RNA2. This suggested the involvement of error-prone RNA replication.
To confirm that marker mutations in the coding regions were stable and that mutations in the noncoding regions were unstable, a new M1345-RNA2-BMV mutant, carrying four marker mutations, was constructed (Fig. 1, Table 2). Sequencing (3 to 4 clones for each mutation) and restriction analysis (24 clones for each mutation) of the progeny RNA2 from plants infected with M1345-RNA2-BMV demonstrated the presence of marker mutations within the coding regions of the molecules but the absence of mutations in the noncoding 3′ and 5′ regions; the latter reverted to the wild-type sequences. Sequencing excluded the possibility that recombination with the remaining BMV RNAs was responsible for 3′ or 5′ reversions.
Similar analyses were conducted for progeny of O1-, O3-, O4-, O6-, O7-, and O8-RNA1-BMV mutants (Table 2). Again, the 3′ and 5′ untranslated region (UTR) mutations reverted to a wt RNA1 sequence, but the remaining RNA1 mutants (O3-, O4-, O6-, and O8-RNA1-BMV) were stable and thus suitable for coinfection experiments.
Coinfection with M2-RNA2-BMV and M34-RNA2-BMV.
Since previous results suggested the role of BMV RNA polymerase (protein 2a) in recombination (13), we first studied recombination frequency on RNA2. The marker mutations in M2- and M34-RNA2-BMV flanked two regions of RNA2 (see Fig. 3A): a 571-nt sequence between BamHI sites at positions 326 and 897 and an 811-nt sequence between BamHI and BstEII sites at positions 897 and 1708, respectively.
FIG. 3.

Analysis of recombinant BMV RNA1 progeny in doubly infected barley host. Two pairs of parental RNA1 mutants designated as P1 through P4, corresponding to O3-RNA1/O6-RNA1 (A) and O6-RNA1/O8-RNA1 (B) pairs, and the projected RNA1 recombinants (designated as R1 through R4) are shown schematically on the left. All the elements are as described in the legends to Fig. 1 and 2. The tables on the right show the distribution of the parental and recombinant cDNA clones in the progeny RNA1 variants as they were identified in total RNA extracts from the whole leaf tissue, as well as the oRF. Parental molecules and recombinants are labeled, and vertical arrows represent marker restriction sites. The experimental methodology used was similar to that for RNA2, as described in the legend to Fig. 2.
The coinoculations were done on either C. quinoa or barley plants to study the recombination in either a limited or a high number of replication cycles, respectively. A restriction analysis of the RT-PCR cDNA products obtained from RNA extracts of 24 separate local lesions on C. quinoa leaves revealed that 50% lesions were infected with only one parental RNA2 variant (data not shown). To obtain more detailed recombinant profiles in a limited number of replication cycles, we analyzed recombinants in doubly infected lesions. The RT-PCR cDNA clones were analyzed by digestion with BamHI and BstEII enzymes, which revealed a recombination frequency of 26% between the M2 and M3 markers. The concentrations of either parental RNA2 variant in the progeny virus were almost equal (13 clones of M2-RNA2 and 16 clones of M34-RNA2; Fig. 2). The recombination frequency within the second region (between the M3 and M4 markers) was 13% and, again, the ratio between both parental RNA2s was nearly equal (19 clones of M2-RNA2 and 15 clones of M34-RNA2; Fig. 2).
FIG. 2.

Analysis of homologous recombinant BMV RNA2 progeny in doubly infected C. quinoa or barley hosts. (A) The location of marker mutations within M-RNA2 is shown schematically. Two pairs of parental RNA2 mutants, designated as P1 through P4, corresponding to M2-RNA2/M34-RNA2 (B) or M34-RNA2/M6-RNA2 (C) pairs, and the projected RNA2 recombinants (designated as R1 through R8) are shown on the left. All the elements are as described in the Fig. 1 legend. The tables on the right show the distribution of the parental and recombinant cDNA clones in the progeny RNA2 variants found in total RNA extracts either from the combined local lesion tissue or from the whole-leaf tissue of the infected C. quinoa or barley tissues, along with the oRF. Parental molecules and recombinants are labeled, while vertical arrows represent marker restriction sites. The progeny RNA2 was amplified by RT-PCR using total RNA extracts, and the resulting cDNA products were cloned and analyzed by restriction enzyme digestion and by sequencing, as described in Materials and Methods. Recombinants R1 through R4, R2 and R3, and R5 through R8 are created by homologous crossovers within regions I, II, and III, respectively, of RNA2. Because only 50% of local lesions (loc. les.) on C. quinoa leaves were doubly infected with both parental RNA1 or RNA2 variants, the recombination frequencies for the whole-leaf samples were approximated by dividing into half the number of the examined clones. n/a, not analyzed.
Secondly, we determined the concentration of recombinants in the combined local lesion (whole leaf of C. quinoa) in order to gain a possibly average recombination frequency. Because earlier we demonstrated that only 50% of the local lesions are infected with both parental variants, the number of the examined clones was divided by two. The RNA2 sequences were amplified by RT-PCR from the total RNA extracts, and the resulting cDNA clones were analyzed by restriction digestion. In the region flanked by M2 and M3 markers, the recombination frequency was 22%, with 21 M2-RNA2 and 11 M34-RNA2 parental clones (Fig. 2). For the region between M3 and M4 markers, the recombination frequency was 22%, and there were 21 M2-RNA2 and 11 M34-RNA2 parental clones (Fig. 2).
To analyze recombination frequency after a high number of replication cycles, M2- and M34-RNA2-BMV mutants were coinfected on barley plants. A restriction analysis of progeny cDNA clones revealed a recombination frequency of 17% between M2 and M3 markers with, respectively, 18 M2-RNA2 and 31 M34-RNA2 parental clones (Fig. 2). In the region flanked by the M3 and M4 markers, recombination frequency reached 21%, and there were 14 M2-RNA2 and 16 M34-RNA2 parental RNA2 clones (Fig. 2). Thus, both parental RNA2 mutants accumulated with a minimal selective advantage compared to each other and to recombinants.
Coinfection with M34-RNA2-BMV and M6-RNA2-BMV.
The second region in RNA2 that was covered by marker sites comprised a central sequence of 811 nucleotides in mutant M34-RNA2-BMV (Fig. 1; Table 2). Yet another mutant, M6-RNA2-BMV, carried one marker (NsiI) site at position 2517, covering a 3′ region of 809 nucleotides. Firstly, the coinfection of M34- and M6-RNA2-BMV was tested on a C. quinoa host. The restriction analysis of the RT-PCR products from eight separate doubly infected local lesions revealed that the M4-M6 region supported recombinants with a frequency of 18% (2 R5 clones and 1 R8 clone from the total number of 17 analyzed clones), and the ratios of parental molecules in the RNA2 progeny were equal (7 clones for each M34-RNA2 and M6-RNA2; Fig. 2), confirming similar availabilities of both parental constructs for recombination.
Secondly, the progeny RNA2 was analyzed after extraction from whole infected C. quinoa leaves in order to round up the recombination frequency (as earlier, the number of the examined clones was divided by two). This revealed that the region between the M4 and M6 markers supported a recombination frequency of 15%; 3 R5 and 1 R8 recombinants were found in the total number of 26 clones (in fact, 53 clones were analyzed) (Fig. 2b). Among the characterized clones, 28 M34-RNA2 and 21 M6-RNA2 parental molecules were found.
Next, similar experiments were performed on barley. Restriction analysis revealed that in the region between the M4 and M6 markers, the recombination frequency reached 14% (4 R5 and 6 R8 recombinants per 71 analyzed clones) (Fig. 2b). The recombination frequency between M3 and M4 markers reached 24% (two R2 and four R3 clones). There were 11 M34-RNA2 and 7 M6-RNA2 parental clones. These analyses demonstrated that recombination frequencies were similar in both the local lesion and systemic hosts.
Coinfections of O3-RNA1-BMV/O6-RNA1-BMV and O6-RNA1-BMV/O8-RNA1-BMV.
In order to figure out how the observed frequency of homologous recombination in RNA2 compared to that in RNA1, we tested O3-, O6-, and O8-RNA1-BMV variants in the coinfection experiments. Marker mutations divided the RNA1 molecule into two regions: the O3 (BglII, position 511) and O6 (BstBI, position 1472) markers flanked the 962-nt-long 5′ region, while O6 and O8 (BglII, position 2933) demarked the 1465-nt-long 3′ region (Fig. 1; Table 2).
Since for the RNA2 segment the frequencies of recombination were similar for C. quinoa and barley hosts, comparable patterns were expected for RNA1, and both regions were tested only on barley plants. First, coinfection of O3- and O6-RNA1-BMV gave 63 RT-PCR cDNA clones from the total RNA extracts, and the BglII and BstBI digestions revealed the presence of 7 recombinant clones, which corresponded to a recombination frequency of 11%. The rest represented the parental (input) RNA1 mutants, where 16 clones carried the O3-RNA1 sequence and 40 clones carried the O6-RNA1 sequence (Fig. 3).
The second region between the O6 and O8 markers supported a recombination frequency of 27%; among 55 clones, 15 represented the recombinants (Fig. 3), whereas 26 clones corresponded to O6-RNA1 and 14 clones to O8-RNA1.
Control RT-PCR amplifications.
To test whether RT-PCRs generated the recombinants, control infections were performed. Barley seedlings were infected separately with M34-RNA2-BMV and M6-RNA2-BMV mutants, and total RNAs were extracted from the infected leaves. Then, both RNA extracts were mixed together and used as templates for RT-PCRs. The resulting cDNA products were cloned and 24 clones were analyzed by restriction enzyme digestion. It turned out that all the examined clones contained either of the parental cDNA sequences (17 M34-RNA2 and 7 M6-RNA2 clones) and none carried the wt sequence. This result demonstrated that RT-PCR amplifications neither generated the recombinants nor further modified the RNA sequence, which would have affected the introduced marker sites.
Comparing the oRF and cRF.
To predict how the ability to accumulate each of the two parental RNA variants influenced recombination, we carried out the following calculations. In an ideal situation, both RNA mutants used in the coinfection experiments would replicate and accumulate to the same level. For simplification, we have designated one mutant as Mut1 (the 5′-side marker mutation) and the second mutant as Mut2 (the 3′-side marker mutation). Four types of template switching events can occur with equal probabilities (Fig. 4). In the first, BMV RdRp starts on Mut1 and switches onto Mut2, producing recombinant Mut1/2 (lacking marker mutations). In the second, the RdRp starts on Mut2 and switches onto Mut1, producing recombinant Mut2/1 (carrying both marker mutations). In the third and fourth, the RdRp starts on Mut1 or Mut2 and then switches onto the same RNA type (Mut1 or Mut2, respectively), regenerating identical molecules Mut1/1 or Mut2/2, respectively. Thus, by analyzing the distribution of marker mutations in the progeny RNA, one can identify only 50% of recombinants. Therefore, the calculated recombination frequency (cRF) equals two times the observed recombination frequency (oRF). Since the final recombination outcome can involve additional secondary crossovers among the initial recombinants, the doubled oRF represents the minimal value for cRF.
FIG. 4.

Four types of theoretical recombinants that may arise during homologous crossovers between two coinfecting RNA molecules carrying two different marker mutations. (a and b) Crossovers between molecules of two markers at different locations; the progeny recombinants carry either no marker mutations (Mut1/2) or two marker mutations (Mut2/1). (c and d) Crossover between two RNAs carrying the same marker mutation, leading to their reconstruction. The grey line represents RNA molecules, and the circles represent marker mutations.
In practice, both parental RNA mutants can replicate and accumulate to different levels. In this case, the probability (P) of each recombination event needs to be estimated based on the known composition of progeny RNA. The probability calculations require a sufficient number (n) of cDNA clones representing the whole population of the studied RNA (n > 30 clones). Then, the numbers of Mut1, Mut2, Mut1/2, and Mut2/1 molecules has to be determined experimentally (n1, n2, n1/2, and n2/1, respectively). In addition, we make two conjectures: (i) the ratio between Mut1 and Mut2 does not change during virus infection and (ii) recombination events involve Mut1 and Mut2 molecules only, so the total number of molecules available for recombination is not n but n1 + n2 (secondary recombinations are not considered).
The probabilities for the formation of each recombinant type are calculated as follows. The probability of Mut1/2 formation (P1/2) reflects the probability that polymerase will select Mut1 [P1 = n1/(n1 + n2), i.e., the number of Mut1 molecules (n1) divided by the total number of Mut1 and Mut2 molecules available for recombination (n1 + n2)] and the probability that polymerase will select Mut2 [P2 = n2/(n1 + n2), i.e., the number of Mut2 molecules (n2) divided by the total number of Mut1 and Mut2 molecules available for recombination (n1 + n2)]. Thus, P1/2 = n1 × n2 / (n1 + n2)2.
Similarly, the probabilities of Mut2/1, Mut1/1, and Mut2/2 formation are calculated with the following equations: P2/1 = P2 × P1 = [n2 / (n1 + n2)] × [n1 / (n1 + n2)] = n2 × [n1 / (n1 + n2)2], P1/1 = P1 × P1 = [n1 / (n1 + n2)] × [n1 / (n1 + n2)] = n12 / (n1 + n2)2, and P2/2 = P2 × P2 = [n2 / (n1 + n2)] × [n2 / (n1 + n2)] = n22 / (n1 + n2)2.
Out of the four different types of expected recombinants that involve Mut1 and Mut2 (Fig. 4), two are detectable (Mut1/2 and Mut2/1) and two are undetectable (Mut1/1 and Mut2/2). The probability of the formation of detectable or undetectable recombinants (Pd or Pu, respectively) is the sum of P1/2 plus P2/1 or P1/1 plus P2/2, respectively. Thus, Pd = 2 × n1n2 / (n1 + n2)2, while Pu = n12 + n22 / (n1 + n2)2.
The ratio of Pd to Pu is equal to the ratio between the number of detectable and undetectable recombinants, Pd/Pu = nd/nu, where
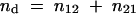 |
(1) |
and
 |
(2) |
Because Pd/Pu = nd/nu, then nu = nd × Pu/Pd = [(n12 + n21) × (n12 + n22) / (n1 + n2)2] / [2 × n1n2 / (n1 + n2)2] and
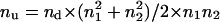 |
(3) |
Based on all known parameters necessary to calculate nu, one can calculate the actual number of recombinants and the recombination frequency, respectively, with the following equations:
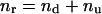 |
(4) |
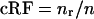 |
(5) |
We used equations 1 to 5 to calculate the cRFs, and in particular their fractional values per 100 nt of length (cRF/100). As shown in Table 3, for the upstream region in RNA2, the value was nearly 9% in the C. quinoa local lesion host, while the middle and the downstream RNA2 portions supported recombination with frequencies of 6 and 4%/100 nt, respectively. Interestingly, the corresponding cRF/100 values for RNA2 in barley reached similar levels. Also for RNA1, the cRF/100 ranged between 3 and 4% (in barley). Overall, these results demonstrated that homologous crossovers were distributed relatively evenly among the RNA1 and RNA2 segments and that they were similar in two different hosts.
TABLE 3.
cRF of homologous crossovers occuring between BMV RNA1 and RNA2 moleculesa
| Region between marker mutations | Host plantb | nc | n1 | n2 | nd | oRFd | nr | cRFe | cRF/100 nt |
|---|---|---|---|---|---|---|---|---|---|
| M2/M3 RNA2 (571 Int) | C. quinoa—local lesion | 39 | 13 | 16 | 10 | 26% | 20.21 | 51% | 8.9% |
| C. quinoa—whole leaf | 36 | 21 | 11 | 4 | 22% | 8.9 | 49% | 8.6% | |
| barley | 59 | 18 | 31 | 10 | 17% | 21.5 | 36% | 6.3% | |
| M3/M4 RNA2 (811 nt) | C. quinoa—local lesion | 39 | 19 | 15 | 5 | 13% | 10.1 | 26% | 3.2% |
| C. quinoa—whole leaf | 36 | 21 | 11 | 4 | 22% | 8.9 | 49% | 6.0% | |
| barleyf | 62 | 27 | 21 | 14 | 22% | 28.4 | 45% | 5.5% | |
| M4/M6 RNA2 (809 nt) | C. quinoa—local lesion | 17 | 7 | 7 | 3 | 18% | 6.0 | 35% | 4.4% |
| C. quinoa—whole leaf | 53 | 28 | 21 | 4 | 15% | 8.2 | 31% | 3.8% | |
| barley | 71 | 39 | 22 | 10 | 14% | 21.7 | 31% | 3.8% | |
| O3/O6 RNA1 (962 nt) | barley | 63 | 40 | 16 | 7 | 11% | 17.1 | 27% | 2.8% |
| O6/O8 RNA1 (1465 nt) | barley | 55 | 26 | 14 | 15 | 27% | 31.25 | 57% | 3.9% |
Recombination frequency was counted as the ratio of recombinants and the total number of analyzed clones.
Because only 50% local lesions on C. quinoa leaves were doubly infected with both parental RNA1 or RNA2 variants, the recombination frequencies for the whole-leaf infections were approximated by dividing into half the number of the examined clones.
oRF = nd/n.
cRF = nr/n.
Recombination frequency within M3/M4 region of RNA2 BMV in barley is the sum of results obtained after coinoculation experiments with M2-/M34-RNA2-BMV and M34-/M6-RNA2-BMV mutants (Fig. 2).
When considering the entire length of RNA1 and RNA2, the cRF values approached or even exceeded 100% (Table 3), suggesting that every progeny BMV RNA molecule represented a product of recombination that has occurred sometime during the amplification of its lineage and emphasizing its importance during the evolution of the viral genome (see Discussion).
DISCUSSION
Previously, we used coinoculations with noncompetitive pairs of BMV RNA3 variants to demonstrate homologous RNA3-RNA3 recombination activity during infection (6). The used methodology involved marker mutations that were introduced at different but functionally neutral locations on the RNA3 molecule. The identified RNA1 and RNA2 recombinants were not the RT-PCR artifacts, nor did they arise from BMV replicase errors, as demonstrated in control in vivo and in vitro experiments.
The basal recombination activity was observed along the entire length of RNA3, and a recombination hot spot was mapped to the intragenic sgp region (39). To determine whether and to what extent the remaining two BMV RNA components supported homologous crossovers, in this paper a similar methodology of pairwise coinoculations was applied. We analyzed homologous recombination in RNAs 1 or 2 in two hosts, the local-lesion C. quinoa host and the systemic barley host. The marker mutations were introduced so that the encoded amino acids were not changed and, indeed, such RNA1 or RNA2 variants coaccumulated in either host with an even ratio.
The use of marker mutations has allowed us to probe recombination activity within selected portions of both BMV RNAs. Although the crossovers were observed in each region studied, there were differences in recombination frequencies. For instance, the average cRF/100 of RNA1 was 3.9% in the 3′ part and 2.8% in the 5′ part in barley host. Among the three regions analyzed in RNA2, the frequency was highest within the 5′ portion (cRF/100 = 6.3%), lower in the middle (5.5%), and lowest at the 3′ side (3.8%). The nearly doubled 5′ recombination on RNA2 suggests elevated crossovers in this region. However, the differences in the calculated frequencies (6.3% versus 3.8% per 100 nt) may not be statistically significant, and more recombinants need to be analyzed before the 5′ recombination hot spot can be claimed for RNA2. Interestingly, the results obtained from barley, where the virus amplified through a higher number of replication cycles than in C. quinoa local lesions, were very similar between the two hosts. Again, a higher frequency occurred at the 5′ side of RNA2 and a lower frequency occurred at the 3′ side. This implies that the host is not the factor affecting homologous recombination, although more studies are required.
How do these new results compare to previously described recombination in BMV RNA3? Homologous RNA3-RNA3 crossovers were observed at a frequency of 10 to 22% (or 2 to 4% per 100 nt) within the 547-nt-long coat protein region (39), comparable to the results observed in this work for RNA1 and RNA2. However, a recombination frequency of 30% was observed for the 100-nt-long subgenomic promoter of RNA3 (39, 40) and one of nearly 70% was observed for the entire intergenic region in RNA3, probably due to the further contribution of the so-called internal replication enhancer (IRE) sequence (6). The high frequency at the sgp sequence was later explained by a mechanism whereby BMV polymerase detaches at the sgp and reinitiates on another RNA3 template (39, 40). The determination of whether the mechanism responsible for the observed crossovers in RNAs 1 and 2 is similar to that of the sgp-mediated crossovers in RNA3 requires further investigation. Most likely, the ability to jump from one template to another within homologous regions is an intrinsic feature characterizing the BMV replicase complex. However, at certain sequence elements the rate of recombination can be increased, e.g., at the RNA3 sgp. Along these lines, other in vivo data on 3′ homologous recombination demonstrated that crossovers between different BMV RNA segments could occur at the AU-rich/GC-rich sequences (27).
We did not observe length variability in the PCR products, and none of the clones carried modified sequences suggestive of a high precision of homologous crossovers. However, this might reflect selection pressure within coding regions of RNA1 or RNA2 similar to that observed in RNA3 (39). In contrast, imprecise crossovers have been observed within the entire RNA3 intergenic recombination hot spot (6, 39, 40), where the selection pressure tolerated certain modifications. Precise homologous recombination was observed between genomic RNAs of other viruses, e.g., those of the closely related CMV and tomato aspermy virus (TAV) (1). In this case, the crossovers occurred only within a short stretch (0.12 kb) of high sequence similarity within the movement protein (3a) open reading frame (ORF). The lack of recombination within the sgp region might be due to the lack of sequence identity between CMV and TAV.
Since certain classes of homologous recombinants were undetectable with the used system of marker mutations, we calculated the recombination frequencies to obtain more-real values. The calculation formula took into account such factors as the detectable versus undetectable crossovers and the variable concentrations of parental BMV RNA variants. As expected, the calculated frequency was higher than the experimental rate (Table 3). However, even with these calculations the number of crossovers was probably underestimated, because the formulas did not take into account secondary crossovers among the primary recombinants.
To determine the possibility of multiple crossovers, the progeny RNA from the M2- RNA2-BMV/M34-RNA2-BMV coinfection was amplified by RT-PCR (Fig. 5), and restriction profiles were determined for the resulting cDNA clones. Both kinds of expected double recombinants, M23 and M4, were identified and included 2 among 39 clones from local lesions of C. quinoa, 3 among 36 clones from a whole-leaf C. quinoa extract, and 4 among 38 clones from barley. Thus, regardless of the host, multiple crossovers were able to occur during BMV infection. Such multiple homologous crossover events were also observed between CMV and TAV during mixed infections (1).
FIG. 5.

Two types of double recombinants found in M2- and M34-RNA2-BMV coinfection. All the elements are as described for Fig. 2 and 3. The table on the right shows the incidence of double recombinants among cDNA clones obtained from C. quinoa and from barley tissue. See the text for more details.
The picture emerging from these studies implies the dynamic nature of the BMV RNA genome. Every region we studied previously (6, 39, 40) and in this work supported detectable homologous recombination rates. Thus, BMV RNA progeny should be considered as mosaic recombinants that are derived from different parental RNA templates. The observed and calculated frequencies imply that each molecule recombined at least once per replication cycle. However, since each assay represents many (probably tens to hundreds) amplification rounds, it is more correct to say that every progeny is the product of a recombination event that occurred at some point in the amplification of its lineage.
There are limited data on homologous recombination in natural populations of other plant or animal RNA viruses. The most recent studies on the dynamics of human immunodeficiency virus 1 recombination in natural host cell populations revealed a frequency of 10% per 100 nt (23), a result similar to that for BMV. Overall, these results demonstrate that viral RNAs recombine frequently. A postulated biological role of recombination is to provide a mechanism complementary to nucleotide mutation, and it further accelerates the diversification of the RNA genome (32). Recombination can introduce blocks of mutated alleles that otherwise would not be possible to assemble, bringing the linkage between alleles and thus preventing a degradation of population fitness so that selection is dominant rather than stochastic. Some results demonstrate that recombination occurs in a manner that preserves the integrity of ORFs or cis elements (42). The observed high recombination rate within the intergenic region of BMV RNA3 may enhance the reshuffling of the blocks of 3a and coat protein ORFs, two genes related to early stages of virus entry and invasion.
The commonality of homologous RNA-RNA crossovers strongly resembles the crossing-over events in DNA organisms. However, despite functional equivalency, the molecular mechanism(s) responsible for viral RNA-RNA crossovers are probably completely different from those responsible for the DNA crossing-over. While the latter occurs postreplicationally, RNA crossovers most likely happen during RNA replication by switching the replicase complex or by means of other mechanism(s).
During these studies, we came across yet another mechanism of variability. We found that marker mutations in noncoding regions in BMV RNAs1 and 2 were replaced with wild-type sequences, most likely due to error-prone replication. We surmise that this is because both the 5′ and 3′ noncoding regions in BMV RNA1 and RNA2 carry promoters of RNA synthesis (12, 14, 18) and because the 3′ noncoding region participates in RNA adenylation (12). From hence proceeds the strong selection pressure against sequence modifications, which must exist to eliminate the mutated sequences from an RNA population. Our observations suggest that both error-prone RNA replication and selection pressure are beneficial for the viral genome in keeping its key sequences functional.
Acknowledgments
This work was supported by a grant from the National Science Foundation (MCB-0317039) and by the Polish government through grants (6 P04C 046 19 and 3 P04A 039 25) from the State Committee for Scientific Studies, both awarded to J.J.B., and by the Plant Molecular Biology Center at Northern Illinois University.
REFERENCES
- 1.Aaziz, R., and M. Tepfer. 1999. Recombination between genomic RNAs of two cucumoviruses under conditions of minimal selective pressure. Virology 263:282-289. [DOI] [PubMed] [Google Scholar]
- 2.Adair, T. L., and C. M. Kearney. 2000. Recombination between a 3-kilobase tobacco mosaic virus transgene and a homologous viral construct in the restoration of viral and nonviral genes. Arch. Virol. 145:1867-1883. [DOI] [PubMed] [Google Scholar]
- 3.Agol, V. I. 1997. Recombination and other genomic rearrangements in picornaviruses. Semin. Virol. 8:77-84. [Google Scholar]
- 4.Allison, R. C., C. Thompson, and P. Ahlquist. 1990. Regeneration of a functional RNA virus genome by recombination between deletion mutants and requirement for cowpea chlorotic mottle virus 3a and coat genes for systemic infection. Proc. Natl. Acad. Sci. USA 87:1820-1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banner, L. R., and M. M. Lai. 1991. Random nature of coronavirus RNA recombination in the absence of selective pressure. Virology 185:441-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruyere, A., M. Wantroba, S. Flasinski, A. Dzianott, and J. J. Bujarski. 2000. Frequent homologous recombination events between molecules of one RNA component in a multipartite RNA virus. J. Virol. 74:4214-4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bujarski, J. J., and A. M. Dzianott. 1991. Generation and analysis of nonhomologous RNA-RNA recombinants in brome mosaic virus: sequence complementarities at crossover sites. J. Virol. 65:4153-4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bujarski, J. J., P. D. Nagy, and S. Flasinski. 1994. Molecular studies of genetic RNA-RNA recombination in brome mosaic virus. Adv. Virus Res. 43:275-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cervera, M. T., J. L. Riechmann, M. T. Martin, and J. A. Garcia. 1993. 3′-terminal sequence of the plum pox virus PS and o6 isolates: evidence for RNA recombination within the potyvirus group. J. Gen. Virol. 74:329-334. [DOI] [PubMed] [Google Scholar]
- 10.Chen, Y. K., R. Goldbach, and M. Prins. 2002. Inter- and intramolecular recombinations in the cucumber mosaic virus genome related to adaptation to alstroemeria. J. Virol. 76:4119-4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devold, M., K. Falk, B. Dale, B. Krossoy, E. Biering, V. Aspehaug, F. Nilsen, and A. Nylund. 2002. Strain variation, based on the hemagglutinin gene, in Norwegian ISA virus isolates collected from 1987 to 2001: indications of recombination. Dis. Aquat. Org. 47:119-128. [DOI] [PubMed] [Google Scholar]
- 12.Dreher, T. W., and T. C. Hall. 1988. Mutational analysis of the sequence and structural requirements in brome mosaic virus RNA for minus strand promoter activity. J. Mol. Biol. 201:31-40. [DOI] [PubMed] [Google Scholar]
- 13.Figlerowicz, M., P. D. Nagy, and J. J. Bujarski. 1997. A mutation in the putative RNA polymerase gene inhibits nonhomologous but not homologous genetic recombination in an RNA virus. Proc. Natl. Acad. Sci. USA 94:2073-2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayes, R. J., V. C. A. Pereira, and K. W. Buck. 1994. Plant proteins that bind to the 3′-terminal sequences of the negative-strand RNA of three diverse positive-strand RNA plant viruses. FEBS Lett. 352:331-334. [DOI] [PubMed] [Google Scholar]
- 15.Hill, K. R., M. Hajjou, J. Y. Hu, and R. Raju. 1997. RNA-RNA recombination in Sindbis virus: roles of the 3′ conserved motif, poly(A) tail, and nonviral sequences of template RNAs in polymerase recognition and template switching. J. Virol. 71:2693-2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirst, G. K. 1962. Genetic recombination with Newcastle disease virus, polioviruses and influenza. Cold Spring Harbor Symp. Quant. Biol. 27:303-309. [DOI] [PubMed] [Google Scholar]
- 17.Hu, W. S., T. Rhodes, Q. Dang, and V. Pathak. 2003. Retroviral recombination: review of genetic analyses. Front. Biosci. 8:143-155. [DOI] [PubMed] [Google Scholar]
- 18.Huntley, C. C., and T. C. Hall. 1993. Interference with brome mosaic virus replication by targeting the minus strand promoter. J. Gen. Virol. 74:2445-2452. [DOI] [PubMed] [Google Scholar]
- 19.Janda, M., R. French, and P. Ahlquist. 1987. High efficiency T7 polymerase synthesis of infectious RNA from cloned brome mosaic virus cDNA and effects of 5′ extensions of the transcripts infectivity. Virology 158:259-262. [DOI] [PubMed] [Google Scholar]
- 20.Kirkegaard, K., and D. Baltimore. 1986. The mechanism of RNA recombination in poliovirus. Cell 47:433-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lai, M. M. 1992. RNA recombination in animal and plant viruses. Microbiol. Rev. 56:61-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lane, L. 1986. Propagation and purification of RNA plant viruses. Methods Enzymol. 118:687-696. [Google Scholar]
- 23.Levy, D. N., G. M. Aldrovandi, O. Kutsch, and G. M. Shaw. 2004. Dynamics of HIV-1 recombination in its natural target cells. Proc. Natl. Acad. Sci. USA 101:4204-4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magiorkinis, G., D. Paraskevis, A. M. Vandamme, E. Magiorkinis, V. Sypsa, and A. Hatzakis. 2003. In vivo characteristics of human immunodeficiency virus type 1 intersubtype recombination: determination of hot spots and correlation with sequence similarity. J. Gen. Virol. 84:2715-2722. [DOI] [PubMed] [Google Scholar]
- 25.Moya, A., E. C. Holmes, and F. Gonzales-Candelas. 2004. The population genetics and evolutionary epidemiology of RNA viruses. Nat. Rev. Microbiol. 2:279-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagy, P. D., and J. J. Bujarski. 1995. Efficient system of homologous RNA recombination in brome mosaic virus: sequence and structure requirements and accuracy of crossovers. J. Virol. 69:131-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagy, P. D., and J. J. Bujarski. 1997. Engineering of homologous recombination hotspots with AU-rich sequences in brome mosaic virus. J. Virol. 71:3799-3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagy, P. D., C. Ogiela, and J. J. Bujarski. 1999. Mapping sequences active in homologous RNA recombination in brome mosaic virus: prediction of the recombination hot-spots. Virology 254:92-104. [DOI] [PubMed] [Google Scholar]
- 29.Nagy, P. D., and A. E. Simon. 1998. In vitro characterization of late steps of RNA recombination in turnip crinkle virus. I. Role of motif1-hairpin structure. Virology 249:379-392. [DOI] [PubMed] [Google Scholar]
- 30.Nagy, P. D., and A. E. Simon. 1998. In vitro characterization of late steps of RNA recombination in turnip crinkle virus. II. The role of the priming stem and flanking sequences. Virology 249:393-405. [DOI] [PubMed] [Google Scholar]
- 31.Palasingam, K., and P. N. Shaklee. 1992. Reversion of Qβ RNA phage mutants by homologous RNA recombination. J. Virol. 66:2435-2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rouzine, I. M., J. Wakely, and J. M. Coffin. 2003. The solitary wave of asexual evolution. Proc. Natl. Acad. Sci. USA 100:587-592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith, D. B., and S. C. Inglis. 1987. The mutation rate and variability of eukaryotic viruses: an analytical review. J. Gen. Virol. 68:2729-2740. [DOI] [PubMed] [Google Scholar]
- 34.Steinhauer, D. A., and J. J. Holland. 1987. Rapid evolution of RNA viruses. Annu. Rev. Microbiol. 41:409-433. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki, Y., T. Gojobori, and O. Nakagomi. 1998. Intragenic recombinations in rotaviruses. FEBS Lett. 427:183-187. [DOI] [PubMed] [Google Scholar]
- 36.van der Kuyl, A. C., L. Neeleman, and J. F. Bol. 1991. Complementation and recombination between alfalfa mosaic virus RNA3 mutants in tobacco plants. Virology 183:731-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Most, R. G., L. Heijnen, W. J. M. Spaan, and R. J. deGroot. 1992. Homologous RNA recombination allows efficient introduction of site-specific mutations into the genome of coronavirus MHV-A59 via synthetic co-replicating RNAs. Nucleic Acids Res. 20:3375-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.White, K. A., and T. J. Morris. 1994. Recombination between defective tombusvirus RNAs generates functional hybrid genomes. Proc. Natl. Acad. Sci. USA 91:3642-3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wierzchosławski, R., A. Dzianott, S. Kunimalayan, and J. J. Bujarski. 2003. A transcriptionally active subgenomic promoter supports homologous crossovers in plus-stranded RNA virus. J. Virol. 77:6769-6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wierzchosławski, R., A. Dzianott, and J. J. Bujarski. 2004. Dissecting the requirement for subgenomic promoter sequences by RNA recombination of brome mosaic virus in vivo: evidence for functional separation of transcription and recombination. J. Virol. 78:8552-8564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang, X., and M. M. Lai. 1994. Unusual heterogeneity of leader-mRNA fusion in a murine coronavirus: implications for the mechanism of RNA transcription and recombination. J. Virol. 68:6626-6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhuang, J., A. E. Jetzt, G. Sun, H. Yu, G. Klarmann, Y. Ron, and J. P. Dougherty. 2002. Human immunodeficiency virus type 1 recombination: rate, fidelity, and putative hot spots. J. Virol. 76:11273-11282. [DOI] [PMC free article] [PubMed] [Google Scholar]