

Key Points
-
•
Venetoclax-R2 is an active regimen in R/R MCL, showing an ORR of 63%.
-
•
Forty-eight percent of patients discontinued treatment of venetoclax-R2 because of molecular remission.
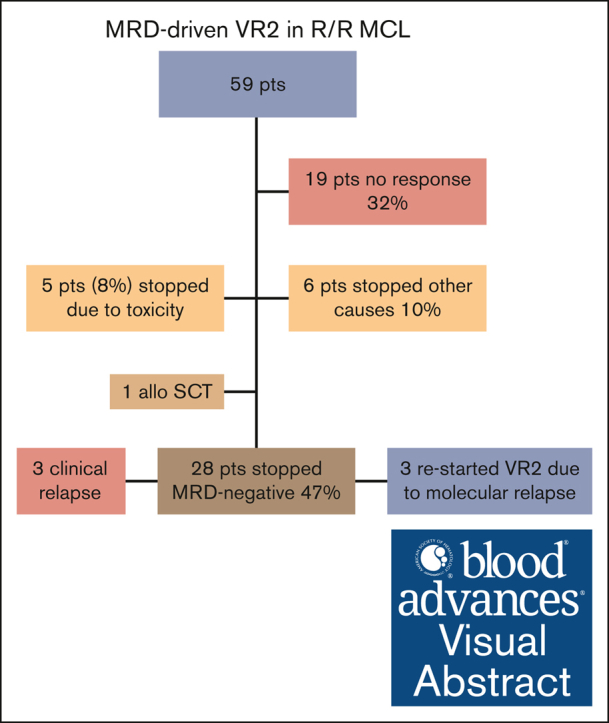
Visual Abstract
Abstract
Despite improvements in treatment of mantle cell lymphoma (MCL), most patients eventually relapse. In this multicenter phase 1b/2 trial, we evaluated safety and efficacy of minimal residual disease (MRD)–driven venetoclax, lenalidomide, and rituximab (venetoclax-R2) in relapsed/refractory (R/R) MCL and explored the feasibility of stopping treatment in molecular remission. The primary end point was overall response rate (ORR) at 6 months. After dose escalation, the recommended phase 2 dose was lenalidomide 20 mg daily, days 1 to 21; venetoclax 600 mg daily after ramp-up; and rituximab 375 mg/m2 weekly for 4 weeks, then every 8 weeks. MRD monitoring by RQ-PCR was performed every 3 months. When MRD-negativity in the blood was reached, treatment was continued for another 3 months; if MRD-negativity was then confirmed, treatment was stopped. In total, 59 patients were enrolled, with a median age of 73 years. At 6 months, the ORR was 63% (29 complete remission [CR], 8 partial remission [PR]), and 40% (4 CR, 2 PR) for patients previously failing a Bruton tyrosine kinase (BTK) inhibitor. Median progression-free survival (PFS) was 21 months, with median overall survival of 31 months. TP53 mutation was associated with inferior PFS (P < .01). Overall, 28 patients (48%) discontinued treatment in molecular remission, and 25 remain MRD negative after a median of 17.4 months. Hematological toxicity was frequent, with 52 of 59 (88%) patients with G3-4 neutropenia and 21 of 59 (36%) patients with G3-4 thrombocytopenia. To conclude, MRD-driven venetoclax-R2 is feasible and tolerable and shows efficacy in R/R MCL, also after BTK inhibitor failure. This trial was registered at www.ClinicalTrials.gov as #NCT03505944.
Introduction
Relapsed or refractory mantle cell lymphoma (R/R MCL) is often associated with poor outcome. The choice of therapy depends on the efficacy of prior lines of treatment. The Bruton tyrosine kinase inhibitor (BTKi) ibrutinib has shown favorable outcome compared with chemoimmunotherapy regimens, particularly in early relapse,1 and BTKis is the preferred therapy at first relapse.2 BTKis other than ibrutinib, particularly acalabrutinib,3 zanubrutinib,4 and pirtobrutinib,5 show comparable efficacy but are associated with a more favorable safety profile. In patients who relapse after BTKi treatment, a chimeric antigen receptor (CAR) T-cell product, brexucabtagene autoleucel is approved, both in the United States and Europe.6 Despite these achievements, patients with MCL continue to relapse.7 Other agents with activity in R/R MCL are bortezomib, temsirolimus, lenalidomide (LEN), and venetoclax (VEN).2, 3, 4, 5, 6
LEN is an immunomodulatory drug, with antiangiogenic and antineoplastic properties. In B-cell malignancies, LEN interacts with the ubiquitin E3 ligase cereblon and enhances its enzymatic activity to degrade the transcription factors IKZF1 (Ikaros) and IKZF3 (Aiolos), leading to reduced activity of interferon regulatory factor 4, a downstream target of cereblon. This leads to proliferation and activation of natural killer (NK) cells, enhancing NK cell–mediated cytotoxicity and antibody-dependent cellular cytotoxicity.7 In this respect, LEN acts as an immunosensitizer, enhancing the activity of rituximab. Hence, the combination of rituximab and LEN has shown to be very active in MCL, in both the relapse and frontline setting.8,9
VEN is an inhibitor of B-cell lymphoma 2 protein, thereby potentiating apoptosis in tumor cells. Early phase 1 data showed this agent to be highly active in R/R MCL and that it could act synergistically with BTKi. Based on the activity and good tolerability of both VEN as a single agent and of the rituximab-LEN combination, the Nordic Lymphoma Group aimed to assess the efficacy and safety of the triplet combination in R/R MCL by simultaneously targeting the CD20 surface antigen, stimulating apoptosis, and modulating the microenvironment. Moreover, we aimed to explore the feasibility of stopping treatment in patients achieving molecular remission, to minimize toxicity and financial burden.
Methods
Study design and participants
Patients with R/R MCL were enrolled in this open-label, single arm, multicenter, Nordic Lymphoma Group phase 1b/2 trial at 14 centers in Sweden, Finland, Norway, and Denmark.
Key eligibility criteria included age of ≥18 years; confirmed MCL diagnosis; ≥1 previous treatments including at least 1 rituximab-containing regimen; Eastern Cooperative Oncology Group performance status score of 0 to 3; measurable disease (long axis of >1.5 cm); absolute neutrophil count of ≥1000/mm3; platelets of ≥100 000/mm3 or ≥50 000/mm3 if bone marrow involvement; alanine amino transferase and aspartate amino transferase lower than 3-times the upper limit of normal; and serum creatinine no higher than 2-times the upper limit of normal. Key exclusion criteria included known central nervous system involvement, and active hepatitis B or C infection. The study was approved by the respective national ethics committees in the 4 countries and conducted in accordance with the Declaration of Helsinki and International Conference on Harmonisation guidelines for Good Clinical Practice. All patients provided written informed consent. The trial was registered at www.ClinicalTrials.gov as #NCT03505944.
Study end points
The primary end point was overall response rate (ORR) at 6 months. Secondary end points included ORR in patients previously treated with a BTKi, ORR in patients with a TP53 mutation, complete response (CR) rate, safety, molecular remission rate, progression-free survival (PFS), and overall survival (OS).
Treatment
Phase 1b portion
Patients received rituximab once weekly for 4 weeks during cycle 1, then every 8 weeks. The initial dose was given IV at a dose of 375 mg/m2. Subsequent doses could then either be given IV at the same dose, or as a subcutaneous injection of 1400 mg. No dose reductions were permitted for rituximab. LEN was given orally on days 1 to 21 in each cycle of 28 days. VEN was given orally daily, the dose was ramped up, 20 mg, 50 mg, 100 mg, 200 mg, for 1 week each, until reaching the target dose. For both VEN and LEN, the phase 1b part of the study followed a sequential dose escalation, “3 + 3” design. Dose-limiting toxicity (DLT) was defined as a grade ≥3 nonhematological toxicity within the first 8 weeks of therapy. Granulocyte colony-stimulating factor was mandatory on days 22 to 28, unless with an absolute neutrophil count of >10 × 109/L.
Initially, 3 patients were treated with dose regimen A (LEN 15 mg, target dose VEN 400 mg). After the third patient completed 8 weeks of treatment, if no DLT occurred, the next group of 3 patients was treated at the next dose level B (LEN 20 mg, target dose VEN 400 mg). If 1 of 3 initial patients experienced a DLT, the cohort of patients would be expanded to 6 patients. If <2 of 6 patients experienced a DLT, then the next higher dose group would be initiated. If ≥2 (of a cohort of up to 6) patients experienced a DLT, no higher dose levels were tested, and the maximum tolerated dose (MTD) had been exceeded. Intrapatient dose escalation was not permitted. If ≥2 patients, of 6, with level A experienced a DLT, the next group of 3 patients would be treated in the deescalation Group X (LEN 10 mg, target dose VEN 400 mg). The maximal level was dose level C (LEN 20 mg, target dose VEN 800 mg). The MTD was defined as the highest dose studied for which the incidence of DLT was <2 of 6 patients during the first 8 weeks of treatment.
Adverse events (AEs) were assessed according to the Common Toxicity Criteria for Adverse Events version 4.03. The following events were deemed sufficient cause to terminate study treatment: progressive disease, grade 4 nonhematological toxicity, the patient’s own wish to terminate study treatment, or if the responsible physician thought a change of therapy would be in the best interest of the patient.
Outcome assessment and response-adapted treatment
Treatment response was assessed according to the Lugano criteria with positron emission tomography (PET) with computed tomography (CT) and bone marrow examination.8
Assessment of minimal residual disease (MRD) by patient-specific RQ-PCR, according to EURO-MRD criteria,9 was performed on blood and BM specimens every 3 months. At baseline, DNA from BM lymphoma cells was extracted and used for PCR primer design and amplification of patient-specific clonally rearranged immunoglobulin heavy chain genes (IGH) and/or CCND1/IGH rearrangement (translocation 11;14). The sensitivity of the MRD assay was 1 in 105 cells, except for 4 patients with a sensitivity of 1 in 104 cells. For patients with <1% tumor cells in the BM or peripheral blood (PB) at baseline in which a quantitative MRD assay was not feasible, a qualitative nested PCR MRD assay was used, as previously described.10
When a patient obtained MRD negativity in blood according to EURO-MRD guidelines for deescalating treatment,9 treatment was continued for another 3 months, when a new evaluation of MRD in the blood and bone marrow was performed. If MRD negativity was confirmed in both the blood and BM, in combination with clinical and radiological CR, treatment was discontinued, and the patient was followed-up for MRD in the PB and with CT every 3 months for up to 24 months. If MRD-negativity was not attained, treatment continued for another 3 months, until later MRD negativity or until clinical progression, for up to 24 months.
Patients without BM/blood involvement by RQ-PCR at baseline or for whom a probe for MRD could not be constructed for other reasons, were followed-up by PET-CT every 3 months. If PET-CT became negative (Deauville score of 1-2), treatment continued for another 3 months followed by a repeated PET-CT. If PET negativity was confirmed, treatment was discontinued, and the patient was followed-up with PET-CT every 3 months for up to 24 months.
When MRD positivity recurred, according to EURO-MRD guidelines for escalating treatment,9 without a clinical relapse, the treatment was restarted.
Genetic analyses
Baseline BM or sections from formalin-fixed paraffin-embedded diagnostic biopsies were subjected to targeted sequencing. Details are provided in supplemental Material.
Statistical analysis
The following assumptions were used to estimate the sample size and the trial duration: a power of 80% to detect an ORR superiority of VEN + LEN + rituximab vs the historical control of LEN + rituximab (77% vs 57%)11; an inclusion period of up to 2 years, and an additional follow-up period up to 2 years; an inclusion rate of 30 per year; and a drop-out rate of 10% of included patients. For this purpose, 44 patients would need to be recruited. The analysis of the primary objective was performed according to the intention to treat. No exclusion or censoring was done in case of protocol violations. As an exploratory substudy, 15 patients with untreated MCL, ineligible for combination chemotherapy were planned to be enrolled. However, because of competing trials in the Nordic area, no patients in this population were found to be eligible, and a decision was made by the Nordic Lymphoma MCL Group, on 4 November 2020, to omit this cohort from the study, and to include an additional 15 patients with R/R MCL.
Descriptive statistics were used to summarize patient demographics and baseline characteristics. The safety analysis included any patient who received the treatment irrespective of eligibility or the duration of treatment received. The Kaplan-Meier method was used to estimate PFS and OS. OS was defined as time from study inclusion until death or censoring. PFS was defined as time from inclusion until progressive disease, death, or censoring. Duration of response was defined as time from reaching CR or PR until progressive disease, death, or censoring. The log-rank test was used to assess the difference in time-to-event end points between patient subgroups. Multivariate analysis was performed by Cox proportional hazards regression.
Results
Between 5 July 2018 and 20 October 2020, 59 patients with R/R MCL were enrolled. Baseline characteristics are presented in Table 1. Most patients (83%) had intermediate or high-risk disease according to the Mantle Cell International Prognostic Index. The median number of previous lines of therapy was 2 (range, 1-7). The most common first-line regimens were the Nordic MCL2 protocol (34 patients, 57%) and R-bendamustine (14 patients, 24%); 6 (10%) patients were primary refractory. In total, 35 (59%) had received previous autologous stem cell transplantation (ASCT), and 15 (25%) had been treated with ibrutinib, 2 as part of first-line treatment. Patients who were BTKi naïve had received a median of 1.5 lines of treatment. At the time of relapse, a TP53 mutation was present in 18 of 46 evaluable cases (39%).
Table 1.
Patient and disease characteristics
| Total (N = 59) | |
|---|---|
| Median age, y | 72 (54-81) |
| Female | 11 (19%) |
| Male | 48 (81%) |
| ECOG performance status score 0-1 | 56 (95%) |
| MCL International Prognostic Index | |
| Low risk | 8 (14%) |
| Intermediate risk | 28 (48%) |
| High risk | 21 (36%) |
| Missing data | 2 (3%) |
| Stage IV | 42 (71%) |
| Median number of previous lines of therapy | 2 (1-7) |
| Previous ASCT | 35 (59%) |
| Previous ibrutinib | 15 (25%) |
| S-IgG < 4 g/L | 11 (19%) |
| CD4 count < 200/mL | 16 (27%) |
| Histology | |
| Classical | 25 (42%) |
| Blastoid | 12 (20%) |
| Pleomorphic | 1 (2%) |
| Unknown | 21 (36%) |
| Ki67 | |
| ≤30% | 40 (68%) |
| >30% | 19 (32%) |
| TP53 mutational status at relapse | |
| Mutated | 18 (30%) |
| Unmutated | 28 (48%) |
| Not evaluable | 13 (22%) |
Data are number (percentage) or median (range).
ECOG, Eastern Cooperative Oncology Group; S-IgG, XXX.
Phase 1b
After evaluating 3 patients each at dose level A, B and C, after 8 weeks of treatment, no DLT were encountered at level A and B. At level C, 2 of 3 patients were hospitalized for grade 3 and 4 infection, and the MTD was considered to have been exceeded. To investigate further dose levels, it was decided to include patients at 1 more dose level, Y (LEN 20 mg, target dose VEN 600 mg). No DLT was seen in the evaluation period for 3 patients at this level, and level Y was recommended as the recommended phase 2 dose.
Outcome
Overall, 42 of 59 (71%) patients were evaluable for response at 6 months, the primary end point. In total, 12 patients progressed before this evaluation point, and 5 patients were not evaluable because of stopping treatment secondary to treatment-related toxicity before response evaluation. The total number of responding patients was 37. When calculating response according to intention to treat, the ORR was 63% (95% confidence interval [CI], 50-74); 29 (49%; 95% CI, 37-62) achieved a CR, and 8 (14%; 95% CI, 7-25) a partial response (PR; Table 2). Response was evaluated as stable disease in 5 patients (8%; 95% CI, 4-18). Among patients (n = 15) previously exposed to ibrutinib, 6 responded (ORR, 40%; 95% CI, 20-64), 4 had CR, and 2 PR.
Table 2.
Maximal response to treatment with VEN, LEN, and rituximab in R/R MCL, in total, and according to exposure to a BTKi (ibrutinib)
| Total (N = 59) | BTKi exposure (n = 15) | |
|---|---|---|
| ORR | 37 (63%; 95% CI, 50-74) | 6 (40%; 95% CI, 20-64) |
| Complete remission | 29 (49%; 95% CI, 37-62) | 4 (27%; 95% CI, 11-50) |
| Partial remission | 8 (14%; 95% CI, 7-25) | 2 (13%; 95% CI, 3-38) |
| Stable disease | 5 (8%; 95% CI, 3-18) | 2 (13%; 95% CI, 3-38) |
| Progressive disease/stopped early | 17 (29%; 95% CI, 19-41) | 7 (47%; 95% CI, 25-70) |
The median PFS was 21 months and median OS was 31 months (Figure 2A). The 24-month PFS was 45% (95% CI, 33-61), and OS 57% (95% CI, 45-74).
Figure 2.

Survival. PFS and OS with VEN-LEN-rituximab (A). PFS and OS according to the (B) presence or (C) absence of a TP53 mutation.
Outcome was inferior in the 18 cases with a TP53 mutation, of whom only 6 patients responded, all with CR (33%). Among patients with TP53 mutation, 12-month OS was 67% (95% CI, 48-92), and PFS 33% (95% CI, 17-64); for patients without TP53 mutation, 12-month OS was 86% (95% CI, 74-100) and PFS was 71% (95% CI, 57-90; Figure 2B-C). The median duration of response was 19 months (95% CI, 6.6-33) in patients with TP53 mutation, compared with not reached in those with wild-type–TP53 disease.
Molecular response and the feasibility of stopping treatment
At baseline, 58 of 59 patients (98%) had an informative MRD assessment in the BM. At 3 months, 29 patients were evaluable for MRD in the BM, of which 28 were negative (97%), corresponding to 47% of the total number of patients. All 36 patients evaluable in the PB (61% of all patients) were negative at this time point. At 6 months, 35 of 37 (94%) patients were MRD negative in the BM, and 39 of 39 in the PB (100%) (Figure 1; Table 3).
Figure 1.

MRD analysis. Sankey plot of MRD assessment in the BM at baseline and at cycles 4, 7, and 10 (C4-C10) with LEN, VEN, and rituximab in R/R MCL. Numbers at the bottom refer to the number of patients with MRD test available at the different cycles/number of patients on treatment.
Table 3.
Molecular remission rate in the PB and BM with VEN, LEN, and rituximab in R/R MCL
| 3 months |
6 months |
9 months |
12 months |
|||||
|---|---|---|---|---|---|---|---|---|
| BM | PB | BM | PB | BM | PB | BM | PB | |
| Negative | 36 | 28 | 33 | 39 | 11 | 31 | 10 | 24 |
| Positive | 1 | 0 | 2 | 0 | 5 | 1 | 1 | 2 |
| Not evaluable | 22 | 31 | 24 | 20 | 43 | 27 | 48 | 33 |
| Molecular remission in evaluable patients (%) | 97 | 100 | 94 | 100 | 69 | 97 | 91 | 92 |
| Molecular remission, intention to treat (%) | 61 | 47 | 56 | 66 | 19 | 53 | 17 | 41 |
After a median of 6.4 months, 28 patients stopped treatment, being MRD negative in both the BM and PB, and in clinical CR. The median time off treatment is currently 17.4 months. In 3 patients a molecular relapse was detected, after between 10 and 18 months, while still in clinical remission. These patients restarted treatment and reversed to MRD negativity. The median time on treatment after restarting is currently 4.6 months. One patient stopped treatment after 3 months because of moving to another city, the other 2 patients are still in molecular and clinical remission (Figures 3; supplemental Figure 1). Three patients experienced a clinical relapse, and went off study, 2 after 8 months, and 1 after 17 months.
Figure 3.

Swimmers plot of 59 patients with R/R MCL, treated with VEN-LEN-rituximab.
Among the 2 patients showing MRD the positivity in BM at 6 months, 1 patient, clinically in PR at 6 months, showed a slower time to response, becoming negative at 9 months, confirmed at 12 months, and then discontinuing treatment. The second patient progressed clinically at 6 months and went off study. Seven patients were MRD negative at 6 months but not in CR (5 PR, 1 stable disease, 1 progressive disease). One patient in PR continued treatment, obtained a CR at 12 months, and was consolidated with allogeneic SCT; the other 6 patients progressed clinically soon after the 6-month evaluation.
Safety
At a median follow-up of 31 months, treatment was discontinued in 31 (53%) patients for reasons other than stopping treatment in remission: progressive disease (n = 19, 32%), allogeneic SCT (n = 1, 2%), treatment-related toxicity (n = 5, 8%), withdrawal of consent (n = 2, 3%), and unspecified reasons (n = 4, 7%). In total, 36 (61%) patients required dose reduction of LEN and 31 patients (52.5%) of VEN because of AEs. Median relative dose intensity (calculated for patients receiving the recommended phase 2 dose) was for LEN 89% (range, 72%-106%), and for VEN 60% (range, 53%-67%).
In general, hematological AEs were the most frequent. Maximal grade of neutropenia was grade 4 in 24 patients (41%), and grade 3 in 28 patients (47%). All patients required granulocyte colony-stimulating factor during treatment. Thrombocytopenia grade 3 or 4 occurred in 8 (14%) and 13 (22%) patients, respectively. In 19 patients (32%) the cumulative number of days with grade 3/4 neutropenia was >30 days; 11 (19%) patients had >30 days with grade 3/4 thrombocytopenia.
The most common nonhematological AEs (>30% of patients) were gastrointestinal toxicity (diarrhea or obstipation) in 35 (59%) patients, of which the majority (n = 24) were grade 1; infections in 23 (39%); and rash in 21 (36%) patients (supplemental Table 1). No cases of tumor lysis syndrome were encountered. There were 55 reports of serious AEs, from 30 individual patients. There were 4 fatal serious AEs: 1 from aspergillosis, 1 from progressive multifocal leukoencephalopathy, and 2 from unspecified infection; 3 of these patients had previously received ASCT, 3 patients had received R-bendamustine, and 2 ibrutinib at first relapse. Five patients (8%) stopped treatment because of toxicity (2 infection, 1 stroke, 1 autoimmune hemolytic anemia, and 1 progressive multifocal leukoencephalopathy).
Overall, 28 patients (47%) died while on the study. The cause of death was MCL in 18 patients (31%); 4 patients died from treatment-related toxicity, 2 patients from concurrent disease, 1 patient because of second primary malignancy (oropharyngeal cancer), 2 from infection while on salvage chemoimmunotherapy, and in 1 patient the cause was unknown.
Discussion
In this study, we show that the triplet of VEN, rituximab, and LEN (VEN-R2) is an active combination in R/R MCL. The study did not meet its primary objective, showing an overall response rate (ORR) of 63%, with a lower bound of 95% CI that included the 57% with R2 alone.11 However, one should note that the patient population in the R2 study was different from that of this study, being from the pre-BTKi era, and with only 11% having received ASCT. These patients were also younger, with a median age of 66 years, compared with 72 years in this series. The median PFS with VEN-R2 was 21 months, compared with 11 months in the R2 trial.
In this trial, we were able to assess the rate of molecular remission with this novel regimen and to test the feasibility of stopping treatment in patients achieving molecular remission. The rate of MRD negativity in the BM after 6 months in evaluable patients was very high (97%). For comparison, in the OAsIs trial, with obinutuzumab-ibrutinib-VEN, the rate of MRD negativity in R/R MCL was 75% (9/12) at the same time point,12 and with ibrutinib-VEN, 67% (12/18) were negative in the BM at 4 months,13 both using similar RQ-PCR methods.
Phillips et al recently reported on the use of VEN-R2 in a cohort of 28 patients with previously untreated MCL.14 The dose of LEN (20 mg) was the same as in our study, but the dose of VEN was lower, 400 mg daily. The treatment duration was notably longer, with 6 cycles at full dose, followed by a maintenance phase of 18 months with reduced dose of LEN, and stopping VEN after 6 cycles of maintenance. Rituximab was given for a total of 36 months. Outcome in the first-line setting was clearly superior compared with the R/R situation, with an ORR of 96%, and an estimated 24-month PFS of 89%. There was also less hematological toxicity, with grade 3/4 neutropenia of 21% and grade 3/4 thrombocytopenia of 17%, possibly because of the lower dose of VEN. No treatment-related deaths were reported in this series, compared with a treatment-related mortality rate of 6.8% because of infections in this report. Most likely, this reflects the cumulative immunosuppressive effect of previous treatment lines, particularly ASCT, bendamustine, and BTKis, and may limit its use in this patient group. This indicates that the VR2 regimen may be more effective and tolerable if used in an earlier line of therapy. In our series, the outcome was inferior in patients who had been exposed to BTKis. However, this population was also more heavily pretreated, and one may hypothesize that the efficacy may be higher in a less pretreated, BTKi-exposed population.
An important aim of this trial was to explore the feasibility of response-adapted therapy, using MRD as a tool for decision making. This strategy was pioneered in MCL by our group 20 years ago in the Nordic MCL2 trial, using preemptive rituximab in patients with molecular relapse.15 A time-limited treatment is attractive for several reasons, both from a patient and a health care provider perspective, to minimize both toxicity and cost. In this trial, we used RQ-PCR for MRD assessment, being well established and highly sensitive but dependent on the presence of lymphoma cells in the PB or BM. In this trial, 98% were evaluable by MRD, and we were able to show that discontinuing treatment after a minimum treatment duration of 6 months was feasible in 48% of the patient population. At this point, 89% remain in molecular remission for a current median of 14.4 months. In 3 patients experiencing a molecular relapse, we were able to reinduce a molecular remission and prevent a clinical relapse. For patients with high-risk features (including TP53 aberrations) an early MRD evaluation may be used to identify patients in need of intensification, such as CAR T cells. In patients at biological low risk, the MRD-guided reinitiation was proven feasible and could be combined with definite therapy depending on the longer-term outcome for patients who whom treatment was reinitiated. Other methods, based on circulating tumor DNA, possibly in combination with RQ-PCR, may improve the applicability of an MRD-guided approach even further. However, although we could show that an MRD-guided approach is feasible, it remains to be proven to be superior in terms of sensitivity compared with the use of clinical assessment only.
One-fifth of the patients in this trial progressed before the 6-month evaluation and may be considered treatment refractory. In contrast, another subpopulation of 48%, obtained deep remissions, and were able to discontinue therapy. Part of the explanation of this almost dichotomous response may be related to TP53 mutational status. Our group and others have reported that mutations of TP53 are found in ∼10% of patients with untreated MCL, and that even with intensive immunochemotherapy and ASCT consolidation, the response to treatment and eventual outcome is dismal for patients with TP53 mutations.16, 17, 18 In the present trial, TP53 mutations were enriched compared with untreated MCL and survival was inferior in patients with TP53-mutated MCL, suggesting that VEN-R2 may not be as active in this population. This is in line with the study by Phillips et al in untreated MCL, also showing clearly inferior outcome with VEN-R2 in TP53-mutated disease.14
Treatment of R/R MCL has rapidly evolved since the initiation of this trial. At that point, BTKis were not generally approved for this indication in the Nordic area, but will likely soon be part of standard first-line treatment.19 For patients who have been exposed to BTKis, several options have become available, including brexucabtagene autoleucel, a CD19-targeting CAR T-cell product6; as well as pirtobrutinib, a noncovalent BTKi.5 VEN as single agent has shown relatively high activity after BTKis, although median PFS is limited.20 In addition, several potent agents are undergoing development in this space, in particular bispecific CD20/CD3 antibodies such as glofitamab21 and epcoritamab.22 VEN-R2 may be another option in this situation, in patients ineligible for CAR T cells, or as a bridging regimen to CAR T-cell or allogeneic SCT, or possibly in CARD11-mutated MCL as an alternative to BTKis. In this context, MRD-guided approaches should be further explored.
Conflict-of-interest disclosure: M.J. has received grants, personal fees and nonfinancial support from AbbVie; grants and nonfinancial support from Celgene, during the conduct of the study; grants and nonfinancial support from Janssen; and has received grants, personal fees, and nonfinancial support from Kite/Gilead, BeiGene, AstraZeneca, Roche, and Pierre Fabre, outside the submitted work. C.U.N. has received research funding and/or consultancy fees outside the current work from AbbVie, AstraZeneca, Janssen, Octapharma, Takeda, CSL Behring, BeiGene, and Genmab. S.L. has received grants and personal fees from Genmab; personal fees from Incyte; grants from Nordic Nanovector; grants and personal fees from Novartis; grants and personal fees from Roche; personal fees from Merck; grants from Bayer; grants and personal fees from Celgene; and personal fees from Orion, outside the submitted work. I.G. has received institutional support from Takeda, Janssen Cilag, and Lokon Pharma. A.P. has received consultation and personal fees outside the current work from Gilead, BeiGene, Roche, and Incyte. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank all of the staff at the Clinical Trial Office in Aarhus, Denmark for excellent trial management and coordination during the trial, as well as the research staff at all participating sites.
Sequencing was funded by grants from the Research Council of Finland and Finnish Cancer Organization (S.L.). The study was supported by a research grant from AbbVie to the Nordic Lymphoma Group. Study drug was supported by AbbVie and Bristol Myers Squibb. The companies were not involved in the protocol writing, in collection, analysis, or interpretation of the data, or in writing of the report but were able to review the manuscript before submission.
Authorship
Contribution: M.J., M.H., A.P., K.F.W., J.H.C., A.K., T.C.E.-G., and I.G. designed the trial and wrote the study protocol; R.R.K.J. performed the statistical analyses; L.M. and S.L. performed the sequencing; L.B.P. and C.U.N. performed the MRD assessment; and all authors had access to raw data, took part in writing, and approved the final version of the manuscript.
Footnotes
Data are available on request from the corresponding author, Mats Jerkeman (mats.jerkeman@med.lu.se). The study protocol is included as a data supplement available with the online version of this article. Individual participant data will not be shared.
The full-text version of this article contains a data supplement.
Supplementary Material
References
- 1.Dreyling M, Jurczak W, Jerkeman M, et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet. 2016;387(10020):770–778. doi: 10.1016/S0140-6736(15)00667-4. [DOI] [PubMed] [Google Scholar]
- 2.Visco C, Di Rocco A, Evangelista A, et al. Outcomes in first relapsed-refractory younger patients with mantle cell lymphoma: results from the MANTLE-FIRST study. Leukemia. 2021;35(3):787–795. doi: 10.1038/s41375-020-01013-3. [DOI] [PubMed] [Google Scholar]
- 3.Wang M, Rule S, Zinzani PL, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet. 2018;391(10121):659–667. doi: 10.1016/S0140-6736(17)33108-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tam CS, Opat S, Simpson D, et al. Zanubrutinib for the treatment of relapsed or refractory mantle cell lymphoma. Blood Adv. 2021;5(12):2577–2585. doi: 10.1182/bloodadvances.2020004074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang ML, Jurczak W, Zinzani PL, et al. Pirtobrutinib in covalent BTK-inhibitor pre-treated mantle cell lymphoma. J Clin Oncol. 2023;41(24):3988–3997. doi: 10.1200/JCO.23.00562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang M, Munoz J, Goy A, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382(14):1331–1342. doi: 10.1056/NEJMoa1914347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eskelund CW, Dimopoulos K, Kolstad A, et al. Detailed long-term follow-up of patients who relapsed after the Nordic Mantle Cell Lymphoma Trials: MCL2 and MCL3. Hemasphere. 2021;5(1) doi: 10.1097/HS9.0000000000000510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32(27):3059–3068. doi: 10.1200/JCO.2013.54.8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Velden VH, Cazzaniga G, Schrauder A, et al. Analysis of minimal residual disease by Ig/TCR gene rearrangements: guidelines for interpretation of real-time quantitative PCR data. Leukemia. 2007;21(4):604–611. doi: 10.1038/sj.leu.2404586. [DOI] [PubMed] [Google Scholar]
- 10.Geisler CH, Kolstad A, Laurell A, et al. Long-term progression-free survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivo-purged stem cell rescue: a nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood. 2008;112(7):2687–2693. doi: 10.1182/blood-2008-03-147025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang M, Fayad L, Wagner-Bartak N, et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: a phase 1/2 clinical trial. Lancet Oncol. 2012;13(7):716–723. doi: 10.1016/S1470-2045(12)70200-0. [DOI] [PubMed] [Google Scholar]
- 12.Le Gouill S, Morschhauser F, Chiron D, et al. Ibrutinib, obinutuzumab, and venetoclax in relapsed and untreated patients with mantle cell lymphoma: a phase 1/2 trial. Blood. 2021;137(7):877–887. doi: 10.1182/blood.2020008727. [DOI] [PubMed] [Google Scholar]
- 13.Tam CS, Anderson MA, Pott C, et al. Ibrutinib plus venetoclax for the treatment of mantle-cell lymphoma. N Engl J Med. 2018;378(13):1211–1223. doi: 10.1056/NEJMoa1715519. [DOI] [PubMed] [Google Scholar]
- 14.Phillips TJ, Bond DA, Takiar R, et al. Adding venetoclax to lenalidomide and rituximab is safe and effective in patients with untreated mantle cell lymphoma. Blood Adv. 2023;7(16):4518–4527. doi: 10.1182/bloodadvances.2023009992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andersen NS, Pedersen LB, Laurell A, et al. Pre-emptive treatment with rituximab of molecular relapse after autologous stem cell transplantation in mantle cell lymphoma. J Clin Oncol. 2009;27(26):4365–4370. doi: 10.1200/JCO.2008.21.3116. [DOI] [PubMed] [Google Scholar]
- 16.Eskelund CW, Dahl C, Hansen JW, et al. TP53 mutations identify younger mantle cell lymphoma patients who do not benefit from intensive chemoimmunotherapy. Blood. 2017;130(17):1903–1910. doi: 10.1182/blood-2017-04-779736. [DOI] [PubMed] [Google Scholar]
- 17.Obr A, Klener P, Furst T, et al. A high TP53 mutation burden is a strong predictor of primary refractory mantle cell lymphoma. Br J Haematol. 2020;191(5):e103–e106. doi: 10.1111/bjh.17063. [DOI] [PubMed] [Google Scholar]
- 18.Ferrero S, Rossi D, Rinaldi A, et al. KMT2D mutations and TP53 disruptions are poor prognostic biomarkers in mantle cell lymphoma receiving high-dose therapy: a FIL study. Haematologica. 2020;105(6):1604–1612. doi: 10.3324/haematol.2018.214056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dreyling M, Doorduijn JK, Gine E, et al. Efficacy and safety of ibrutinib combined with standard first-line treatment or as substitute for autologous stem cell transplantation in younger patients with mantle cell lymphoma: results from the Randomized Triangle Trial by the European MCL Network. Blood. 2022;140(suppl 1):1–3. [Google Scholar]
- 20.Eyre TA, Walter HS, Iyengar S, et al. Efficacy of venetoclax monotherapy in patients with relapsed, refractory mantle cell lymphoma after Bruton tyrosine kinase inhibitor therapy. Haematologica. 2019;104(2):e68–e71. doi: 10.3324/haematol.2018.198812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phillips TJ, Dickinson M, Morschhauser F, et al. Glofitamab monotherapy induces high complete response rates in patients with heavily pretreated relapsed or refractory mantle cell lymphoma. Blood. 2022;140(suppl 1):178–180. [Google Scholar]
- 22.Hutchings M, Mous R, Clausen MR, et al. Dose escalation of subcutaneous epcoritamab in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an open-label, phase 1/2 study. Lancet. 2021;398(10306):1157–1169. doi: 10.1016/S0140-6736(21)00889-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.