

Abstract
Pankreatische neuroendokrine Neoplasien (PanNEN) sind eher selten. Die Morphologie hilft in der Zusammenschau mit der Immunhistochemie bei der Typisierung und weiteren Einteilung des jeweiligen Tumortyps. Je nach Tumorstadium und Differentialdiagnose variiert das diagnostische Panel. Die vorliegende Arbeit fasst die obligaten diagnostischen, prognostischen und prädiktiven Marker bei PanNEN zusammen.
Marker der Wahl zum Nachweis eines neuroendokrinen Phänotyps sind Synaptophysin, Chromogranin A sowie INSM1. Die Proliferationsfraktion Ki67 ist zur Graduierung unabdingbar, während p53 und Rb1 in der Abgrenzung zum neuroendokrinen Karzinom (NEC) helfen können. Transkriptionsfaktoren, wie beispielsweise CDX2, TTF‑1, Islet‑1 geben Hinweise auf die Lokalisation eines Primarius in der Cancer-of-unknown-primary(CUP)-Situation. Die DAXX/ATRX-Immunhistochemie hat vor allem prognostischen Wert. Molekularpathologische Untersuchungen haben bisher einen geringen Stellenwert in der Diagnostik der PanNEN.
Wichtiger Fallstrick in der Routinediagnostik ist das breite Spektrum an Differentialdiagnosen, welche neuroendokrine Neoplasien imitieren. Ein erweitertes immunhistochemisches Panel ist im Zweifelsfall empfohlen.
Schlüsselwörter: Neuroendokrin, Immunhistochemie, PanNET, Pankreas, Fallstricke
Abstract
Pancreatic neuroendocrine neoplasms (PanNEN) are rather rare entities. Morphology, combined with immunohistochemistry, allows typing and grading, thereby leading therapeutic decisions. Depending on tumor stage and differential diagnosis, a broad diagnostic panel may be required. The present work summarizes the minimal diagnostic, prognostic, and predictive markers in PanNEN.
Markers of choice for defining a neuroendocrine phenotype are synaptophysin, chromogranin A, and INSM1. The proliferation fraction Ki67 is indispensable for grading, while p53 and Rb1 can help in the differentiation from neuroendocrine carcinoma (NEC). Transcription factors, such as cdx2, TTF‑1, and Islet‑1, can indicate the site of a primary tumor in the setting of a cancer of unknown primary (CUP). DAXX/ATRX immunohistochemistry has mainly prognostic value. Molecular pathology studies currently have little practical value in the diagnosis of PanNEN.
An important pitfall in routine diagnostics is the wide spectrum of differential diagnoses mimicking neuroendocrine neoplasms. An expanded immunohistochemical panel is strongly recommended in case of doubt.
Keywords: Neuroendocrine, Immunohistochemistry, PanNET, Pancreas, Pitfalls
Immunhistochemische Marker neuroendokriner Differenzierung
Gut und schlecht differenzierte pankreatische neuroendokrine Neoplasien (PanNEN) zeichnen sich durch die Expression der neuroendokrinen Marker Synaptophysin, Chromogranin A und INSM1 aus. Gut differenzierte PanNEN sind meist stark und durchgehend positiv für Synaptophysin, Chromogranin A und INSM1 (Abb. 1). Die Expression der neuroendokrinen Marker nimmt in schlecht differenzierten PanNEN ab. Chromogranin A ist häufig negativ, INSM1 und Synaptophysin können nur sehr fokal exprimiert sein [1]. Es gibt noch weitere Marker, die den neuroendokrinen Phänotyp erkennen, aber nicht spezifisch (CD56) oder schwierig einzustellen sind (NSE). Daher sollten sie nicht für die Diagnose neuroendokriner Neoplasien verwendet werden. Zudem können neuroendokrine Marker in konventionellen Adenokarzinomen exprimiert sein. Gemischte neuroendokrine-nichtneuroendokrine Neoplasien können erst dann diagnostiziert werden, wenn auch eine typische, morphologisch distinkte Komponente konventionell histologisch erkennbar ist [2–4].

Proliferationsindex „Ki67“
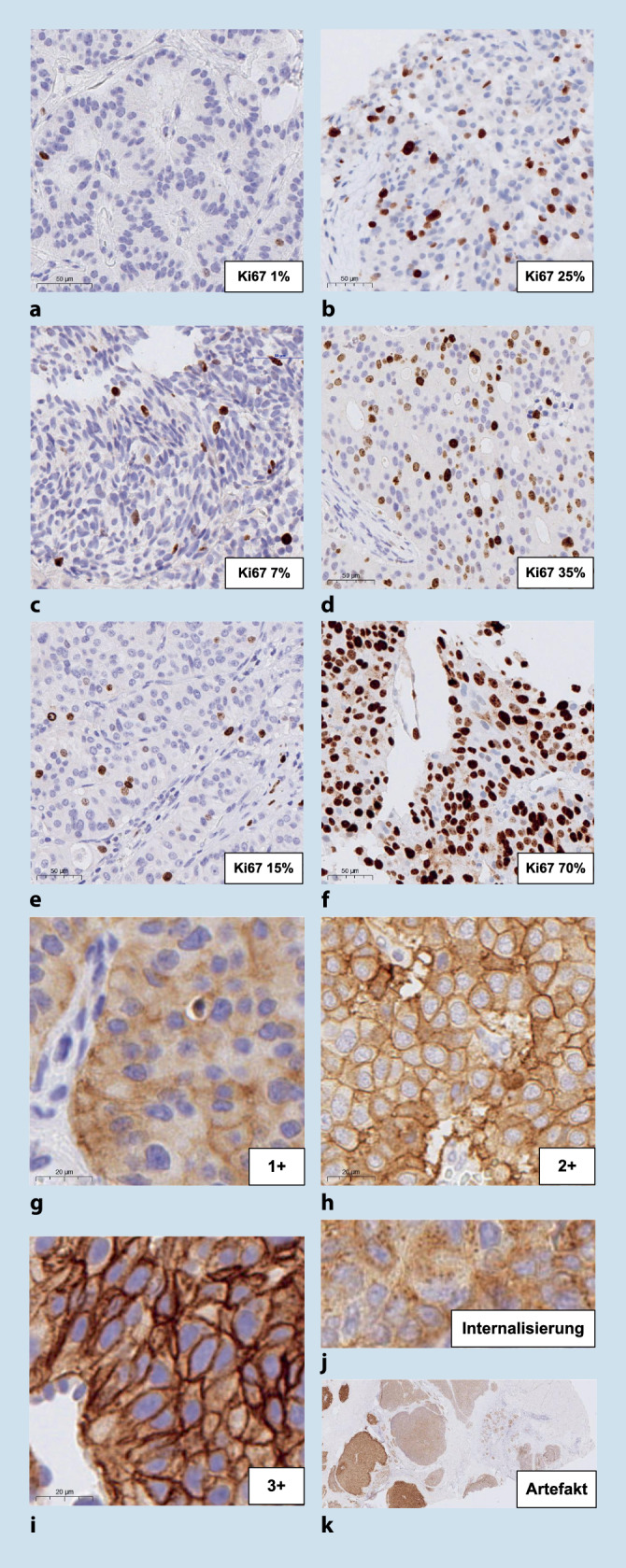
Die Proliferationsfraktion Kiel 67 (Ki67) ist ein seit Langem etablierter Biomarker zur Gradierung pankreatischer neuroendokriner Neoplasien [5]. Wichtige Cut-offs sind 3 % und 20 %. Die Quantifizierung mittels Auszählung auf einem Ausdruck ist verlässlich und kostengünstig [6].
Ki67 wird im „Hotspot“ (mind. 500 Zellen) gezählt (Abb. 2) und kann auf Schnittpräparaten heterogen sein [7]. Eine PHH3-Immunhistochemie detektiert Mitosefiguren und ist weniger gut untersucht als Mitosefiguren in der HE-Färbung oder Ki67. Gastroenteropankreatische neuroendokrine Karzinome (NEC), G3, zeigen bei einem Ki67 < 55 % ein reduziertes Ansprechen auf platinbasierte Therapie [8].
Somatostatinrezeptor 2A (SSTR2A)
Neuroendokrine Tumoren können Somatostatinrezeptoren (SSTR) exprimieren, die diagnostisch, prognostisch [9] und therapeutisch als Zielmoleküle relevant sind [10]. Somatostatinanaloga werden an radioaktive Isotope gekoppelt und binden so an SSTR (interne Peptidradiorezeptortherapie).
Der Nachweis von SSTR2A erfolgt immunhistochemisch. Die Expression ist membranär, allerdings heterogen und teils deutlich fixierungsabhängig (Cave: Fixierungsgradient als Artefakt, Abb. 2; [11]). Endothel kann als interne Positivkontrolle genutzt werden. Nichtfunktionelle pankreatische neuroendokrine Tumoren (PanNET) zeigen häufig eine starke Expression, während Insulinome häufig negativ sind. Eine Intensitätseinteilung in 3 Stufen hat sich bewährt (Abb. 2). Die Intensität und der prozentuale Anteil positiver Zellen (> 10 % membranäre Positivität) korrelieren mit der realen Rezeptordichte [12]. Zu beachten ist eine dosisabhängige SSTR2-Internalisierung bei Pankreasoperationen unter Einsatz von Somatostatinanaloga, bspw. Octreotid [13]. Dies ist nicht zu verwechseln mit einer schwachen Expression von SSTR2A.
p53 und Rb1
In der differentialdiagnostischen Abgrenzung von NET G3 zu NEC haben sich die beiden Marker p53 und Rb1 als hilfreich erwiesen. RB1 und TP53 sind Tumorsuppressorgene, die in PanNET ganz überwiegend einen Wildtypstatus aufweisen und in bis zu 70 % der NEC alteriert sein können (Abb. 3). Das Expressionsmuster von Rb1 gilt im Falle eines kompletten nukleären Verlustes als pathologisch, die Endothelien der intratumoralen Blutgefäße dienen als interne Positivkontrolle. Dieser Rb1-Verlust ist häufig epigenetisch induziert. Ein Rb1-Verlust kann einen prädiktiven Wert für eine platinbasierte Therapie haben [14].

Die Interpretation des Expressionsmusters von p53 ist komplexer. Hier sind sowohl der komplette nukleäre Verlust als auch die nukleäre Überexpression pathologisch und mit inaktivierender Mutation vergesellschaftet. Allerdings muss man in einigen Fällen zum einen zur Abgrenzung eines kompletten nukleären Verlustes positive Tumorzellkerne regelrecht suchen, zum anderen existiert kein verlässlicher Cut-off zur Definition einer nukleären Überexpression, was in einigen Fällen zu diagnostischer Unsicherheit führen kann. Eine starke nukleäre Expression von p53 in mehr als 20 % der Tumorzellen korreliert gut mit einer TP53-Mutation. Die pathologische Expression von Rb1 und p53 ist ein indirekter Hinweis auf eine Genalteration, der Mutationsstatus ist allerdings nicht vorhersagbar, da auch Veränderungen der Genkopienzahl und epigenetische Regulationen eine entscheidende Rolle spielen. Die definitive Einordnung von NET G3 und NEC muss allerdings in erster Linie aufgrund histologischer Wachstumsmuster erfolgen, da auch wenige NET G3 eine pathologische Expression von p53 (10 %) und Rb1 (selten) zeigen können [1, 3, 15, 16].
DAXX/ATRX und Menin
DAXX und ATRX codieren für Proteine, die die genomische Stabilität unterstützen, insbesondere in den Telomerregionen des Genoms. Somatische Mutationen in diesen Genen schließen sich gegenseitig aus und führen zu alternativer Telomerverlängerung (ALT). DAXX- und ATRX-Mutationen treten meist erst in größeren (> 2 cm) PanNET auf und sind ein Progressionsmechanismus.
Immunhistochemisch ist der nukleäre Expressionsverlust eines dieser Proteine mit einer schlechteren Prognose und einer höheren Rezidivrate bei PanNET assoziiert [15]. Endothel, Stroma oder Immunzellen können als interne Kontrollen verwendet werden, um falsch negative Befunde zu vermeiden.
MEN1-Mutationen sind die häufigsten Mutationen bei PanNET, nicht nur im Rahmen genetischer Syndrome, sondern auch bei sporadischen Tumoren. Der immunhistochemische Verlust der Menin-Expression, der auch im Zellkern zu beobachten ist, kann die Diagnose unterstützen. MEN1-Mutationen sind nicht mit schlechterer Prognose assoziiert.
Bedeutung von Transkriptionsfaktoren
Transkriptionsfaktoren helfen bei der Bestimmung des Ursprungs einer neuroendokrinen Neoplasie. Islet‑1 ist spezifisch für einen pankreatischen Primarius (kann im Duodenum oder Rektum ebenfalls positiv sein) [17, 18]. PDX1 und ARX als Transkriptionsfaktoren für β‑ und α‑Zellen können im Zweifel einen pankreatischen Ursprung weiter erhärten (wie auch ein Expressionsverlust von DAXX/ATRX). Während „thyroid transcription factor 1“ (TTF-1) in erster Linie einen pulmonalen oder thyreoidalen (medulläres Schilddrüsenkarzinom) Ursprung anzeigt, deutet CDX2 auf einen gastroenteropankreatischen Primarius. PITX2 wurde kürzlich als nützlicher Adjunkt für neuroendokrine Primarien des Mitteldarms beschrieben [19]. Falls es sich bei dem Primarius um ein neuroendokrines Karzinom handelt, sind diese Transkriptionsfaktoren nicht aussagekräftig.
Eine Erweiterung der diagnostischen Immunhistochemie ist insbesondere bei zweifelhafter Morphologie und klinisch-bildgeberischer Korrelation geboten. Dies gilt speziell für Differentialdiagnosen, welche eine neuroendokrine Morphologie imitieren (siehe Tab. 1 sowie Beitrag im vorliegenden Themenheft).
| Fragestellung | Immunhistochemische Marker |
|---|---|
| Neuroendokrine Neoplasie? | Chromogranin A, CK, INSM1, Synaptophysin |
| DD Neuroendokrines Karzinom | DAXX/ATRX/MEN1, p16, p53, Rb1 |
| Ursprung der neuroendokrinen Neoplasie | CDX2, DAXX/ATRX, Islet‑1, TTF‑1, PITX2, (ARX, PDX1) |
| MiNEN | BCL10, CA19.9, CEA, EMA, MUC1, Trypsin, ggf. weitere |
| Solid-pseudopapilläre Neoplasie (SPN) | β‑Catenin, Chromogranin A, CK |
| Metastase klarzelliges Nierenzellkarzinom | Chromogranin A, RCC (Islet-1/PAX8 können positiv sein) |
| Paragangliom | Zytokeratin negativ, GATA3 |
| Azinuszellkarzinom | BCL10, Trypsin |
| Adrenokortikales Karzinom | SF1 |
CK Zytokeratin, DD Differentialdiagnose, MiNEN gemischt neuroendokrin-nichtneuroendokrine Neoplasien, PAX8 „paired box gene 8“, RCC „renal cell carcinoma“, SF1 „steroidogenic factor 1“
Relevanz der Hormone
PanNETs können eine Vielzahl unterschiedlicher Peptidhormone produzieren, die neben den Hormonen der Langerhans-Inseln (Insulin, Glukagon, Somatostatin, pankreatisches Polypeptid) auch ektope Hormone einschließen (z. B. Serotonin, Gastrin, VIP, ACTH). In einigen Fällen ist die Hormonproduktion mit einem klinischen Syndrom assoziiert. Dann werden die Tumoren nach Ihrem produzierten Hormon benannt (u. a. Insulinom, Glukagonom, Gastrinom). Für die Routinediagnostik primärer PanNET ist der immunhistochemische Hormonnachweis nur in Ausnahmefällen relevant, da hormonaktive PanNET meist eine charakteristische hormonspezifische Klinik (Flush, Diarrhö, u. a.) zeigen, die den klinischen Kolleg*innen in Kombination mit laborchemischen Hinweisen in der Regel genügen. In der metastatischen Situation kann der immunhistochemische Nachweis von Peptidhormonen bei unbekanntem Primarius differentialdiagnostisch allerdings in vielen Fällen hilfreich sein [3, 20].
Immunhistochemische Tricks bei Pitfalls
NET G3 versus NEC
Die Unterscheidung zwischen NET G3 und NEC ist für die Wahl der Systemtherapie und die Prognose entscheidend. NET G3 entstehen im Pankreas häufiger als in anderen Organen des Gastrointestinaltrakts. Sowohl Histomorphologie, immunhistochemische und molekulare Untersuchungen als auch klinische Geschichte helfen bei der Unterscheidung ([14, 21]; Abb. 4, Tab. 2). Bei einem NET in der Vorgeschichte ist Vorsicht bei der Diagnosestellung eines NEC angebracht. In der seltenen Situation einer Transformation schlagen wir eine Bezeichnung als NET G3 mit „NEC-artigen Eigenschaften“, die klinische Bedeutung ist noch nicht geklärt, vor.

| NET G3 | NEC | |
|---|---|---|
| Wachstumsmuster | Organoid | Expansive solide/kribriforme Areale |
| Stroma | Hyalinisiert | Desmoplastisch |
| Kapilläre Gefäße | Eng mit Nestern assoziiert | Entfernt von Gefäßen |
| Nekrose | Fehlend oder punktförmig | Geografisch |
| Synaptophysin/Chromogranin A | Homogen stark | Fleckförmig |
| p53 | Pathologisch in 10 % | Pathologisch in 50 % |
| Rb1-Verlust | < 5 % | 70 % |
| p16-Überexpression | Nicht vorhanden | 70 % stark positiv (Achtung Zervix bei HPV) |
| Verlust DAXX/ATRX/MEN1 | 70 % von PanNET, in anderen NET weniger relevant | < 10 % |
| Ki-67 | Häufig < 55 % | Häufig > 55 % |
PanNET pankreatische neuroendokrine Tumoren
NEC vs. MiNEN vs. Adenokarzinom mit neuroendokriner Differenzierung
Die Histomorphologie ist entscheidend für die Unterscheidung zwischen NEC, gemischten neuroendokrinen Neoplasien (MiNEN) und Adenokarzinomen mit neuroendokriner Differenzierung (ADNE).
Morphologische Adenokarzinome, die diffus Synaptophysin exprimieren, sind im Kolon gut untersucht und als ADNE beschrieben. Sie kommen selten auch im Pankreas vor. Neben der morphologisch erkennbaren drüsigen Komponente kann der fehlende Nachweis einer relevanten Expression von Chromogranin A und INSM1 helfen. Da zur Diagnose eines MiNEN mindestens 30 % beider Komponenten vorliegen müssen, empfehlen wir bei Biopsaten eine beschreibende Diagnose einer gemischten Neoplasie (Adenokarzinom und großzelliges NEC) mit Kommentierung, dass formell 30 % notwendig sind. Das gemischte azinär-neuroendokrine Karzinom als Subtyp einer MiNEN wird insbesondere als metastatische Läsion nicht selten als NET G3 verkannt.
Anwendung von Molekularpathologie in pankreatischen neuroendokrinen Neoplasien
Molekulare Schlüsselalterationen in pankreatischen neuroendokrinen Neoplasien sind MEN1-Mutationen als initiierende Veränderung sowie DAXX- oder ATRX-Mutationen im Rahmen der Progression [22]. Eine Anwendung von DAXX/ATRX-Immunhistochemie hat sich als prognostischer Marker zusätzlich zu Ki67 bewährt. Die immunhistochemische Untersuchung von Menin sowie die Anwendung der Fluoreszenz-in-situ-Hybridisierung zur Detektion alternativer Telomerverlängerung (ALT) haben bislang keinen Eingang in die Routine gefunden. Next Generation Sequencing (NGS) kann in Einzelfällen zur Unterscheidung von NET G3 versus NEC bei inkonklusiver Morphologie und Immunhistochemie hilfreich sein, da NEC genetische Alterationen aufweisen können, die dem pankreatischen duktalen Adenokarzinom näherstehen.
In kolorektalen neuroendokrinen Neoplasien wurden Mikrosatelliteninstabilität sowie BRAF-Mutationen [23, 24] als therapeutisch relevant beschrieben. In PanNEN dagegen gibt es diesbezüglich keine klare Datenlage. Mausmodelle zeigen bei pankreatischen neuroendokrinen Tumoren verschiedene molekularpathologische Subtypen mit heterogenem metabolischen Profil und unterschiedlicher biologischer Aggression [25]. Reproduzierbare Indikatormoleküle für diese Subtypen sind bisher nicht publiziert.
Fazit für die Praxis
Synaptophysin, Chromogranin A und Insulinoma-assoziiertes Protein 1 (INSM1) sind etablierte Marker zur Determinierung einer neuroendokrinen Neoplasie.
Die Proliferationsfraktion Ki67 ist unerlässlich zur WHO-konformen Graduierung pankreatischer neuroendokriner Neoplasien und hat eine hohe prognostische Bedeutung.
Somatostatinrezeptoren sind potenzielle therapeutische Targets und können immunhistochemisch detektiert werden.
p53 und Rb1 helfen häufig in der Differenzierung zwischen pankreatischen neuroendokrinen Tumoren (PanNET) G3 und neuroendokrinen Karzinomen (NEC). Morphologische Kriterien sind obligat zu beachten.
Die Differentialdiagnose zur pankreatischen neuroendokrinen Neoplasie ist breit. Immunhistochemische Untersuchungen helfen in der Typisierung.
Molekularpathologische Untersuchungen haben aktuell einen geringen diagnostischen Stellenwert.
Funding
Open access funding provided by University of Bern
Einhaltung ethischer Richtlinien
Interessenkonflikt
K. Bräutigam, A. Chouchane, B. Konukiewitz und A. Perren geben an, dass kein Interessenkonflikt besteht.
Für diesen Beitrag wurden von den Autor/-innen keine Studien an Menschen oder Tieren durchgeführt. Für die aufgeführten Studien gelten die jeweils dort angegebenen ethischen Richtlinien.
Footnotes
Konstantin Bräutigam und Aziz Chouchane teilen die Erstautorenschaft, Björn Konukiewitz und Aurel Perren die Letztautorenschaft.

QR-Code scannen & Beitrag online lesen
Hinweis des Verlags
Der Verlag bleibt in Hinblick auf geografische Zuordnungen und Gebietsbezeichnungen in veröffentlichten Karten und Institutsadressen neutral.
Literatur
- 1.Nagtegaal ID, Odze RD, Klimstra D, Paradis V, Rugge M, Schirmacher P, et al. The 2019 WHO classification of tumours of the digestive system. Histopathology. 2020;76(2):182–188. doi: 10.1111/his.13975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Litmeyer AS, Konukiewitz B, Kasajima A, Foersch S, Schicktanz F, Schmitt M, et al. High expression of insulinoma-associated protein 1 (INSM1) distinguishes colorectal mixed and pure neuroendocrine carcinomas from conventional adenocarcinomas with diffuse expression of synaptophysin. J Pathol Clin Res. 2023 doi: 10.1002/cjp2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konukiewitz B, Jesinghaus M, Kasajima A, Klöppel G. Neuroendocrine neoplasms of the pancreas: diagnosis and pitfalls. Virchows Arch. 2022;480(2):247–257. doi: 10.1007/s00428-021-03211-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Konukiewitz B, Kasajima A, Schmitt M, Schwamborn K, Groll T, Schicktanz F, et al. Neuroendocrine differentiation in conventional colorectal adenocarcinomas: incidental finding or prognostic biomarker? Cancers. 2021;13(20):5111. doi: 10.3390/cancers13205111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pelosi G, Bresaola E, Bogina G, Pasini F, Rodella S, Castelli P, et al. Endocrine tumors of the pancreas: Ki-67 immunoreactivity on paraffin sections is an independent predictor for malignancy: A comparative study with proliferating-cell nuclear antigen and progesterone receptor protein immunostaining, mitotic index, and other clinicopathologic variables. Hum Pathol. 1996;27(11):1124–1134. doi: 10.1016/S0046-8177(96)90303-2. [DOI] [PubMed] [Google Scholar]
- 6.Reid MD, Bagci P, Ohike N, Saka B, Erbarut Seven I, Dursun N, et al. Calculation of the Ki67 index in pancreatic neuroendocrine tumors: a comparative analysis of four counting methodologies. Mod Pathol. 2015;28(5):686–694. doi: 10.1038/modpathol.2014.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sobecki M, Mrouj K, Colinge J, Gerbe F, Jay P, Krasinska L, et al. Cell-cycle regulation accounts for variability in Ki-67 expression levels. Cancer Res. 2017;77(10):2722–2734. doi: 10.1158/0008-5472.CAN-16-0707. [DOI] [PubMed] [Google Scholar]
- 8.Sorbye H, Welin S, Langer SW, Vestermark LW, Holt N, Osterlund P, et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): The NORDIC NEC study. Ann Oncol. 2013;24(1):152–160. doi: 10.1093/annonc/mds276. [DOI] [PubMed] [Google Scholar]
- 9.Nielsen K, Binderup T, Langer SW, Kjaer A, Knigge P, Grøndahl V, et al. P53, Somatostatin receptor 2a and Chromogranin A immunostaining as prognostic markers in high grade gastroenteropancreatic neuroendocrine neoplasms. Bmc Cancer. 2020;20(1):27. doi: 10.1186/s12885-019-6498-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caplin ME, Pavel M, Ćwikła JB, Phan AT, Raderer M, Sedláčková E, et al. Anti-tumour effects of lanreotide for pancreatic and intestinal neuroendocrine tumours: the CLARINET open-label extension study. Endocr Relat Cancer. 2016;23(3):191–199. doi: 10.1530/ERC-15-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Volante M, Brizzi MP, Faggiano A, Rosa SL, Rapa I, Ferrero A, et al. Somatostatin receptor type 2A immunohistochemistry in neuroendocrine tumors: a proposal of scoring system correlated with somatostatin receptor scintigraphy. Mod Pathol. 2007;20(11):1172–1182. doi: 10.1038/modpathol.3800954. [DOI] [PubMed] [Google Scholar]
- 12.Körner M, Waser B, Schonbrunn A, Perren A, Reubi JC. Somatostatin receptor subtype 2A Immunohistochemistry using a new monoclonal antibody selects tumors suitable for in vivo Somatostatin receptor targeting. Am J Surg Pathol. 2012;36(2):242. doi: 10.1097/PAS.0b013e31823d07f3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reubi JC, Waser B, Cescato R, Gloor B, Stettler C, Christ E. Internalized somatostatin receptor subtype 2 in neuroendocrine tumors of octreotide-treated patients. J Clin Endocrinol Metab. 2010;95(5):2343–2350. doi: 10.1210/jc.2009-2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Venizelos A, Elvebakken H, Perren A, Nikolaienko O, Deng W, Lothe IMB, et al. The molecular characteristics of high-grade gastroenteropancreatic neuroendocrine neoplasms. Endocr Relat Cancer. 2022;29(1):1–14. doi: 10.1530/ERC-21-0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konukiewitz B, Schlitter AM, Jesinghaus M, Pfister D, Steiger K, Segler A, et al. Somatostatin receptor expression related to TP53 and RB1 alterations in pancreatic and extrapancreatic neuroendocrine neoplasms with a Ki67-index above 20. Mod Pathol. 2017;30(4):587–598. doi: 10.1038/modpathol.2016.217. [DOI] [PubMed] [Google Scholar]
- 16.Kasajima A, Konukiewitz B, Schlitter AM, Weichert W, Klöppel G. An analysis of 130 neuroendocrine tumors G3 regarding prevalence, origin, metastasis, and diagnostic features. Virchows Arch. 2022;480(2):359–368. doi: 10.1007/s00428-021-03202-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graham RP, Shrestha B, Caron BL, Smyrk TC, Grogg KL, Lloyd RV, et al. Islet-1 is a sensitive but not entirely specific marker for pancreatic neuroendocrine neoplasms and their metastases. Am J Surg Pathol. 2013;37(3):399. doi: 10.1097/PAS.0b013e31826f042c. [DOI] [PubMed] [Google Scholar]
- 18.Schmitt AM, Riniker F, Anlauf M, Schmid S, Soltermann A, Moch H, et al. Islet 1 (Isl1) Expression is a Reliable Marker for Pancreatic Endocrine Tumors and Their Metastases. Am J Surg Pathol. 2008;32(3):420. doi: 10.1097/PAS.0b013e318158a397. [DOI] [PubMed] [Google Scholar]
- 19.Soukup J, Manethova M, Faistova H, Krbal L, Vitovcova B, Hornychova H, et al. Pitx2 is a useful marker of midgut-derived neuroendocrine tumours—an immunohistochemical study of 224 cases. Histopathology. 2022;81(6):799–807. doi: 10.1111/his.14789. [DOI] [PubMed] [Google Scholar]
- 20.Konukiewitz B, von Hornstein M, Jesinghaus M, Steiger K, Weichert W, Detlefsen S, et al. Pancreatic neuroendocrine tumors with somatostatin expression and paraganglioma-like features. Hum Pathol. 2020;102:79–87. doi: 10.1016/j.humpath.2020.07.004. [DOI] [PubMed] [Google Scholar]
- 21.Elvebakken H, Perren A, Scoazec JY, Tang LH, Federspiel B, Klimstra DS, et al. A consensus-developed morphological re-evaluation of 196 high-grade gastroenteropancreatic neuroendocrine neoplasms and its clinical correlations. Neuroendocrinology. 2021;111(9):883–894. doi: 10.1159/000511905. [DOI] [PubMed] [Google Scholar]
- 22.Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331(6021):1199–1203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klempner SJ, Gershenhorn B, Tran P, Lee TK, Erlander MG, Gowen K, et al. BRAFV600E mutations in high-grade colorectal neuroendocrine tumors May predict responsiveness to BRAF-MEK combination therapy. Cancer Discov. 2016;6(6):594–600. doi: 10.1158/2159-8290.CD-15-1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sahnane N, Furlan D, Monti M, Romualdi C, Vanoli A, Vicari E, et al. Microsatellite unstable gastrointestinal neuroendocrine carcinomas: a new clinicopathologic entity. Endocr Relat Cancer. 2015;22(1):35–45. doi: 10.1530/ERC-14-0410. [DOI] [PubMed] [Google Scholar]
- 25.Sadanandam A, Wullschleger S, Lyssiotis CA, Grötzinger C, Barbi S, Bersani S, et al. A cross-species analysis in pancreatic neuroendocrine tumors reveals molecular subtypes with distinctive clinical, metastatic, developmental, and metabolic characteristics. Cancer Discov. 2015;5(12):1296–1313. doi: 10.1158/2159-8290.CD-15-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]