

Abstract
The soluble [NiFe]-hydrogenase (SH) of the facultative lithoautotrophic proteobacterium Ralstonia eutropha H16 has up to now been described as a heterotetrameric enzyme. The purified protein consists of two functionally distinct heterodimeric moieties. The HoxHY dimer represents the hydrogenase module, and the HoxFU dimer constitutes an NADH-dehydrogenase. In the bimodular form, the SH mediates reduction of NAD+ at the expense of H2. We have purified a new high-molecular-weight form of the SH which contains an additional subunit. This extra subunit was identified as the product of hoxI, a member of the SH gene cluster (hoxFUYHWI). Edman degradation, in combination with protein sequencing of the SH high-molecular-weight complex, established a subunit stoichiometry of HoxFUYHI2. Cross-linking experiments indicated that the two HoxI subunits are the closest neighbors. The stability of the hexameric SH depended on the pH and the ionic strength of the buffer. The tetrameric form of the SH can be instantaneously activated with small amounts of NADH but not with NADPH. The hexameric form, however, was also activated by adding small amounts of NADPH. This suggests that HoxI provides a binding domain for NADPH. A specific reaction site for NADPH adds to the list of similarities between the SH and mitochondrial NADH:ubiquinone oxidoreductase (Complex I).
Hydrogenases (reaction H2 ↔ H− + H+ ↔ 2H+ + 2e−) are the key enzymes in the H2 metabolism of many microorganisms. All hydrogenases are metalloenzymes. Presently, three main classes are known. Although these classes are phylogenetically unrelated (16, 76, 77), it is most amazing to note that the active sites of hydrogenases have two properties in common: (i) all contain Fe and most contain Ni as well, and (ii) all contain CO as ligand to Fe and most contain CN as ligand as well. Most enzymes belong to the class of [NiFe]-hydrogenases, which have a (CysS)2Ni(μ-′O′)(μ-CysS)2Fe(CN)2(CO) active site in the aerobically isolated form (5, 6, 26, 50, 78, 79). Very recent crystallographic studies indicated that the oxygen species in the ′O′ bridge can be a di-oxo species (peroxide) or a mono-oxy species (hydroxide) (A. Volbeda, personal communication). When the oxygen bridge is present, the enzymes are inactive. Reduction with H2 removes this ligand and replaces it with a hydride, resulting in active enzymes (11, 22, 64). The second class of hydrogenases, the [FeFe]-hydrogenases (previously called Fe-only or [Fe]-hydrogenases) contain a (CN)(CO)(′O′)Fe(μ-CO)(μ-SRS)Fe(CysS)(CN)(CO) group as the active site [R = NH(CH2-)2] (45, 46, 49, 51, 74). Also, here, the ′O′ species is present only in the inactive state of these enzymes. The [Fe]-hydrogenases form the third class and contain a Fe(CO)2 group bound to an organic cofactor (38, 39, 62). No crystal structure of a member of this class is available yet.
The facultative chemolithoautotrophic proteobacterium Ralstonia eutropha H16 (Table 1) (formerly Alcaligenes eutrophus H16 [18]) is able to use hydrogen as the sole energy source in an oxic environment. Energy-yielding H2 oxidation in this bacterium is catalyzed by two [NiFe]-hydrogenases: (i) a membrane-bound enzyme (MBH) which is associated with the respiratory chain via a b-type cytochrome and (ii) a cytoplasmic enzyme (soluble [NiFe]-hydrogenase [SH]) which couples oxidation of H2 to the reduction of NAD+. The SH can also mediate the reverse reaction, the production of H2 from NADH, albeit at a low rate (7, 54, 58).
TABLE 1.
Strains and plasmids
| Strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| Ralstonia eutropha | ||
| H16 | SH+MBH+ | DSM 428, ATCC 17699 |
| HF160 | SH−MBH+HoxI−hoxF::Tn5 | 35, 47 |
| HF412 | SH+MBH+hoxIΔ | 67 |
| HF424 | SH−MBH−hoxFUYHWΔ hoxGΔ | 41 |
| Escherichia coli | ||
| S17-1 | Tra+recA pro thi hsdR chr::RP4-2 | 63 |
| JM109 | F′ traD36 lacIq, Δ(lacZ)M15 proA+B+/e14− (McrA−) Δ(lac-proAB) thi gyrA96 (Nalr) endA1 hsdR17(rK−mK+) relA1 supE44 recA1 | 82 |
| XL1-Blue | recA thi hsdR1 supE44 relA1 lac(F′ proAB lacqlacZΔM15 Tn10[Tet]) | 13 |
| Plasmids | ||
| pEDY309 | Tcr RK2 ori Mob+; promoterless lacZ gene | 31 |
| pVK101 | Kmr Tcr RP4ori | 37 |
| pCH234 | 1.3-kb KpnI fragment of pGE15 in pTZ18R | 67 |
| pCH291 | 2.9-kb HindIII-BamHI fragment of pGE15 in pTZ18R | A. Tran-Betcke and B. Friedrich |
| pCH455 | 15-kb HindIII of pGE15 in pBlueKS+ | 41 |
| pCH552 | hoxHYΔ in pCH455 | 41 |
| pCH781 | 0.9-kb XhoI fragment of pCH234 | This study |
| pCH1084 | Derivative of pCH234 with a 937-bp SacII-EcoRI PCR product containing the Strep-tag II sequence at the 5′ end of hoxI | This study |
| pCH1085 | Derivative of pCH291 with a 818-bp HindIII-NcoI PCR product containing the Strep-tag II sequence at the 5′ end of hoxF | This study |
| pCH1086 | 11.8-kb BamHI fragment of pCH455 in pCH1085 | This study |
| pCH1087 | 10.6-kb BamHI fragment of pCH552 in pCH1085 | This study |
| pGE15 | 14.6-kb HindIII fragment of pHG1 in pVK101 | 69 |
| pGE151 | Derivative of pRK404 | 34 |
| pGE346 | SH−MBH+hoxFUΔ | 41 |
| pGE371 | SH−MBH+hoxYHΔ | 41 |
| pGE436 | 0.9-kb KpnI-HindIII fragment of pCH781 in pGE151 | This study |
| pGE549 | Derivative of pGE15 containing the 1.4-kb KpnI fragment of pCH1084 | This study |
| pGE550 | Derivative of pGE371 containing the 1.4-kb KpnI fragment of pCH1084 | This study |
| pGE551 | Derivative of pGE346 containing the 1.4-kb KpnI fragment of pCH1084 | This study |
| pGE552 | 14.6-kb HindIII fragment of pCH1086 in pEDY309 | This study |
| pGE553 | 13.4-kb HindIII fragment of pCH1087 in pVK101 | This study |
The SH of R. eutropha, which is the subject of this study, is a member of the heteromultimeric [NiFe]-hydrogenases and the first representative of the subgroup of bidirectional NAD-linked hydrogenases to be isolated (58, 77). Related hydrogenases are present in Rhodococcus opacus (24, 56), in several species of cyanobacteria (65), and in Thiocapsa roseopersicina (53). The SH consists of four heterologous subunits forming two functional modules: a hydrogenase dimer encoded by the hoxH and hoxY genes and an NADH-dehydrogenase dimer encoded by hoxF and hoxU. The latter moiety provides the NAD+-binding site and harbors several Fe-S clusters in addition to one flavin mononucleotide (FMN) cofactor (called FMN-b [41, 69, 72]). The HoxH subunit of the SH contains six conserved motifs, which are typical of [NiFe] hydrogenases. The amino acids identified as conserved signatures, among which are two pairs of cysteines, surround the Ni-Fe active site (15, 40). The HoxY subunit of the SH is a truncated version of the small subunit of standard [NiFe]-hydrogenases and presumably accommodates only the proximal [4Fe-4S] cluster. Recently, a second FMN (called FMN-a) was identified in the SH (72). It was postulated that the hydride formed upon H2 cleavage at the Ni-Fe site is transferred to FMN-a, from where the electrons proceed via the Fe-S clusters to FMN-b and finally to NAD+.
In addition to the four hox genes mentioned above, the SH operon contains two additional genes, hoxW and hoxI, downstream of the hoxF, hoxU, hoxY, and hoxH genes. The hoxW gene encodes an SH-specific endopeptidase that removes 24 amino acids from the C-terminal end of the HoxH precursor prior to subunit assembly. The role of hoxI (formerly designated orf2) has up to now been elusive (67).
In this study, we have investigated the relationship between HoxI and the SH. We demonstrate that HoxI is identical with the so-called B protein which has been shown to be coexpressed with the SH at high levels (30). HoxI harbors a putative cyclic nucleotide-binding site in its central domain. The routine purification procedure used in this field since 1983 (57) has focused on the isolation of an enzyme which displays H2-NAD+ activity, finally yielding a heterotetrameric SH. We have modified the purification protocol and characterized the enzyme as a hexameric [NiFe] hydrogenase. Whereas rapid activation of the tetrameric SH is achieved only with NADH but not with NADPH, the hexameric SH is activated by addition of both forms of the nucleotide. This suggests that HoxI provides a specific binding domain for NADPH.
MATERIALS AND METHODS
Strains and plasmids.
The strains and plasmids used are listed in Table 1. Strains with the initials HF were derived from R. eutropha H16 (ATCC 17699) harboring the endogenous megaplasmid pHG1. Escherichia coli JM109 (82) and XL1-Blue (13) were used for standard cloning procedures. For conjugative transfer, E. coli S17-1 served as donor strain (63).
Recombinant DNA techniques.
The 8-amino-acid peptide Strep-tag II (WSHPQFEK [33]) specifically interacts with an immobilized variant of streptavidin called StrepTactin allowing the one-step purification of proteins under mild conditions. The Strep-tag II sequence was fused to the N terminus of the HoxF and HoxI proteins. To construct a Strep-tag-II-HoxI fusion protein, plasmid pCH234 was amplified by inverse PCR with Pfx polymerase (Invitrogen, Carlsbad, California) using the forward primer 5′-ATGGCTAGCTGGAGCCACCCGCAGTTCGAAAAAGGCGCCAAAGAGCAGGAAATCGCAGGATCGCAACGATGATC-3′ and the reverse primer 5′-CTAGCTAGCCATCGCGTTCTCCTTCTAACTG-3′. The forward primer contained the Strep-tag II sequence and a 2-amino acid spacer at both ends (NH2-AS, GA-COOH [33]). The 1.4-kb PCR product was digested with NheI (this site was introduced via the N-terminal spacer) and religated. A 937-bp SacII-EcoRI fragment was then isolated and cloned into SacII-EcoRI-digested pCH234, resulting in plasmid pCH1084. The 1.4-kb KpnI fragment of pGE15 was replaced with the appropriate fragment of pCH1084. Plasmid pGE549 was transferred from E. coli S17-1 to R. eutropha HF160 by agar-spot mating (19). Due to a Tn5 insertion, HF160 is SH− and HoxI− (47). The 1.4-kb KpnI fragment with the Strep-tag II sequence was also introduced into pGE371 (HoxYH−) and pGE346 (HoxFU−), yielding plasmids pGE550 and pGE551, respectively.
Inverse PCR was used to introduce the Strep-tag II sequence into pCH291 harboring hoxF (forward primer, 5′-ATGGCTAGCTGGAGCCACCCGCAGTTCGAAAAAGGCGCCGATAGTCGTATCACGACAATACTCGAGCGCTACCGC-3′; reverse primer, 5′-TACGCTAGCCATGTTGTCTCCTCCTTACTA-3′). The 2.8-kb PCR product was digested with NheI and ligated. An 818-bp HindIII-NcoI fragment was isolated and used to replace the unmodified fragment of pCH291, resulting in plasmid pCH1085. Plasmid pCH1086 was obtained after cloning an 11.8-kb BamHI fragment of pCH455 into BamHI-digested pCH1085. A 14.6-kb HindIII fragment of pCH1086 was ligated with pEDY309 (HindIII) to give plasmid pGE552. To produce the Strep-tagged HoxF derivative in a HoxHY− background, the 10.6-kb BamHI hoxYHΔ fragment of pCH552 was cloned into pCH1085. The 13.4-kb HindIII fragment of the resulting plasmid pCH1087 was ligated with pVK101 (yielding pGE553). The HoxF derivatives were introduced into HF424 (SH−MBH−) or HF160 (SH−HoxI−).
Cell growth and preparation of soluble extracts for immunoblot analysis.
Escherichia coli strains were cultivated in Luria-Bertani medium (43). For isolation of the SH, R. eutropha cells were heterotrophically grown in fructose-glycerol minimal medium (FGN medium [60]) in a Biostat D fermentor (Braun, Melsungen, Germany) at a 50-l scale at 30°C under hydrogenase-derepressing conditions. Cells were harvested at an optical density at 436 nm of 9 to 11, rapidly frozen in liquid nitrogen, and stored at −30°C. The Strep-tagged SH derivatives were cultivated in 200 ml FGN medium in 1-liter baffled Erlenmeyer flasks and were harvested after 48 h at an optical density at 436 nm of 10 to 12.
Soluble extracts for immunoblot analysis were prepared from 50-ml FGN cultures. The cell pellets were washed once and resuspended into 2 ml 50 mM potassium phosphate (KPi) buffer (pH 7.0). After two passages through a chilled French pressure cell at 6.2 MPa, the crude extracts were subjected to ultracentrifugation for 30 min at 100,000 × g. Proteins were separated by electrophoresis in sodium dodecyl sulfate (SDS)-12.5% or 15% polyacrylamide gels and transferred to nitrocellulose membranes (Biotrace NT; Pall, Michigan) according to a standard protocol (68). Protein standards were purchased from Invitrogen or New England Biolabs (Beverly, Massachussetts). Protein concentrations were determined by the method of Bradford (10).
Purification procedures.
The tetrameric SH was routinely purified at 4°C according to a standard protocol (20) but omitting the cethyltrimethyl-ammoniumbromide treatment. The enzyme was dissolved in 50 mM Tris-HCl (pH 8.0) and stored in liquid nitrogen.
The alternative procedure for SH purification was as follows. Cells (60 g [wet weight]) were washed with 50 mM KPi buffer (pH 7) and resuspended in 40 ml of the same buffer containing DNaseI and 0.1 mM phenylmethylsulfonyl fluoride. After two passages through a chilled French pressure cell at 7.6 MPa, cell debris and membrane fraction were removed by ultracentrifugation at 100,000 × g twice for 30 min. The supernatant was brought to a 30% ammonium sulfate saturation and centrifuged at 10,000 × g for 20 min. Enzyme was precipitated by 60% ammonium sulfate saturation. After centrifugation, the pellet was dissolved in 30 ml 50 mM KPi buffer, pH 7. The protein solution was dialyzed twice for 2 h against 2 liters of fresh buffer containing 0.05 mM phenylmethylsulfonyl fluoride. The dialyzed extract was applied to a DEAE Sephacel column (350 ml; Amersham Biosciences, Uppsala, Sweden). After washing with one column volume of 50 mM KPi buffer (pH 7), a linear gradient of 3.5 column volumes of 0 to 300 mM potassium chloride (in KPi buffer) was applied. Fractions containing SH were combined, concentrated by ammonium sulfate precipitation, dialyzed against 50 mM KPi buffer (pH 7), and subjected to gel filtration (Superdex 200; Amersham Biosciences). This purification resulted in the hexameric SH (see Results). To convert the hexameric SH into the tetrameric form, a second gel filtration column was run in 50 mM Tris-HCl buffer (pH 8). Isolation of tetrameric SH was also achieved by using a hydrophobic-interaction column instead of gel filtration. Fractions after DEAE were precipitated with 60% ammonium sulfate, redissolved in 200 mM KPi buffer, and applied to a phenyl Sepharose column (Phenyl Sepharose 6 Fast Flow, 100 ml; Amersham Biosciences). After two washing steps with 200 and 50 mM KPi buffer, pH 7, the enzyme was eluted using a linear gradient ranging from 10 to 0 mM KPi buffer (three column volumes). SH-containing fractions were immediately dialyzed against fresh 50 mM KPi buffer (pH 7). For affinity chromatography, the cells were resuspended in 2 ml 50 mM KPi buffer (pH 7) or 100 mM Tris-HCl with 150 mM NaCl (pH 8.0) containing DNaseI. After two passages through a chilled French pressure cell at 6.2 MPa, the suspension was centrifuged at 100,000 × g for 30 min. The soluble extract was applied to a 1-ml Streptactin Superflow column (IBA, Göttingen, Germany), washed six times with 500 μl resuspension buffer, and eluted with the same buffer containing 5 mM desthiobiotin (six times with 500 μl). Afterward, the column was washed and regenerated according to the manufacturer's manual.
Activity assays and activation procedures.
NAD(P)H oxidation with K3Fe(CN)6 as electron acceptor was measured aerobically at room temperature in a 1-ml cuvette. The absorption decrease at 420 nm was monitored using a Varian Cary 50 UV-visible spectrophotometer [ɛ = 1 mM−1 cm−1 for K3Fe(CN)6]. The reaction mixture contained 1.25 mM NAD(P)H and enzyme (17 to 100 nM) in 50 mM Tris-HCl buffer (pH 8.0). The reaction was started by the addition of 1 mM K3Fe(CN)6. Corrections were made for the slow direct reduction of K3Fe(CN)6 by NAD(P)H. The oxidation of H2 in 50 mM Tris-HCl buffer (pH 8.0) with benzyl viologen (BV), NAD(P)+, or K3Fe(CN)6 as electron acceptors was measured amperometrically at 30°C in a cell (2.15 ml) equipped with a Clark-type electrode (YSI 5331) (17). Enzyme (17 to 100 nM) and H2 (36 μM to 90 μM), in the form of H2-satured water, were added. The enzyme was activated with 5 μM NADH before the reaction was started by the addition of 2.5 mM BV, 5 mM NAD(P)+, or 2 mM K3Fe(CN)6.
To study the activation of the enzyme by NAD(P)H, either NADH (5 μM) or varying concentrations of NADPH were added; NAD+ or K3Fe(CN)6 was used as an acceptor. The NAD(P)H oxidation with K3Fe(CN)6 as electron acceptor was also performed anaerobically. In this case, all solutions were flushed with Ar, and glucose (50 mM) plus glucose oxidase (1.5 mg/ml) was added to remove traces of oxygen. Three minutes later, enzyme, NAD(P)H, H2, and K3Fe(CN)6 were added, in that order. All specific activities were based on Bradford protein determinations (10).
Cross-linking reaction.
The concentration of the SH in the cross-linking reactions was adjusted to 1 mg/ml in 20 mM sodium phosphate buffer (pH 8.0). Cross-linker bis-sulfosuccinimidylsuberate (BS3) was used at a concentration of 0.5 mM. The cross-linking reaction was performed at room temperature for 30 min. The cross-linked protein was analyzed by SDS-10% polyacrylamide gel electrophoresis (PAGE) (36). Cross-linked bands were cut out and subjected to mass spectrometric analysis.
Mass spectrometry, Edman degradation, and protein identification.
For matrix-assisted laser desorption ionization (MALDI) analysis, protein-containing gel slices were S-alkylated, digested with trypsin (Roche, Basel, Switzerland), and extracted according to the protocol of Shevchenko et al. (61). Peptides were collected and desalted on ZipTip μC18 pipette tips (Millipore, Bedford, MA) according to the manufacturer's instructions and eluted into 10 μl 60% acetonitrile-0.1% HCOOH. A sample (0.5 μl) of the resultant eluate was mixed (1:1, vol/vol) with 10 mg/ml cyanohydroxycinnaminic acid and spotted onto MALDI target plates. Reflectron MALDI-time of flight (TOF) spectra were acquired on a TOFSPEC 2E-C mass spectrometer (Micromass, Wythenshawe, United Kingdom) and used to query the ABCC nonredundant protein database (release 01042001). Determination of the subunit stoichiometry was performed with the aid of a Procise 494A protein sequencer (Applied Biosystems, Foster City, CA). Ten cycles of automated Edman degradation of the intact enzyme preparation were performed, and peak heights of the separated PTH amino acids were used for quantification of the relative protein amounts.
RESULTS
Identification of HoxI as a product of the SH-specific gene cluster.
An in-frame deletion in hoxI had, at first glance, no detectable effect on the SH activity (67). However, a major protein species usually coexpressed with the R. eutropha SH was missing in SDS-PAGE gels (S. Thiemermann and B. Friedrich, unpublished data). Earlier studies by Kärst et al. (30) showed that several proteins are specifically synthesized under hydrogenase-derepressing conditions in R. eutropha H16. One of them, a 19-kDa species called the B protein, had been purified. As the hoxI gene encodes a protein of similar size (18.6 kDa), we reinvestigated the genetic origin of the B protein. An immunoblot using an antibody against the B protein revealed the absence of this protein in the hoxI deletion mutant HF412. Genetic complementation with pGE436 containing the hoxI gene cloned in the broad-host-range vector pGE151 restored the B-protein-specific signal (Fig. 1). This result unambiguously identified the B protein as the product of the hoxI gene.
FIG. 1.
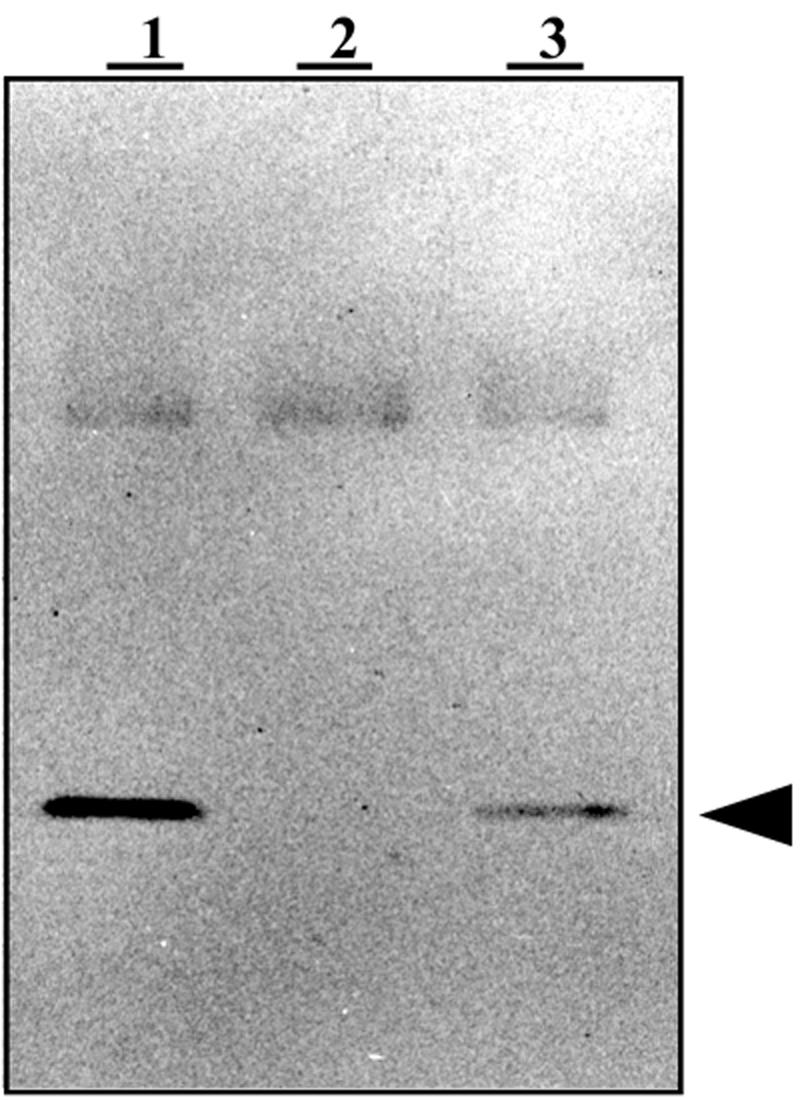
The B protein is the product of hoxI. Soluble extracts (15 μg) from wild-type H16 (pGE151) (lane 1), the HoxI− mutant HF412 (pGE151) (lane 2), and the HoxI+ transconjugant HF412 (pGE436) (lane 3) were subjected to SDS-PAGE separation and blotted against an antibody that had been raised against the B protein. The vector pGE151 was used as a control. The signal corresponding to the HoxI protein is indicated by an arrowhead.
HoxI copurifies with the SH.
The fact that the B protein coeluted with the SH on different chromatographic media (30) prompted us to reinvestigate the purification conditions. After anion exchange chromatography on DEAE, a 19-kDa protein coeluted with the SH. This complex could be further purified by gel filtration in 50 mM KPi buffer (pH 7.0) (Fig. 2A, lane 1). The fast moving band corresponded to the HoxI protein, as identified by immunological analysis (Fig. 2B, lane 1). A second gel filtration step in 50 mM Tris-HCl (pH 8) led to the isolation of the heterotetrameric SH (Fig. 2A, lane 2) and the release of HoxI (Fig. 2A, lane 3, and B, lane 2). Pure SH tetramer was also obtained after hydrophobic interaction on phenyl Sepharose or affinity chromatography on Procion-Red. The latter method has routinely been used for SH purification, since an improved yield and specific activity were reported (57).
FIG. 2.

Purification of the SH and release of HoxI. (A) The Coomassie blue-stained SDS-12.5% PAGE gel shows the SH subunits HoxF, HoxH, HoxU, and HoxY and the accessory protein HoxI. Purified proteins (5 μg) were applied to each lane. Enzyme after anionic-exchange chromatography and gel filtration in 50 mM KPi buffer (pH 7) is shown in lane 1. After purification, the enzyme was subjected to additional gel filtration in 50 mM Tris-HCl (pH 8.0). Two protein peaks were observed: the high-molecular-mass peak contained tetrameric SH (lane 2), and the low-molecular-mass peak contained the accessory protein HoxI (lane 3). (B) The SH-HoxI complex after gel filtration at pH 7 (see panel A, lane 1) and the HoxI fraction after dissociation of the complex during additional gel filtration in 50 mM Tris-HCl buffer at pH 8 (Fig. 2A, lane 3) were subjected to immunoblot analysis using the Anti-HoxI antibody. Purified proteins (5 μg) were applied to each lane. Lane 1, SH-HoxI complex; lane 2, purified HoxI.
Thus, careful analysis revealed that HoxI copurified with the SH only under certain purification conditions. The stability of the HoxI-containing enzyme complex strongly depended on both the ionic strength and the pH of the buffer (Table 2). HoxI could be maintained only in a complex with the SH with 50 mM KPi buffer between pH 6 and 7. At higher or lower ionic strength or at an alkaline pH, HoxI was released from the complex.
TABLE 2.
Stability of the SH-HoxI complex in different buffer systems
| Buffer | pH | SH-HoxI complex |
|---|---|---|
| 50 mM KPi | 7.0 | + |
| 200 mM KPi | 7.0 | − |
| 50 mM KPi | 6.2 | + |
| 50 mM KPi | 8.0 | − |
| 50 mM Tris-HCl | 8.0 | − |
| 50 mM Tris-HCl | 7.2 | − |
| 200 mM Tris-HNO3 | 7.4 | − |
Protein composition of the SH high-molecular-weight complex.
To firmly establish that the extra subunit was indeed HoxI, the 19-kDa band (Fig. 3, lane 1) was cut out of the gel and digested with trypsin. A mass fingerprint of the eluted peptide was obtained with MALDI-TOF as described in the Materials and Methods section. The resultant spectrum (Fig. 4A) was used to search the nonredundant database. In this way, the extra band was unequivocallly identified as HoxI. The 18 peptides marked with asterisks represent 71% of the amino acid sequence of HoxI.
FIG. 3.
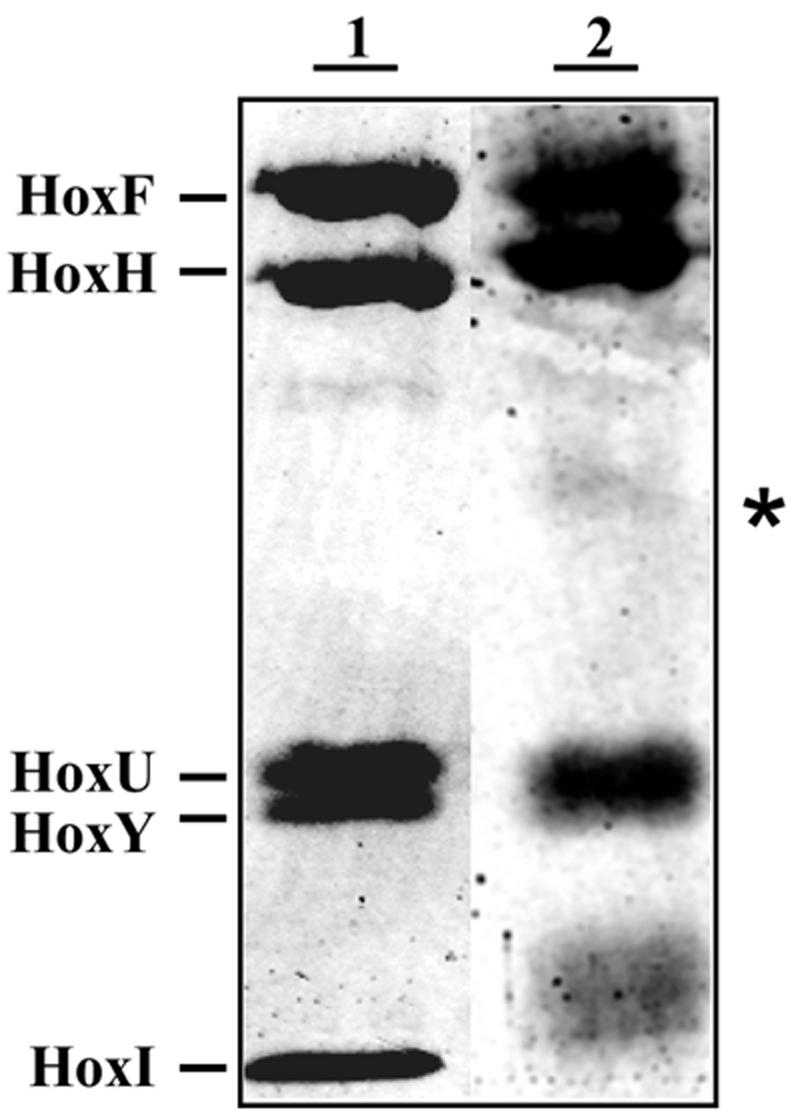
Proximity of the two HoxI subunits as detected by cross-linking. SDS-PAGE of the as-isolated (lane 1) and cross-linked SH-HoxI complex (lane 2). Upon incubation with the cross-linker BS3, a new band (marked with asterisks) appeared, corresponding to an apparent molecular mass of 38 kDa.
FIG. 4.

MALDI-TOF spectra from tryptic digests of HoxI. (A) Spectrum of the 19-kDa protein band from untreated SH-HoxI complex (Fig. 3, lane 1). (B) Spectrum of the 38-kDa band (Fig. 3, lane 2) obtained after BS3 cross-linking. Peptides marked with asterisks are derived from HoxI. Peaks marked with circles are tryptic autolysis products
To establish the subunit stoichiometry of the complex, the as-isolated SH high-molecular-weight preparation was subjected to 10 cycles of Edman degradation (Table 3). All subunits had unblocked amino termini and could be sequenced. Simultaneous quantification of the five different sequences was slightly complicated by the fact that in some cycles, two or more subunits had the same amino acid (e.g., both HoxH and HoxY have an arginine at position 2). Nevertheless, the unique amino acids in four cycles (3, 4, 5, and 8) enabled us to establish a clear ratio of subunits: one copy each of HoxH, -Y, -F, and -U and two copies of HoxI. The proline present at position 4 of HoxY decreased the sequencing yield and therefore led to an underestimation of the HoxY content. These results clearly indicate that the SH is in fact a hexamer containing two HoxI subunits.
TABLE 3.
Yields of Edman degradation of the untreated hexameric SH
| Cycle | HoxF
|
HoxH
|
HoxU
|
HoxY
|
HoxI
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aaa | pmolb | Relative amtc | aa | pmol | Relative amt | aa | pmol | Relative amt | aa | pmol | Relative amt | aa | pmol | Relative amt | |
| 3 | S | 2.29 | 1 | K | 3.01 | 1.31 | Q | 3.9 | 1.70 | A | 3.74 | 1.63 | E | 6.99 | 3.05 |
| 4 | R | 3.37 | 1 | L | 3.25 | 0.96 | I | 3.17 | 0.94 | P | 2.27 | 0.67 | Q | 6.91 | 2.05 |
| 5 | I | 3.42 | 1 | V | 3.19 | 0.93 | T | 2.68 | 0.78 | H | 1.55 | 0.45 | E | 5.82 | 1.70 |
| 8 | I | 3.62 | 1 | P | 2.44 | 0.67 | G | 4.8 | 1.32 | E | 2.6 | 0.72 | R | 6.22 | 1.72 |
| Mean | 1 | 0.97 | 1.19 | 0.87 | 2.13 | ||||||||||
aa, amino acid present at that position.
Amount normalized against the standard PTH mixture, not corrected for the sequencing yield.
Relative amount of that amino acid normalized against the amino acid of HoxF at the same position.
To further investigate the spatial relationship between the HoxI subunits, cross-links were introduced into the SH with the bifunctional lysine-reactive cross-linker BS3. Analysis of the BS3 cross-linked SH on SDS-PAGE gels revealed the formation of an additional weak protein band migrating with an apparent molecular mass of about 38 kDa (Fig. 3, lane 2). The MALDI-TOF spectrum of the tryptic digest of the 38-kDa band proved to be identical to that of the HoxI subunit (Fig. 4B). This demonstrates that the two HoxI subunits in the SH are in close proximity.
Enzymatic and infrared-spectroscopic characterization of the hexameric SH.
The tetrameric SH consists of two cooperating enzymatic modules (58, 41). Aside from the overall reaction, the H2-dependent reduction of NAD+, several module-specific enzymatic activities can be measured, e.g., the H2-dependent reduction of BV or K3Fe(CN)6 and the oxidation of NAD(P)H with K3Fe(CN)6 as electron acceptor. Table 4 gives an overview of these activities for both the tetrameric and the hexameric SH. All assays were carried out in 50 mM Tris-HCl buffer (pH 8) since SH activities are maximal under these conditions (58). The SH-HoxI complex dissociated after prolonged incubation at pH 8 but remained stable during the activity measurements. To compensate for the difference in molecular mass, turnover numbers were calculated as well. Since the specific activities of routine preparations can vary over a relatively wide range, the differences in turnover number observed for the two forms of the SH are not really significant. Remarkably, the hexameric SH showed a low but clearly detectable NADPH-K3Fe(CN)6 activity (0.6 U/mg), whereas the tetrameric form did not. Hydrogen oxidation with NADP+ as electron acceptor was absent in both preparations. Moreover, no H2 production from NADPH could be detected whereas both forms coupled the oxidation of NADH to proton reduction (data not shown).
TABLE 4.
Enzymatic activities of the purified tetrameric and hexameric SH
| Activity assay | sp act (U/mg)
|
Turnover no. (s−1)
|
||
|---|---|---|---|---|
| Tetramer | Hexamer | Tetramer | Hexamer | |
| NADH-K3Fe(CN)6 | 61.6a | 64.9a | 171 | 222 |
| NADPH-K3Fe(CN)6 | NDc | 0.6b | 2 | |
| H2-BV | 25.2 | 35.1 | 70 | 120 |
| H2-NAD+ | 39.0 | 41.9 | 109 | 143 |
| H2-K3Fe(CN)6 | 102.1 | 108.8 | 284 | 372 |
| H2-NADP+ | NDc | NDc | ||
Both tetrameric and hexameric SH exhibited NADH-H+ activity (around 1 % of the H2-NAD+ activity).
No NADPH-H+ activity could be detected.
ND, no activity could be determined.
The infrared spectra of the tetrameric and hexameric forms of the SH showed peaks at identical positions in the 2150 to 1850 cm−1 spectral region (data not shown) indicating the presence of four CN− and one CO at the Ni-Fe active site, as it was described previously for the tetrameric SH (25, 73).
The hexameric SH shows a different activation behavior.
Like many [NiFe]-hydrogenases, the aerobically purified SH is inactive and requires a reductive treatment for activation. The removal of an oxygen species from the active site by a reductive treatment may be a prerequisite for catalytic activity of hydrogenases in general (23, 25, 44). In the case of the SH, a catalytic amount of NADH (5 μM) is sufficient to obtain rapid activation. In an earlier publication, Schneider and Schlegel (58) described that NADPH (50 μM) could also activate the enzyme. In order to clarify this point, we studied activation by NADH and NADPH for both the tetrameric and the hexameric SH by monitoring H2-dependent NAD+ reduction. Both forms could easily be activated by small amounts of NADH (2 to 3 μM) in the presence of H2. When using NADPH, however, a clear difference was apparent (Fig. 5).
FIG. 5.

The effect of NADPH on the H2-NAD+ activity of the tetrameric and hexameric forms of the SH. The reaction cell was filled with aerobic 50 mM Tris-HCl buffer (pH 8.0). Enzyme (28.5 nM), NADPH (100 μM) (A, D, and E), and hydrogen (36 μM) were added in that order. The reaction was started (arrow) by the addition of 5 mM NAD+ (A through D) or 2.5 mM of K3Fe(CN)6 (E). (A) Tetrameric SH with NADPH; (B) tetrameric enzyme (control); (C) hexameric SH (control); (D) hexameric SH with NADPH; (E) hexameric SH with NADPH [H2-K3Fe(CN)6 assay].
Without prior activation with NADH, the tetrameric enzyme started with a significant lag phase after the addition of excess NAD+ (Fig. 5B). This behavior was not altered when 100 μM NADPH was present (Fig. 5A). Without prior activation, the hexameric enzyme also showed a lag phase (Fig. 5C), but when NADPH was present, it immediately started to consume hydrogen after the addition of excess NAD+ (Fig. 5D). We found that addition of 25 μM was sufficient to fully activate the hexameric SH. In sharp contrast to this, the tetrameric form could be activated only by NADPH at a concentration 3 orders of magnitude higher (40 mM) (data not shown).
Similar results were obtained using K3Fe(CN)6 as electron acceptor. When NADPH (100 μM) was used to activate the tetrameric SH, no activity was observed within 30 min (data not shown). For the hexameric SH, NADPH caused an immediate hydrogen consumption after the addition of K3Fe(CN)6. Unexpectedly, the reaction came to a halt before the hydrogen was completely used up (25 μM H2 remained in the experiment shown in Fig. 5E). The extent of hydrogen consumption was dependent on the amount of NADPH; the more NADPH that was added, the longer the reaction proceeded. Under anaerobic conditions, H2 was completely consumed (data not shown), indicating that NADPH was consumed and subsequently that the enzyme was inactivated by oxygen (Fig. 5E). When NADH (5 μM) was used instead of NADPH, hydrogen was completely consumed (aerobically or anaerobically). The difference can be explained by the fact that NADH, in contrast to NADPH, can be regenerated via the H2-NAD+ reaction.
HoxI is associated with the NADH-dehydrogenase moiety of the SH.
The cross-linking experiments indicated that the two HoxI subunits are in close proximity. To investigate the localization of the HoxI dimer within the hexameric complex, we used an affinity-chromatography-based approach. A Strep-tag II sequence (33) was fused to the N terminus of the HoxI protein. This construct, designated HoxIStrep, allowed a one-step affinity purification of the hexameric SH (Fig. 6, lane 1). The modified complex behaved like the nonmodified form (data not shown). Moreover HoxIStrep was released under the same conditions as HoxI from the native complex. Thus, only one additional washing step with 50 mM Tris-HCl (pH 8.0) was required to obtain the purified tetrameric SH (Fig. 6, lane 3), followed by elution of HoxI (Fig. 6, lane 4).
FIG. 6.

Purification of the SH by affinity chromatography with Strep-tagged HoxI and HoxF derivatives. SDS-15% PAGE of: lane 1, SH-HoxIStrep complex; lane 2, SH-HoxFStrep; lane 3, tetrameric SH dissociated from the SH-HoxIStrep complex after an additional washing step with 50 mM Tris-HCl (pH 8.0); lane 4, HoxIStrep after elution at pH 8.0. To obtain the samples depicted in lanes 1 and 2, all washing and elution steps were performed in 50 mM KPi buffer (pH 7). HoxIStrep derivatives were isolated from HF160(pGE549). Cells of HF160(pGE552) were used for the purification of HoxFStrep SH samples. Purified proteins (5 μg) were applied to each lane.
Likewise, a second construct containing the Strep-tagII at the N terminus of HoxF, the large subunit of the NADH-dehydrogenase module, was successfully used for the isolation of either the hexameric (Fig. 6, lane 2) or the tetrameric form (results not shown) of the SH. These results indicate that the N termini of HoxF and HoxI are probably not involved in complex formation within the enzyme.
In order to determine if HoxI forms a complex with one of the distinct functional modules, the Strep-tagged derivatives were introduced into different SH deletion mutants. Deletion of the hydrogenase moiety HoxHY resulted in a HoxFUI complex that could be isolated either with the N-terminally tagged HoxF or the HoxI protein (Fig. 7). Deleting the NADH-dehydrogenase part HoxFU, however, prevented complex formation. HoxI did not copurify with the hydrogenase dimer. These results indicate that HoxI interacts primarily with the HoxFU module of this hydrogenase.
FIG. 7.

Affinity of HoxI for the NADH-dehydrogenase module (HoxFU) of the SH. SDS-15% PAGE of HoxFStrep and HoxIStrep subcomplexes of the SH purified in 50 mM KPi buffer (pH 7). Protein (5 μg) was applied to each lane. (A) Lane 1, complex isolated from HF424(pGE553) harboring HoxFStrep in an HoxYH− background. (B) Lane 1, complex obtained from H160(pGE550) where HoxIStrep is produced in a HoxHY− background; lane 2, HoxIStrep purified from HF160(pGE551), with pGE551 carrying a deletion in hoxF and hoxU.
DISCUSSION
The SH from lithoautotrophic bacteria was up to now considered a heterotetrameric enzyme consisting of the hydrogenase moiety HoxHY and the NADH-dehydrogenase module HoxFU. Our results show that the enzyme can be purified as a well-defined, functional heterohexameric protein, HoxFUYHI2, provided that 50 mM KPi (pH 7.0) is used throughout the purification and no hydrophobic interaction or affinity chromatography steps are used. The HoxI subunits are evidently removed during the routinely used purification procedure involving affinity chromatography on Procion Red (57). Stability of the purified hexamer was dependent on ionic strength and pH. The hexameric enzyme was stable in 50 mM KPi at pH 7.0. At a higher ionic strength or alkaline pH dissociation occurred, releasing the HoxI subunits. In crude fractions, the enzyme could apparently withstand the very high-ionic-strength conditions during the (NH4)2SO4 fractionation. The hexameric form of the SH is the first multimeric enzyme of this type to be described.
Catalytic properties of the hexameric SH.
The SH has a number of catalytic properties that are different from those described for the standard [NiFe]-hydrogenases. Although it requires reductive activation, the active enzyme is not sensitive to oxygen or carbon monoxide. Infrared spectroscopic studies, chemical analyses, and recent X-ray absorption studies suggest that the inactive SH has a (CysSO)2(CN)(OOH)Ni(μ-CysS)2Fe(CN)3(CO) site where both metal ions are six coordinated (14, 25, 72, 73). It has been suggested that the oxygen ligand to nickel is removed upon activation of the SH (25, 44, 72). For reasons reported elsewhere (14), we favor a peroxide group as ligand to nickel. The specific activation of the SH by NADPH would be consistent with the idea that activation involves the reduction of this peroxide group to water by the two reducing equivalents provided by NADPH. The two extra cyanides, together with an environment dominated by oxo ligands, have been proposed as the basis for the lack of redox transitions of the Ni-Fe active site and for the insensitivity toward oxygen and carbon monoxide (14, 25, 73). Under reducing conditions, the SH is unstable. One of the two flavin cofactors, FMN-a, is rapidly lost, and subsequently, the extra CN− on the Ni is released, resulting in catalytically active but O2-sensitive SH (72, 73).
The FMN-a group is indispensable for reductive activation; when absent, H2 oxidation with NAD+ was no longer detectable, whereas the H2-BV and the NADH-BV activities were not affected. The H2-NAD+ activity could be fully restored by the addition of FMN under reducing conditions. These results indicate that this FMN-a group is located in the hydrogenase moiety of the SH enzyme (72). Due to the presence of a flavodoxin fold in the small hydrogenase subunit, FMN-a was assigned to HoxY (2).
Reductive activation of the SH can be achieved by incubation with small amounts of NADH in the presence of H2. The present study shows that this holds for both the tetrameric and the hexameric forms of the SH. Whereas the hexameric form could also be activated by 25 μM NADPH, the tetrameric form required 40 mM NADPH for activation, which is presumably mediated via reaction of NADPH at the regular site for NADH. We therefore propose that the two HoxI subunits in the hexameric form provide a specific binding site for NADPH.
The present study also shows that in the absence of the hydrogenase module, HoxI copurifies with the diaphorase part of the SH enzyme. Conversely, no copurification of HoxI was observed with the hydrogenase module. We postulate that HoxI is associated with HoxF in close proximity to HoxY, where FMN-a and the proximal [4Fe-4S] cluster are located (Fig. 8). Electrons from NADPH are transferred to FMN-a, and this is proposed to lead to the reduction of the peroxide group to water, allowing the binding and activation of H2. The low NADPH- K3Fe(CN)6 activity shows that NADPH can be slowly oxidized by the hexameric SH. Although the turnover number is very low, it is sufficient to achieve rapid activation of the enzyme. The absence of H2-NADP+ activity is the reason that NADPH cannot be regenerated by hexameric SH and thus seems to not be involved in the catalytic cycle.
FIG. 8.

Model for the soluble hydrogenase of R. eutropha. The two HoxI subunits form the binding site for one NADPH molecule. NADPH specifically supplies reducing equivalents to the FMN-a group. The [2Fe-2S] and [4Fe-4S] clusters are indicated as 2Fe and 4Fe, respectively.
Activation of the SH by NADPH may be an advantage for the enzyme, which directly supplies reducing equivalents from H2 to the intracellular NAD+ pool. Under lithoautotrophic conditions, large amounts of NADH are required for CO2 fixation via the Calvin-Benson-Bassham reductive pentose-phosphate cycle (9). Thus, access to an alternative reducing agent such as NADPH might be useful under these conditions. For some chemolithoautotrophic bacteria, it has been shown that during adaptation to autotrophic conditions, the NADPH to NADP ratio increases (32, 75). In the case of E. coli, it has been reported that, under oxidative stress conditions, the reducing equivalents are present mainly as NADPH in order to reduce DNA damage. Exposure to H2O2 induces the activity of NADH/NADP+ transhydrogenase and glucose-6-phosphate dehydrogenase, leading to the production of NADPH (12). In general, rapid activation, which is indispensable for the efficient use of small amounts of H2, is more likely if the enzyme can utilize either of two alternative nucleotides.
Conserved motifs in the HoxI protein.
HoxI homologues were found in Ralstonia metallidurans and Rhodococcus opacus cells that contain a soluble hydrogenase with high similarity to the R. eutropha SH (72% identity; the sequence of R. opacus HoxI is incomplete). For both the N- and C-terminal regions of R. eutropha HoxI, no homology to other proteins with known function could be detected. In the central domain of HoxI, a cyclic nucleotide-binding site similar to those of the catabolite gene activator protein/cyclic AMP (cAMP) receptor protein family of regulatory proteins (27) was found (26 and 24% identity to the cAMP-binding proteins from Desulfitobacterium hafniense and Thermobifida fusca, repectively) (Fig. 9). Cyclic nucleotide-binding domains generally comprise 133 amino acid residues and have been described for the regulatory subunit of eukaryotic protein kinases (66) and for the cyclic nucleotide-gated ion channels (42). They all harbor two sequence signatures that are also conserved in the R. eutropha HoxI protein. The first pattern contains two invariant glycine residues. The second pattern that forms the phosphate-binding loop comprises a glycine and three other invariant residues (PROSITE entry PDOC00691 [21]). Dinucleotide-binding sites, like the Rossmann fold, show a very low overall sequence homology (8). Binding of a dinucleotide to such a fold involves two mononucleotide-binding motifs that together form a six-stranded parallel β-sheet flanked by α-helices. However, secondary structure predictions for the HoxI protein did not show such a pattern (results not shown). We speculate that two adjacent HoxI subunits, each with a mononucleotide-binding site, could build a dinucleotide-binding domain for the binding of NADPH, but this remains to be verified by future studies.
FIG. 9.

The putative cyclic nucleotide-binding domain in the R. eutropha HoxI protein. (A) Schematic representation; (B) alignment of the central HoxI domain with cyclic nucleotide-binding proteins. Conserved regions are boxed as follows: 1, PROSITE motif PS00888; 2, PS0089 (21). Invariant residues are highlighted. The total length of the proteins is given in parentheses. Db., Desulfitobacterium; E., Escherichia; R., Ralstonia; Rh., Rhodococcus; T., Thermobifida.
Homology of the SH to other multimeric [NiFe]-hydrogenases and Complex I.
Cytoplasmic multiheteromeric hydrogenases with extensive homology to the R. eutropha SH tetramer have been found in phototrophic organisms including cyanobacteria and Thiocapsa. These so-called bidirectional [NiFe]-hydrogenases were originally described as heterotetrameric proteins. However, in various cases, e.g., Synechocystis sp. PCC 6803, Synechococcus sp. PCC 6301, and Thiocapsa roseopersicina (53, 55), enzyme purification has subsequently uncovered an additional hydrogenase-related subunit designated HoxE. A HoxE homologue is predicted from the genome sequence of Nostoc sp. PCC 7120 (29). HoxE proteins show homology to the NuoE subunit of NADH:ubiquinone oxidoreductase (Complex I) and probably host a [2Fe-2S] cluster (4). Thus, HoxE seems to be a constituent of the NADH-dehydrogenase module. The sequence of the R. eutropha HoxI protein has no homology with HoxE. Sequence motifs indicative of Fe-S clusters are absent in HoxI.
The tetrameric form of the SH has a number of properties in common with the bacterial and mitochondrial Complex I. These include amino acid sequence similarities between the HoxFUYH subunits and five subunits of complex I, a related pattern of Fe-S clusters, and the occurrence of two functionally different FMN groups (1, 2, 3, 52, 69). The presence of a specific site for NADPH in the hexameric form of the SH documented in this study is yet another property which is shared with Complex I (28, 70, 71). This raises the question whether Complex I contains subunits with amino acid sequence homology to HoxI.
Photoaffinity labeling studies with submitochondrial particles have uncovered five nucleotide-binding subunits in bovine Complex I. Two of them could be labeled with [32P]NADP(H) but not with NAD(H) (80). The 39-kDa (NUEM) subunit (as well as its homologue, ND4, in Neurospora crassa) forms a stable complex with NADPH (81, 59). However, there is no apparent similarity between this subunit and HoxI on the sequence level. The other candidate is the 18-kDa subunit, which has about the same mass as HoxI. The 18-kDa subunits of the Complex I family contain a carboxyterminal Ser residue in an RVS consensus motif that can be phosphorylated by a cAMP-dependent protein kinase, resulting in an enhancement of NAD-linked mitochondrial respiration (48). The triplet RVS is absent from the carboxy-terminal part of HoxI. Instead, we find the motif RVH. However, no other sequence similarities were apparent.
Acknowledgments
We thank C. G. Friedrich and U. Kärst for providing antibodies against the B protein and Ed Schwartz for critical reading of the manuscript.
B.F. was funded by the Deutsche Forschungsgemeinschaft (SPP 1070) and the Fonds der Chemischen Industrie. S.P.J.A. and B.F. acknowledge the European Union Cooperation in the field of Scientific and Technical Research (COST), Action-841 for funding an Expert Meeting and a Short Term Scientific Mission. The protein sequencer and the MALDI-TOF mass spectrometer have largely been funded by the Council for Medical Sciences of The Netherlands Organization for Scientific Research (NWO-ZonMW).
REFERENCES
- 1.Albracht, S. P. J., E. van der Linden, and B. W. Faber. 2003. Quantitative amino-acid analysis of bovine NADH:ubiquinone oxidoreductase (Complex I) and related enzymes. Consequences for the number of prosthetic groups. Biochim. Biophys. Acta 1557:41-49. [DOI] [PubMed] [Google Scholar]
- 2.Albracht, S. P. J., and R. Hedderich. 2000. Learning from hydrogenases: location of a proton pump and of a second FMN in bovine NADH-ubiquinone oxidoreductase (Complex I). FEBS Lett. 485:1-6. [DOI] [PubMed] [Google Scholar]
- 3.Albracht, S. P. J. 1993. Intimate relationships of the large and the small subunits of all nickel hydrogenases with two nuclear-encoded subunits of mitochondrial NADH: ubiquinone oxidoreductase. Biochim. Biophys. Acta 1144:221-224. [DOI] [PubMed] [Google Scholar]
- 4.Appel, J., and R. Schulz. 1996. Sequence analysis of an operon of a NAD(P)-reducing nickel hydrogenase from the cyanobacterium Synechocystis sp. PCC 6803 gives additional evidence for direct coupling of the enzyme to NAD(P)H-dehydrogenase (complex I). Biochim. Biophys. Acta 1298:141-147. [DOI] [PubMed] [Google Scholar]
- 5.Bagley, K. A., E. C. Duin, W. Roseboom, S. P. J. Albracht, and W. H. Woodruff. 1995. Infrared-detectable groups sense changes in charge density on the nickel center in hydrogenase from Chromatium vinosum. Biochemistry 34:5527-5535. [DOI] [PubMed] [Google Scholar]
- 6.Bagley, K. A., C. J. Van Garderen, M. Chen, E. C. Duin, S. P. J. Albracht, and W. H. Woodruff. 1994. Infrared studies on the interaction of carbon monoxide with divalent nickel in hydrogenase from Chromatium vinosum. Biochemistry 33:9229-9236. [DOI] [PubMed] [Google Scholar]
- 7.Bernhard, M., E. Schwartz, J. Rietdorf, and B. Friedrich. 1996. The Alcaligenes eutrophus membrane-bound hydrogenase gene locus encodes functions involved in maturation and electron transport coupling. J. Bacteriol. 178:4522-4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bottoms, C. A., P. E. Smith, and J. J. Tanner. 2002. A structurally conserved water molecule in Rossmann dinucleotide-binding domains. Protein Sci. 11:2125-2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowien, B., and B. Kusian. 2002. Genetics and control of CO2 assimilation in the chemoautotroph Ralstonia eutropha. Arch. Microbiol. 178:85-93. [DOI] [PubMed] [Google Scholar]
- 10.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye-binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 11.Brecht, M., M. van Gastel, T. Buhrke, B. Friedrich, and W. Lubitz. 2003. Direct detection of a hydrogen ligand in the [NiFe] center of the regulatory H2-sensing hydrogenase from Ralstonia eutropha in its reduced state by HYSCORE and ENDOR spectroscopy. J. Am. Chem. Soc. 125:13075-13083. [DOI] [PubMed] [Google Scholar]
- 12.Brumaghim, J. L., Y. Li, E. Henle, and S. Linn. 2003. Effects of hydrogen peroxide upon nicotinamide nucleotide metabolism in Escherichia coli: changes in enzyme levels and nicotinamide nucleotide pools and studies of the oxidation of NAD(P)H by Fe(III). J. Biol. Chem. 278:42495-42504. [DOI] [PubMed] [Google Scholar]
- 13.Bullock, W. O., J. M. Fernandez, and J. M. Short. 1987. XL1-blue: a high efficiency plasmid transforming recA− Escherichia coli strain with betagalactosidase selection. BioTechniques 5:376-379. [Google Scholar]
- 14.Burgdorf, T., S. Löscher, P. Liebisch, E. Van der Linden, M. Galander, F. Lendzian, W. Meyer-Klaucke, S. P. J. Albracht, B. Friedrich, H. Dau, and M. Haumann. 2005. Structural and oxidation-state changes at its nonstandard Ni-Fe site during activation of the NAD-reducing hydrogenase from Ralstonia eutropha detected by X-ray absorption, EPR, and FTIR spectroscopy. J. Am. Chem. Soc. 127:576-592. [DOI] [PubMed] [Google Scholar]
- 15.Burgdorf, T., A. L. De Lacey, and B. Friedrich. 2002. Functional analysis by site-directed mutagenesis of the NAD+-reducing hydrogenase from Ralstonia eutropha. J. Bacteriol. 184:6280-6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cammack, R., M. Frey, and R. Robson (ed.). 2001. Hydrogen as a fuel: learning from nature. Taylor & Francis, London, United Kingdom.
- 17.Coremans, J. M. C. C., J. W. van der Zwaan, and S. P. J. Albracht. 1989. Redox behavior of nickel in hydrogenase from Methanobacterium thermoautotrophicum (strain Marburg)—correlation between the nickel valence state and enzyme activity. Biochim. Biophys. Acta 997:256-267. [Google Scholar]
- 18.Davis, D. H., M. Doudoroff, R. Y. Stanier, and M. Mandel. 1969. Proposal to reject the genus Hydrogenomonas: Taxonomic implications. Int. J. Syst. Bacteriol. 19:375-390. [Google Scholar]
- 19.Eberz, G., C. Hogrefe, C. Kortlüke, A. Kamienski, and B. Friedrich. 1986. Molecular cloning of structural and regulatory hydrogenase (hox) genes of Alcaligenes eutrophus H16. J. Bacteriol. 168:636-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erkens, A., K. Schneider, and A. Müller. 1996. The NAD-linked soluble hydrogenase from Alcaligenes eutrophus H16: detection and characterization of EPR signals derived from nickel and flavin. J. Biol. Inorg. Chem. 1:99-110. [Google Scholar]
- 21.Falquet, L., M. Pagni, P. Bucher, N. Hulo, C. J. Sigrist, K. Hofmann, and A. Bairoch. 2002. The PROSITE database, its status in 2002. Nucleic Acids Res. 30:235-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foerster, S., M. Stein, M. Brecht, H. Ogata, Y. Higuchi, and W. Lubitz. 2003. Single crystal EPR studies of the reduced active site of [NiFe] hydrogenase from Desulfovibrio vulgaris Miyazaki F. J. Am. Chem. Soc. 125:83-93. [DOI] [PubMed] [Google Scholar]
- 23.Garcin, E., X. Vernede, E. C. Hatchikian, A. Volbeda, M. Frey, and J. C. Fontecilla-Camps. 1999. The crystal structure of a reduced [NiFeSe] hydrogenase provides an image of the activated catalytic center. Struct. Fold. Des. 7:557-566. [DOI] [PubMed] [Google Scholar]
- 24.Grzeszik, C., M. Lübbers, M. Reh, and H. G. Schlegel. 1997. Genes encoding the NAD-reducing hydrogenase of Rhodococcus opacus MR11. Microbiology 143:1271-1286. [DOI] [PubMed] [Google Scholar]
- 25.Happe, R. P., W. Roseboom, G. Egert, C. G. Friedrich, C. Massanz, B. Friedrich, and S. P. J. Albracht. 2000. Unusual FTIR and EPR properties of the H2-activating site of the cytoplasmic NAD-reducing hydrogenase from Ralstonia eutropha. FEBS Lett. 466:259-263. [DOI] [PubMed] [Google Scholar]
- 26.Happe, R. P., W. Roseboom, A. J. Pierik, S. P. J. Albracht, and K. A. Bagley. 1997. Biological activation of hydrogen. Nature 385:126. [DOI] [PubMed] [Google Scholar]
- 27.Harman, J. G. 2001. Allosteric regulation of the cAMP receptor protein. Biochim. Biophys. Acta 1547:1-17. [DOI] [PubMed] [Google Scholar]
- 28.Hatefi, Y., and W. G. Hanstein. 1973. Interactions of reduced and oxidized triphosphopyridine nucleotides with the electron-transport system of bovine heart mitochondria. Biochemistry 12:3515-3522. [DOI] [PubMed] [Google Scholar]
- 29.Kaneko, T., Y. Nakamura, C. P. Wolk, et al. 2001. Complete genomic sequence of the filamentous nitrogen-fixing cyanobacterium Anabaena sp. strain PCC 7120. DNA Res. 8:205-213, 227-253. [DOI] [PubMed] [Google Scholar]
- 30.Kärst, U., S. Suetin, and C. G. Friedrich. 1987. Purification and properties of a protein linked to the soluble hydrogenase of hydrogen-oxidizing bacteria. J. Bacteriol. 169:2079-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kleihues, L., O. Lenz, M. Bernhard, T. Buhrke, and B. Friedrich. 2000. The H2 sensor of Ralstonia eutropha is a member of the subclass of regulatory [NiFe] hydrogenases. J. Bacteriol. 182:2716-2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knight, M., L. Dijkhuizen, and W. Harder. 1978. Metabolic regulation in Pseudomonas oxalaticus OX1. Enzyme and coenzyme concentration changes during substrate transition experiments. Arch. Microbiol. 116:85-90. [DOI] [PubMed] [Google Scholar]
- 33.Korndörfer, I. P., and A. Skerra. 2002. Improved affinity of engineered streptavidin for the Strep-tag II peptide is due to a fixed open conformation of the lid-like loop at the binding site. Protein Sci. 11:883-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kortlüke, C., K. Horstmann, E. Schwartz, M. Rohde, R. Binsack, and B. Friedrich. 1992. A gene complex coding for the membrane-bound hydrogenase of Alcaligenes eutrophus H16. J. Bacteriol. 174:6277-6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kortlüke, C., C. Hogrefe, G. Eberz, A. Pühler, and B. Friedrich. 1987. Genes of lithoautotrophic metabolism are clustered on the megaplasmid pHG1 in Alcaligenes eutrophus. Mol. Gen. Genet. 210:122-128. [Google Scholar]
- 36.Laemmli, U. K. 1970. Cleavage of structural proteins during the asssembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 37.Lenz, O., E. Schwartz, J. Dernedde, M. Eitinger, and B. Friedrich. 1994. The Alcaligenes eutrophus H16 hoxX gene participates in hydrogenase regulation. J. Bacteriol. 176:4385-4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lyon, E. J., S. Shima, R. Boecher, R. K. Thauer, F. W. Grevels, E. Bill, W. Roseboom, and S. P. J. Albracht. 2004. Carbon monoxide as an intrinsic ligand to iron in the active site of the iron-sulfur-cluster-free hydrogenase H2-forming methylenetetrahydromethanopterin dehydrogenase as revealed by infrared spectroscopy. J. Am. Chem. Soc. 126:14239-14248. [DOI] [PubMed] [Google Scholar]
- 39.Lyon, E. J., S. Shima, G. Buurman, S. Chowdhuri, A. Batschauer, K. Steinbach, and R. K. Thauer. 2004. UV-A/blue-light inactivation of the ‘metal-free’ hydrogenase (Hmd) from methanogenic archaea. Eur. J. Biochem. 271:195-204. [DOI] [PubMed] [Google Scholar]
- 40.Massanz, C., and B. Friedrich. 1999. Amino acid replacements at the H2-activating site of the NAD-reducing hydrogenase from Alcaligenes eutrophus. Biochemistry 38:14330-14337. [DOI] [PubMed] [Google Scholar]
- 41.Massanz, C., S. Schmidt, and B. Friedrich. 1998. Subforms and in vitro reconstitution of the NAD-reducing hydrogenase of Alcaligenes eutrophus. J. Bacteriol. 180:1023-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matulef, K., and W. N. Zagotta. 2003. Cyclic nucleotide-gated ion channels. Annu. Rev. Cell. Dev. Biol. 19:23-44. [DOI] [PubMed] [Google Scholar]
- 43.Miller, J. H. 1972. Experiments in molecular genetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 44.Müller, A., A. Erkens, K. Schneider, H.-F. Nolting, V. A. Solé, and G. Henkel. 1997. NADH-induzierte Einflüsse auf die Koordination des Nickels im aktiven Zentrum der löslichen Hydrogenase aus Alcaligenes eutrophus: XAFS-Untersuchungen an drei ESR-spektroskopisch unterscheidbaren Zuständen. Angew. Chem. 109:1812-1815. [Google Scholar]
- 45.Nicolet, Y., A. L. de Lacey, X. Vernede, V. M. Fernandez, E. C. Hatchikian, and J. C. Fontecilla-Camps. 2001. Crystallographic and FTIR spectroscopic evidence of changes in Fe coordination upon reduction of the active site of the Fe-only hydrogenase from Desulfovibrio desulfuricans. J. Am. Chem. Soc. 123:1596-1601. [DOI] [PubMed] [Google Scholar]
- 46.Nicolet, Y., C. Piras, P. Legrand, C. E. Hatchikian, and J. C. Fontecilla-Camps. 1999. Desulfovibrio desulfuricans iron hydrogenase: the structure shows unusual coordination to an active site Fe binuclear center. Struct. Fold. Des. 7:13-23. [DOI] [PubMed] [Google Scholar]
- 47.Oelmüller, U., H. G. Schlegel, and C. G. Friedrich. 1990. Differential stability of mRNA species of Alcaligenes eutrophus soluble and particulate hydrogenases. J. Bacteriol. 172:7057-7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Papa, S. 2002. The NDUFS4 nuclear gene of complex I of mitochondria and the cAMP cascade. Biochim. Biophys. Acta 1555:147-153. [DOI] [PubMed] [Google Scholar]
- 49.Peters, J. W., W. N. Lanzilotta, B. J. Lemon, and L. C. Seefeldt. 1998. X-ray crystal structure of the Fe-only hydrogenase (CpI) from Clostridium pasteurianum to 1.8 angstrom resolution. Science 282:1853-1858. [DOI] [PubMed] [Google Scholar]
- 50.Pierik, A. J., W. Roseboom, R. P. Happe, K. A. Bagley, and S. P. J. Albracht. 1999. Carbon monoxide and cyanide as intrinsic ligands to iron in the active site of [NiFe]-hydrogenases. NiFe(CN)2CO: biology's way to activate H2. J. Biol. Chem. 274:3331-3337. [DOI] [PubMed] [Google Scholar]
- 51.Pierik, A. J., M. Hulstein, W. R. Hagen, and S. P. J. Albracht. 1998. A low-spin iron with CN and CO as intrinsic ligands forms the core of the active site in [Fe]-hydrogenases. Eur. J. Biochem. 258:572-578. [DOI] [PubMed] [Google Scholar]
- 52.Pilkington, S. J., J. M. Skehel, R. B. Gennis, and J. E. Walker. 1991. Relationship between mitochondrial NADH-ubiquinone reductase and a bacterial NAD-reducing hydrogenase. Biochemistry 30:2166-2175. [DOI] [PubMed] [Google Scholar]
- 53.Rákhely, G., A. T. Kovács, G. Maróti, B. D. Fodor, G. Csanádi, D. Latinovics, and K. L. Kovács. 2004. Cyanobacterial-type, heteropentameric, NAD+-reducing NiFe hydrogenase in the purple sulfur photosynthetic bacterium Thiocapsa roseopersicina. Appl. Environ. Microbiol. 70:722-728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schink, B., and H. G. Schlegel. 1979. The membrane-bound hydrogenase of Alcaligenes eutrophus. I. Solubilization, purification, and biochemical properties. Biochim. Biophys. Acta 567:315-324. [DOI] [PubMed] [Google Scholar]
- 55.Schmitz, O., G. Boison, H. Salzmann, H. Bothe, K. Schutz, S. H. Wang, and T. Happe. 2002. HoxE—a subunit specific for the pentameric bidirectional hydrogenase complex (HoxEFUYH) of cyanobacteria. Biochim. Biophys. Acta 1554:66-74. [DOI] [PubMed] [Google Scholar]
- 56.Schneider, K., R. Cammack, and H. G. Schlegel. 1984. Content and localization of FMN, Fe-S clusters and nickel in the NAD-linked hydrogenase of Nocardia opaca 1b. Eur. J. Biochem. 142:75-84. [DOI] [PubMed] [Google Scholar]
- 57.Schneider, K., M. Pinkwart, and K. Jochim. 1983. Purification of hydrogenases by affinity chromatography on Procion Red-agarose. Biochem. J. 213:391-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schneider, K., and H. G. Schlegel. 1976. Purification and properties of the soluble hydrogenase from Alcaligenes eutrophus H16. Biochim. Biophys. Acta 452:66-80. [DOI] [PubMed] [Google Scholar]
- 59.Schulte, U., V. Haupt, A. Abelmann, W. Fecke, B. Brors, T. Rasmussen, T. Friedrich, and H. Weiss. 1999. A reductase/isomerase subunit of mitochondrial NADH:ubiquinone oxidoreductase (complex I) carries an NADPH and is involved in the biogenesis of the complex. J. Mol. Biol. 292:569-580. [DOI] [PubMed] [Google Scholar]
- 60.Schwartz, E., U. Gerischer, and B. Friedrich. 1998. Transcriptional regulation of Alcaligenes eutrophus hydrogenase genes. J. Bacteriol. 180:3197-3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shevchenko, A., M. Wilm, O. Vorm, and M. Mann. 1996. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal. Chem. 68:850-858. [DOI] [PubMed] [Google Scholar]
- 62.Shima, S., E. J. Lyon, M. Sordel-Klippert, M. Kauss, J. Kahnt, R. K. Thauer, K. Steinbach, X. Xie, L. Verdier, and C. Griesinger. 2004. The cofactor of the iron-sulfur cluster free hydrogenase hmd: structure of the light-inactivation product. Angew. Chem. Int. Engl. Ed. 43:2547-2551. [DOI] [PubMed] [Google Scholar]
- 63.Simon, R., U. Priefer, and A. Pühler. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram-negative bacteria. Bio/Technology 1:784-791. [Google Scholar]
- 64.Stein, M., and W. Lubitz. 2004. Relativistic DFT calculation of the reaction cycle intermediates of [NiFe] hydrogenase: a contribution to understanding the enzymatic mechanism. J. Inorg. Biochem. 98:862-877. [DOI] [PubMed] [Google Scholar]
- 65.Tamagnini, P., R. Axelsson, P. Lindberg, F. Oxelfelt, R. Wünschiers, and P. Lindblad. 2002. Hydrogenases and hydrogen metabolism of cyanobacteria. Microbiol. Mol. Biol. Rev. 66:1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Taylor, S. S., J. A. Buechler, and W. Yonemoto. 1990. cAMP-dependent protein kinase: framework for a diverse family of regulatory enzymes. Annu. Rev. Biochem. 59:971-1005. [DOI] [PubMed] [Google Scholar]
- 67.Thiemermann, S., J. Dernedde, M. Bernhard, W. Schroeder, C. Massanz, and B. Friedrich. 1996. Carboxy-terminal processing of the cytoplasmic NAD-reducing hydrogenase of Alcaligenes eutrophus requires the hoxW gene product. J. Bacteriol. 178:2368-2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Towbin, H., T. Staehelin, and J. Gordon. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA 76:4350-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tran-Betcke, A., U. Warnecke, C. Böcker, C. Zaborosch, and B. Friedrich. 1990. Cloning and nucleotide sequences of the genes for the subunits of NAD-reducing hydrogenase of Alcaligenes eutrophus H16. J. Bacteriol. 172:2920-2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van Belzen, R., M. C. van Gaalen, P. A. Cuypers, and S. P. J. Albracht. 1990. New evidence for the dimeric nature of NADH:Q oxidoreductase in bovine-heart submitochondrial particles. Biochim. Biophys. Acta 101:152-159. [DOI] [PubMed] [Google Scholar]
- 71.van Belzen, R., and S. P. J. Albracht. 1989. The pathway of electron transfer in NADH:Q oxidoreductase. Biochim. Biophys. Acta 974:311-320. [DOI] [PubMed] [Google Scholar]
- 72.van der Linden, E., B. W. Faber, B. Bleijlevens, T. Burgdorf, M. Bernhard, B. Friedrich, and S. P. Albracht. 2004. Selective release and function of one of the two FMN groups in the cytoplasmic NAD+-reducing [NiFe]-hydrogenase from Ralstonia eutropha. Eur. J. Biochem. 271:801-808. [DOI] [PubMed] [Google Scholar]
- 73.Van der Linden, E., T. Burgdorf, M. Bernhard, B. Bleijlevens, B. Friedrich, and S. P. J. Albracht. 2004. The soluble [NiFe]-hydrogenase from Ralstonia eutropha contains four cyanides in its active site, one of which is responsible for the insensitivity towards oxygen. J. Biol. Inorg. Chem. 9:616-626. [DOI] [PubMed] [Google Scholar]
- 74.van der Spek, T. M., A. F. Arendsen, R. P. Happe, S. Yun, K. A. Bagley, D. J. Stufkens, W. R. Hagen, and S. P. J. Albracht. 1996. Similarities in the architecture of the active sites of Ni-hydrogenases and Fe-hydrogenases detected by means of infrared spectroscopy. Eur. J. Biochem. 237:629-634. [DOI] [PubMed] [Google Scholar]
- 75.van Keulen, G., L. Dijkhuizen, and W. G. Meijer. 2000. Effects of the Calvin cycle on nicotinamide adenine dinucleotide concentrations and redox balances of Xanthobacter flavus. J. Bacteriol. 182:4637-4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vignais, P. M., and A. Colbeau. 2004. Molecular biology of microbial hydrogenases. Curr. Issues Mol. Biol. 6:159-188. [PubMed] [Google Scholar]
- 77.Vignais, P. M., B. Billoud, and J. Meyer. 2001. Classification and phylogeny of hydrogenases. FEMS Microbiol. Rev. 25:455-501. [DOI] [PubMed] [Google Scholar]
- 78.Volbeda, A., E. Garcin, C. Piras, A. L. De Lacey, V. M. Fernandez, E. C. Hatchikian, M. Frey, and J. C. Fontecilla-Camps. 1996. Structure of the [NiFe] hydrogenase active site: evidence for biologically uncommon Fe-ligands. J. Am. Chem. Soc. 118:12989-12996. [Google Scholar]
- 79.Volbeda, A., M.-H. Charon, C. Piras, E. C. Hatchikan, M. Frey, and J. C. Fontecilla-Camps. 1995. Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature 373:580-587. [DOI] [PubMed] [Google Scholar]
- 80.Yamaguchi, M., G. I. Belogrudov, A. Matsuno-Yagi, and Y. Hatefi. 2000. The multiple nicotinamide nucleotide-binding subunits of bovine heart mitochondrial NADH:ubiquinone oxidoreductase (complex I). Eur. J. Biochem. 267:329-336. [DOI] [PubMed] [Google Scholar]
- 81.Yamaguchi, M., G. I. Belogrudov, and J. Hatefi. 1998. Mitochondrial NADH-ubiquinone oxidoreductase (Complex I). Effect of substrates on the fragmentation of subunits by trypsin. J. Biol. Chem. 273:8094-8098. [DOI] [PubMed] [Google Scholar]
- 82.Yanisch-Perron, C., J. Vieira, and J. Messing. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103-119. [DOI] [PubMed] [Google Scholar]