

Abstract
Nicotinamide riboside (NR) increases blood levels of NAD+, a cofactor central to energy metabolism, and improves brain function in some rodent models of neurodegeneration. We conducted a placebo-controlled randomized pilot study with the primary objective of determining safety of NR in older adults with mild cognitive impairment (MCI). Twenty subjects with MCI were randomized to receive placebo or NR using dose escalation to achieve, and maintain, a final dose of 1 g/day over a 10-week study duration. The primary outcome was post-treatment change from baseline measures of cognition (Montreal Cognitive Assessment, MoCA). Predefined secondary outcomes included post-treatment changes in cerebral blood flow (CBF); blood NAD+ levels; and additional neurocognitive, psychometric, and physical performance tests. DNA methylation was assessed in peripheral blood mononuclear cells (PBMCs) as an exploratory outcome. The target NR dose was safely achieved as evidenced by a 2.6-fold increase in blood NAD+ in the NR group (p < 0.001, 95% CI [17.77, 43.49]) with no between-group difference in adverse event reporting. MoCA and other neurocognitive and psychometric metrics remained stable throughout the study. NR reduced CBF in the default mode network (DMN) with greatest differences observed in the left inferior parietal lobe (IPL) (DMN p = 0.013, μ = 0.92, 95% CI [0.23, 1.62]; left IPL p = 0.009, μ = 1.66, 95% CI [0.5, 2.82]). Walking speed in the placebo group significantly improved across the study duration suggestive of a practice effect but did not change in the NR group (p = 0.0402 and p = 0.4698, respectively). Other secondary outcome measures remained stable. Global methylation analyses indicated a modest NR-associated increase in DNA methylation and concomitant reduction in epigenetic age as measured by PhenoAge and GrimAge epigenetic clock analyses. In summary, NR significantly increased blood NAD+ concentrations in older adults with MCI. NR was well tolerated and did not alter cognition. While CBF was reduced by NR treatment, statistical significance would not have withstood multiple comparisons correction. A larger trial of longer duration is needed to determine the potential of NR as a strategy to improve cognition and alter CBF in older adults with MCI. ClinicalTrials.gov NCT02942888
Supplementary Information
The online version contains supplementary material available at 10.1007/s11357-023-00999-9.
Keywords: Nicotinamide riboside, Placebo-controlled trial, Mild cognitive impairment, Geroscience, NAD, Dementia
Introduction
Mild cognitive impairment (MCI) refers to an intermediate cognitive state between changes that occur with normal aging and dementia. The incidence of MCI is 14–18% in older adults ≥ 70 years old [1, 2]. Individuals with MCI remain independent despite recognizable and measurable deficits in at least one cognitive domain. A small percentage of people with MCI improve to normal [3] or remain clinically stable [4]. However, MCI progresses to dementia at a rate of 10–15% in clinical samples [5, 6] and 3.8–6.3% in community-based samples [7–10]. The primary cause of dementia in people with MCI is Alzheimer’s disease (AD) [11], the most common form of dementia in the USA [12].
The underlying pathogenic processes that initiate MCI and/or accelerate progression to dementia are not well understood. Advanced chronological age is the greatest risk factor for developing dementia. Growing evidence indicates that biological aging processes may contribute to brain dysfunction [13]. Therapies that target biological aging pathways extend healthy lifespan in animal models of normal aging and improve brain structure and function in mouse models of AD pathology [14–22]. These interventions have significant translational appeal and are moving toward clinical studies [13, 23, 24]. Toward this end, strategies to increase nicotinamide adenine dinucleotide (NAD+) are gaining attention [25]. NAD+ is critical to cellular function as it is an essential substrate for dozens of enzymes including sirtuins, poly(ADP-ribose) polymerases (PARPs), and cyclic ADP-ribose synthetases and as a co-enzyme for hundreds of redox reactions [26]. Through these cofactor function(s), NAD+ regulates numerous cellular processes including energy metabolism, transcriptional regulation, cellular viability, DNA damage repair, and calcium signaling. In mammalian cells, NAD+ synthesis occurs de novo from the amino acid tryptophan or through the salvage pathway from its precursors (nicotinamide (NAM), nicotinic acid (NA), nicotinamide riboside (NR), and nicotinamide mononucleotide (NMN)). Studies using brain imaging techniques to measure intracellular NAD+ and associated metabolites have demonstrated an age-dependent decrease in NAD+ in human brain [27]. Sub-physiological NAD+ levels manifest various symptoms, including brain dysfunction in mice; pharmacological or genetically increasing NAD+ effectively delays or mitigates rodent-modeled AD pathogenesis [28–32]. These preclinical studies support advancing NAD+ therapy to older adults with cognitive impairment.
Increasing NAD+ levels can be achieved through oral supplementation of its precursor molecules, which has been used in clinical practice used to treat disorders of niacin (vitamin B3) deficiency such as pellagra and neuropsychiatric disorders (reviewed, [33, 34]). Healthy individuals have self-reported improved memory and concentration and enhanced athletic performance after supplementing with NAD+ [35]. These potential health benefits, and the increase in antioxidant consumption due to increasing healthy-living trend [36], have led to an increased interest in NAD+ boosting supplements. However, the extent of absorption, action, and effectiveness of its supplementation is unclear. While presumably safe, i.e., no reports of overdose or toxicity, high doses of NADH (> 10 mg/day) may cause agitation, anxiety, and sleeplessness [37]. There is a need for more controlled clinical trials to test safety and efficacy of NAD+ precursors on aging and cognitive outcomes.
The most recently identified precursor, NR, is a pyridine nucleoside form of vitamin B3 [38]. It is naturally found in milk and is available as a nutraceutical. Prior studies suggest that oral supplementation of NR is safe and well tolerated and boosts NAD+ and metabolites in adults [39–41]. Recent work has demonstrated NR brain penetrance [42]. A separate study reported an NR-associated reduction in Aβ and tau in neuronally derived extracellular vesicles in a cohort of older adults to suggest potential alterations in AD relevant pathogenic processes [43]. Our goal was to test the hypothesis that NR therapy would be safe and well tolerated in older adults with MCI, which would be evidenced through stable brain function measures of cognition and cerebral blood flow (CBF). Furthermore, we hypothesized that increasing NAD+ may modulate other outcomes relevant to older adults such as physical function and DNA methylation. To test this, we conducted a phase 2 randomized clinical trial to investigate the effects of NR (4-week dose escalation to reach 1 g/day followed by 6 weeks of maximum tolerated dose) on cognition, CBF, and brain volume using magnetic resonance imaging (MRI), and physical function in older adults with MCI. Here, we report the clinical outcomes of the 20 study subjects that completed this clinical trial.
Methods
Study participants
We enrolled men and women aged ≥ 65 years old with mild cognitive impairment (MCI) recruited from the Texas Alzheimer’s Research and Care Consortium (TARCC), the community events, and the Barshop Institute for Longevity and Aging Studies’ Call Center Database. All participants recruited from the TARCC had been previously diagnosed with MCI based on inclusion criteria of the TARCC study. Participants recruited from other sites were clinically diagnosed by the PI. Diagnosis criteria included Montreal Cognitive Assessment (MoCA [44]) of < 26 with no instrumental activities of daily living (IADL) or activities of daily living (ADL) impairments and no depression, anxiety, learning disability, vision loss, or hearing loss. Participants taking opiates or niacin (> 200 mg) required a 2-week washout period before being allowed to participate. Participants were excluded if they had previously been considered healthy individuals without an MCI or Alzheimer’s disease diagnosis based on exclusion criteria of the TARCC study, or a clinical diagnosis of normal cognition by the principal investigator. Additional exclusions included neurological, psychiatric, or active systemic medical disease; diabetes taking insulin (oral agents acceptable) or HgbA1c > 7.5 mmol/L; moderate or severe depression or anxiety as determined by the Geriatric Depression Scale (GDS) and the Geriatric Anxiety Scale (GAS [45]), respectively; diagnosis of dementia; vision, motor, or language deficits or hearing loss impairing ability to follow commands; alcohol or drug use; implantation of metal devices; and administration of Alzheimer’s disease drugs, anticholinergics, anticonvulsants, opiates, systemic steroids, benzodiazepines, tricyclic antidepressants, monoamine oxidase inhibitors, anti-psychotics and mood stabilizers.
Study design, randomization, and intervention
This was a 10 week, double-blind, randomized, placebo-controlled study (Fig. 1). Twenty (20) MCI subjects were randomized (1:1) to receive either placebo (n=10) or nicotinamide riboside (n=10) (NR, NIAGEN, ChromaDex Inc.) two 250 mg twice a day, orally, for a total of 1 g daily as tolerated.
Fig. 1.

Study design and timeline. The primary objective of the study was to assess safety and tolerability of NR in older adults with MCI. The primary outcome was change in cognition as measured by MoCA. CBC, complete blood count; ADLs, activities of daily living; IADLs, instrumental activities of daily living; MoCA, Montreal Cognitive Assessment; GDS, Geriatric Depression Scale; GAS, Geriatric Anxiety Scale; EXIT, executive interview; TAPS, test of auditory processing skills; CLOX, executive clock drawing task; MRI, magnetic resonance imaging; NR, nicotinamide riboside. Shaded cells indicate procedures were conducted during the study visit. Dose escalation strategy: 250 mg × 1 week, 500 mg × 1 week, 750 mg × 1 week, and 1 g to end of study (6 weeks)
Evaluation of safety and tolerability
Adherence to the intervention was assessed by pill count. Subjects reported to the laboratory halfway through the study (after week 5) to receive a new bottle of capsules and to discuss any issues with tolerability or treatment-emergent adverse events (AEs) with a member of the research team who was not involved in data collection or analysis in order to ensure blinding of the investigators. Standard clinical markers of hematology, liver and kidney function, and blood lipids were analyzed using standardized clinical assays at the Audie Murphy VA Hospital Clinical Laboratory, and any abnormal blood results were reviewed by the study physician.
Blood collection for NAD+ quantification
Blood collection for analyzing NAD+ and metabolites was drawn under fasting conditions in the morning. The pre-blood draw was taken during the screening visit, and the post-blood draw was taken within 1–7 days of discontinuation of NR. One hundred microliter aliquots of whole blood were separated into microcentrifuge tubes containing 0.1 mM forodesine hydrochloride. The 100 μL blood containing a final concentration of 10 μM forodesine hydrochloride was snap frozen in liquid nitrogen and stored at −80°C and shipped to the University of Iowa to extract and quantify NAD+ and associated metabolites.
Materials
Nicotinamide riboside and placebo were obtained from ChromaDex Inc. (Irvine, CA). Forodesine hydrochloride was purchased from BOCSCI Inc. (Shirley, NY).
Extraction of NAD+ metabolites and nucleosides/nucleotides
The measurement of NAD+ related metabolites in whole blood was conducted after extracting as recently described [39, 46]. For analysis of NAD+, NADP, NMN, NAR, NAAD, ADPR, AMP, ADP, and ATP, 20 μL of a working solution of internal standard prepared from a 13C-yeast extract [46] and also containing 10 μM d3-Me4Py was prepared.
To quantify NAD+, NADP, NMN, NAR, NAAD, ADPR, AMP, ADP, and ATP, 100 μL of whole blood was added to 20 μL of the above working internal standard prepared in water and mixed with 500 μL of 3:1 4% trichloroacetic acid (TCA):acetonitrile with vortexing in sets of four. After resting on ice until 24 samples were processed, the samples were centrifuged as at 4°C, 16.2 rcf for 12 min. Next, supernatant was removed and dried on a SpeedVac overnight at room temperature and reconstituted in 2% acetonitrile/water immediately prior to the start of the analytical run. Standards, controls, and blank zeros were prepared by combining internal standard solution and the appropriate working standard solution. Aged whole blood was added, the sample vortexed, and TCA/acetonitrile added as above. The remainder of the processing was identical to that for analytical samples. Because most of the analytes are found in significant quantities in aged blood, six blank zero samples were prepared containing only internal standard and blood to serve as the baseline for the standard additions to make the calibration curve.
NAD+ quantification (LC-MS)
To assess the effects of NR supplementation on NAD+ metabolism, we used a targeted liquid chromatography-mass spectrometry (LC-MS/MS) method as previously described [39, 46]. Briefly, separation was performed on a Hypercarb (Thermo Scientific) 2.1 × 100 mm column using a mobile phase of A (7.5 mM ammonium acetate, 0.05% NH4OH) and B (acetonitrile with 0.05% NH4OH). Samples were eluted on a gradient using multiple reaction monitoring (MRM) transitions. Response factors (Rf) were calculated; the calibration curve was recalculated by a least squares fit after subtracting the mean Rf for the blanks from the Rf for each sample and control.
Epigenetic analyses
EPIC array methylation data was preprocessed with the R package Minfi [47] and normalized using the prequantile process. Probes were filtered for low quality (p-value greater than 0.01) and known single nucleotide polymorphisms. Probes belonging to the sex chromosomes were excluded. Cumulative distribution function (CDF) plots and PCA plots of methylation profiles for samples were created with ggplot2 [48] and limma [49], respectively.
Differential probe methylation was determined with limma (lmFit(object=mVals, design=design, block=targets$Patient_ID, correlation=corfit$consensus.correlation)) using M values. Sample IDs were treated as blocked. A consensus correlation was calculated using patient ID as a blocking parameter. The model design matrix was ~0+treatment_group+sex. Statistically significant changes were designated as values with an unadjusted p-value of less than 1×10−6. Differentially methylated regions were determined with the mCSEA [50] package using default parameters.
Epigenetic age was estimated with the Horvath DNA Methylation Age Calculator website (https://dnamage.genetics.ucla.edu/home) according to the Horvath recommended parameters. Beta values were first quantile normalized and then normalized with Horvath BMIQ normalization. DNA methylation age and various age acceleration values were obtained from the age calculator.
Mean values of epigenetic age were then compared between placebo- and NR-treated individuals via paired t-test. Estimation statistics were used to calculate the bootstrap 95% confidence interval (bootstrap = 5000) of the mean paired difference between pre- and post-intervention in the placebo and NR groups for AgeAcceleration values of four different epigenetic clocks: Horvath-IEAA, Hannum-EEAA [51], PhenoAge [52], and GrimAge [53]. Estimation graphs and statistics were created with the dabestr package [54].
Functional status
Katz activities of daily living (ADL [55]) assessment scale and the Lawton instrumental activities of daily living (IADL [56]) assessment scale were administered during the screen visit and at the completion of the study.
Assessments of cognition
Assessment of MCI was performed using the MoCA [44] with a cutoff score of < 26. Further cognitive testing included the executive function interview (EXIT [57]) and executive clock drawing (CLOX 1 [58]). Possible cognitive cofounders were screened for including depression using the Geriatric Depression Scale (GDS), anxiety using the Geriatric Anxiety Scale (GAS [45]), and hearing loss using a pure tone average of air conduction thresholds at 0.5, 1, 2, and 4 kHz. These tests were performed prior to participants beginning study medication and at completion of the study.
Magnetic resonance imaging
Two MRI imaging modalities were used to acquire distinct data for each participant in pre-treatment and post-treatment sessions. They included a 7-min structural T1-weighted MRI scan used to determine changes in gray matter density pre- and post-treatment using voxel-based morphometry values and two pseudocontinuous arterial spin labeling (pcASL) scans per session to measure radio-labeled arterial blood flow pre- and post-treatment. Two scans were performed per individual per session to accommodate for field of view limitations superiorly and inferiorly. All MRI acquisitions were done on a 3T Siemens TIM-Trio MRI scanner at the University of Texas Health Science Center at San Antonio’s Research Imaging Institute. An 8-element high-resolution phase array head coil equipped with foam padding was used to comfortably restrict head motion. A standard localizer image was obtained for each participant for determining head placement, followed by a standard shim sequence. T1-weighted structural image processing was performed using the FMRIB Software Library’s (FSL) brain extraction [59], voxel-based morphometry [60, 61]), brain segmentation, and non-linear registration to MNI space protocol tools [62]. pcASL image processing was performed using the FSL’s Bayesian inferencing for ASL (BASIL) [63]. More detailed information can be found in the supplementary methods section.
Two sets of regions of interest were selected prior to scanning sessions. The first involved the gross structures of the hippocampus obtained from the Harvard-Oxford structural probability atlas [64]. The second involved the default mode network (DMN) collectively and its nine constituent regions in the form of 12-mm spherical nodes centered on maxima coordinates. The DMN and its comprising nodes were derived from an activation likelihood estimation and meta-analytic connectivity modeling analysis by Laird et. al. [65]. The DMN nodes include precuneus (pC), left middle frontal gyrus (l. MFG), left inferior parietal lobule (l. IPL), right inferior parietal lobule (r. IPL), posterior cingulate cortex (PCC), middle prefrontal gyrus (m. PFG), left middle temporal gyrus (l. MTG), right middle temporal gyrus (r. MTG), and ventral anterior cingulate cortex (v. ACC). All nodes and ROIs are provided as supplementary material in the Montreal Neurological Institute (MNI) brain space.
Physical function assessments
Upper body extremity function was assessed using a handheld dynamometer to measure grip strength, and lower extremity function was assessed using the short physical performance battery (SPPB [66]). These tests were performed prior to participants beginning study medication and at completion of the study.
Statistical analyses
The primary objective was to estimate the pre-/post-difference in the NR group rather than hypothesis testing. The primary outcome was the MoCA score. The pre-/post-difference relative baselines were estimated using the paired t-test and 95% confidence intervals, and the between-group differences were evaluated with the Welch t-test. All p-values were two-sided with an alpha of 0.05. Given the exploratory nature of this study, p-values were not corrected for multiple comparisons unless stated in the text.
Sample size considerations
Assuming that the between-subject standard deviation of MoCA score change is 3.11 and a correlation of 0.5 between pre- and post-intervention measurements, 10 participants will allow the pre-/post-difference to be estimated with a 95% confidence interval with a half width of ~ 2.2.
Ethical approval, informed consent, and study location
All procedures were approved by the UT Health San Antonio Institutional Review Board. The nature, benefits, and risks of the study were explained to all subjects, and their written informed consent was obtained prior to participation. All measurements were performed at the Clinical Research Unit of the Audie Murphy VA Hospital in San Antonio, TX. All brain imaging was performed at UT Health San Antonio within the Research Imaging Institute. The study was registered on ClinicalTrials.gov under the identifier NCT02942888.
Results
Study population
A total of 150 potential participants were screened for eligibility; 36 were enrolled and 20 were randomized between August 2017 and August 2019. Of the 36 enrolled subjects, 1 withdrew prior to randomization due to study time commitment and 15 failed to meet eligibility criteria including 2 that were unable to perform MRI and were excluded after randomization. A total of 10 MCI participants were randomized to the NR group and 10 were randomized to the placebo group (Fig. 2); demographics and baseline characteristics are described in Table 1.
Fig. 2.
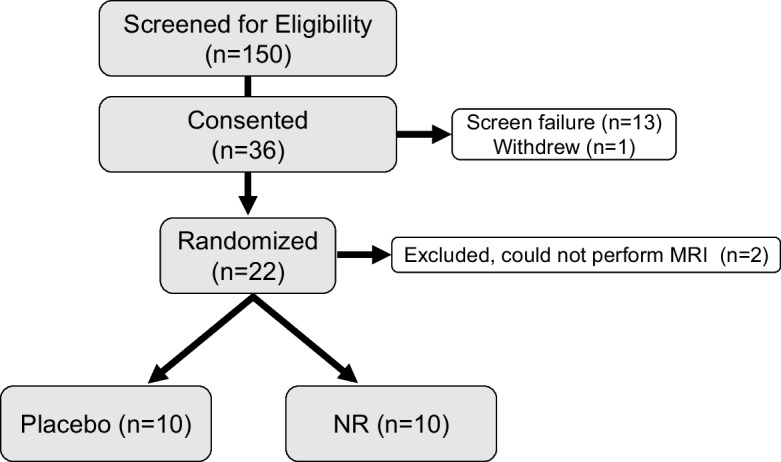
Study flow diagram. Of the 150 screened individuals, we present data from the 20 individuals that completed the study
Table 1.
Subject demographics and baseline characteristics
| Characteristic | Placebo (N=10) | NR (N=10) |
|---|---|---|
| Sex (M/F) | 2/8 | 5/5 |
| Age, years: average (min, max) | 75.2 (67, 86) | 77.1 (71, 83) |
| Ethnicity (Hispanic/not Hispanic) | 9/1 | 8/2 |
| Race (White/Black or African American) | 10/0 | 8/2 |
| MoCA score (Montreal Cognitive Assessment) | 24.20 ± 1.81 | 23.10 ± 2.77 |
| GAS (Geriatric Anxiety Scale) | 2.30 ± 2.41 | 2.20 ± 2.20 |
| GDS (Geriatric Depression Score) | 1.40 ± 1.43 | 1.00 ± 0.94 |
Data presented as mean ± standard error
Course of treatment
Enrolled subjects began a dose escalation of NR to reach 1 g/day. The dosing strategy consisted of 250 mg/day for the first week, 500 mg/day for the second week, and 750 mg/day for the third week. By week 4, all subjects began taking 1 g/day (Fig. 1), which was well tolerated and maintained throughout the 10-week study. One subject experienced severe nausea which resolved by temporarily reducing the NR dose; the final 1 g/day dose was re-achieved by dose escalation with no complaints.
Safety and tolerability
Adherence to the study treatment was excellent, with all subjects consuming greater than 95% of all NR and placebo capsules administered. NR was well tolerated at the dose tested, and no serious adverse events (AEs) occurred. One subject that received placebo suffered a stroke. A total of 18 AEs were reported by 7 of the 10 participants randomized to NR and 21 AEs reported by 7 of the 10 participants randomized to placebo. These included a procedure-related bruise on the wrist (0/10 NR; 1/10 placebo), heartburn/acid reflux (2/10 NR; 0/10 placebo), nausea (1/10 NR; 0/10 placebo), fatigue (1/10 NR; 0/10 placebo), hot flash/light-headed/vertigo (1/10 NR; 0/10 placebo), unsteady on feet (0/10 NR; 1/10 placebo), gastrointestinal complaints (2/10 NR; 2/10 placebo), and headache (1/10 NR; 0/10 placebo) (Supplementary Table 1). The self-reported AEs associated with the study ranged in severity from mild to moderate, except for one participant enrolled in the NR study arm that reported severe nausea and heartburn. Both symptoms were resolved by reducing the NR dose from 1 g to 750 mg for 1 week; the participant was then able to return to 1 g for the remainder of the study. No subjects dropped out during the study.
Clinical laboratory values were derived from blood samples collected before (pre) and after (post) taking oral supplementation of either NR or placebo from all participants (20 of 20 subjects completed the study). No meaningful differences were observed between treatment conditions for hematology (Supplementary Table 2), blood chemistry (Supplementary Table 3), or blood lipid profiles (Supplementary Table 4).
NR increased NAD+ and associated metabolites
NAD+ levels in peripheral blood mononuclear cells (PBMCs) peak 8 h after the administration of NR [39] in a dose-dependent manner [40]. The upper limit, 1 g/day, previously has been shown to be well tolerated and increase NAD+ levels in PBMCs by 142% [40]. Similarly, we observed an average 139% increase in NAD+ (mean change = 30.63 pmol/μL blood). NR supplementation also significantly increased NAAD, NMN, and Me4Py (6, 1.2, and > 10-fold increases, respectively) (Fig. 3). These results are in agreement with the previous reports [39, 67].
Fig. 3.

Oral supplementation with NR alters NAD+ blood metabolism. a Whole blood levels of NAD+ and associated metabolites from placebo- and NR-treated subjects before and after treatment. The pre-/post-difference relative baselines were estimated using the paired t-test and 95% confidence intervals, and the between-group differences were evaluated with the Welch t-test. b Diagram of NAD+ metabolism depicting analytes that significantly changed during the study. Data are plotted as within subject pre- vs post-levels. Open circles, baseline levels; gray circles, post-treatment values. Lines connect individual subjects. n = 10/group. *p < 0.05; **p < 0.01; ***p < 0.001. NR, nicotinamide riboside; NaM, nicotinamide; NA, nicotinic acid; NAR, nicotinic acid riboside; NAMN, nicotinic acid mononucleotide; NMN, nicotinamide mononucleotide; NAAD, nicotinic acid adenine dinucleotide; NAD+, nicotinamide adenine dinucleotide; ADPR, ADP-ribose; AMP, adenosine monophosphate; Me-4-Py, N-methyl-4-pyridone-5-carboxamide
Effects of NR on indicators of cognitive function
To evaluate cognition, we compared scores from MoCA, CLOX, CLOX2, and EXIT before and after 10 weeks of treatment and compared pre- vs post-differences within study arms. The primary cognitive outcome measure, MoCA, decreased in the placebo arm from 24 ± 1.8 at screening to 23.11 ± 2.47 at the end of the 10-week study (post-/pre-difference, μ, −0.89, 95% CI [−2.19, 0.41], Table 2). In the active treatment arm, we observed a decrease in MoCA score from 23.1 ± 2.77 at screening to 22.8 ± 3.46 after 10 weeks of NR (post-/pre-difference, μ= −0.3, 95% CI [−2.21, 1.61]). No appreciable differences in CLOX, CLOX2, or EXIT were observed in either treatment arm. All subjects in both arms scored normal on the GAS and GDS at both pre- and post-study visits. Collectively, these results indicate that depression, anxiety, and cognitive performance in short-term memory; visuospatial abilities; executive functions; attention, concentration, and working memory; language; and orientation to time and place remained stable during the 10-week study.
Table 2.
NR effects on arterial blood flow (pcASL) within the default mode network. Each structure is represented as a 12-mm-diameter spherical region of interest centered at the activation maxima coordinates from [68]
| Region of interest | Placebo | NR | Group difference | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DMN structures | Pre | Post | Pre-post (95% CI) | p-value | Pre | Post | Pre-post (95% CI) | p-value | Group differences (95% CI) | p-value |
| Total DMN | 9.7 ± 1.75 | 10.05 ± 2.59 | −0.36 [−2.03, 1.32] | 0.642 | 9.51 ± 2.25 | 8.59 ± 1.73 | 0.92 [0.23, 1.62] | 0.013 | −1.28 [−3.03, 0.47] | 0.138 |
| Precuneus | 11.88 ± 2.93 | 12.74 ± 3.43 | −0.86 [−3.4, 1.67] | 0.46 | 11.21 ± 3.12 | 9.81 ± 2.52 | 1.4 [−0.13, 2.92] | 0.069 | −2.26 [−5.07, 0.54] | 0.107 |
| PCC | 13.28 ± 3.23 | 13 ± 4.74 | 0.28 [−2.09, 2.64] | 0.798 | 11.39 ± 4.1 | 9.85 ± 3.55 | 1.54 [0.14, 2.94] | 0.033 | −1.26 [−3.88, 1.35] | 0.32 |
| r. IPL | 11.43 ± 2.86 | 12.27 ± 2.91 | −0.84 [−2.7, 1.01] | 0.33 | 11.76 ± 2.48 | 10.4 ± 2.42 | 1.36 [−0.1, 2.81] | 0.066 | −2.2 [−4.42, 0.02] | 0.052 |
| v. ACC | 8.2 ± 1.78 | 8.83 ± 2.42 | −0.63 [−3.06, 1.81] | 0.574 | 7.37 ± 2.82 | 6.59 ± 1.64 | 0.79 [−0.45, 2.02] | 0.192 | −1.42 [−4.03, 1.2] | 0.266 |
| m. PFC | 7.31 ± 2.23 | 7.64 ± 2.22 | −0.33 [−1.81, 1.15] | 0.626 | 7.59 ± 2.34 | 6.86 ± 2.16 | 0.73 [−0.25, 1.71] | 0.131 | −1.06 [−2.74, 0.62] | 0.2 |
| r. MTG | 9.16 ± 2.82 | 8.19 ± 2.93 | 0.97 [−1.28, 3.23] | 0.349 | 8.54 ± 2.96 | 8.69 ± 2.91 | −0.15 [−2.11, 1.8] | 0.867 | 1.12 [−1.65, 3.9] | 0.406 |
| l. MFG | 7.34 ± 2.42 | 8.1 ± 2.53 | −0.76 [−3.05, 1.53] | 0.474 | 8.21 ± 2.86 | 7.33 ± 1.99 | 0.87 [−0.19, 1.94] | 0.1 | −1.63 [−4.06, 0.8] | 0.171 |
| l. IPL | 10.21 ± 2.66 | 10.36 ± 2.32 | −0.14 [−1.9, 1.62] | 0.858 | 10.77 ± 3.08 | 9.11 ± 2.98 | 1.66 [0.5, 2.82] | 0.009 | −1.8 [−3.8, 0.19] | 0.074 |
| l. MTG | 8.3 ± 2.44 | 7.85 ± 2.56 | 0.45 [−1.89, 2.8] | 0.669 | 9.02 ± 3.02 | 8.51 ± 2.14 | 0.51 [−1.22, 2.24] | 0.531 | −0.06 [−2.78, 2.66] | 0.965 |
| r. Hippo | 6.15 ± 1.93 | 5.36 ± 1.95 | 0.79 [−0.76, 2.34] | 0.274 | 6.44 ± 1.82 | 6.51 ± 1.4 | −0.07 [−1.28, 1.14] | 0.902 | 0.86 [−0.97, 2.69] | 0.337 |
| l. Hippo | 6.13 ± 1.86 | 5.24 ± 1.83 | 0.89 [−1.11, 2.88] | 0.334 | 6.35 ± 1.66 | 6.27 ± 1.1 | 0.08 [−1, 1.16] | 0.877 | 0.81 [−1.34, 2.96] | 0.43 |
NR nicotinamide riboside, DMN default mode network, PCC posterior cingulate cortex, r. IPL right inferior parietal lobe, v. ACC ventral anterior cingulate cortex, m. PFC medial prefrontal cortex, r. MTG right mid temporal gyrus, l. MFG left mid frontal gyrus, l. IPL left inferior parietal lobe, l. MTG left mid temporal gyrus, r. Hippo right hippocampus, l. Hippo left hippocampus
Effects of NR on brain function and volume as assessed by MRI
We did not observe pre- vs post-differences in gray matter volume in either treatment arm as assessed by structural MRI analyses (Supplementary Table 5). CBF was measured by pseudocontinuous arterial spin labeling (pcASL). Subjects randomized to the placebo group showed no pre- vs post-treatment changes. In contrast, NR-treated individuals showed decreased blood flow in the nine DMN nodes collectively post-treatment (p = 0.013). More specifically, the left inferior parietal lobe and posterior cingulate cortex showed statistical significance (p = 0.009 and p = 0.033, respectively), and the right inferior parietal lobe and precuneus showed similar trends (p = 0.066 and p = 0.069, respectively; Fig. 4 and Table 3). Differences in other brain regions were not observed to indicate brain region specificity of the NR-mediated change in CBF.
Fig. 4.

Effects of NR treatment in specific regions of the default mode network (DMN) and hippocampus reported as uncorrected p-values. a Medial prefrontal cortex (m. PFC), ventral anterior cingulate cortex (v. ACC), left mid frontal gyrus (l. MFG), left hippocampus (l. HC), right hippocampus (r. HC), left inferior parietal lobe (l. IPL), right inferior parietal lobe (r. IPL), posterior cingulate cortex (PCC), left mid temporal gyrus (l. MTG), and right mid temporal gyrus (r. MTG). b The change in precuneus cerebral blood flow (CBF) significantly, inversely correlated with post-treatment blood NAD+ levels. N=19 subjects; one subject was excluded due to poor image quality on post-visit. p = 0.0448; R2 = 0.22. Change in CBF reflects normalized signal intensity values from MRI scans
Table 3.
Intervention effects on neuropsychiatric and cognitive performance
| Instrument | Placebo | NR | Group difference | ||||
|---|---|---|---|---|---|---|---|
| Pre | Post | p-value | Pre | Post | p-value | p-value | |
| MoCA | 24 ± 1.8 | 23.11 ± 2.47 | 0.154 | 23.1 ± 2.77 | 22.8 ± 3.46 | 0.730 | 0.57 |
| CLOX | 13.22 ± 1.79 | 14 ± 1.12 | 0.193 | 13.38 ± 2.67 | 14 ± 0.76 | 0.582 | 0.902 |
| CLOX2 | 14.44 ± 0.73 | 14.56 ± 0.53 | 0.760 | 14.75 ± 0.46 | 14.88 ± 0.35 | 0.598 | 0.974 |
| EXIT | 9.7 ± 3.95 | 9 ± 5.12 | 0.612 | 10.88 ± 3.18 | 11.38 ± 4.66 | 0.705 | 0.524 |
| GAS | 2.3 ± 2.41 | 1.4 ± 1.78 | 0.384 | 2.44 ± 2.19 | 2.78 ± 3.19 | 0.631 | 0.315 |
| GDS | 1.4 ± 1.43 | 0.9 ± 1.29 | 0.138 | 1 ± 0.94 | 1.7 ± 2.11 | 0.132 | 0.035 |
NR nicotinamide riboside, MoCA Montreal Cognitive Assessment, CLOX executive clock drawing task, EXIT executive interview, GAS Geriatric Anxiety Scale, GDS Geriatric Depression Scale
Effects of NR on indicators of physical function
To assess physical function, we administered the short physical performance battery (SPPB) [66] and grip strength before and after 10 weeks of treatment and compared pre- vs post-differences within study arms (Supplementary Table 6). The placebo arm showed a modest, statistically significant, improvement in the SPPB total score (pre, 8.67 ± 2.12; post, 10.11 ± 1.62; p = 0.044) while the NR group did not (pre, 10.4 ± 1.51; post, 9.5 ± 2.12; p = 0.134), which resulted in a statistically significant between-group difference, p = 0.011 (Supplementary Table 6). No changes were observed in balance or grip strength in either group. Similar to SPPB, we observed that subjects in the placebo arm, but not the NR arm, performed modestly better in the five times sit-to-stand time in their post-visit than their pre-visit (placebo pre 17.54 ± 4.57 s vs post 13.35 ± 2.66 s, p = 0.008; NR pre 14.47 ± 4.67 s vs post 14.21 ± 2.97 s, p = 0.864), with a significant group effect (p = 0.03, Supplementary Table 6). Group differences were also observed in gait speed, a more robust measure of frailty, whereby the placebo arm increased their walking speed as evidenced by a reduced amount of time to complete a 4-m walk (pre 4.05 ± 0.63 s vs post 3.67 ± 0.63 s) but the NR group did not (pre 4.57 ± 1.1 s vs post 4.83 ± 0.88 s). These small between-group patterns resulted in a subtle, but statistically significant, group difference for improvement in the placebo group which may reflect a practice effect (p = 0.044; Supplementary Table 6).
Effect of NR on other domains of physiological function
Other physiological measures monitored during the study included body temperature, weight, blood pressure, heart rate, respiratory rate, body mass index, and hearing. No changes in any of these measurements were observed in either treatment arm during the course of the study, Supplementary Table 7.
Effect of NR on epigenetics
Peripheral blood mononuclear cells (PBMC) were collected from each subject at baseline and at the end of study. Illumina EPIC methylation arrays were used to measure PBMC DNA methylation profiles. Analyses with cumulative distribution function (CDF) plots indicated similar global methylation profiles of the placebo- and NR-treated participants. We observed subtle differences in methylation between treatment groups in CpG sites with baseline methylation frequency between 0.4 and 0.7 (Fig. 5a). Pre- to post-treatment comparisons in each treatment group revealed an insignificant trend toward reduced methylation in individuals in the placebo arm and trend toward increased methylation in individuals treated with NR (Fig. 5a). Differential methylation analysis between randomization groups, while accounting for sex, revealed little statistically significant changes between placebo and NR study arms (Fig. 5b).
Fig. 5.

Effects of NR on DNA methylation profiles in peripheral blood mononuclear cells. a Cumulative distribution function plots showing methylation profiles of the most variable CpG sites in the range of 0.4–0.7 methylation frequency. b CpG volcano plots of differences between the placebo and NR intervention. X-axis, log2 fold-change of methylation value; Y-axis, −log10 of unadjusted p-values. Red horizontal line, p-value = 1 × 10−6. c Estimation plot of rate of aging using AgeAccelPheno (above) and paired mean difference (below) shows treatment-associated differences in rate of epigenetic aging between study arms. Dot, average paired mean difference; black vertical line, 95% CI; and distribution in placebo and NR groups
We also calculated epigenetic age, which is a proposed biomarker for biological age (for review [69]), across samples. The epigenetic ages (DNAmAge) in our samples were below that of their corresponding chronological ages, and we did not detect significant changes in paired mean differences between the treatment groups. We also calculated AgeAcceleration values of four different epigenetic clocks, Horvath-IEAA, Hannum-EEAA, PhenoAge, and GrimAge. We did not detect statistically significant changes across the study in either arm using paired t-tests. As an alternative, estimation statistics were used to calculate the bootstrap 95% confidence interval (bootstrap = 5000) of the mean paired difference between pre- and post-intervention in the NR and placebo groups (Supplementary Figure 1). Both AgeAccelPheno and AgeAccelGrim detected a subtle decrease in paired mean difference values when comparing pre- vs post-NR treatment (Fig. 5c and Supplementary Figure 1a, respectively). In participants treated with placebo, the mean difference in AgeAccelPheno had negligible change, while the AgeAccelGrim identified an increase in “biological age” over the course of placebo treatment. These findings are in contrast to quantification of epigenetic age using the EEAA, which identified markers of accelerated aging upon NR treatment but not placebo (Supplementary Figure 1b). IEAA clock analysis did not detect changes between groups (Supplementary Figure 1c).
Discussion
Previous studies have evaluated the safety, tolerability, and pharmacology of NR [39, 41, 67] but not in the context of cognitive impairment. We conducted a randomized phase 2, 10-week pilot study comparing NR to placebo for pre- vs post-study differences in cognition, CBF and brain volume, and measures of physical frailty in older adults with MCI. Consistent with prior studies, NR was well tolerated with no differences in AEs, blood chemistry, lipid, or other physiological measures reported between NR and placebo treatment arms (Supplementary Tables 1–4, 6). Dosing with NR effectively increased levels of NAD+ and associated metabolites in the blood (Fig. 3). The changes in our study were consistent with that reported using 1 g/day in previous studies [40]. The most common complaint of niacin, a historically used strategy to increase NAD+, is flushing, which was not reported here indicating that NR provides a safe and tolerable alternative to increasing NAD+. A prior study infused NAD+ intravenously for 8 h and achieved a 398% increase in plasma NAD+ levels which coincided with a low magnitude, but significant rise in bilirubin possibly suggesting red blood cell turnover consistent with infusion induced hemolysis [70]. We did not observe evidence of hemolysis with NR despite a 139% increase in NAD+ (Supplementary Tables 2, 3). These findings suggest that indirectly increasing NAD+ through its precursor molecules may be a safer approach than directly infusing it; alternatively, the magnitude of increase may be an important consideration to control and monitor when using NAD+ boosting therapies.
The establishment of a favorable safety profile for NR has motivated investigators to begin testing its therapeutic potential, primarily on metabolic outcomes. The initial efficacy studies revealed an NR-dependent reduction in blood pressure in healthy middle-aged and older adults [41] and enhanced NAD+ metabolism in skeletal muscle [71, 72]. Studies on muscle samples from 12 older men that completed a 3-week trial of 1 g/day NR indicated downregulated energy metabolism and mitochondrial pathways, but not mitochondrial bioenergetics. The trial also reported a reduction in circulating levels of IL-6 which has prompted the investigation of anti-inflammatory effects of NR [71]. A 6-week trial of 2 g/day NR in overweight or obese men and women failed to meet the primary endpoint of increased insulin sensitivity, but the trial reported an increase in sleeping metabolic rate and statistically significant reduction in fat mass [72]. When tested in obese insulin-resistant men, NR did not improve insulin sensitivity or whole-body glucose metabolism [73] underscoring a need to evaluate disease-modifying benefits of NAD+ therapies.
To our knowledge, our study was the first attempt to directly determine the safety profile of NR on cognition and brain volume and function in older adults with MCI in a randomized, double-blind placebo-controlled trial. Changes in the MoCA over time are subtle with an annual 0.52-point decrease in individuals with MCI and a 0.17 point decrease in the cognitively intact adults aged 58–77 years [74]. In our 10-week study, we observed declines in both groups that were greater than expected for the time interval, and NR may have modestly reduced the decline. On average, the placebo group experienced a 0.89-point decrease whereas the NR decreased by 0.3 points. We are cautious to not overinterpret these results due to the small sample size. Moreover, our study population was 85% Hispanic; the rate of decline is not as well defined/stratified by ethnicity. Evidence suggests that Hispanics are at greater risk of developing dementia due to risk factors including lower level of education, vascular disease, diabetes, and hypertension [75–77], thus may decline more rapidly than other ethnic groups. The subtle between-group differences observed in our pilot study are encouraging, i.e., NR did not worsen cognition in a primarily Hispanic population However, a larger study with a longer trial duration is needed to confidently infer treatment effects.
MRI analyses have identified changes in brain volume and function associated with aging and pathologic cognitive decline. Similar to rate of decline in cognition, the annual changes in brain volume are subtle. A systematic review with meta-analysis evaluated annual cerebral atrophy rates in healthy controls and MCI. They estimated whole brain atrophy rates of 0.57%/year for controls and 1.02%/year for MCI [78]. We did not observe significant changes in brain volume indicating that NR did not improve or worsen disease trajectory over the course of our 10-week study period. Functional brain imaging studies have shown age-associated changes that differ between normal aging and disease in several brain regions, particularly structures within the DMN [68]. The DMN is comprised of a set of brain regions that exhibit increased activity during resting state and decreased activity during task-related paradigms, with the exception of social cognition tasks. Normal aging deficit patterns have been associated with anterior regions of the DMN whereas posterior regions show greater susceptibility to MCI and AD [79–81]. In our study, NR decreased blood flow in the nine DMN nodes (collectively) post-treatment (p = 0.013) compared to placebo controls where no change was observed (p = 0.642). More specifically, the left inferior parietal lobe (p = 0.009) and posterior cingulate cortex (p = 0.033) showed statistical significance, and the right inferior parietal lobe (p = 0.066) and precuneus (p = 0.069) showed similar trends. The left inferior parietal lobe (also known as Gerschwind’s territory) is an important brain region for processing perspective differences and episodic memory [82]. Increased CBF in MCI/early AD has been hypothesized to reflect compensatory cellular and vascular processes to counteract pathogenesis [83]. Specifically, the nodal stress hypothesis proposes that decreases in blood flow in high metabolically active brain regions, such as the DMN, would decrease oxidative stress [84, 85]. Therefore, the NR-associated decrease in blood flow may prevent these regions from experiencing degradation from high metabolic cost. However, it is important to note that other studies have found reduced CBF in MCI [86] suggesting that a further reduction with NR may be detrimental. Although the level of significance in this study did not meet the thresholds after correction for multiple comparisons, a power analysis showed that a similar study conducted with 40 participants could yield statistically significant results across these brain regions. These data also indicate that for future trials, instruments of social cognition may be more sensitive at detecting effects from NR.
We included multiple physiological outcome measures to evaluate effects of NR in older adults with MCI. We did not observe significant NR-associated changes across most systems analyzed (Supplementary Tables 2–5, 7). The SPPB increased in the placebo group and modestly decreased in the NR group resulting in a statistically significant group difference (Supplementary Table 6). While the between-group difference is intriguing (i.e., lack of improvement in NR), the pre-visit group differences indicate that individuals in the placebo group performed slightly worse at baseline in the time to stand test (pre-visit measures, 17.54 s ± 4.57 vs 14.47 s ± 4.67 in placebo and NR, respectively). A “practice” effect was observed in the placebo group in this test; however, the NR study arm did not improve which may reflect a ceiling effect in this group. Walking speed, a measure of frailty and mortality [87], showed a significant group difference. Specifically, we observed an improvement in time to complete a 4-m walk in the placebo arm, but not in the NR arm; this improvement in the placebo group may also be a practice effect. The stable time to stand and walking speed in the NR arm support a positive safety profile for the intervention, although the blunting of the practice effects seen with placebo warrants further investigation.
NAD+ is critical to cellular function across tissues. Prior studies indicate an inverse correlation between cellular NAD+ levels and DNA methylation [88, 89]. Despite the NR-induced increase in NAD+, we did not detect large changes in DNA methylation in either group. To investigate more subtle changes, we generated CDF plots of the methylation profiles for the groups. These analyses indicated that NR treatment resulted in methylation levels 40–70% as that observed in placebo. We found that participants in the NR arm seemed to gain methylation while the placebo-treated individuals lost methylation over the course of the study. This finding is supported by analyzing unadjusted p-values (no multiple test correction), which suggests that the most significant CpG methylation sites were more methylated in the NR-treated group than the placebo group. These results contrast with prior work showing that NAD+ reduces methylation [88] and increased DNA methylation observed in the presence of low NAD+ [89]. The discrepancies may reflect cell-type-specific responses to NAD+ as shown in vitro, whereby HEK 293 cells have a greater demethylation response than Jurkat cells [88]. Our analysis of epigenetic aging using four different methods revealed slowing of epigenetic aging in the NR-treated group using epigenetic clocks that are based on lifespan and healthspan measures (AgeAccelPheno and AgeAccelGrim). On the other hand, epigenetic clocks that are based on chronological aging (EEAA and IEAA) did not reveal a beneficial effect of NR treatment. While this trial was not powered to detect significant changes in DNA methylation, our analyses suggest that this NAD+ intervention may subtly alter methylation.
Strengths and limitations
While our study provides critical information on the safety and efficacy of NR in older adults with MCI, there are several important limitations that must be considered. First, our study was designed to evaluate safety and tolerability. Therefore, it was not powered to assess outcomes related to cognition or disease modification. Another limitation is the lack of measures for target engagement in the brain; ongoing studies are working to address this (e.g., NCT04430517). While the small sample size precludes concrete conclusions, strong trends with differences specifically in CBF in brain regions affected by the progression of MCI to dementia warrant further investigation. Follow-up testing in Hispanics, the majority of our study population, is best supported by our pilot study; nonetheless, understanding whether or not potential benefits of NR translate to other ethnicities also should be pursued.
Conclusions
A 10-week oral administration of NR at a dose of 1 g/day is safe in older adults with MCI. This dose and duration significantly increased blood levels of NAD+ and associated metabolites. NR supplementation was associated with alterations in CBF in the posterior regions of the DMN implicated in MCI and AD as assessed by MRI. These data support a larger RCT to test NR therapy as a strategy to modulate the progression of MCI to AD; a sample size of n = 40 subjects is predicted to provide statistically significant results in CBF measures. Additionally, future studies may consider including neuropsychiatric measures for social cognition and other more subtle paradigms relevant to the DMN and IPL.
Supplementary information
(DOCX 219 kb)
Acknowledgements
We would like to thank Beverly Orsak and Terry Romo for assisting with study coordination and patient visits, Rozmin Jiwani for assisting with study visits and Meghana Koleti for data entry; Crystal Franklin in the Research Imaging Institute staff for data curation and essential preprocessing steps. We also thank Dr. Brian Kennedy for offering valuable insights on experimental design.
Author contribution
Research study design: MEO, PTF, NM, and BP; subject enrollment: DBK; conducted experiments: EK, DBK, MSS, YW, JG, and BP; provided reagents: CB; data analyses and interpretation: MEO, EK, PR, CB, PTF, JG, SE, BF, NM, and BP; drafted the manuscript: MEO, EK, and BP; critical revision: MEO, EK, CB, PTF, JG, SE, NM, and BP.
Funding
Some authors were supported in part by funds from the National Institutes of Health (NIH South Texas Medical Scientist Training Program: T32GM113898, NIMH: R01MH074457-11S1, NIMH: R01MH074457, NCATS Translational Scientist Training Program: TL1TR002647-01). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Data Availability
The minimum datasets necessary to interpret, verify and extend the research in the article are available within the paper and its Supplementary Information. The trial was registered on ClinicalTrials.gov: NCT02942888.
Declarations
Conflict of interest
ChromaDex Inc. provided the investigators with NR and placebo. Charles Brenner, a co-author, is an inventor of intellectual property on uses of nicotinamide riboside. He serves as Chief Scientific Advisor of ChromaDex and owns ChromaDex stock. Dr. Brenner’s contributions included data interpretation regarding NAD+ metabolites and manuscript editing. He was not involved in study design or execution. Dr. Orr reports a grant from US Department of Veterans Affairs during the conduct of the study. In addition, Dr. Orr has a patent biosignature and therapeutic approach for neuronal senescence pending outside the scope of this work. No other co-authors have COIs to declare.
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Miranda E. Orr, Nicolas Musi, and Becky Powers contributed equally to this study.
References
- 1.Petersen RC, et al. Mild cognitive impairment: ten years later. Arch Neurol. 2009;66(12):1447–1455. doi: 10.1001/archneurol.2009.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts RO, et al. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology. 2008;30(1):58–69. doi: 10.1159/000115751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Busse A, et al. Mild cognitive impairment: long-term course of four clinical subtypes. Neurology. 2006;67(12):2176–2185. doi: 10.1212/01.wnl.0000249117.23318.e1. [DOI] [PubMed] [Google Scholar]
- 4.Panza F, et al. Current epidemiology of mild cognitive impairment and other predementia syndromes. Am J Geriatr Psychiatry. 2005;13(8):633–644. doi: 10.1097/00019442-200508000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Petersen RC, et al. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56(3):303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- 6.Bozoki A, et al. Mild cognitive impairments predict dementia in nondemented elderly patients with memory loss. Arch Neurol. 2001;58(3):411–416. doi: 10.1001/archneur.58.3.411. [DOI] [PubMed] [Google Scholar]
- 7.Morris JC, et al. Mild cognitive impairment represents early-stage Alzheimer disease. Arch Neurol. 2001;58(3):397–405. doi: 10.1001/archneur.58.3.397. [DOI] [PubMed] [Google Scholar]
- 8.Ritchie K, Artero S, Touchon J. Classification criteria for mild cognitive impairment: a population-based validation study. Neurology. 2001;56(1):37–42. doi: 10.1212/WNL.56.1.37. [DOI] [PubMed] [Google Scholar]
- 9.Daly E, et al. Predicting conversion to Alzheimer disease using standardized clinical information. Arch Neurol. 2000;57(5):675–680. doi: 10.1001/archneur.57.5.675. [DOI] [PubMed] [Google Scholar]
- 10.Grober E, et al. Memory impairment on free and cued selective reminding predicts dementia. Neurology. 2000;54(4):827–832. doi: 10.1212/WNL.54.4.827. [DOI] [PubMed] [Google Scholar]
- 11.Jicha GA, et al. Neuropathologic outcome of mild cognitive impairment following progression to clinical dementia. Arch Neurol. 2006;63(5):674–681. doi: 10.1001/archneur.63.5.674. [DOI] [PubMed] [Google Scholar]
- 12.2023 Alzheimer's disease facts and figures. Alzheimers Dement. 2023;19(4):1598–1695. [DOI] [PubMed]
- 13.Gonzales MM, et al. Biological aging processes underlying cognitive decline and neurodegenerative disease. J Clin Invest. 2022;132(10). [DOI] [PMC free article] [PubMed]
- 14.Tong JJ, et al. Chronic acarbose treatment alleviates age-related behavioral and biochemical changes in SAMP8 mice. Behav Brain Res. 2015;284:138–152. doi: 10.1016/j.bbr.2015.01.052. [DOI] [PubMed] [Google Scholar]
- 15.Dunn HC, et al. Restoration of lipoxin A4 signaling reduces Alzheimer’s disease-like pathology in the 3xTg-AD mouse model. J Alzheimers Dis. 2015;43(3):893–903. doi: 10.3233/JAD-141335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chandra S, Jana M, Pahan K. Aspirin induces lysosomal biogenesis and attenuates amyloid plaque pathology in a mouse model of Alzheimer’s disease via PPARalpha. J Neurosci. 2018;38(30):6682–6699. doi: 10.1523/JNEUROSCI.0054-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Medeiros R, et al. Aspirin-triggered lipoxin A4 stimulates alternative activation of microglia and reduces Alzheimer disease-like pathology in mice. Am J Pathol. 2013;182(5):1780–1789. doi: 10.1016/j.ajpath.2013.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caccamo A, et al. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285(17):13107–13120. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Majumder S, et al. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One. 2011;6(9):e25416. doi: 10.1371/journal.pone.0025416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang L, et al. Evaluating the effectiveness of GTM-1, rapamycin, and carbamazepine on autophagy and Alzheimer disease. Med Sci Monit. 2017;23:801–808. doi: 10.12659/MSM.898679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orr ME, et al. Mammalian target of rapamycin hyperactivity mediates the detrimental effects of a high sucrose diet on Alzheimer’s disease pathology. Neurobiol Aging. 2014;35(6):1233–1242. doi: 10.1016/j.neurobiolaging.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Musi N, et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell. 2018;17(6):e12840. doi: 10.1111/acel.12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gonzales MM, et al. A geroscience motivated approach to treat Alzheimer’s disease: senolytics move to clinical trials. Mech Ageing Dev. 2021;200:111589. doi: 10.1016/j.mad.2021.111589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gonzales MM, et al. Senolytic therapy in mild Alzheimer’s disease: a phase 1 feasibility trial. Nat Med. 2023;29(10):2481–2488. doi: 10.1038/s41591-023-02543-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajman L, Chwalek K, Sinclair DA. Therapeutic potential of NAD-boosting molecules: the in vivo evidence. Cell Metab. 2018;27(3):529–547. doi: 10.1016/j.cmet.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ansari HR, Raghava GP. Identification of NAD interacting residues in proteins. BMC Bioinform. 2010;11:160. doi: 10.1186/1471-2105-11-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu XH, et al. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci U S A. 2015;112(9):2876–2881. doi: 10.1073/pnas.1417921112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Long AN, et al. Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer’s disease-relevant murine model. BMC Neurol. 2015;15:19. doi: 10.1186/s12883-015-0272-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klaidman LK, et al. Nicotinamide as a precursor for NAD+ prevents apoptosis in the mouse brain induced by tertiary-butylhydroperoxide. Neurosci Lett. 1996;206(1):5–8. doi: 10.1016/0304-3940(96)12446-0. [DOI] [PubMed] [Google Scholar]
- 30.Donmez G, et al. SIRT1 suppresses beta-amyloid production by activating the alpha-secretase gene ADAM10. Cell. 2010;142(2):320–332. doi: 10.1016/j.cell.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Fang EF, et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci. 2019;22(3):401–412. doi: 10.1038/s41593-018-0332-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hou Y, et al. NAD(+) supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc Natl Acad Sci U S A. 2018;115(8):E1876–E1885. doi: 10.1073/pnas.1718819115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu XJ, Jiang GS. Niacin-respondent subset of schizophrenia - a therapeutic review. Eur Rev Med Pharmacol Sci. 2015;19(6):988–997. [PubMed] [Google Scholar]
- 34.Holubiec P, et al. Pathophysiology and clinical management of pellagra - a review. Folia Med Cracov. 2021;61(3):125–137. doi: 10.24425/fmc.2021.138956. [DOI] [PubMed] [Google Scholar]
- 35.Mero A, et al. Effects of nicotinamide adenine dinucleotide hydride on physical and mental performance. J Sports Sci. 2008;26(3):311–319. doi: 10.1080/02640410701474200. [DOI] [PubMed] [Google Scholar]
- 36.Arenas-Jal M, et al. Trends in the food and sports nutrition industry: a review. Crit Rev Food Sci Nutr. 2020;60(14):2405–2421. doi: 10.1080/10408398.2019.1643287. [DOI] [PubMed] [Google Scholar]
- 37.DS W, et al. Drugbank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–D672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell. 2004;117(4):495–502. doi: 10.1016/S0092-8674(04)00416-7. [DOI] [PubMed] [Google Scholar]
- 39.Trammell SA, et al. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun. 2016;7:12948. doi: 10.1038/ncomms12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Conze D, Brenner C, Kruger CL. Safety and metabolism of long-term administration of NIAGEN (nicotinamide riboside chloride) in a randomized, double-blind, placebo-controlled clinical trial of healthy overweight adults. Sci Rep. 2019;9(1):9772. doi: 10.1038/s41598-019-46120-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Airhart SE, et al. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS One. 2017;12(12):e0186459. doi: 10.1371/journal.pone.0186459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brakedal B, et al. The NADPARK study: a randomized phase I trial of nicotinamide riboside supplementation in Parkinson’s disease. Cell Metab. 2022;34(3):396–407 e6. doi: 10.1016/j.cmet.2022.02.001. [DOI] [PubMed] [Google Scholar]
- 43.Vreones M, et al. Oral nicotinamide riboside raises NAD+ and lowers biomarkers of neurodegenerative pathology in plasma extracellular vesicles enriched for neuronal origin. Aging Cell. 2022;22:e13754. doi: 10.1111/acel.13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nasreddine ZS, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005;53(4):695–699. doi: 10.1111/j.1532-5415.2005.53221.x. [DOI] [PubMed] [Google Scholar]
- 45.Segal DL, et al. Development and initial validation of a self-report assessment tool for anxiety among older adults: the Geriatric Anxiety Scale. J Anxiety Disord. 2010;24(7):709–714. doi: 10.1016/j.janxdis.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 46.Trammell SA, Brenner C. Targeted, LCMS-based metabolomics for quantitative measurement of NAD(+) metabolites. Comput Struct Biotechnol J. 2013;4:e201301012. doi: 10.5936/csbj.201301012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aryee MJ, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ram V, ZJ C. ggplot2: elegant graphics for data analysis. 2. Meas Interdiscip Res Perspect; 2019. [Google Scholar]
- 49.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martorell-Marugan J, Gonzalez-Rumayor V, Carmona-Saez P. mCSEA: detecting subtle differentially methylated regions. Bioinformatics. 2019;35(18):3257–3262. doi: 10.1093/bioinformatics/btz096. [DOI] [PubMed] [Google Scholar]
- 51.Hannum G, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49(2):359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Levine ME, et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY). 2018;10(4):573–591. doi: 10.18632/aging.101414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu AT, et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY). 2019;11(2):303–327. doi: 10.18632/aging.101684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ho J, et al. Moving beyond P values: data analysis with estimation graphics. Nat Methods. 2019;16(7):565–566. doi: 10.1038/s41592-019-0470-3. [DOI] [PubMed] [Google Scholar]
- 55.Katz S, et al. Studies of illness in the aged. The index of Adl: a standardized measure of biological and psychosocial function. JAMA. 1963;185:914–919. doi: 10.1001/jama.1963.03060120024016. [DOI] [PubMed] [Google Scholar]
- 56.Lawton MP, Brody EM. Assessment of older people: self-maintaining and instrumental activities of daily living. Gerontologist. 1969;9(3):179–186. doi: 10.1093/geront/9.3_Part_1.179. [DOI] [PubMed] [Google Scholar]
- 57.Royall DR, Mahurin RK, Gray KF. Bedside assessment of executive cognitive impairment: the executive interview. J Am Geriatr Soc. 1992;40(12):1221–1226. doi: 10.1111/j.1532-5415.1992.tb03646.x. [DOI] [PubMed] [Google Scholar]
- 58.Royall DR, Cordes JA, Polk M. CLOX: an executive clock drawing task. J Neurol Neurosurg Psychiatry. 1998;64(5):588–594. doi: 10.1136/jnnp.64.5.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Douaud G, et al. Anatomically related grey and white matter abnormalities in adolescent-onset schizophrenia. Brain. 2007;130(Pt 9):2375–2386. doi: 10.1093/brain/awm184. [DOI] [PubMed] [Google Scholar]
- 60.Good CD, et al. A voxel-based morphometric study of ageing in 465 normal adult human brains. Neuroimage. 2001;14(1 Pt 1):21–36. doi: 10.1006/nimg.2001.0786. [DOI] [PubMed] [Google Scholar]
- 61.Smith SM, et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage. 2004;23(Suppl 1):S208–S219. doi: 10.1016/j.neuroimage.2004.07.051. [DOI] [PubMed] [Google Scholar]
- 62.Andersson JL, Jenkinson M, Smith S. Non-linear registration aka spatial normalisation. Oxford, United Kingdom: FMRIB Centre; 2007. [Google Scholar]
- 63.Ma C, et al. Variational Bayesian inference for a nonlinear forward model. IEEE Trans Signal Proc. 2009;57(1):223–236. doi: 10.1109/TSP.2008.2005752. [DOI] [Google Scholar]
- 64.Jenkinson M, et al. Fsl. Neuroimage. 2012;62(2):782–790. doi: 10.1016/j.neuroimage.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 65.Laird AR, et al. Investigating the functional heterogeneity of the default mode network using coordinate-based meta-analytic modeling. J Neurosci. 2009;29(46):14496–14505. doi: 10.1523/JNEUROSCI.4004-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guralnik JM, et al. A short physical performance battery assessing lower extremity function: association with self-reported disability and prediction of mortality and nursing home admission. J Gerontol. 1994;49(2):M85–M94. doi: 10.1093/geronj/49.2.M85. [DOI] [PubMed] [Google Scholar]
- 67.Martens CR, et al. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD(+) in healthy middle-aged and older adults. Nat Commun. 2018;9(1):1286. doi: 10.1038/s41467-018-03421-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li K, et al. Progressive bidirectional age-related changes in default mode network effective connectivity across six decades. Front Aging Neurosci. 2016;8:137. doi: 10.3389/fnagi.2016.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018;19(6):371–384. doi: 10.1038/s41576-018-0004-3. [DOI] [PubMed] [Google Scholar]
- 70.Grant R, et al. A pilot study investigating changes in the human plasma and urine NAD+ metabolome during a 6 hour intravenous infusion of NAD. Front Aging Neurosci. 2019;11:257. doi: 10.3389/fnagi.2019.00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Elhassan YS, et al. Nicotinamide riboside augments the aged human skeletal muscle NAD(+) metabolome and induces transcriptomic and anti-inflammatory signatures. Cell Rep. 2019;28(7):1717–1728 e6. doi: 10.1016/j.celrep.2019.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Remie CME, et al. Nicotinamide riboside supplementation alters body composition and skeletal muscle acetylcarnitine concentrations in healthy obese humans. Am J Clin Nutr. 2020;112. [DOI] [PMC free article] [PubMed]
- 73.Dollerup OL, et al. A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: safety, insulin-sensitivity, and lipid-mobilizing effects. Am J Clin Nutr. 2018;108(2):343–353. doi: 10.1093/ajcn/nqy132. [DOI] [PubMed] [Google Scholar]
- 74.Krishnan K, et al. Changes in Montreal Cognitive Assessment scores over time. Assessment. 2017;24(6):772–777. doi: 10.1177/1073191116654217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jagust WJ, et al. Brain function and cognition in a community sample of elderly Latinos. Neurology. 2002;59(3):378–383. doi: 10.1212/WNL.59.3.378. [DOI] [PubMed] [Google Scholar]
- 76.Haan MN, et al. Prevalence of dementia in older Latinos: the influence of type 2 diabetes mellitus, stroke and genetic factors. J Am Geriatr Soc. 2003;51(2):169–177. doi: 10.1046/j.1532-5415.2003.51054.x. [DOI] [PubMed] [Google Scholar]
- 77.Mulrow CD, et al. Function and medical comorbidity in south Texas nursing home residents: variations by ethnic group. J Am Geriatr Soc. 1996;44(3):279–284. doi: 10.1111/j.1532-5415.1996.tb00914.x. [DOI] [PubMed] [Google Scholar]
- 78.Tabatabaei-Jafari H, Shaw ME, Cherbuin N. Cerebral atrophy in mild cognitive impairment: a systematic review with meta-analysis. Alzheimers Dement (Amst). 2015;1(4):487–504. doi: 10.1016/j.dadm.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Damoiseaux JS, et al. Reduced resting-state brain activity in the “default network” in normal aging. Cereb Cortex. 2008;18(8):1856–1864. doi: 10.1093/cercor/bhm207. [DOI] [PubMed] [Google Scholar]
- 80.Jones DT, et al. Age-related changes in the default mode network are more advanced in Alzheimer disease. Neurology. 2011;77(16):1524–1531. doi: 10.1212/WNL.0b013e318233b33d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Greicius MD, et al. Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci U S A. 2004;101(13):4637–4642. doi: 10.1073/pnas.0308627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Arora A, et al. Left inferior-parietal lobe activity in perspective tasks: identity statements. Front Hum Neurosci. 2015;9:360. doi: 10.3389/fnhum.2015.00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dai W, et al. Mild cognitive impairment and Alzheimer disease: patterns of altered cerebral blood flow at MR imaging. Radiology. 2009;250(3):856–866. doi: 10.1148/radiol.2503080751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhou J, et al. Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron. 2012;73(6):1216–1227. doi: 10.1016/j.neuron.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fatokun AA, Stone TW, Smith RA. Oxidative stress in neurodegeneration and available means of protection. Front Biosci. 2008;13:3288–3311. doi: 10.2741/2926. [DOI] [PubMed] [Google Scholar]
- 86.Zhang H, Wang Y, Lyu D, Li Y, Li W, Wang Q, Li Y, Qin Q, Wang X, Gong M, Jiao H, Liu W, Jia J. Cerebral blood flow in mild cognitive impairment and Alzheimer’s disease: A systematic review and meta-analysis. Ageing Res Rev. 2021;71. [DOI] [PubMed]
- 87.Studenski S, et al. Gait speed and survival in older adults. JAMA. 2011;305(1):50–58. doi: 10.1001/jama.2010.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ummarino S, et al. NAD modulates DNA methylation and cell differentiation. Cells. 2021;10(11). [DOI] [PMC free article] [PubMed]
- 89.Chang J, et al. Nicotinamide adenine dinucleotide (NAD)-regulated DNA methylation alters CCCTC-binding factor (CTCF)/cohesin binding and transcription at the BDNF locus. Proc Natl Acad Sci U S A. 2010;107(50):21836–21841. doi: 10.1073/pnas.1002130107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 219 kb)
Data Availability Statement
The minimum datasets necessary to interpret, verify and extend the research in the article are available within the paper and its Supplementary Information. The trial was registered on ClinicalTrials.gov: NCT02942888.