

Abstract
Using mouse models and high-throughput proteomics, we conducted an in-depth analysis of the proteome changes induced in response to seven interventions known to increase mouse lifespan. This included two genetic mutations, a growth hormone receptor knockout (GHRKO mice) and a mutation in the Pit-1 locus (Snell dwarf mice), four drug treatments (rapamycin, acarbose, canagliflozin, and 17α-estradiol), and caloric restriction. Each of the interventions studied induced variable changes in the concentrations of proteins across liver, kidney, and gastrocnemius muscle tissue samples, with the strongest responses in the liver and limited concordance in protein responses across tissues. To the extent that these interventions promote longevity through common biological mechanisms, we anticipated that proteins associated with longevity could be identified by characterizing shared responses across all or multiple interventions. Many of the proteome alterations induced by each intervention were distinct, potentially implicating a variety of biological pathways as being related to lifespan extension. While we found no protein that was affected similarly by every intervention, we identified a set of proteins that responded to multiple interventions. These proteins were functionally diverse but tended to be involved in peroxisomal oxidation and metabolism of fatty acids. These results provide candidate proteins and biological mechanisms related to enhancing longevity that can inform research on therapeutic approaches to promote healthy aging.
Supplementary Information
The online version contains supplementary material available at 10.1007/s11357-023-00917-z.
Keywords: Longevity, Proteomics, Mass spectrometry, Mouse model
Introduction
Multiple pharmaceutical, dietary, and genetic interventions have been identified as having longevity-promoting effects in model organisms [1], but there is limited understanding of the specific biological mechanisms by which these interventions promote longevity. This reflects both the multifaceted effects of these interventions as well as the technical limitations involved in measuring biological responses at a molecular scale across large numbers of features, such as the entire suite of expressed proteins, or proteome, in an organism or organ. These challenges limit our ability to identify mechanisms and subsequently develop targets for therapeutic interventions aimed at promoting longevity.
Some of the best-documented longevity-promoting interventions in mice and other animals involve factors that regulate growth hormone production or response. This includes two genetically mutated mouse strains that lower mTORC1 signaling, one bearing a growth hormone receptor knockout (GHRKO) and another with a mutation in the Pit-1 locus (henceforth referred to as “Snell dwarf” or “SnellDW” mice) that results in a reduction of multiple downstream hormonal factors [2, 3]. Multiple drugs with diverse physiological effects and mechanisms of action have also been identified as having longevity-promoting effects, including rapamycin, acarbose, canagliflozin, and 17-α estradiol [4–7]. Finally, chronic caloric restriction has repeatedly been shown to promote longevity in mice and other animals [1]. Notably, many of these treatments, namely acarbose, canagliflozin, and 17-α estradiol, have sex-specific effects such that longevity is promoted more strongly in males than females [5–7]. Others, such as GHRKO, Snell dwarf, calorie restriction, and rapamycin, have strong longevity-promoting effects in both sexes [8].
The degree to which these alterations and treatments affect longevity by targeting the same or different biological pathways is still not fully understood. However, we have previously described biological alterations that were shared among several lifespan-enhancing interventions. For example, expression of GPLD1 in plasma and liver and BDNF and DCX in brain tissues were upregulated in GHRKO and Snell dwarf mice, and the GPLD1 increase was also seen in mice treated with rapamycin, acarbose, and 17-α estradiol or in mice fed a calorie-restricted diet [9]. A set of proteins whose mRNAs are preferentially translated by cap-independent translation is higher in Snell, GHRKO, and PAPPA KO mice, as well as in mice treated with rapamycin, acarbose, and 17-α estradiol [10, 11]. METTL3, an enzyme that promotes cap-independent translation, is higher in the liver and kidney of rapamycin, acarbose, and 17-α estradiol [10], as well as in Snell and GHRKO mice [11]. Rapamycin, acarbose, and 17-α estradiol also lead to the elevation of 4EBP1, a target of mTORC1 that plays a role in the control of mRNA translation [10]. A set of acute phase proteins, including serum amyloid protein, hemoxygenase-2, and caspase-6, was decreased in the liver of mice treated with rapamycin, acarbose, and 17-α estradiol [12]. A separate set of changes, including increases of muscle FNDC5, plasma irisin, and adipocyte UCP1, is seen in Snell and GHRKO mice [13], as well as in mice on the calorie restriction diet and those treated with rapamycin, acarbose, and 17-α estradiol [14]. This growing collection of changes in specific proteins, noted in multiple varieties of mice rendered long-lived by mutations, calorie restriction, or anti-aging drugs, suggests that unbiased proteomic searches might produce evidence for additional shared phenotypes.
Identifying the mechanisms by which such interventions affect longevity would contribute to our understanding of how to promote healthy aging in humans. While there have been studies that examined the changes in mRNA expression profiles in response to many of the interventions that extend lifespan in mice [8], similar studies of the changes at the protein level are not available. The use of high-throughput mass spectrometry-based proteomics allows the assessment of a comprehensive library of proteins to enable the identification of such targets. Adopting a comprehensive strategy to measure proteome-wide effects of longevity treatments while accounting for these challenges will allow robust comparisons across treatments to identify high-value targets, at both the protein and pathway level.
We carried out a study to identify the repertoire of proteins and pathways that are affected by genetic, dietary, and drug interventions known to promote longevity in mice, including two genetic manipulations (GHRKO and Snell dwarf), four drug treatments, specifically rapamycin (“rapa”), acarbose (“aca”), canagliflozin (“cana”), and 17-α estradiol (“17aE2”), and calorie restriction (“CR”). We adopted a balanced experimental design sampling 264 mice of both male and female mice across three genetic backgrounds (UM-HET3 for drug/diet treated mice and untreated controls, C57BL/6 for GHRKO mice and wildtype controls, and DW×C3H for Snell dwarf mice and wildtype controls) using controls matched for sex, age, and genetic background (Table 1). Using high-throughput mass-spectrometry, we measured protein abundances in mouse livers, kidneys, and gastrocnemius muscle to gain insight into proteome changes that might either be shared across metabolically active organs or perhaps be organ-specific. We examined the quantitative proteome responses to these interventions using a modeling approach specifically tailored to proteomics data. While we characterized and present the responses of all quantifiable proteins measured, we specifically sought to identify responses that are shared across interventions under the assumption that if these interventions promoted longevity through common mechanisms, then the proteins related to the longevity-promoting effects are more likely to respond similarly across interventions. We further inferred the molecular and functional pathways that were activated by each intervention to investigate whether longevity-promoting interventions act through similar mechanisms even in cases where the individual protein responses differ.
Table 1.
Experimental design
| Intervention | Background | Sex | Number of mice | % increase in median survival | Reference |
|---|---|---|---|---|---|
| GHRKO | C57BL/6 | Male | 9 | 30 | Bonkowski et al. (2006) [15] |
| Female | 9 | 30 | |||
| Wild type (control) | Male | 10 | - | ||
| Female | 10 | - | |||
| Snell DW | DW×C3H | Male | 10 | 42 | Flurkey et al. (2001) [16] |
| Female | 9 | 42 | |||
| Wild type (control) | Male | 12 | - | ||
| Female | 9 | - | |||
| Calorie restriction | UM-HET3 | Male | 14 | 32 | Flurkey et al. (2010) [17] |
| Female | 12 | 40 | |||
| Rapamycin | Male | 14 | 13 | Miller et al. (2014) [18] | |
| Female | 13 | 21 | |||
| Acarbose | Male | 14 | 22 | Harrison et al. (2014) [5] | |
| Female | 13 | 5 | |||
| Canagliflozin | Male | 14 | 14 | Miller et al. (2020) [7] | |
| Female | 13 | 1 | |||
| 17α-estradiol | Male | 14 | 12 | Harrison et al. (2014) [5] | |
| Female | 13 | 0 | |||
| Untreated (control) | Male | 26 | - | ||
| Female | 26 | - |
Methods
Mouse husbandry and interventions
All the mouse samples were prepared at the University of Michigan in a specific pathogen-free colony. Genetic background and husbandry conditions for the Snell dwarf and GHRKO mice were described previously [19]. Mice used for rapamycin (encapsulated, used at 14 ppm), canagliflozin (180 ppm), 17α-estradiol (14.4 ppm), acarbose (1000 ppm), and calorie-restricted samples were of the UM-HET3 stock, produced as the offspring of CByB6F1/J mothers and C3D2F1/J fathers, as described in [18]. This is the stock used by the NIA Interventions Testing Program; the lifespan effects of the four drugs are reviewed, with references, in [20]. The calorie-restricted diet has been shown to extend the lifespan in UM-HET3 mice [17]; in the current study, calorie-restricted mice received 60% of the amount of food consumed by age-matched controls. The base diet was Purina 5LG6. All four drug treatments, as well as calorie restriction, were begun when mice were 4 months of age. To monitor specific-pathogen status, sentinel mice were exposed to spent bedding for 2 weeks prior to testing for a set of anti-viral antibodies and ecto-parasites; these tests were done quarterly, and all tests were negative for the entire aging colony during the experimental period. The protocols were reviewed and approved by the University of Michigan’s Institutional Animal Care and Use Committee.
Sample harvesting and preparation
All mice were humanely euthanized at 12 months of age. Euthanasia occurred between 8 am and 11 am (lights cycled on at 6 am and off at 6 pm) by rapid asphyxiation in a bag containing CO2 gas. Mice were unconscious within 5 s and stopped breathing at about 10 s. The interval between removal from the home cage to death was less than 1 min. Mice were removed from the bag as soon as breathing ceased, and blood was harvested by closed-chest cardiac puncture. Tissue sections from the liver, kidney, and gastrocnemius muscle were then dissected, flash frozen in liquid N2 immediately after removal, and then stored at −80°C until shipment to the proteomics laboratory for assessment. Tissue samples stored for long periods of time (≥ 9 years) at −80°C have been shown to provide at least the same quality of RNA and protein (as judged by microcapillary electrophoresis measurements of total RNA integrity and mass spectrometry for proteins) and in the case of RNA a superior quality, as storage in vapor phase liquid nitrogen [21]. The integrity of proteins during storage is of utmost importance and we have adhered to guidelines to ensure our storage conditions met internationally proposed guidelines [22]. In our experiments, tissues were kept at −80°C throughout, and when it came time to section and lyse the tissue samples, each was then kept on a floating marble tray on dry ice to transmit cold and sectioned expeditiously before being immediately placed in lysis buffer and processed for deep quantitative proteomics mass spectrometry-based analysis.
These frozen tissue samples were then processed for comprehensive proteome analysis by data-independent analysis (DIA) liquid chromatography and tandem mass spectrometry (LC-MS/MS). Frozen tissue was lysed in 50mM ammonium bicarbonate buffer pH 7.55 with 5% SDS and homogenized on a Precellys Evolution (Bertin Instruments, France) with 2.8-mm ceramic beads (Qiagen) for 9 rounds of 20 s at 8500rpm with 30-s rest in between. The protein concentration of the lysate was determined using the BCA protein assay (Pierce, Cat# 23227). Lysate containing 200μg protein was aliquoted out and denatured for 2 min at 90°C. After denaturation, samples were reduced with 5mM TCEP (Sigma Cat# 4706) for 30 min at 60°C and then alkylated with 10mM α-iodoacetamide (Millipore Cat# 407710) for 30 min at room temperature in the dark. Samples were acidified with phosphoric acid to a final concentration of 1.2%, and S-Trap buffer (100mM ammonium bicarbonate buffer pH 7.1 + 90% methanol) was added at a 1:7 ratio prior to loading on a S-Trap 96-well plate (ProtiFi, USA). Using a positive pressure, manifold (Resolvex M10, Tecan, Switzerland) samples were washed twice with S-Trap buffer before overnight digestion at 37°C with Trypsin (Promega Cat# V511X) in digestion buffer (50mM ammonium bicarbonate pH8.0) at 1:50 ratio. Digested peptides were eluted as per the manufacturer’s instructions (S-Trap™ 96-well plate protocol version 1.4). Importantly for normalization, all plates contained an even mix of samples from all tissues, arranged systematically to avoid bias across either rows or columns, and in nearly all cases, all three tissue samples from the same donor were loaded onto the same plate.
Liquid chromatography and mass spectrometry
Digested peptides were lyophilized and resuspended in 0.1% formic acid (FA). Each sample was spiked with iRT standard peptides [23, 24] (Biognosys AG, Schlieren, Switzerland) and loaded on individual Evotips for desalting and washing as per the instructions (Evosep, Odense, Denmark). Peptides were fractionated by RP-HPLC on a PepSep column, (200 μm internal diameter (ID) × 25 cm) packed with ReproSil C18 1.5 μm, 120Å resin using a predefined 30 samples per day method (~48 min) (Evosep RC.Net 1.3 plugin). Mobile phases A and B were 0.1% FA in Milli-Q water (Millipore USA) and 0.1% FA in HPLC grade Acetonitrile (ACN) (Aventor, USA), respectively, and coupled to a timsTOF Pro ion mobility time-of-flight mass spectrometer (Bruker Daltonics, Bremen, Germany) via a CaptiveSpray nano-electrospray ion source. Mass spectrometry measurements were acquired in data-independent acquisition Parallel Accumulation Serial Fragmentation (DIA-PASEF) mode. For ion mobility settings, the inversed mobilities from 1/K0 0.57 to 1.47 Vs/cm2 were analyzed with ion accumulation and ramp time of 100 ms, respectively. One survey TIMS-MS scan was followed by 32 DIA-PASEF scans of 25 m/z isolation windows (timsControlPy5 schema) covering 400–1200 m/z precursor spectra and 100–1700 m/z fragment ion spectra. Singly charged ions were excluded using the polygon filter mask.
Hybrid spectral assay library generation and quality control
A mouse hybrid spectra assay library was generated by combining the project-specific in-house DIA run files of three different mouse tissues (kidney, liver, and gastrocnemius muscle) with a published comprehensive mouse reference spectral library [25] (available at http://www.swathatlas.org/PASS01569). We implemented a search archive methodology to combine the spectral library from multiple sources while maintaining FDR control and homogeneous protein inference. Spectra were searched using the Spectronaut Pulsar search engine (version 15.4.210913.50606, Biognosys AG, Schlieren, Switzerland) against the full non-redundant, canonical mouse reference proteome from UniProtKB/Swiss-Prot (Proteome ID UP000000589, September 2021) with 16,998 reviewed proteins and 11 iRT peptides appended. Fixed modification mass differences were included for cysteine carbamidomethylation (57.0214 Da) and methionine oxidation (15.9949 Da) set as variable modifications. Up to two missed tryptic cleavages were allowed. For the search identifications, a library-wide FDR was controlled at <1% at PSM, peptide, and protein level. A filter of a minimum and maximum of 6 fragments per precursor was applied to generate a T6 version of the hybrid spectral library.
Spectral assay library quality control using DIALib-QC
The mouse hybrid T6 spectral assay library was evaluated using DIALib-QC (http://www.swathatlas.org/DIALibQC.php), a freely available software tool by the authors that highlights library’s characteristics, completeness, and correctness across 64 parameters of compliance [26]. The DIALib-QC assessment report for the hybrid spectral library is provided in Supplemental Table 8.
DIA-PASEF data analysis
All mouse tissue DIA mass spectrometry data were analyzed with Spectronaut (version 15.4.210913.50606, Biognosys AG, Schlieren, Switzerland). The hybrid library was used directly as generated and described above. The HTRMS converter was used to convert the raw .d folders into HTRMS files which were then imported into Spectronaut. A nonlinear iRT calibration strategy was used for both mass tolerance and XIC RT extraction window. Preprocessing of MS1 and MS2 calibration strategy was enabled. Decoy assays were generated using the scambled decoy method with a set size as a fraction of the input spectral library size. For identification, a normal distribution estimator with precursor and protein identification results filtered with a q value of <0.01 was used. The peptide quantification was estimated by summing theMS2 ion peak areas.
Data processing and normalization
To estimate protein abundances from peptide intensities, we first summed intensity values across all charge states for each unique peptide amino acid sequence and then log2-transformed the totals after adding the constant 1 to each sum to enforce positive values. This transformation was done to help ensure near-normal distributions for subsequent model fitting. After matching peptides to proteins proteotypically (i.e., discarding degenerate peptides matching to more than one protein), we removed proteins with fewer than three proteotypic peptides, as these were deemed insufficiently represented given the level of inter-peptide variation within proteins. Within each tissue, we fit a random, crossed-effects, peptides-by-sample model to each remaining protein using the expectation–maximization (EM) algorithm. To obtain an abundance measure for each protein within each sample, we calculated the sum of the overall mean plus the best linear unbiased predictor (BLUP) for the sample based on the estimated random effects. This is equivalent to conditioning the sample’s abundance estimate on the observation from a typical peptide by subtracting the peptide BLUP from the model prediction. In cases where the crossed-effects model could not be fit because of insufficient overlap in the peptides that were observed for which samples, a peptide fixed-effects estimator (i.e., the overall mean plus the average residual across peptides observed for the sample) was used instead.
Prior to statistical analysis, we normalized these protein abundance measurements to minimize technical biases across all samples and organs. We first log2-transformed the protein measurements to reduce heteroscedasticity and skew and help enforce near-normal distributions. Since abundance measures obtained using mass spectrometry technology are unitless, such transformations do not remove or alter any biological meaning. We then residualized these protein abundance measures by subtracting tissue-specific group-by-sex means for each protein, subtracted tissue-specific overall mean residuals for each protein, and then fit a hierarchical mixed-effects linear model in the log-transformed abundance. For this model, we entered cubic splines of run order and indicators for dissection batch/team as fixed effects and included crossed random effects of cage (~100 levels) and plate (10 levels) in addition to a random effect of run position (i.e., well position) nested within the plate. We then fit the model on the residual abundance as described above, employing REML estimation (maximization of the residual quasi-likelihood) by the EM algorithm. Model predictions were subtracted from the sample protein abundances to generate normalized abundances that are comparable between samples across proteins and tissues.
Variation in protein abundances due to tissue, sex, genetic background, and intervention
To measure the correlation between pairs of tissues within mice while accounting for repeated measures (each protein was potentially measured three times per mouse, once for each tissue), we calculated a repeated measures correlation coefficient as per Bland (1995) [27] using the rmcorr R package [28]. For measuring the correlations in protein abundances among genetic background or between sexes, we calculated the mean abundance for each protein in the controls of each group in the comparison (e.g., UM-HET3 untreated controls of both sexes for the genetic background comparison or male controls of all three backgrounds for the sex comparison) and estimated the correlation coefficient from a linear model fit to these values.
To partition variance in protein abundances among mouse-specific factors (sex, genetic background, and intervention), we fit a linear mixed effects model with sex, background, and intervention nested within the background as random effects to the abundances of each protein separately to obtain estimates of the fraction of the total variation explained by each factor. We then averaged these fractions across proteins to compare the overall variation in protein abundances explained by sex, genetic background, and intervention.
Measuring the protein responses to interventions
We developed a custom differential abundance analysis pipeline that we designed to account for many of the unique problems and challenges associated with proteomics data. First, because there are biases in quantification that may differ among peptides/proteins, we applied our pipeline to each protein individually, rather than combining information across proteins as is often done in pipelines developed for gene expression data. Second, our pipeline separately accounts for protein detection and abundance. Proteomics data is characterized by a high degree of “missingness” or unmeasured quantities. This is made further problematic since due to the technology used, a peptide may not be detected in a sample either due to biological reasons (e.g., it is either absent from the specimen or its abundance is below the detection limit) or technological reasons related to its specific chemistry or its lack of representation in a reference [29]. Misidentification also occurs at a very small but still nonzero rate, and stochastic (i.e., unexplained) dropout can occur as well, even in DIA pipelines [30]. The technical factors influencing detection likely differ from the ones that influence the abundance quantification.
To account for both differential missingness and abundance, we used an exponential hurdle model that separately models changes in presence/absence and abundance, as implemented in Stata software [31, 32]. The hurdle model is a two-part model consisting of both a selection model, which models the probability of zero values, and a second that models the non-zero values. This model was fit to the normalized log2-transformed protein abundances across samples within each tissue using mouse intervention and sex as independent variables for both parts of the model (including an intervention-sex interaction term in the abundance model) for each protein that is not detected in every sample in the comparison. For those proteins that were detected in all samples (i.e., no “missingness”), we fit a standard linear model instead, also using intervention and sex as fully interacted independent variables. Proteins detected in less than three samples in either intervention or control groups were dropped from the analysis.
This modeling framework was used to estimate the log2 fold-change and its standard error for each protein in each tissue for each contrast of interest: each drug/diet treatment or genetic manipulation compared to its relevant control in a sex-specific manner (i.e., within males or females). We calculated a p-value associated with this change from the standard errors assuming a normal distribution and subsequently approximated Bayes factors from these p-values as an estimate of the odds against a null effect [33]. We then used the log2 fold-change and Bayes factor to highlight proteins of large effect. Given the exploratory nature of the study and considering sample size constraints, we adopted moderately liberal criteria in categorizing the responses of proteins in each intervention: unless otherwise noted, we considered a protein to be “significantly” affected if it exhibited an absolute log2 fold-change greater than or equal to 0.5 associated with a Bayes factor greater than or equal to 10. An approximate Bayes factor of 10 for a protein suggests at most ten to one odds that an intervention affected the abundance of that protein, so the selection of significant proteins based on the cutoff ≥ 10 captures all proteins with evidence at least this good, while the selection of +/− 0.5 as a minimum log2 fold-change cutoff was selected in part based on the observation that just under 5% of the estimated protein responses in the liver were above this threshold.
Identifying genetic and functional pathways affected by longevity-promoting treatment.
To provide biological insight into the genetic pathways and processes that could be involved in the generation of the proteomic patterns observed in our data, we performed both a gene set over-representation analysis and a pathway impact analysis [34]. To identify the biological processes that were overrepresented, or enriched, among the proteins that exhibited shared responses across multiple longevity-promoting interventions, we first assigned gene ontology (GO) terms to each protein in our dataset using the Bioconductor package org.Mm.eg.db (version 3.17) genome-wide annotation for mouse [35]. We then performed a Fisher’s exact test comparing the GO terms in the focus set of proteins with the GO terms represented in all other proteins in the dataset using the “enrichGO” function in the clusterProfiler package in R [36]. To determine which pathways were most impacted by interventions, we performed a pathway impact analysis on KEGG pathways using the Pathway-Express algorithm [37] as implemented in the ROntoTools package in R [38]. This analysis was performed using release 105 of the KEGG database [39]. We performed this analysis separately for each intervention-sex combination inputting all proteins, regardless of effect size or Bayes factor, into the analysis but weighting each protein’s contribution to the perturbation score by the p-value of its response. The resulting p-values comparing the perturbation scores to the null were FDR-adjusted, and perturbation scores were normalized by the number of downstream genes in the pathway.
Results
Variation of mouse proteomes across liver, kidney, and gastrocnemius muscle
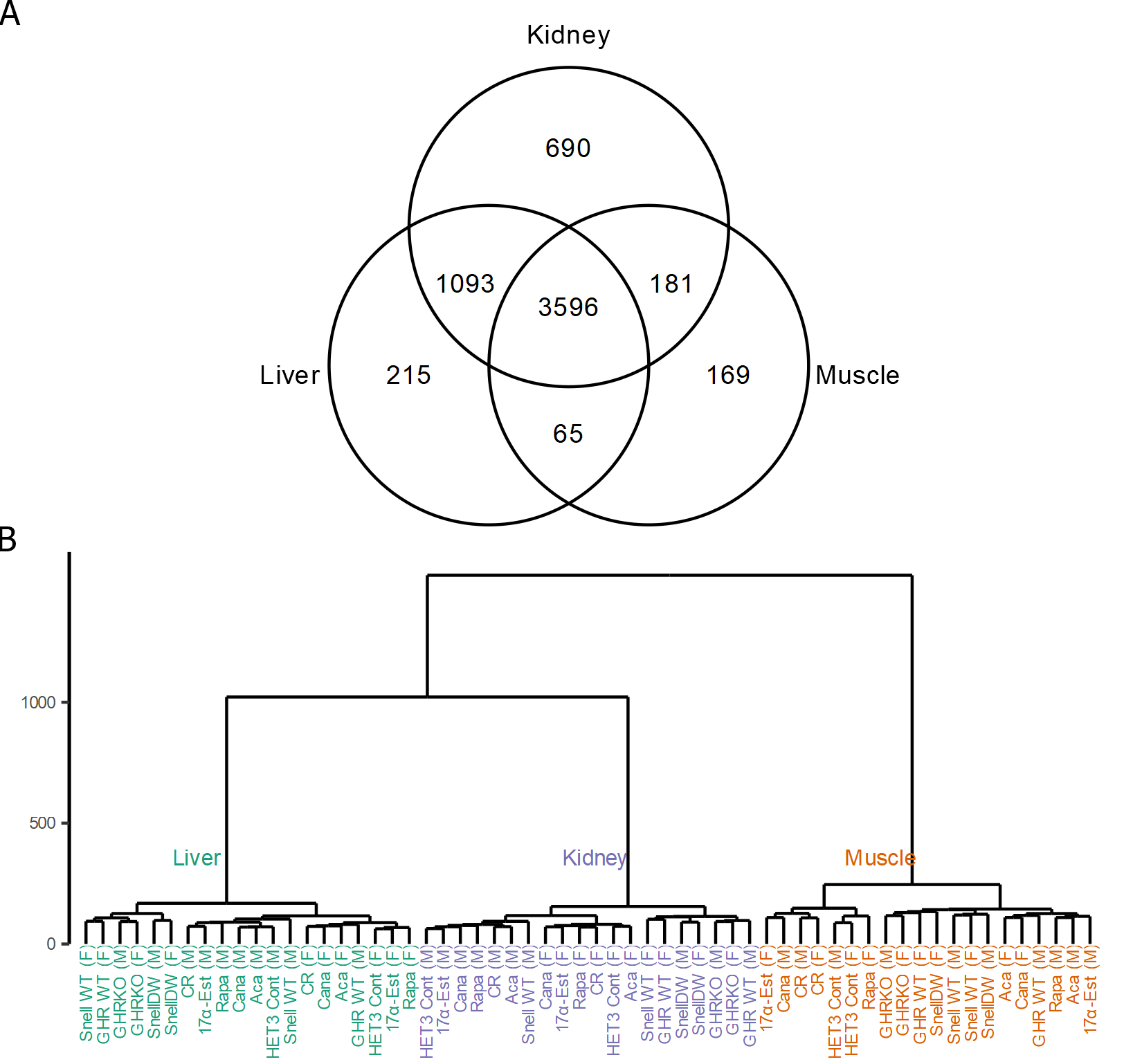
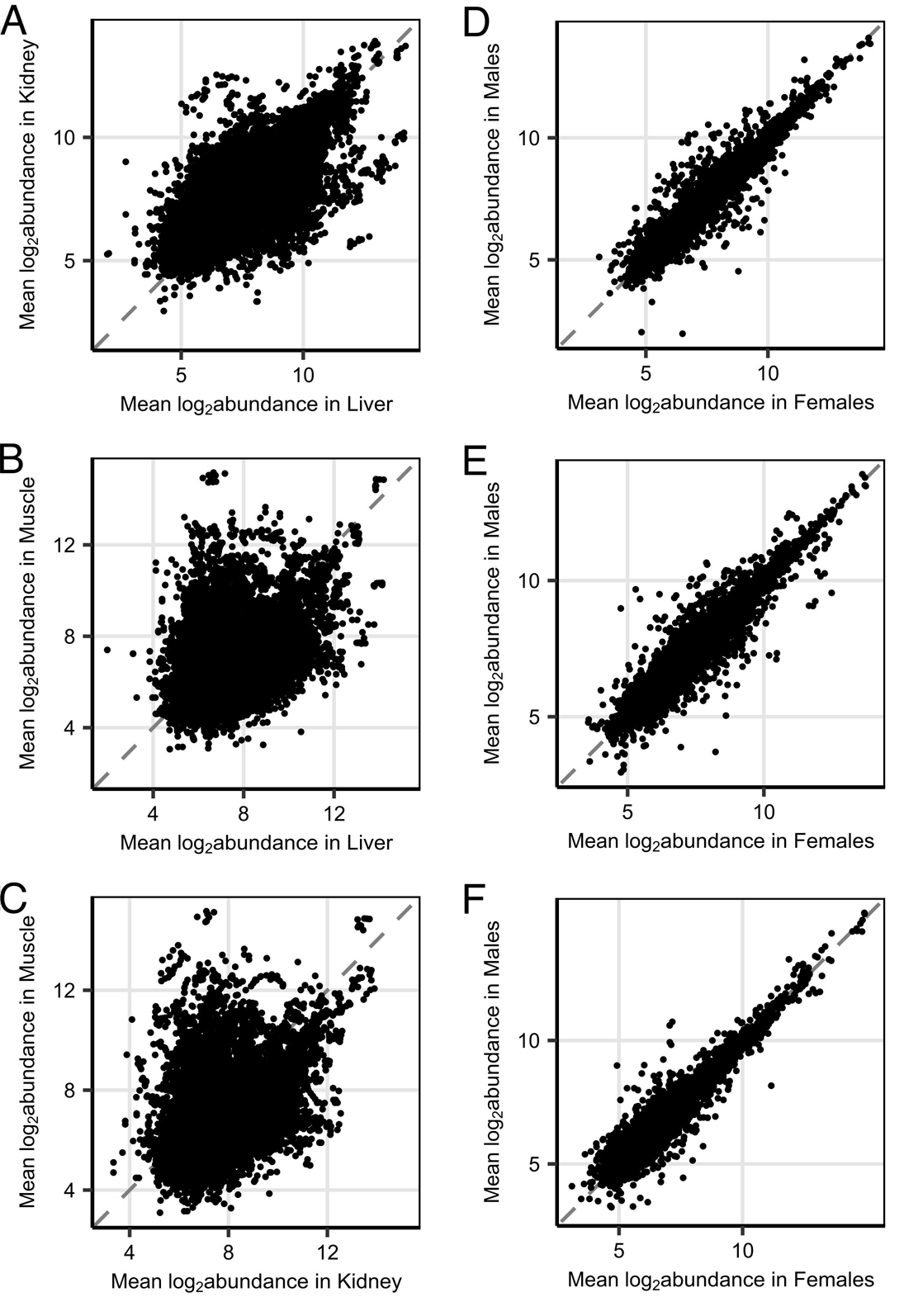
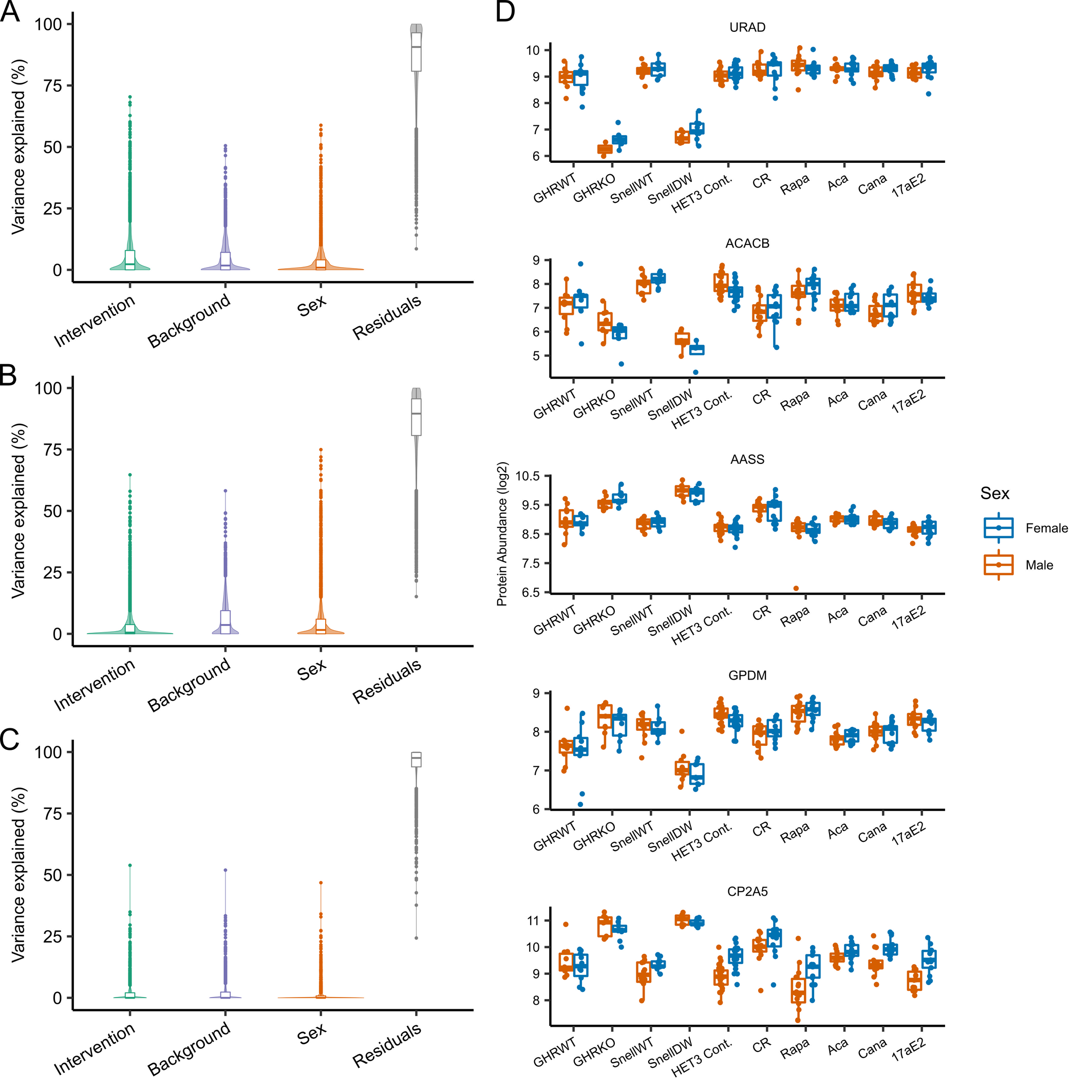
We first investigated the composition of mouse proteomes across individuals and tissues to identify sources of variation and how they relate to the effects of longevity-promoting interventions. In total, we quantified 6009 proteins after quality filtering (which included the removal of proteins with fewer than 3 peptides detected—see “Methods”). Of these 6009 proteins, 4969 were detected in the liver, 5560 in the kidney, 4011 in muscle, and 3596 in all three tissues (Supplemental Fig. 1A). For those proteins detected in multiple tissues, relative abundances (hereafter referred to as “abundances” for simplicity) within mice were correlated most strongly between liver and kidney tissues (repeated measures correlation coefficient, r = 0.779, p < 0.001) and more weakly between muscle and either kidney (r = 0.532, p < 0.001) or liver (r = 0.497, p < 0.001) (Supplemental Fig. 2A-C). Likewise, the correlation between sexes in mean protein abundances among control mice across backgrounds was high within each tissue (r = 0.98, 0.97, and 0.96 for liver, kidney, and muscle tissue respectively; Supplemental Fig. 2D-F). Consistent with the noisy nature of proteomics and other omics data in general as well as the inherent biological variability of the system, there was a large amount of variability in protein measurements across mice. The high correlation in abundances among backgrounds and between sexes left little of this variation in protein abundances to be explainable by these factors (Supplemental Fig. 3A-C). The impact of the intervention was of a similar scale, explaining on average only about 2% of the variation in protein abundances in the liver and less than 1% in either kidney or muscle. However, there were a small handful of proteins for which the variation in abundances due to intervention was high relative to residual variation, including five for which the variation explained by intervention was greater than 60%, including acetyl-CoA carboxylase 2 (ACACB), alpha-aminoadipic semialdehyde synthase (AASS), and cytochrome P450 2A5 (CP2A5) which were also highlighted by subsequent analyses (Supplemental Fig. 3D). Overall, this resulted in a general pattern in which the protein abundance profiles averaged by intervention-by-sex group clustered in similarity first by tissue, then secondarily by background, sex, and intervention (Supplemental Fig. 1B).
General patterns in protein responses to longevity-promoting interventions
For each protein in each tissue, we estimated the fold-change in log2-transformed protein abundances and the associated approximate Bayes factor in response to each intervention compared to its respective control group in a sex-specific manner (see “Methods”). A full list of protein responses to each intervention, including their log2 fold-change and Bayes factor, for each tissue can be found in Supplemental Tables 1-3. We considered a protein to be significantly affected by an intervention if it exhibited an estimated absolute log2 fold-change ≥ 0.5 with a Bayes factor ≥ 10; an approximate Bayes factor of 10 for a protein suggests at most ten to one odds that an intervention affected the abundance of that protein, so the selection of significant proteins based on the cutoff ≥ 10 captures all proteins with evidence at least this good, while the selection of +/− 0.5 as a minimum log2 fold-change cutoff was chosen in part based on the observation that just under 5% of the estimated protein responses in liver were above this threshold. Intervention-by-sex groups clustered in terms of the similarity of their protein responses first by tissue, then by genetic background followed by sex (Fig. 1). There was relatively low concordance in protein responses across tissues for any given intervention, and responses were more similar within backgrounds and sexes. For the drug and diet interventions within UM-HET3 mice, the similarity in protein responses was separated by sex but consistent within sex; for example, protein changes in acarbose-treated mice were more similar to canagliflozin-treated mice of the same sex than acarbose-treated mice of the opposite sex. These patterns were largely consistent across liver, kidney, and muscle tissues with one notable exception: in livers, the response of proteins to calorie restriction was similar in both male and female mice.
Fig. 1.

Dendrograms of co-sine distances in protein log2 fold-changes among intervention-sex groups (top; the suffixes “_F” and “_M” indicate female and male groups respectively) and heatmaps of protein fold-changes compared to respective controls by intervention-sex group (bottom) for liver (A), kidney (B), and muscle samples (C). The color scale for protein fold-changes is capped at +/- log2(1) to enhance legibility of modest fold-changes. Proteins are arranged on the y-axis separately for each tissue based on mean fold-changes across groups
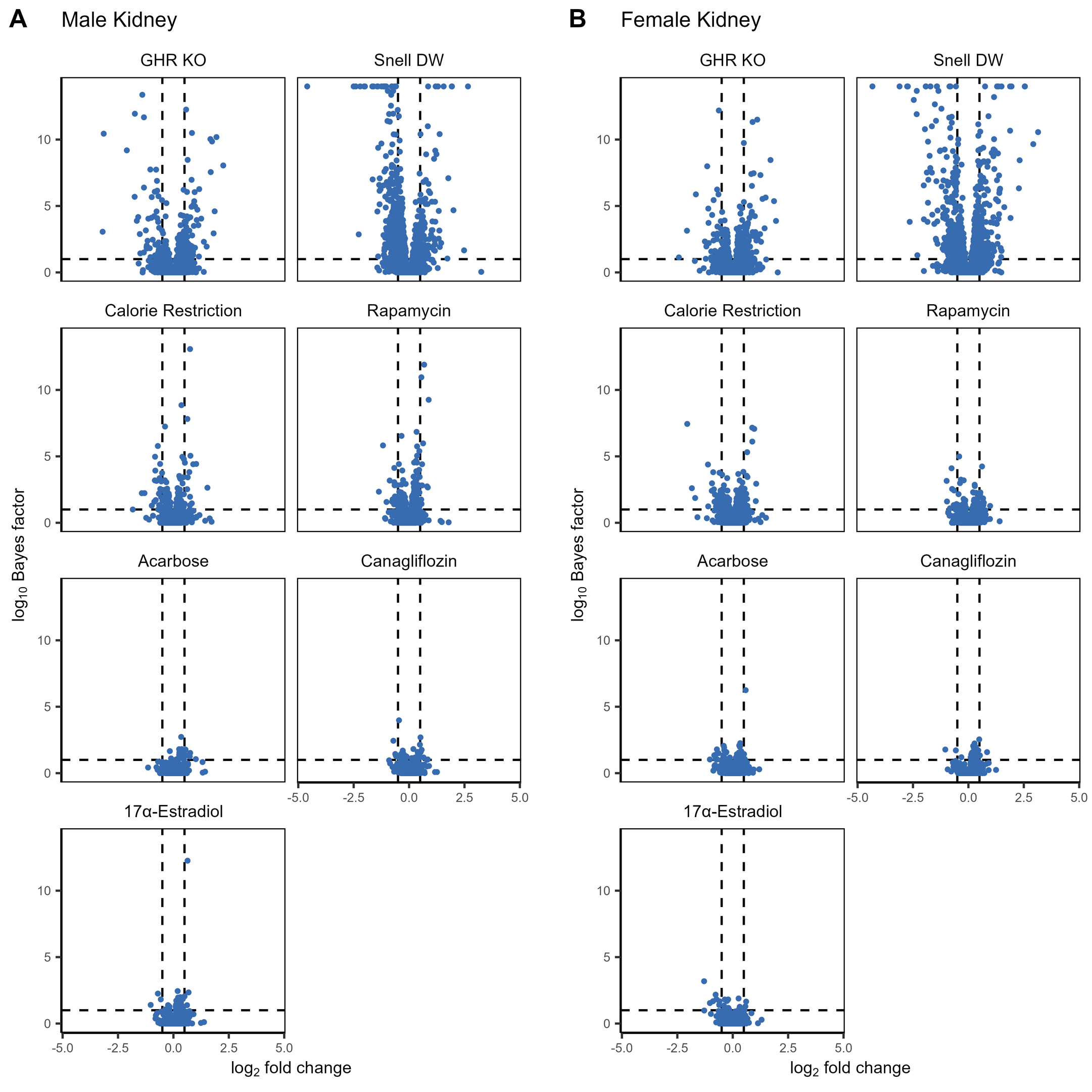
Both the number of significantly affected proteins and the magnitude of the changes varied by intervention, both within and across tissue and sex (Fig. 2, Supplemental Figs. 4, 5). Within tissues, liver samples had a much larger number of proteins that responded significantly to at least one intervention (N = 842), followed by the kidney (N = 672) and then muscle samples (N = 337). GHRKO and Snell dwarf genetic interventions resulted in the greatest effects on the proteome, both in terms of the magnitude of overall changes and the number of affected proteins, while among the drug and diet treatments, the largest changes were associated with caloric restriction. Acarbose, canagliflozin, and 17α-estradiol interventions in females resulted in the lowest number of affected proteins, consistent with these treatments having either little or no longevity-promoting effect on females. Notably, these patterns were qualitatively true for liver, kidney, and muscle tissues, albeit much less prominent in muscle where the overall effects were much smaller across all interventions.
Fig. 2.

The log2 fold-change and log10 Bayes factor for proteins within each treatment compared to controls for male (A) and female (B) liver samples. Dashed vertical lines correspond to a log2 fold-change of +/- 0.5 while the dashed horizontal line corresponds to a Bayes factor of 10
Intervention-by-sex interactions in protein responses
The combined observations that virgin UM-HET3 female mice live longer than virgin UM-HET3 male mice, and many of the drug treatments in our study—namely acarbose, canagliflozin, and 17α-estradiol—are known to promote longevity primarily in males and not females prompted us to identify proteins whose response to those interventions differed by sex. In each tissue, we identified a set of proteins that exhibited significant responses that had sex interaction effects with a Bayes factor ≥ 10 (Supplemental Fig. 6; Supplemental Tables 4-6). These sex interactions took multiple forms. In most cases, the response to an intervention differed between sexes only in the magnitude of the response but not the direction. However, we were primarily interested in interactions by sex in response to acarbose, canagliflozin, and 17α-estradiol, as these interventions exhibited sex-specific effects on longevity. There were 20, 8, and 15 proteins in liver, kidney, and muscle respectively with significant sex interactions in their response to these three treatments (Supplemental Table 7). Notably, none of these proteins exhibited such a response to more than one of the three treatments that have sex-specific effects on lifespan, and only one did so in multiple tissues (LIFR in response to 17α-estradiol in liver and kidney). In contrast to the other interventions that did not have a sex-specific effect on longevity, only 10 of these 42 proteins exhibited sex-specific responses in the same direction but with different magnitudes. Over half of the proteins (26 out of 42) responded positively in males but negatively in females in acarbose, canagliflozin, or 17α-estradiol, though not all of these responses met our threshold for significance.
Shared protein responses to longevity-promoting interventions
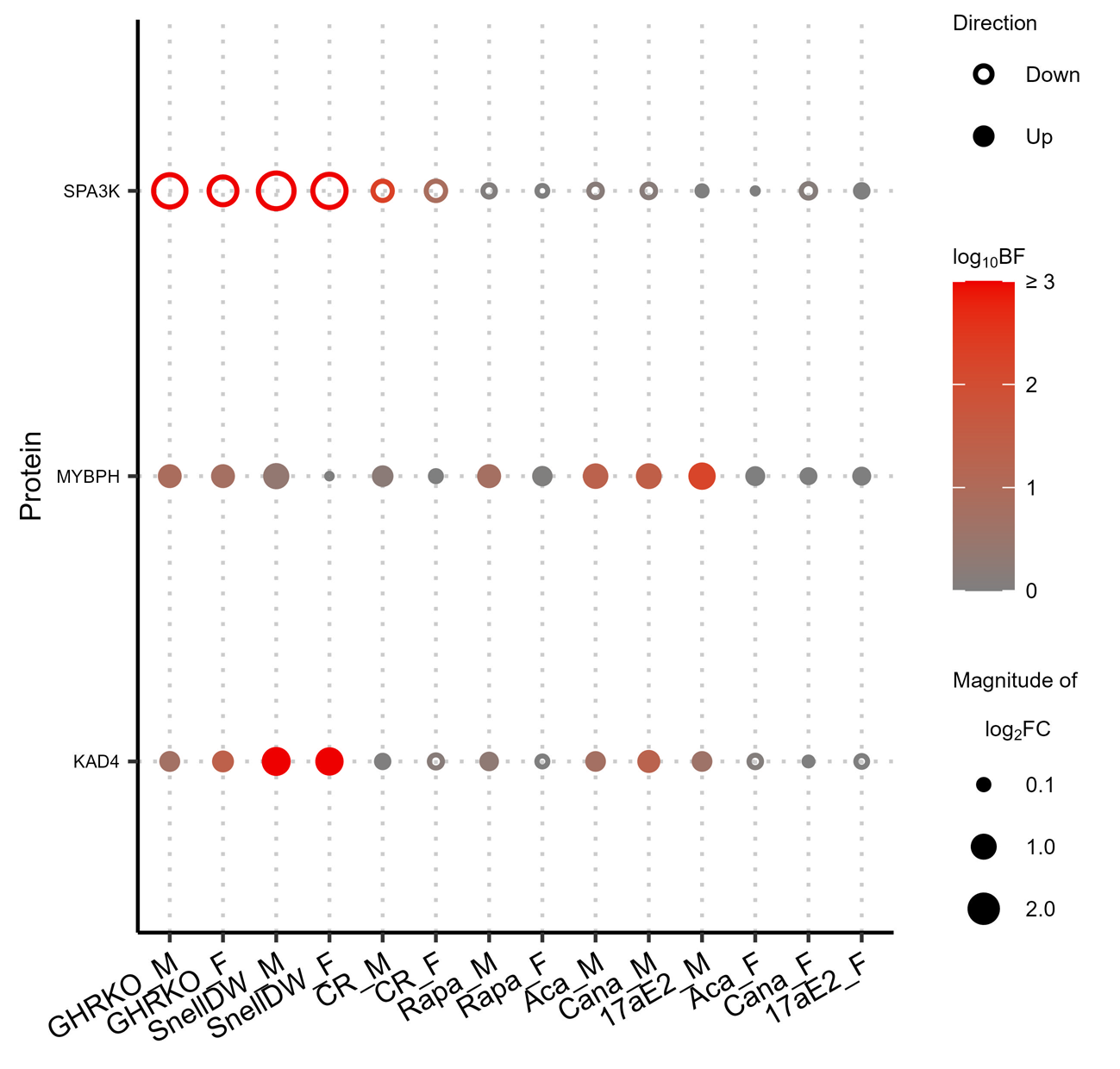
We were especially interested in proteins that were similarly affected by multiple interventions. While most significant protein responses were unique to a single intervention, there were 242 proteins in liver, 174 in kidney, and 33 in muscle that were significantly affected by two or more interventions (Fig. 3). The largest number of shared responses involved GHRKO and/or Snell dwarf mice of either sex. Only three proteins, one in each tissue, responded significantly to at least three interventions not including GHRKO or Snell dwarf: glycerophosphocholine phosphodiesterase (GPCP1) in the liver, complement factor D (CFAD) in the kidney, and myosin-binding protein H (MYBPH) in muscle (Fig. 4A, Supplemental Figs. 7A & 8). In the liver, calorie restriction induced many protein changes in common with Snell dwarf and to some extent GHRKO. This pattern was qualitatively similar in kidney and muscle, although the numbers of shared protein changes were fewer in these tissues as were the number of pairs of interventions that shared at least one significantly responding protein, particularly in muscle.
Fig. 3.

A–C Networks of shared protein responses across interventions in male and female mouse liver (A), kidney (B), and muscle (C) samples. Node size corresponds to the number of proteins with log-fold changes ≥ 0.5 associated with a Bayes factor ≥ 10 in each longevity intervention, while edge width represents the number of those proteins shared between two interventions. Nodes in yellow represent interventions expected to promote longevity while those in blue represent interventions in which the longevity-promoting effect is negligible or absent. D–F The number of proteins that are significantly affected by multiple interventions, regardless of sex in mouse liver (D), kidney (E), and muscle (F) samples. The x-axis gives the largest number of interventions a protein is affected by. The blue area represents the number of proteins that are significantly affected by GHRKO and/or Snell dwarf, while the gray area quantifies proteins only affected only by drug/diet treatments
Fig. 4.

A Changes in protein abundances across multiple longevity treatments in liver. Proteins shown are limited to those with an absolute log2 fold-change ≥ 0.5 and a Bayes factor ≥ 10 in GHRKO, Snell dwarf, and at least one other drug or diet treatment contrasts of either sex. The color of the points indicates the associated Bayes factor and the size is proportional to the magnitude of the fold-change with closed points indicating a positive change and open points indicating a negative change. B GO-Terms that are enriched (with an associated adjusted p-value ≤ 0.01) among proteins with shared responses across longevity-promoting interventions in liver (shown in A)
While there were numerous examples of proteins that were altered by at least two interventions, there were no proteins that were significantly affected by every longevity-promoting intervention (i.e., all intervention-by-sex groups except acarbose, canagliflozin, and 17α-estradiol females). We expanded our focus to those proteins that were significantly affected in the same direction by at least three interventions, resulting in a total of 66 proteins in the liver (Fig. 4A), 31 in the kidney (Supplemental Fig. 7), and 3 in muscle samples (Supplemental Fig. 8). Most of these exhibited responses in the same direction in both genetic interventions (GHRKO and Snell dwarf) and often calorie restriction as well. In the liver, protein responses to rapamycin tended to be the most divergent from responses that were common across other interventions. In the kidney, however, protein responses to rapamycin were much more likely to be shared with other interventions.
A single protein, serine protease inhibitor A3K (SPA3K), was altered by three interventions in all tissues (liver, kidney, and muscle). SPA3K exhibited a significant negative response to GHRKO, Snell dwarf, and calorie restriction interventions in both sexes and no significant response to any other intervention. In addition to SPA3K, 4 more proteins met the shared response criteria in both liver and kidney (but not muscle): alpha-aminoadipic semialdehyde synthase (AASS), beta-ureidopropionase (BUP1), cytochrome P450 2A5 (CP2A5), and glutathione S-transferase A2 (GSTA2). In both the liver and kidney, each of these proteins responded positively to GHRKO, Snell dwarf, and calorie restriction in at least one sex. They also responded positively to acarbose and canagliflozin in males in the liver but not the kidney, while AASS also responded positively to rapamycin in the kidney but not the liver.
The protein with the highest degree of overlap in response to longevity-promoting intervention-by-sex groups was the aforementioned CP2A5, which exhibited a significant, positive response in both male and female GHRKO, Snell dwarf, and calorie-restricted mice as well as in male acarbose and canagliflozin–treated mice, and also responded significantly to multiple interventions in the kidney. In the opposite direction, acetyl-CoA carboxylase 2 (ACACB) and pyruvate kinase (PKLR) both exhibited significant and negative responses to seven intervention-by-sex groups in liver: male and female GHRKO, Snell dwarf, and calorie-restricted mice as well as male acarbose and canagliflozin–treated mice with rapamycin males trending in the same direction. Sialidase-1 (NEUR1), a protein involved in carbohydrate and lipid metabolism, was also notable in that it was one of the few proteins that responded positively to a relatively high number of interventions (six), including rapamycin in males, whereas most other proteins that shared a response across interventions did not respond similarly to rapamycin.
Proteins affected by multiple interventions implicate shared functional pathways
The proteins that significantly responded to multiple interventions tended to belong to similar metabolic pathways and functional groups. Among all the proteins that exhibited a shared response across at least three interventions the biological processes most enriched compared to all other proteins measured in the dataset were those relating to metabolism and oxidation, especially of fatty acids, lipids, and other exogenous molecules (Fig. 4)B. This enrichment was largely driven by the multitude of cytochrome p450 proteins. For example, 11 of the 66 proteins that responded significantly to at least three interventions in the liver, including the protein that exhibited a significant response to the largest number of intervention-sex groups (CP2A5), belonged to the cytochrome p450 family of proteins. These proteins are involved in the oxidation and metabolism of xenobiotics, fatty acids, steroids, and other exogenous compounds in the liver. They have also been shown to be controlled by growth-hormone pulses [40, 41] and thus are expected to be altered by mutations that alter growth hormone production or response. Many of the other proteins with a shared response are involved in fatty acid transport and regulating the balance between fatty acid synthesis and metabolism, such as acetyl-CoA carboxylases ACACA and ACACB, which act as major rate-limiting steps in fatty acid synthesis and oxidation, fatty acid synthase (FAS), fatty acid binding protein (FABPL), acetyl-CoA acyltransferase (THIKB), ATP citrate synthase (ACLY), and acyl-CoA and acyl-CoA thioesterase (AACS and ACOT3).
Functional pathways predicted to be broadly affected by longevity-promoting interventions
The previous observations represent the processes enriched among the relatively limited number of proteins that exhibit shared responses to multiple longevity-promoting interventions, but they do not account for common pathways enriched through increased expression of different proteins. This prompted us to investigate whether there was a more consistent response across interventions at the pathway level. To do so, we performed a pathway impact analysis to infer which biological pathways were most perturbed by the longevity-promoting interventions from the observed protein abundances and changes. The differential abundance results for all proteins in response to all interventions were included in this analysis, but the relative impact of proteins to each pathway was weighted by the p-values associated with their fold-changes. Doing so enables a whole-proteome view of the pathways affected by each intervention and helps ensure that arbitrary cutoffs for assigning significance to protein responses do not influence our ability to characterize patterns emerging from many proteins of small effect across the proteome.
There was no pathway that was predicted to be significantly affected (with an adjusted p-value ≤ to 0.01) consistently by all the longevity-promoting interventions (Fig. 5). Furthermore, even for pathways that were most perturbed by an intervention, there was often a lack of consistency in the predicted changes to different components of a pathway. However, a handful of pathways showed a consistent response across multiple interventions. In particular, the peroxisome proliferator-activated receptor (PPAR) signaling and peroxisome KEGG pathways exhibited the highest total normalized perturbation scores across interventions in the liver and kidney, both showing evidence of upregulation (Fig. 5). A similar increase in peroxisomal proteins was previously shown in the hearts of mice upon acarbose intervention [42]. These patterns were driven by many of the same proteins that exhibited strong responses across interventions (as shown in Fig. 4A) and related proteins, including multiple proteins from the cytochrome p450 family, proteins involved in fatty acid transport, FABPL and FABPH, and numerous proteins involved in fatty acid oxidation (e.g., THIKA, THIKB). The AMPK and PI3K-akt signaling pathways, both of which play large roles in regulating metabolism and cell growth [43], as well as the complement and coagulation cascade (primarily in the kidney) also showed evidence of being modulated by multiple interventions.
Fig. 5.

Normalized perturbation scores for KEGG pathways from a pathway impact analysis per intervention-by-sex group. Shown are pathways that were significantly (adjusted p ≤ 0.05; indicated by an asterisk) predicted to be impacted by at least three interventions regardless of sex in A liver and B kidney tissues (no pathways met these criteria in muscle samples)
Discussion
By characterizing the responses of thousands of proteins to multiple longevity-promoting interventions in three different mouse tissues, this study not only provides a comprehensive description of the effect of each intervention on mouse proteomes but also identifies unique and shared alterations that call attention to mechanisms by which they might promote longevity. A motivating hypothesis for these experiments was that there are shared biological bases for the longevity-promoting effects of the interventions studied and that the discovery of common mechanisms could underlie the development of therapeutic approaches to extend health span in humans. However, our interrogation of 6009 proteins in mouse liver, kidney, and muscle revealed that most of the protein and pathway alterations induced by each intervention were either unique to that invention or shared by only one or two others. Furthermore, we did not detect any proteins that were commonly affected by all longevity-promoting interventions, including those related to the shared mechanisms previously reported to be common to several interventions (see “Introduction”). It is possible that shared mechanisms may be identified by studying additional tissues [13, 44], larger numbers of experimental animals, or by utilizing alternative proteomic methods or experimental designs. Additionally, shared pathways that involve differences in post-translational modifications, including, for example, the altered phosphorylation state of specific proteins [12] or proteolytic cleavage of proteins to produce signaling peptides [13], were beyond the scope of the methods we used in this study. Thus, our findings do not preclude the possibility that there are core mechanisms that are related to lifespan extension in mice.
Our findings are most consistent with the idea that there are multiple mechanisms that underlie longevity, and that the various interventions act, at least in part, through different means. Specifically, the shared protein responses we observed hint at potentially three broad groups of mechanisms, one common to the GHRKO, Snell dwarf, and calorie restriction, another that additionally includes acarbose, canagliflozin, and—to a lesser extent—17α-estradiol treatment in males, and a third more specific to rapamycin treatment in both sexes. Patterns of protein changes implicate peroxisomal oxidation metabolism of fatty acids and xenobiotic metabolism as common processes increased by multiple interventions. These pathways have previously been implicated as having important roles in aging and longevity primarily at the level of gene expression, and our results identify them at the protein level [8, 40, 41, 45]. We highlight several cytochrome P450, acetyl-CoA carboxylase, and glutathione S-transferase proteins that exhibited shared responses across multiple interventions. For the most part, these proteins responded similarly to the two genetic modifications and calorie restriction with acarbose and canagliflozin treatment in males often eliciting concordant behavior. The responses of these proteins to 17α-estradiol treatment in males were weaker in magnitude and less certain but otherwise tended to be in the same direction as the other interventions. In contrast, rapamycin treatment often significantly affected these proteins in the opposite direction of the other interventions. This discrepancy was evident in the liver but not observed in the kidney, supporting the likelihood that longevity-promoting mechanisms of some interventions are tissue specific.
There are some notable similarities between the findings in these experiments and some other attempts to identify shared longevity-associated mechanisms. Tyshkovskiy et al. (2019) examined the transcriptional changes induced by most of the same interventions studied here (excluding canagliflozin) and observed qualitatively similar results [8]. Like our observations at the protein level, they did not observe any gene whose expression was commonly affected by all interventions. Nevertheless, several did respond to multiple interventions, and the pathways commonly affected included those involved in fatty acid oxidation and metabolism by cytochrome P450 proteins and glutathione. Furthermore, they also found that changes in gene expression in response to rapamycin showed limited overlap with responses to other longevity-promoting treatments, a pattern observed both here and elsewhere [8, 18]. Despite the similarity of findings at the pathway level, the specific genes whose expression Tyshkovskiy et al. identified as commonly affected by multiple interventions did not necessarily match the proteins we identified as exhibiting shared responses. This discrepancy may be the result of differences in the sensitivity and bias of the different technologies used to measure transcriptomes and proteomes, but it may also reflect differences in the biological mechanisms that regulate mRNA levels and those that are responsible for protein homeostasis [46, 47].
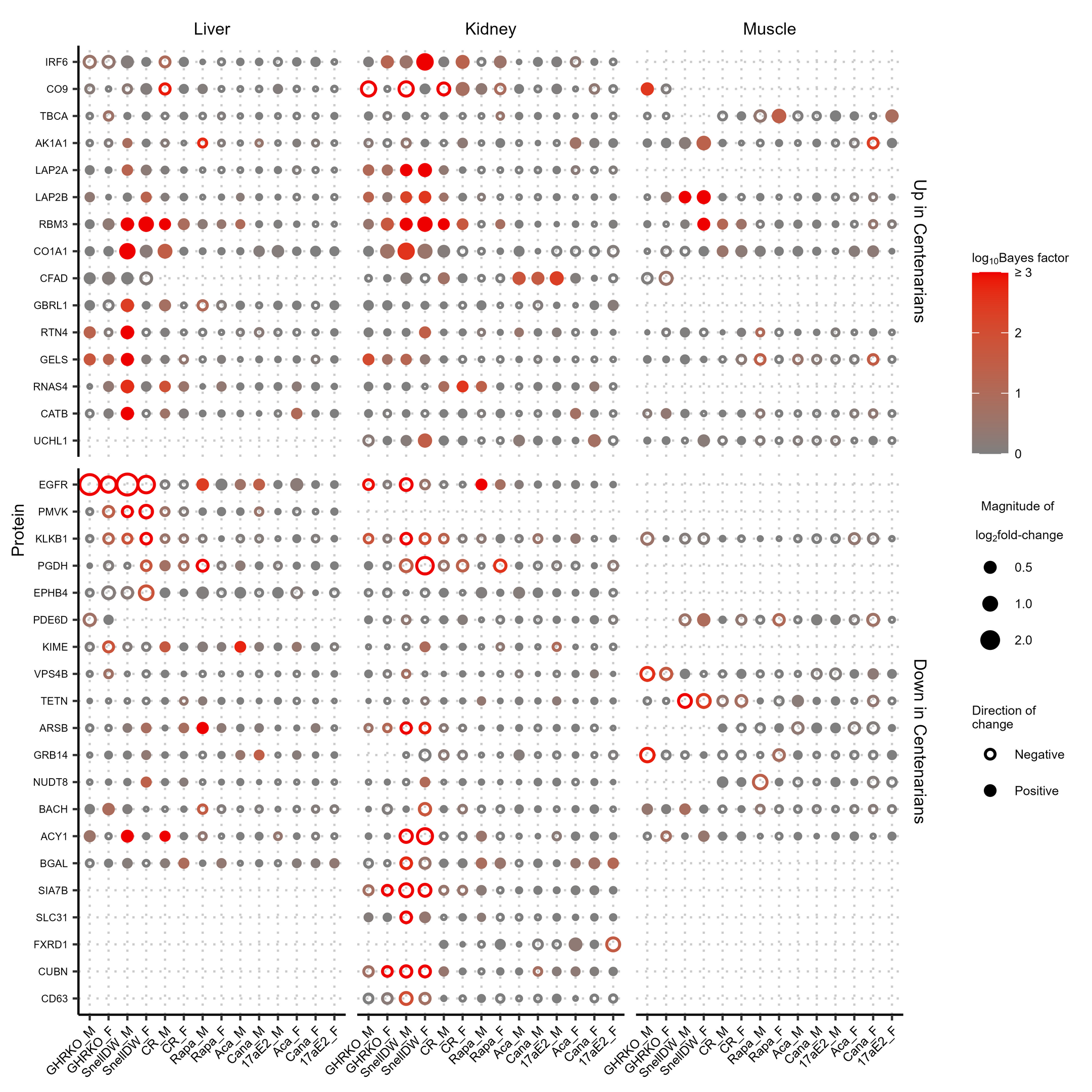
We also identified multiple proteins involved in the immune response that, though they only responded to a small number of interventions in mice, have orthologs that have been associated with longevity-related phenotypes in humans. The immunoglobulin proteins IGHM, IGHG3, complement C9 (CO9), and C-reactive protein (CRP) responded negatively to GHRKO, Snell dwarf, calorie restriction, and/or rapamycin in mice while their human orthologs discriminated older (over 65 years old) males destined to live to the 90th percentile age of their birth cohort [48] (Supplemental Fig. 9). Additionally, CFAD responded positively to acarbose, canagliflozin, and 17α-estradiol in male mice, while human orthologs exhibited increased abundance in centenarian humans over 100 years old compared to controls [49] (Supplemental Fig. 10). This implication of immune-related pathways is further supported by the shared impact of multiple interventions, notably rapamycin, on the complement and coagulation cascades in the liver and kidney. While the substantial differences both among the human studies and between the human studies and our mouse experiment (e.g., species differences, human serum or plasma compared to mouse liver, kidney, or muscle, human subjects were individuals of widely differing degrees of advanced age with uncontrolled drug and diet exposures, while the mice in our study were experimentally manipulated young adults, etc.) mean that the overlap in proteins associated with longevity-related phenotypes should be interpreted cautiously, they also mean that any such overlaps are surprising and, as such, worthy of note.
Our ability to characterize the shared effects of these interventions across the mouse proteome was facilitated by combining a broad experimental design with novel technological and analytical approaches. Simultaneously characterizing the effects of multiple interventions within the same experiment enabled us to identify shared responses that would otherwise be obfuscated by differences across separate experiments. The scale of this design was made possible by exploiting advances in mass spectrometry proteomics to quantify the abundances of thousands of proteins in three tissues across nearly three hundred mice. This further required developing a comprehensive statistical pipeline to address the analytical challenges related to the nature of protein quantification. However, there remain important limitations. While the number of proteins quantified was large, it was not exhaustive and does not include a full complement of protein post-translational modifications. Therefore, it would not be expected to represent a complete characterization of the mouse proteome. Thus, important effects of the interventions may not have been detected. Furthermore, despite characterizing the proteomes of nearly three hundred mice, they were divided across seven interventions, their respective controls, and two sexes, potentially limiting the statistical power to detect small but meaningful changes in protein abundances. In addition, each of the interventions imposes numerous physiological effects, complicating the identification of protein responses that are associated with longevity. Our strategy of focusing on shared protein responses was meant to circumvent this issue, but it is possible that some of the shared responses represent common physiological effects of the interventions unrelated to longevity. Given these limitations, the proteins and pathways implicated in this study require further validation before any confident conclusions can be made regarding their role in longevity. Longitudinal study designs may offer more opportunities to evaluate the biological effects of antiaging interventions. Using mouse plasma may allow for longitudinal sampling of mice as they age at the cost of a loss in the information provided from metabolically active tissues like the liver and kidney. While the present study was explicitly focused on identifying proteins in early adulthood that are associated with anticipated longevity extension in later life, additional mechanisms may be identified by repeating the analysis as mice age.
A major goal of longevity research has been to identify specific proteins or pathways as targets for therapeutical interventions. From this perspective, the findings of this study highlight a narrowed list of potential candidates and contribute a better mechanistic understanding of the effects of various individual longevity-promoting interventions on vertebrate proteomes. Though we predicted that these experiments would uncover protein alterations that were common across all interventions, thus revealing essential underlying biological bases for longevity, we found limited universally shared effects. These results do not eliminate the possibility that there are common underpinnings of interventions that extend lifespan, but they suggest that it is equally plausible that there are multiple mechanisms by which these interventions promote longevity. Overall, the changes in protein abundance we found to be associated with interventions known to extend lifespan should form the basis for more extended investigation.
Supplementary information
{kind=link}
Supplemental Figure 1: (A) Venn diagram of the number of proteins detected in each tissue. (B) Dendrogram of similarity in protein abundances across all tissues. (PNG 108 kb)
{kind=link}
Supplemental Figure 2: (A-C) Scatterplots comparing mean protein abundances between liver and kidney (A), liver and muscle (B), and kidney and muscle (C) samples across mice of both sexes. (D-F) Scatterplot comparing mean protein abundances between female and male samples in liver (D), kidney (E), and muscle (F) tissues. (PNG 342 kb)
{kind=link}
Supplemental Figure 3: Variation in protein abundances explained by mouse background, sex, and longevity intervention for each quantified protein in liver (A), kidney (B), and muscle (C) tissues. Results from fitting a random effects model with intervention nested within background for each protein individually. (D) The abundances by intervention and sex of proteins for which the percent variation explained by intervention was greater than 60% in liver samples. (PNG 746 kb)
{kind=link}
Supplemental Figure 4: The log2 fold-change and log10 Bayes factor for proteins within each treatment compared to controls for male (A) and female (B) kidney samples. Dashed vertical lines correspond to a log2 fold-change of +/- 0.5 while the dashed vertical line corresponds to a Bayes factor of 10. (PNG 569 kb)
{kind=link}
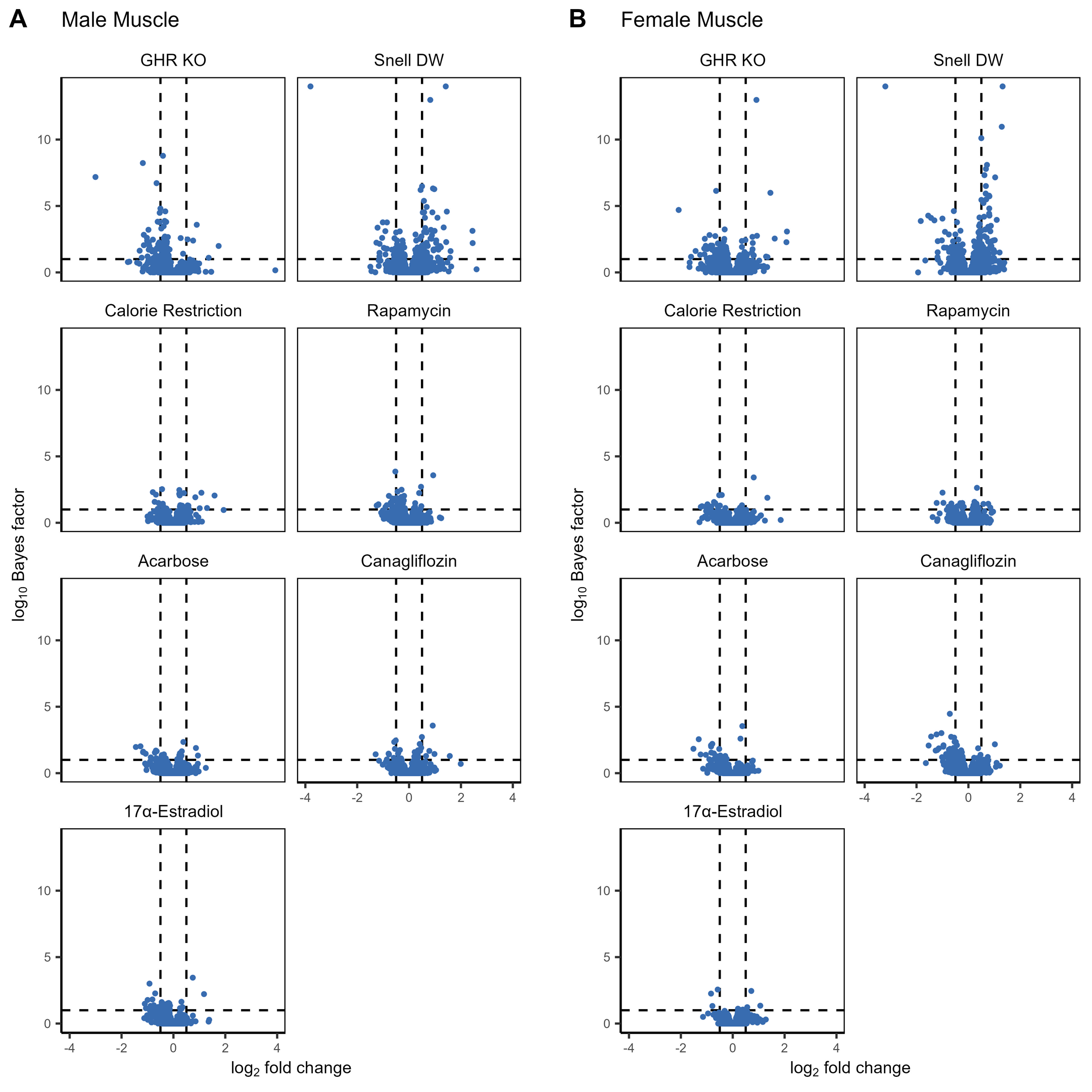
Supplemental Figure 5: The log2 fold-change and log10 Bayes factor for proteins within each treatment compared to controls for male (A) and female (B) muscle samples. Dashed vertical lines correspond to a log2 fold-change of +/- 0.5 while the dashed vertical line corresponds to a Bayes factor of 10. (PNG 464 kb)
{kind=link}
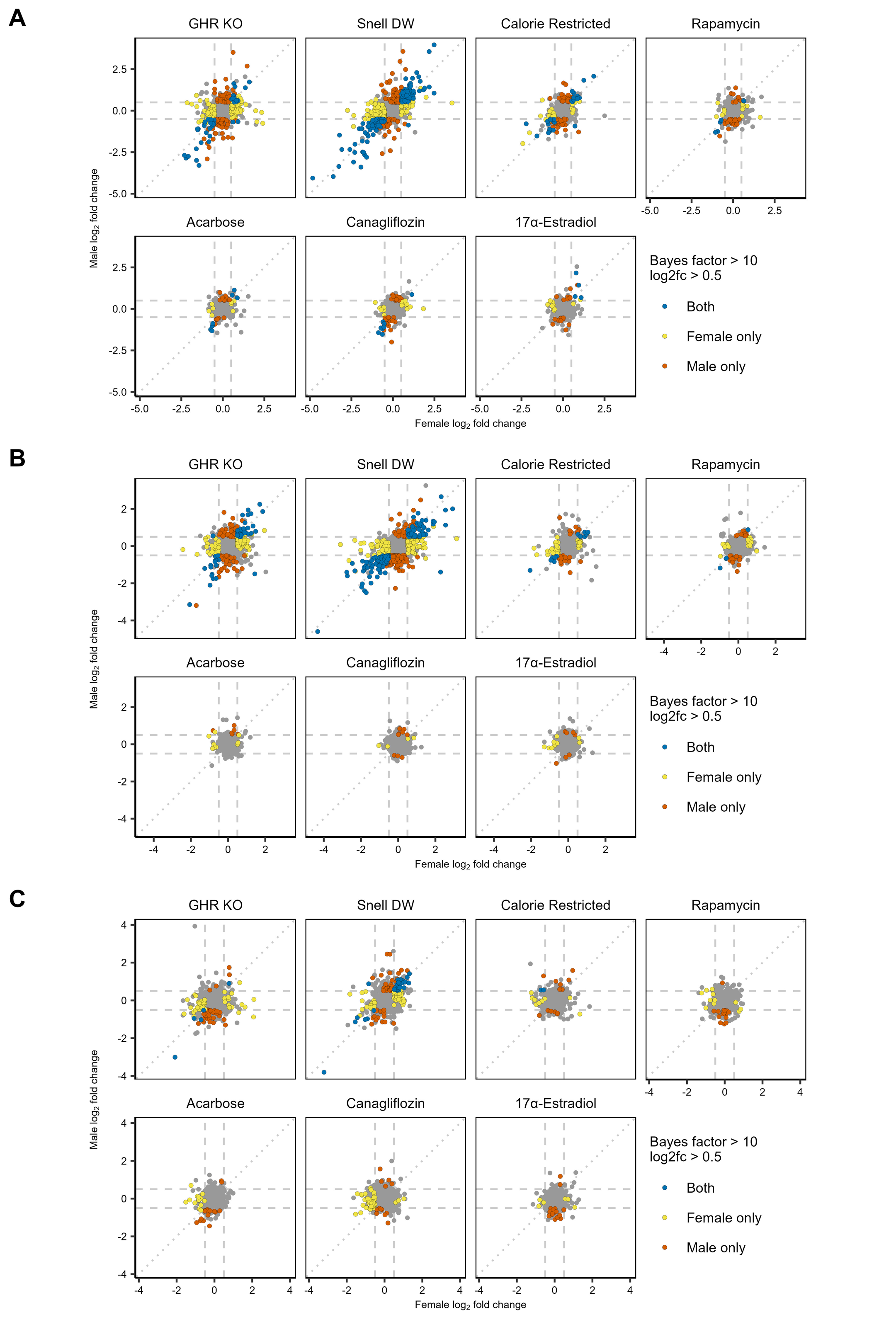
Supplemental Figure 6: Fold-changes in response to each longevity intervention in females (x-axis) compared to males (y-axis) in liver (A), kidney (B), and muscle (C) tissues. Dashed lines indicate log2 fold-changes of +/- 0.5 in males (horizontal) and females (vertical), while the diagonal dotted line marks a 1:1 relationship. Points are colored blue if the protein exhibited an absolute log2 fold-change greater than 0.5 with a Bayes factor greater than 10 in both sexes, red if in males only, yellow if in females only, and grey if in neither. (PNG 1040 kb)
{kind=link}
Supplemental Figure 7: (A) Changes in protein abundances across multiple longevity treatments in kidney. Proteins shown are limited to those exhibiting an absolute log2 fold-change ≥ 0.5 and a Bayes factor ≥ 10 in response to at least three interventions of either sex. The color of the points indicates the associated Bayes factor and the size is proportional to the magnitude of the fold-change with closed points indicating a positive change and open points indicating a negative change. (B) GO-Terms that are enriched (with an associated adjusted p-value ≤ 0.01) among proteins with shared responses across longevity promoting interventions in kidney. (PNG 1022 kb)
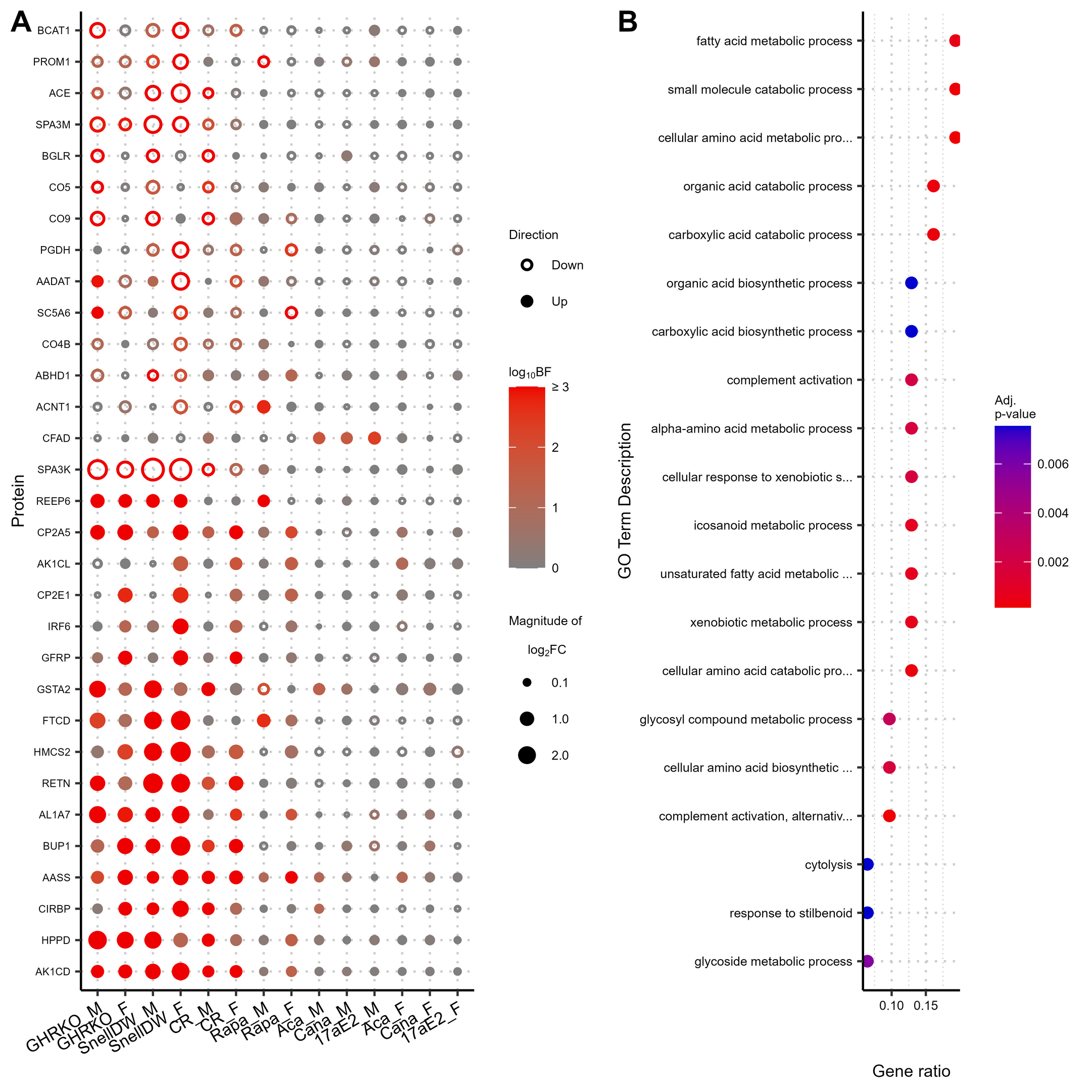
{kind=link}
Supplemental Figure 8: Changes in protein abundances across multiple longevity treatments in muscle. Proteins shown are limited to those exhibiting an absolute log2 fold-change greater than ≥ 0.5 and a Bayes factor ≥ 10 in response to at least three interventions of either sex. The color of the points indicates the associated Bayes factor and the size is correlated with the magnitude of the fold-change with closed points indicating a positive change and open points indicating a negative change. No GO-Terms where are enriched with an associated adjusted p-value ≤ 0.01 among proteins with shared responses across longevity promoting interventions in muscle. (PNG 218 kb)
{kind=link}
Supplemental Figure 9: Comparison of protein responses to longevity interventions in mouse livers to protein differences between long-lived humans and not in the MrOS study for proteins detected in both datasets for liver, kidney, and muscle tissues. The color of the points indicates the associated Bayes factor and the size is proportional to the magnitude of the fold-change with closed points indicating a positive change and open points indicating a negative change. (PNG 1008 kb)
{kind=link}
Supplemental Figure 10: Protein responses to longevity-promoting intervention-by-sex groups in mouse liver, kidney, and muscle for proteins also differentially abundant in centenarians in NECS. Proteins shown are limited to those that exhibit both differential abundance in centenarian humans compared to controls with an adjusted p-value less than 0.001 in NECS and exhibited an absolute log2 fold-change ≥ 0.5 and a Bayes factor ≥ 10 in response to at least one interventions of either sex in mice. The color of the points indicates the associated Bayes factor and the size is proportional to the magnitude of the fold-change with closed points indicating a positive change and open points indicating a negative change. (PNG 1382 kb)
(CSV 4602 kb)
(CSV 5249 kb)
(CSV 2807 kb)
(CSV 119 kb)
(CSV 87 kb)
(CSV 36 kb)
(CSV 6 kb)
(PDF 644 kb)
Acknowledgements
We thank Kelly Crebs for excellent technical assistance.
Funding
This work was funded in part by the National Institutes of Health grants, from the National Institute of General Medical Sciences R01 GM087221, the Office of the Director S10OD026936, the National Institute on Aging U19AG023122, and the National Science Foundation award 1920268.
Data availability
All sample analysis run order files, mass spectrometry dia-PASEF raw folders corresponding to mouse tissue experiments (.d), FASTA database used to generate the assay libraries (.fasta), mouse hybrid spectral assay library (.txt) and its DIALib-QC report (.tsv), and three unnormalized peptide quantitation data files (.csv) for each tissue have been deposited with the ProteomeXchange Consortium via the PRIDE partner repository [50, 51] with the data set identifier PXD040497 (http://www.ebi.ac.uk/pride). Code describing our statistical data analysis that can be used to reproduce our main results can be found at https://github.com/longevity-consortium/M005MouseLongevityPaper2023.
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bartke A, Wright JC, Mattison JA, et al. Extending the lifespan of long-lived mice. Nature. 2001;414:412. doi: 10.1038/35106646. [DOI] [PubMed] [Google Scholar]
- 2.Drake JC, Bruns DR, Peelor FF, et al. Long-lived Snell dwarf mice display increased proteostatic mechanisms that are not dependent on decreased mTORC1 activity. Aging Cell. 2015;14:474–482. doi: 10.1111/acel.12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gesing A, Masternak MM, Lewinski A, et al. Decreased levels of proapoptotic factors and increased key regulators of mitochondrial biogenesis constitute new potential beneficial features of long-lived growth hormone receptor gene–disrupted mice. J Gerontol: Series A. 2013;68:639–651. doi: 10.1093/gerona/gls231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harrison DE, Strong R, Allison DB, et al. Acarbose, 17-α-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging Cell. 2014;13:273–282. doi: 10.1111/acel.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harrison DE, Strong R, Alavez S, et al. Acarbose improves health and lifespan in aging HET3 mice. Aging Cell. 2019;18:e12898. doi: 10.1111/acel.12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller RA, Harrison DE, Allison DB, et al. Canagliflozin extends life span in genetically heterogeneous male but not female mice. JCI Insight. 2020;5 10.1172/jci.insight.140019 [DOI] [PMC free article] [PubMed]
- 8.Tyshkovskiy A, Bozaykut P, Borodinova AA, et al. Identification and application of gene expression signatures associated with lifespan extension. Cell Metab. 2019;30:573–593.e8. doi: 10.1016/j.cmet.2019.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X, Shi X, McPherson M, et al. Cap-independent translation of GPLD1 enhances markers of brain health in long-lived mutant and drug-treated mice. Aging Cell. 2022;21:e13685. doi: 10.1111/acel.13685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen Z, Hinson A, Miller RA, Garcia GG. Cap-independent translation: a shared mechanism for lifespan extension by rapamycin, acarbose, and 17α-estradiol. Aging Cell. 2021;20:e13345. doi: 10.1111/acel.13345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ozkurede U, Kala R, Johnson C, et al. Cap-independent mRNA translation is upregulated in long-lived endocrine mutant mice. J Mol Endocrinol. 2019;63:123–138. doi: 10.1530/JME-19-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wink L, Miller RA, Garcia GG. Rapamycin, Acarbose and 17α-estradiol share common mechanisms regulating the MAPK pathways involved in intracellular signaling and inflammation. Immun Ageing. 2022;19:8. doi: 10.1186/s12979-022-00264-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li X, Frazier JA, Spahiu E, et al. Muscle-dependent regulation of adipose tissue function in long-lived growth hormone-mutant mice. Aging. 2020;12:8766–8789. doi: 10.18632/aging.103380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X, McPherson M, Hager M, et al. Four anti-aging drugs and calorie-restricted diet produce parallel effects in fat, brain, muscle, macrophages, and plasma of young mice. Geroscience. 2023; 10.1007/s11357-023-00770-0 [DOI] [PMC free article] [PubMed]
- 15.Bonkowski MS, Rocha JS, Masternak MM, et al. Targeted disruption of growth hormone receptor interferes with the beneficial actions of calorie restriction. Proc Natl Acad Sci. 2006;103:7901–7905. doi: 10.1073/pnas.0600161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flurkey K, Papaconstantinou J, Miller RA, Harrison DE. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc Natl Acad Sci. 2001;98:6736–6741. doi: 10.1073/pnas.111158898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flurkey K, Astle CM, Harrison DE. Life extension by diet restriction and N-acetyl-L-cysteine in genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2010;65:1275–1284. doi: 10.1093/gerona/glq155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller RA, Harrison DE, Astle CM, et al. Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell. 2014;13:468–477. doi: 10.1111/acel.12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dominick G, Berryman DE, List EO, et al. Regulation of mTOR activity in Snell dwarf and GH receptor gene-disrupted mice. Endocrinology. 2015;156:565–575. doi: 10.1210/en.2014-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macchiarini F, Miller RA, Strong R, et al. Chapter 10 - NIA interventions testing program: a collaborative approach for investigating interventions to promote healthy aging. In: Musi N, Hornsby PJ, et al., editors. Handbook of the Biology of Aging (Ninth Edition) Academic Press; 2021. pp. 219–235. [Google Scholar]
- 21.Auer H, Mobley J, Ayers L, et al. The effects of frozen tissue storage conditions on the integrity of RNA and protein. Biotech Histochem. 2014;89:518–528. doi: 10.3109/10520295.2014.904927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.LaBaer J. Improving international research with clinical specimens: 5 achievable objectives. J Proteome Res. 2012;11:5592–5601. doi: 10.1021/pr300796m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Escher C, Reiter L, MacLean B, et al. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics. 2012;12:1111–1121. doi: 10.1002/pmic.201100463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bruderer R, Bernhardt OM, Gandhi T, Reiter L. High-precision iRT prediction in the targeted analysis of data-independent acquisition and its impact on identification and quantitation. Proteomics. 2016;16:2246–2256. doi: 10.1002/pmic.201500488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krasny L, Bland P, Burns J, et al. A mouse SWATH-mass spectrometry reference spectral library enables deconvolution of species-specific proteomic alterations in human tumour xenografts. Dis Model Mech. 2020;13:dmm044586. doi: 10.1242/dmm.044586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Midha MK, Campbell DS, Kapil C, et al. DIALib-QC an assessment tool for spectral libraries in data-independent acquisition proteomics. Nat Commun. 2020;11:5251. doi: 10.1038/s41467-020-18901-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bland JM, Altman DG. Statistics notes: calculating correlation coefficients with repeated observations: Part 1—correlation within subjects. BMJ. 1995;310:446. doi: 10.1136/bmj.310.6977.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bakdash JZ, Marusich LR (2022) rmcorr: repeated measures correlation [DOI] [PMC free article] [PubMed]
- 29.Lazar C, Gatto L, Ferro M, et al. Accounting for the multiple natures of missing values in label-free quantitative proteomics data sets to compare imputation strategies. J Proteome Res. 2016;15:1116–1125. doi: 10.1021/acs.jproteome.5b00981. [DOI] [PubMed] [Google Scholar]
- 30.McGurk KA, Dagliati A, Chiasserini D, et al. The use of missing values in proteomic data-independent acquisition mass spectrometry to enable disease activity discrimination. Bioinformatics. 2020;36:2217–2223. doi: 10.1093/bioinformatics/btz898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cragg JG. Some statistical models for limited dependent variables with application to the demand for durable goods. Econometrica. 1971;39:829–844. doi: 10.2307/1909582. [DOI] [Google Scholar]
- 32.StataCorp (2021) Stata statistical software
- 33.Sellke T, Bayarri MJ, Berger JO. Calibration of ρ values for testing precise null hypotheses. Am Stat. 2001;55:62–71. doi: 10.1198/000313001300339950. [DOI] [Google Scholar]
- 34.Draghici S, Khatri P, Tarca AL, et al. A systems biology approach for pathway level analysis. Genome Res. 2007;17:1537–1545. doi: 10.1101/gr.6202607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carlson, M (2019) org.Mm.eg.db: Genome wide annotation for Mouse
- 36.Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Voichita C, Donato M, Draghici S. 2012 11th International Conference on Machine Learning and Applications. 2012. Incorporating gene significance in the impact analysis of signaling pathways; pp. 126–131. [Google Scholar]
- 38.Voichita C, Ansari S, Draghici S (2022) ROntoTools: R onto-tools suite.
- 39.Kanehisa M, Furumichi M, Sato Y, et al. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51:D587–D592. doi: 10.1093/nar/gkac963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steinbaugh MJ, Sun LY, Bartke A, Miller RA. Activation of genes involved in xenobiotic metabolism is a shared signature of mouse models with extended lifespan. Am J Physiol Endocrinol Metab. 2012;303:E488–E495. doi: 10.1152/ajpendo.00110.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li X, Bartke A, Berryman DE, et al. Direct and indirect effects of growth hormone receptor ablation on liver expression of xenobiotic metabolizing genes. Am J Physiol Endocrinol Metab. 2013;305:E942–E950. doi: 10.1152/ajpendo.00304.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herrera JJ, Louzon S, Pifer K, et al. Acarbose has sex-dependent and -independent effects on age-related physical function, cardiac health, and lipid biology. JCI Insight. 5:e137474. 10.1172/jci.insight.137474 [DOI] [PMC free article] [PubMed]
- 43.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sadagurski M, Cady G, Miller RA. Anti-aging drugs reduce hypothalamic inflammation in a sex-specific manner. Aging Cell. 2017;16:652–660. doi: 10.1111/acel.12590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mitchell SJ, Madrigal-Matute J, Scheibye-Knudsen M, et al. Effects of sex, strain, and energy intake on hallmarks of aging in mice. Cell Metab. 2016;23:1093–1112. doi: 10.1016/j.cmet.2016.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takemon Y, Chick JM, Gerdes Gyuricza I, et al. Proteomic and transcriptomic profiling reveal different aspects of aging in the kidney. Elife. 2021;10:e62585. doi: 10.7554/eLife.62585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gerdes Gyuricza I, Chick JM, Keele GR, et al. Genome-wide transcript and protein analysis highlights the role of protein homeostasis in the aging mouse heart. Genome Res. 2022;32:838–852. doi: 10.1101/gr.275672.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orwoll ES, Wiedrick J, Nielson CM, et al. Proteomic assessment of serum biomarkers of longevity in older men. Aging Cell. 2020;19:e13253. doi: 10.1111/acel.13253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sebastiani P, Federico A, Morris M, et al. Protein signatures of centenarians and their offspring suggest centenarians age slower than other humans. Aging Cell. 2021;20:e13290. doi: 10.1111/acel.13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vizcaíno JA, Côté R, Reisinger F, et al. A guide to the proteomics identifications database proteomics data repository. Proteomics. 2009;9:4276–4283. doi: 10.1002/pmic.200900402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perez-Riverol Y, Bai J, Bandla C, et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022;50:D543–D552. doi: 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: (A) Venn diagram of the number of proteins detected in each tissue. (B) Dendrogram of similarity in protein abundances across all tissues. (PNG 108 kb)
Supplemental Figure 2: (A-C) Scatterplots comparing mean protein abundances between liver and kidney (A), liver and muscle (B), and kidney and muscle (C) samples across mice of both sexes. (D-F) Scatterplot comparing mean protein abundances between female and male samples in liver (D), kidney (E), and muscle (F) tissues. (PNG 342 kb)
Supplemental Figure 3: Variation in protein abundances explained by mouse background, sex, and longevity intervention for each quantified protein in liver (A), kidney (B), and muscle (C) tissues. Results from fitting a random effects model with intervention nested within background for each protein individually. (D) The abundances by intervention and sex of proteins for which the percent variation explained by intervention was greater than 60% in liver samples. (PNG 746 kb)
Supplemental Figure 4: The log2 fold-change and log10 Bayes factor for proteins within each treatment compared to controls for male (A) and female (B) kidney samples. Dashed vertical lines correspond to a log2 fold-change of +/- 0.5 while the dashed vertical line corresponds to a Bayes factor of 10. (PNG 569 kb)
Supplemental Figure 5: The log2 fold-change and log10 Bayes factor for proteins within each treatment compared to controls for male (A) and female (B) muscle samples. Dashed vertical lines correspond to a log2 fold-change of +/- 0.5 while the dashed vertical line corresponds to a Bayes factor of 10. (PNG 464 kb)
Supplemental Figure 6: Fold-changes in response to each longevity intervention in females (x-axis) compared to males (y-axis) in liver (A), kidney (B), and muscle (C) tissues. Dashed lines indicate log2 fold-changes of +/- 0.5 in males (horizontal) and females (vertical), while the diagonal dotted line marks a 1:1 relationship. Points are colored blue if the protein exhibited an absolute log2 fold-change greater than 0.5 with a Bayes factor greater than 10 in both sexes, red if in males only, yellow if in females only, and grey if in neither. (PNG 1040 kb)
Supplemental Figure 7: (A) Changes in protein abundances across multiple longevity treatments in kidney. Proteins shown are limited to those exhibiting an absolute log2 fold-change ≥ 0.5 and a Bayes factor ≥ 10 in response to at least three interventions of either sex. The color of the points indicates the associated Bayes factor and the size is proportional to the magnitude of the fold-change with closed points indicating a positive change and open points indicating a negative change. (B) GO-Terms that are enriched (with an associated adjusted p-value ≤ 0.01) among proteins with shared responses across longevity promoting interventions in kidney. (PNG 1022 kb)
Supplemental Figure 8: Changes in protein abundances across multiple longevity treatments in muscle. Proteins shown are limited to those exhibiting an absolute log2 fold-change greater than ≥ 0.5 and a Bayes factor ≥ 10 in response to at least three interventions of either sex. The color of the points indicates the associated Bayes factor and the size is correlated with the magnitude of the fold-change with closed points indicating a positive change and open points indicating a negative change. No GO-Terms where are enriched with an associated adjusted p-value ≤ 0.01 among proteins with shared responses across longevity promoting interventions in muscle. (PNG 218 kb)
Supplemental Figure 9: Comparison of protein responses to longevity interventions in mouse livers to protein differences between long-lived humans and not in the MrOS study for proteins detected in both datasets for liver, kidney, and muscle tissues. The color of the points indicates the associated Bayes factor and the size is proportional to the magnitude of the fold-change with closed points indicating a positive change and open points indicating a negative change. (PNG 1008 kb)
Supplemental Figure 10: Protein responses to longevity-promoting intervention-by-sex groups in mouse liver, kidney, and muscle for proteins also differentially abundant in centenarians in NECS. Proteins shown are limited to those that exhibit both differential abundance in centenarian humans compared to controls with an adjusted p-value less than 0.001 in NECS and exhibited an absolute log2 fold-change ≥ 0.5 and a Bayes factor ≥ 10 in response to at least one interventions of either sex in mice. The color of the points indicates the associated Bayes factor and the size is proportional to the magnitude of the fold-change with closed points indicating a positive change and open points indicating a negative change. (PNG 1382 kb)
(CSV 4602 kb)
(CSV 5249 kb)
(CSV 2807 kb)
(CSV 119 kb)
(CSV 87 kb)
(CSV 36 kb)
(CSV 6 kb)
(PDF 644 kb)
Data Availability Statement
All sample analysis run order files, mass spectrometry dia-PASEF raw folders corresponding to mouse tissue experiments (.d), FASTA database used to generate the assay libraries (.fasta), mouse hybrid spectral assay library (.txt) and its DIALib-QC report (.tsv), and three unnormalized peptide quantitation data files (.csv) for each tissue have been deposited with the ProteomeXchange Consortium via the PRIDE partner repository [50, 51] with the data set identifier PXD040497 (http://www.ebi.ac.uk/pride). Code describing our statistical data analysis that can be used to reproduce our main results can be found at https://github.com/longevity-consortium/M005MouseLongevityPaper2023.