

Abstract
The Mycobacterium tuberculosis gene Rv2747 encodes a novel 19-kDa ArgA that catalyzes the initial step in l-arginine biosynthesis, namely the conversion of l-glutamate to α-N-acetyl-l-glutamate. Initial velocity studies reveal that Rv2747 proceeds through a sequential kinetic mechanism, with Km values of 280 mM for l-glutamine and 150 μM for acetyl-coenzyme A and with a kcat value of 200 min−1. Initial velocity studies with l-glutamate showed that even at concentrations of 600 mM, saturation was not observed. Therefore, only a kcat/Km value of 125 M−1 min−1 can be calculated. Inhibition studies reveal that the enzyme is strongly regulated by l-arginine, the end product of the pathway (50% inhibitory concentration, 26 μM). The enzyme was completely inhibited by 500 μM arginine, with a Hill coefficient of 0.60, indicating negatively cooperative binding of l-arginine.
In many microorganisms, l-arginine is used as source of both carbon and nitrogen, and in several organisms the anabolic and catabolic pathways of metabolism have been well defined (12). The transport and metabolism of l-arginine has been shown to be essential for a number of pathogens, including Mycobacterium tuberculosis (13). Although the arginine biosynthetic gene cluster (argCJBDFRGH) has been identified in M. tuberculosis (8), the argF-encoded ornithine carbamoyltransferase cloned and purified from M. bovis BCG (26), and an argF auxotroph made in M. tuberculosis (13), knowledge of the mechanistic detail of l-arginine biosynthesis and its regulation in mycobacteria are limited.
In prokaryotes, the arginine biosynthetic pathway proceeds from glutamate via initial N-acetylation of l-glutamate (Fig. 1). Acetylation acts as a protecting group strategy, preventing the spontaneous cyclization of the glutamate semialdehyde intermediate, thus precluding the formation of the proline precursor pyrroline-5-carboxylate (4). Two alternative pathways have evolved for the subsequent removal of the acetyl group and regulation of metabolic flow through the pathway. The “linear pathway” found in Enterobacteriaceae (12), Vibrionaceae (31), Myxococcus xanthus (16), and the archaeobacterium Sulfolobus solfataricus (28) includes an N-acetylornithine deacetylase, encoded by argE, to catalyze the metal-dependent hydrolysis of the acetamide group to form the arginine precursor l-ornithine and acetate (18). In these organisms, the first enzyme in the pathway, N-acetylglutamate synthase, is the target of feedback inhibition by arginine (21). In contrast, in other prokaryotes and simple eukaryotes such as yeast, the pathway has evolved to be energetically more economic, using an “acetyl recycling” pathway in which the acetyl group is recycled through transfer from N-acetylornithine to l-glutamate, yielding N-acetylglutamate and ornithine (12). This reaction is catalyzed by an α-N-acetyl-ornithine-l-glutamate acetyltransferase encoded by the argJ gene. In such reactions, once α-N-acetylornithine is synthesized the action of the N-acetylglutamate synthase (argA) becomes superfluous and regulation of the metabolic pathway is achieved by arginine feedback inhibition of the second enzyme in the pathway, N-acetylglutamate 5-phosphotransferase (27) (encoded by argB), or by feedback inhibition of ArgJ by ornithine (24).
FIG. 1.

Arginine biosynthetic pathways in microorganisms. The “linear pathway” found in Enterobacteriaceae and in S. solfataricus (28) and the alternative “acetyl recycling” pathway found in B. stearothermophilus (23), S. coelicolor (17), and T. thermophilus (6). Dashed lines indicate further biosynthesis via intermediates.
In Bacillus stearothermophilus, the argJ gene product is bifunctional, possessing both ornithine acetyltransferase and N-acetylglutamate synthase activity (24). This accounts for the fact that no argA has been identified in the genome of this organism. However, in Actinomycetes such as Streptomyces coelicolor and the extreme thermophilic bacterium Thermus thermophilus, the argJ gene product has been shown to encode a monofunctional enzyme displaying only ornithine acetyltransferase activity (6, 17). Interestingly, no readily identifiable ortholog of argA can be identified by sequence analysis of such organisms, thus posing the question of how N-acetylornithine is synthesized.
We have attempted to functionally identify genes involved in amino acid biosynthesis, including l-arginine biosynthesis, in M. tuberculosis. A potential l-glutamate α-N-acetyltransferase, encoded by Rv2747, is a member of the GCN5-related N-acetyltransferase superfamily, as identified by sequence alignment. This gene has been shown to be an essential gene by Himar1-based transposon mutagenesis (25). Rv2747 shows homology (29% identity) to the C terminus of argA of Pseudomonas aeruginosa. However, argA from P. aeruginosa codes for a 48-kDa protein (14), whereas Rv2747 encodes a much smaller, 19-kDa protein. Herein we report the cloning, expression, purification, and characterization of Rv2747 and delineation of its function as a novel α-N-acetylglutamate synthase.
MATERIALS AND METHODS
All chemicals, acetyl-coenzyme A (CoA), and amino acids were purchased from Sigma-Aldrich Chemical Co. Peptides were obtained from Bachem. Escherichia coli strain Rosetta 2 (DE3) pLysS cells and pET-23a(+) plasmid were purchased from Novagen. All restriction enzymes and T4 DNA ligase were obtained from New England Biolabs. PCR primers and pCR-Blunt plasmid kit were obtained from Invitrogen. Ni-NTA Superflow resin was purchased from QIAGEN. Pfu DNA polymerase was purchased from Stratagene.
General methods.
Solution pH values were measured at 25°C with an Accumet model 20 pH meter and Accumet combination electrode standardized at pH 7.0 and 4.0 or 10.0. Protein purification was performed at 4°C using a fast protein liquid chromatography system (Amersham-Pharmacia Biotech). Spectrophotometric assays were performed using a UVIKON XL double beam UV-vis spectrophotometer (BIO-TEK Instruments). 1H nuclear magnetic resonance (NMR) spectra were recorded on a Bruker DRX300 spectrometer at 300 MHz, 1H NMR spectra. Electrospray ionization mass spectra were recorded on a ABI QSTAR Pulsar qQTOF spectrometer.
Cloning, expression, and purification of Rv2747.
The M. tuberculosis gene Rv2747 was amplified by PCR using the primers 5-tactgtttcacatatgaccgaacgtccacgggat-3 and 5-gtcttctcgccaagcttcagcaccagcagcat-3, which were designed to create NdeI and HindIII restriction endonuclease sites (underlined). The PCR product was digested with NdeI and HindIII and was subjected to agarose (0.8%) gel electrophoresis. The amplified DNA product was ligated into the pCR-Blunt plasmid and transformed into One Shot TOP10 cells. Plasmid DNA isolated from these cells was then digested with NdeI and HindIII, and the purified insert was ligated into purified plasmid pET-23a(+) previously linearized with the same restriction enzymes, yielding an expression plasmid for M. tuberculosis Rv2747 with a C-terminal His6 tag.
The plasmid pET-23a(+):Rv2747 was then isolated, sequenced, and used to transform E. coli Rosetta 2 (DE3) pLysS cells. Transformed cells were grown overnight in 50 ml of LB broth containing 100 μg ampicillin and 34 μg of chloramphenicol. One-liter cultures were then inoculated to an A600 of ∼0.05. The cells were grown at 37°C to an A600 of 0.8. Cells were cooled for 1 h at 4°C, and protein expression was subsequently induced by the addition of 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and left to grow for a further 16 h at 20°C. Expression of the protein was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
All protein purification steps were carried out at 4°C. The cell pellet (30 g) was resuspended in 50 ml 20 mM triethanolamine (TEA), 100 mM ammonium sulfate, 10 mM imidazole, pH 7.8, containing two tablets of Complete protease inhibitor cocktail (Roche). The cells were disrupted on ice by sonication using a Branson Sonifier 450. The suspension was then centrifuged (10,000 × g for 30 min) to remove cell debris. The supernatant was then filtered using a 0.2-μm-pore-size syringe filter. The filtered supernatant was applied to a preequilibrated (Buffer A; 20 mM TEA, 100 mM ammonium sulfate, 10 mM imidazole, pH 7.8) QIAGEN Ni-NTA Superflow Column (20 by 1,200 mm). The column was washed at 1 ml min−1 with 4 column volumes of the same buffer and then eluted with a linear gradient of imidazole (10 mM to 500 mM over 10 column volumes) in Buffer A. Protein was detected with an on-line detector monitoring A280, and column fractions were collected and analyzed by SDS-PAGE. Fractions containing the ca. 20-kDa protein were pooled and dialyzed twice against 4 liters of 20 mM TEA, 100 mM ammonium sulfate, 1 mM EDTA, pH 7.8 and concentrated using a YM10 Amicon ultrafiltration membrane to a final concentration of 20 mg/ml and stored in 50% glycerol at −20°C.
Determination of protein concentration.
The enzyme concentration was determined from ɛ 280 nm = 17780 M−1 cm−1 for native Rv2747, and turnover numbers are based on an enzyme monomer. The concentration of enzyme was also determined using the bicinchoninic acid (BCA) protein assay (Pierce) with bovine serum albumin as a standard, which agreed favorably with the value obtained by A280.
Measurement of enzyme activity.
Reaction rates were determined spectrophotometrically by measuring the increase in absorbance at 412 nm due to the formation of 5-thio-2-nitrobenzoate resulting from the reaction between the free sulfhydryl group of CoASH, generated by the amino acid acetylating activity, and 5,5-dithio-bis(2-nitrobenzoic acid) (DTNB). The reaction was continuously monitored spectrophotometrically. Assay mixtures contained 40 mM TEA, 100 mM ammonium sulfate, 500 mM NaCl (NaCl was included in order to maintain a constant ionic strength at high substrate concentrations), pH 8, and 0.2 mM DTNB in addition to substrates and inhibitors. Reactions were initiated by the addition of 30 nM enzyme in a total volume of 1 ml and were maintained at 25°C. Initial velocity kinetic data were fitted using SigmaPlot. Equations 1 and 2 were used to fit initial velocity patterns, where the concentrations of both substrates were varied. Equation 3 was used to determine 50% inhibitory concentration (IC50) values:
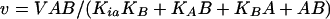 |
(1) |
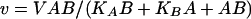 |
(2) |
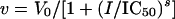 |
(3) |
where V is the maximum velocity, A and B and Ka and Kb are the concentrations and Michaelis constants, respectively, for each of the substrates, IC50 is the concentration of inhibitor necessary to cause 50% loss in activity, s is the slope factor, and I is the concentration of inhibitor.
Analytical gel filtration.
Analytical gel filtration was preformed using a Pharmacia Superose 12 1.4/30 cm gel filtration column, calibrated with Bio-Rad gel filtration standards. The column was run in 20 mM TEA, 100 mM ammonium sulfate, pH 8.0, at a flow rate of 0.5 ml/min.
Product purification and analysis.
l-glutamine (8 mg, 54 μmol) and acetyl-CoA (20 mg, 25 μmol) were dissolved in buffer (1,000 μl of 50 mM TRIS, pH 8.0) and Rv2747 (10 mg) was added, the solution was flushed with nitrogen, and the reaction was stirred at 30°C for 8 h. The reaction mixture was passed through an Amicon YM10 spin filter (13,000 rpm, 30 min at 4°C). The flowthrough was applied to a Poros HQ50 anion exchange column (3 ml; Perceptive Biosystems) and eluted with linear gradient of ammonium bicarbonate from 5 to 250 mM over 45 column volumes (135 ml) at a flow rate of 10 ml min−1. Amino acids were detected at 230 nm. Fractions containing α-N-acetylglutamine typically eluted around 125 mM bicarbonate. Relevant samples were concentrated, and ammonium bicarbonate was removed by lyophilization to yield the required compounds as their ammonium salts.
RESULTS
Cloning, expression, and purification of Rv2747.
To obtain large quantities of Rv2747 for mechanistic studies, the Rv2747 gene was cloned into plasmid pET-23a(+) and Rv2747 was expressed in E. coli Rosetta 2 (DE3) pLysS cells. Metal affinity column chromatography was sufficient to achieve 99% homogeneity (as judged by SDS-PAGE), with the protein being eluted with 400 mM imidazole. Approximately 500 mg of purified enzyme was obtained from 30 g of cell paste. Protein electrospray ionization mass/spectrometry was performed on the protein sample, revealing a single species with a molecular mass of 20,978 Da, compared to 21,109 Da expected for full-length Rv2747, indicating that the N-terminal N-formyl-methionine has been posttranslationally removed. Analytical gel filtration was preformed on the protein sample revealing that the protein existed as a mixture of equal amounts of a dimer and a tetramer.
Sequence analysis.
Orthologues of Rv2747 are present in a number of actinomycetes, including S. coelicolor (60% amino acid identity) and the thermophilic bacterium T. thermophilus (40% amino acid identity). Sequence alignment studies of functionally characterized ArgA proteins from E. coli and P. aeruginosa, Rv2747, and its orthologue from S. coelicolor show an alignment of the actinomyces sequences with the C-terminal region of the Enterobacteriaceae sequences (Fig. 2). A further domain search of the E. coli ArgA revealed that the protein consists of two separate domains. The N-terminal region of the protein shows homology (60% aligned over 234 residues) to the amino acid kinase family of proteins. The C-terminal region of the protein shows significant homology (98% over 83 residues) to the GNAT family of proteins (Fig. 3). This appears to indicate that the C-terminal portion of the E. coli ArgA protein is in fact the catalytic acetyltransferase domain.
FIG. 2.

Multiple alignment of ArgA proteins using the program Multalin (10). The first column lists the organism. Species name abbreviations are TB, M. tuberculosis (NP_217263); SC, S. coelicolor (NP_627584); EC, E. coli (NP_417295); PA, P. aeruginosa (NP_253891). Residues in boldface indicate similarity, residues in black indicate conserved residues. National Center for Biotechnology Information accession numbers are shown in brackets.
FIG. 3.

Schematic representation of the E. coli argA gene reveals that the gene consists of two separate domains. The N-terminal region of the protein shows homology (60% aligned over 234 residues) to the amino acid kinase family of proteins. The C-terminal region of the protein shows significant homology (98% over 83 residues) to the GNAT family of proteins, with the relative position of Rv2747 overlaid at the C terminus (3).
Catalytic properties.
N-acetylglutamate synthases from a number of sources, including both eukaryotes and prokaryotes, display a high degree of specificity for acetyl-CoA and l-glutamate as substrates (22). Similarly, Rv2747 showed a high degree of amino acid specificity, with none of the 20 proteinogenic l-amino acids except glutamine and glutamate serving as substrates. In addition, l-ornithine was not a substrate. The stereospecificity of the substrate was investigated, and neither d-glutamate nor d-glutamine were substrates. Simple dipeptides including l-Gln-l-Gln, l-Glu-δ-l-Gly, l-Glu-l-Gln, and l-Gln-l-Glu were assayed and shown not to function as substrates.
Reactions catalyzed by acetyl-CoA-dependent acetyltransferases are known to proceed through two distinct mechanisms (29). One is a ping-pong mechanism, which involves the transfer of the acetyl group to a cysteinyl residue of the enzyme and the formation of an acetylthioenzyme intermediate. The other is a sequential mechanism, where a ternary complex of enzyme, acetyl-CoA, and substrate forms and the acetyl group of acetyl-CoA is directly transferred to a substrate. The reactions catalyzed by the members of the GNAT superfamily studied so far, including ArgA (5), proceed through a sequential kinetic mechanism (29), with the notable exception of ESA1 histone acetyltransferase, which proceeds through a ping-pong kinetic mechanism (32).
Although l-glutamate and l-glutamine served as substrates for Rv2747, they were relatively poor substrates compared to the kinetic parameters exhibited by other ArgA enzymes (E. coli Glu Km, 3.7 mM; kcat, 100 s−1) (21). Initial velocity studies of Rv2747 with l-glutamate showed that even at concentrations of 600 mM, saturation was not observed. Therefore, only a kcat/Km value of 125 ± 2 M−1 min−1 can be calculated. Interestingly, l-glutamine served as a significantly better substrate compared to l-glutamate. The following steady-state parameters were obtained by fitting the data of Fig. 4 to equation 1; Km of l-glutamine, 280 ± 40 mM; of acetyl-CoA, 150 ± 30 μM; kcat, 200 ± 15 min−1; KiacetylCoA, 15 ± 15 μM; and Kiglutamine, 30 ± 30 mM. The kcat/Km value was 700 ± 70 M−1 min−1 for l-glutamine (5.5-fold better than l-glutamate). The initial velocity plots revealed, on the basis of the standard errors for the fitted parameters as well as visual inspection of the two fits, that the data best fits equation 1 for intersecting lines, diagnostic of a sequential mechanism (Fig. 4) in which both substrates must bind to the enzyme before chemistry can take place, rather than equation 2 for parallel lines, indicative of a ping pong mechanism.
FIG. 4.

Reciprocal initial velocity pattern of l-glutamine N-acetylation by Rv2747. The symbols are experimentally determined values, while the solid lines are fits of the data to equation 1 (sequential) and the dotted lines are fitted to equation 2 (Ping Pong) using SigmaPlot. l-glutamine was varied (50 mM, •; 100 mM, ○; 200 mM, ▾; 500 mM, ▿) at five fixed levels (0.05, 0.100, 0.200, 0.400, and 1.00 mM) of acetyl-CoA.
Regulatory properties.
N-Acetylglutamate synthase from a number of sources, including E. coli (21), Salmonella enterica serovar Typhimurium (2), P. aeruginosa (15), and Saccharomyces cerevisiae (30), are subject to strong feedback inhibition by the end product of the pathway, l-arginine. E. coli ArgA is especially susceptible to this form of feedback inhibition, with an IC50 value of 20 μM (21). Inhibition studies of Rv2747 with the 20 proteinogenic l-amino acids and ornithine showed that only l-arginine functioned as an inhibitor. The IC50 value determined for arginine was 26 ± 3 μM, similar to the value observed for the E. coli enzyme. The enzyme was completely inhibited by 500 μM arginine, and the Hill coefficient was calculated to be 0.60, indicating negatively cooperative binding of l-arginine (Fig. 5).
FIG. 5.

IC50 determination of Rv2747 by l-arginine. l-glutamine and acetyl-CoA were fixed at 10 mM and 1 mM, respectively, and with variable levels (0.01, 0.02, 0.05, 0.1, 0.2, 0.5, 1, 5, 10, 20, 50, 100, 500, 1,000 μM) of l-arginine. The experimental points were fitted to equation 3 to determine the IC50 value.
Product characterization.
In order to confirm the observed kinetic parameters were as a result of product formation and not amino acid-induced acetyl-CoA hydrolysis, the reaction product was purified by high-performance liquid chromatography and characterized by NMR. The purified enzymatic reaction product was shown by 1H NMR to be N-α-acetylglutamine by comparison of the spectral data with those of authentic N-α-acetylglutamine.
DISCUSSION
The arginine biosynthetic pathway begins with an acetylation step, involving acetyl-CoA and l-glutamate (12). In many organisms, except Enterobacteriaceae and S. solfataricus, a second acetylation step can occur where the acetyl group from N-acetylornithine is transferred back to l-glutamate (12). However, for this pathway to function, N-acetylglutamate must first be synthesized in order to synthesize N-acetylornithine (Fig. 1), which can then be used in this “acetyl recycling” pathway. The mechanism by which this occurs is understood in a number of organisms. In B. stearothermophilus, where no argA can be identified in the genome (20), the argJ gene product is bifunctional, possessing both α-N-acetyl-ornithine-l-glutamate acetyltransferase and N-acetylglutamate synthase activity, thus allowing the initial synthesis of N-acetylglutamate (23). However, in organisms such as S. coelicolor and T. thermophilus, the pathway is not fully understood as no readily identifiable ortholog of argA can be identified by sequence analysis in such organisms, thus posing the question of how N-acetylornithine is synthesized.
The M. tuberculosis genome encodes a putative argJ, which is 56% identical to the monofunctional S. coelicolor argJ (17) and only 37% identical to the bifunctional B. stearothermophilus enzyme (23). Although the M. tuberculosis argJ-encoded enzyme has not been functionally characterized, the high degree of amino acid identity between the S. coelicolor and M. tuberculosis enzymes suggests that M. tuberculosis argJ is monofunctional. In organisms with monofunctional ArgJ enzymes, such as S. coelicolor (17) and T. thermophilus (6), orthologues of Rv2747 appear to be present, posing the question of whether Rv2747 or its orthologues catalyze the initial acetylation of glutamate in order to “prime” the pathway with N-acetyl-glutamate for subsequent arginine synthesis. Rv2747 has been shown to be an essential gene by Himar1-based transposon mutagenesis (25), and furthermore orthologues of the enzyme are found in all mycobacterial species, including M. leprae (9), providing further evidence that the gene is essential.
Amino acid sequence alignment studies of Rv2747 with E. coli ArgA indicates that Rv2747 appears to be missing ∼150 amino acids from its N terminus and could be considered an N-terminally truncated form of the E. coli ArgA enzyme. A domain search of the E. coli gene showed some sequence similarity between the N terminus of ArgA and amino acid kinase proteins (ArgB). The N-terminal region of the E. coli protein thus might be involved in regulation or may function in the second biosynthetic reaction, the phosphorylation of the 5-carboxyl group. The C-terminal region of the E. coli ArgA is clearly a GNAT domain that functions as the acetyltransferase.
M. tuberculosis Rv2474 was shown to be able to α-N-acetylate l-glutamine and l-glutamate, although it is unclear why glutamate was a poorer substrate, as logically this should be the physiological substrate for the reaction. However, it is possible that N-acetyl-glutamine could be converted to N-acetyl glutamate either enzymatically by a glutaminase, which can selectively hydrolyze the amide group from N-acetyl-glutamine to yield N-acetyl glutamate, similar to that observed in Bacillus circulans (19), or by a nonenzymatic deamidation (7). The enzyme was strongly inhibited by arginine with an IC50 value of 26 μM, a result arguing for a bona fide physiological role of the enzyme in l-arginine biosynthesis. However, one of the key questions posed by the results of this study was why the Km value for acyl acceptor (l-glutamate) was so high. Typical Km values for l-glutamate for reactions catalyzed by ArgA enzymes range from ∼1 to 10 mM (22), which is 100-fold lower than the observed value for Rv2747. If Rv2747 is functioning as an α-N-acetylglutamate synthase as we propose, the high Km value could be due to a number of reasons. Although as yet functionally uncharacterized, the presence of an argJ homologue in M. tuberculosis might suggest that once even a small amount of α-N-acetyl-l-glutamate was formed, the “acetyl recycling” pathway would provide sufficient biosynthetic flux to provide l-arginine for protein synthesis. Thus, the ArgA enzyme would not need to bind the relatively abundant amino acid glutamate tightly to generate the small amount of N-acetylglutamate that would suffice to “prime” the pathway. Another reason might be that Rv2747 requires a protein partner for efficient and more selective acetylation of l-glutamate. This has been observed for ArgA from S. cerevisiae, where the α-N-acetylglutamate synthase activity requires complex formation with α-N-acetylglutamate kinase (1), and in the case of Synechococcus elongates, where complex formation between the PII signaling protein and α-N-acetyglutamate kinase is required for catalytic activation (11). Therefore, a similar mechanism may have evolved in M. tuberculosis, where a protein partner is required for Rv2747 to function as an efficient catalysis, potentially explaining the high Km value observed. This is currently under investigation.
Acknowledgments
We thank Jordan Kriakov and William R. Jacobs, Jr. (AECOM), for the generous donation of M. tuberculosis H37Rv genomic DNA. We also wish to thank Eddie Nieves for the mass spectroscopy experiments. Finally, the authors wish to thank Argyrides Argyrou for thoughtful discussions.
This work was supported by NIH grant AI33696.
REFERENCES
- 1.Abadjieva, A., K. Pauwels, P. Hilven, and M. Crabeel. 2001. A new yeast metabolon involving at least the two first enzymes of arginine biosynthesis: acetylglutamate synthase activity requires complex formation with acetylglutamate kinase. J. Biol. Chem. 276:42869-42880. [DOI] [PubMed] [Google Scholar]
- 2.Abdelal, A. T., and O. V. Nainan. 1979. Regulation of N-acetylglutamate synthesis in Salmonella typhimurium. J. Bacteriol. 137:1040-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 4.Aral, B., and P. Kamoun. 1997. The proline biosynthesis in living organisms. Amino Acids 13:189-217. [Google Scholar]
- 5.Bachmann, C., S. Krahenbuhl, and J. P. Colombo. 1982. Purification and properties of acetyl-CoA:L-glutamate N-acetyltransferase from human liver. Biochem. J. 205:123-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baetens, M., C. Legrain, A. Boyen, and N. Glansdorff. 1998. Genes and enzymes of the acetyl cycle of arginine biosynthesis in the extreme thermophilic bacterium Thermus thermophilus HB27. Microbiology 144:479-492. [DOI] [PubMed] [Google Scholar]
- 7.Bergana, M. M., J. D. Holton, I. L. Reyzer, M. K. Snowden, J. H. Baxter, and V. L. Pound. 2000. NMR and MS analysis of decomposition compounds produced from N-acetyl-L-glutamine at low pH. J. Agric. Food Chem. 48:6003-6010. [DOI] [PubMed] [Google Scholar]
- 8.Cole, S. T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Eiglmeier, S. Gas, C. E. Barry, 3rd, F. Tekaia, K. Badcock, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, B. G. Barrell, et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537-544. [DOI] [PubMed] [Google Scholar]
- 9.Cole, S. T., K. Eiglmeier, J. Parkhill, K. D. James, N. R. Thomson, P. R. Wheeler, N. Honore, T. Garnier, C. Churcher, D. Harris, K. Mungall, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. M. Davies, K. Devlin, S. Duthoy, T. Feltwell, A. Fraser, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, C. Lacroix, J. Maclean, S. Moule, L. Murphy, K. Oliver, M. A. Quail, M. A. Rajandream, K. M. Rutherford, S. Rutter, K. Seeger, S. Simon, M. Simmonds, J. Skelton, R. Squares, S. Squares, K. Stevens, K. Taylor, S. Whitehead, J. R. Woodward, and B. G. Barrell. 2001. Massive gene decay in the leprosy bacillus. Nature 409:1007-1011. [DOI] [PubMed] [Google Scholar]
- 10.Corpet, F. 1988. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16:10881-10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cowley, S., M. Ko, N. Pick, R. Chow, K. J. Downing, B. G. Gordhan, J. C. Betts, V. Mizrahi, D. A. Smith, R. W. Stokes, and Y. Av-Gay. 2004. The Mycobacterium tuberculosis protein serine/threonine kinase PknG is linked to cellular glutamate/glutamine levels and is important for growth in vivo. Mol. Microbiol. 52:1691-1702. [DOI] [PubMed] [Google Scholar]
- 12.Cunin, R., N. Glansdorff, A. Pierard, and V. Stalon. 1986. Biosynthesis and metabolism of arginine in bacteria. Microbiol. Rev. 50:314-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordhan, B. G., D. A. Smith, H. Alderton, R. A. McAdam, G. J. Bancroft, and V. Mizrahi. 2002. Construction and phenotypic characterization of an auxotrophic mutant of Mycobacterium tuberculosis defective in l-arginine biosynthesis. Infect. Immun. 70:3080-3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haas, D., B. W. Holloway, A. Schambock, and T. Leisinger. 1977. The genetic organization of arginine biosynthesis in Pseudomonas aeruginosa. Mol. Gen. Genet. 154:7-22. [DOI] [PubMed] [Google Scholar]
- 15.Haas, D., V. Kurer, and T. Leisinger. 1972. N-acetylglutamate synthetase of Pseudomonas aeruginosa. An assay in vitro and feedback inhibition by arginine. Eur. J. Biochem. 31:290-295. [DOI] [PubMed] [Google Scholar]
- 16.Harris, B. Z., and M. Singer. 1998. Identification and characterization of the Myxococcus xanthus argE gene. J. Bacteriol. 180:6412-6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hindle, Z., R. Callis, S. Dowden, B. A. Rudd, and S. Baumberg. 1994. Cloning and expression in Escherichia coli of a Streptomyces coelicolor A3(2) argCJB gene cluster. Microbiology 140:311-320. [DOI] [PubMed] [Google Scholar]
- 18.Javid-Majd, F., and J. S. Blanchard. 2000. Mechanistic analysis of the argE-encoded N-acetylornithine deacetylase. Biochemistry 39:1285-1293. [DOI] [PubMed] [Google Scholar]
- 19.Kikuchi, M., H. Hayashida, E. Nakano, and K. Sakaguchi. 1971. Peptidoglutaminase. Enzymes for selective deamidation of gamma-amide of peptide-bound glutamine. Biochemistry 10:1222-1229. [DOI] [PubMed] [Google Scholar]
- 20.Marc, F., P. Weigel, C. Legrain, Y. Almeras, M. Santrot, N. Glansdorff, and V. Sakanyan. 2000. Characterization and kinetic mechanism of mono- and bifunctional ornithine acetyltransferases from thermophilic microorganisms. Eur. J. Biochem. 267:5217-5226. [DOI] [PubMed] [Google Scholar]
- 21.Marvil, D. K., and T. Leisinger. 1977. N-acetylglutamate synthase of Escherichia coli: purification, characterization, and molecular properties. J. Biol. Chem. 252:3295-3303. [PubMed] [Google Scholar]
- 22.Powers-Lee, S. G. 1985. N-acetylglutamate synthase. Methods Enzymol. 113:27-35. [DOI] [PubMed] [Google Scholar]
- 23.Sakanyan, V., D. Charlier, C. Legrain, A. Kochikyan, I. Mett, A. Pierard, and N. Glansdorff. 1993. Primary structure, partial purification and regulation of key enzymes of the acetyl cycle of arginine biosynthesis in Bacillus stearothermophilus: dual function of ornithine acetyltransferase. J. Gen. Microbiol. 139:393-402. [DOI] [PubMed] [Google Scholar]
- 24.Sakanyan, V., P. Petrosyan, M. Lecocq, A. Boyen, C. Legrain, M. Demarez, J. N. Hallet, and N. Glansdorff. 1996. Genes and enzymes of the acetyl cycle of arginine biosynthesis in Corynebacterium glutamicum: enzyme evolution in the early steps of the arginine pathway. Microbiology 142:99-108. [DOI] [PubMed] [Google Scholar]
- 25.Sassetti, C. M., D. H. Boyd, and E. J. Rubin. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48:77-84. [DOI] [PubMed] [Google Scholar]
- 26.Timm, J., I. Van Rompaey, C. Tricot, M. Massaer, F. Haeseleer, A. Fauconnier, V. Stalon, A. Bollen, and P. Jacobs. 1992. Molecular cloning, characterization and purification of ornithine carbamoyltransferase from Mycobacterium bovis BCG. Mol. Gen. Genet. 234:475-480. [DOI] [PubMed] [Google Scholar]
- 27.Udaka, S. 1965. Pathway-specific pattern of control of arginine biosynthesis in bacteria. J. Bacteriol. 91:617-621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van De Casteele, M., M. Demarez, C. Legrain, N. Glansdorff, and A. Pierard. 1990. Pathways of arginine biosynthesis in extreme thermophilic archaeo- and eubacteria. J. Gen. Microbiol. 136:1117-1183.2166770 [Google Scholar]
- 29.Vetting, M. W., L. P. S. de. Carvalho, M. Yu, S. S. Hegde, S. Magnet, S. L. Roderick, and J. S. Blanchard. 2005. Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys. 433:212-226. [DOI] [PubMed] [Google Scholar]
- 30.Wipe, B., and T. Leisinger. 1979. Regulation of activity and synthesis of N-acetylglutamate synthase from Saccharomyces cerevisiae. J. Bacteriol. 140:874-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu, Y., Z. Liang, C. Legrain, H. J. Ruger, and N. Glansdorff. 2000. Evolution of arginine biosynthesis in the bacterial domain: novel gene-enzyme relationships from psychrophilic Moritella strains (Vibrionaceae) and evolutionary significance of N-α-acetyl ornithinase. J. Bacteriol. 182:1609-1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan, Y., S. Harper, D. W. Speicher, and R. Marmorstein. 2002. The catalytic mechanism of the ESA1 histone acetyltransferase involves a self-acetylated intermediate. Nat. Struct. Biol. 9:862-869. [DOI] [PubMed] [Google Scholar]