

Abstract
Background
VPS16 pathogenic variants have been recently associated with inherited dystonia. Most patients affected by dominant VPS16‐related disease display early‐onset isolated dystonia with prominent oromandibular, bulbar, cervical, and upper limb involvement, followed by slowly progressive generalization.
Cases
We describe six newly reported dystonic patients carrying VPS16 mutations displaying unusual phenotypic features in addition to dystonia, such as myoclonus, choreoathetosis, pharyngospasm and freezing of gait. Response to bilateral Globus Pallidus Internus Deep Brain Stimulation (GPi‐DBS) is reported in three of them, associated with significant improvement of dystonia but only minor effect on other hyperkinetic movements. Moreover, five novel pathogenic/likely pathogenic variants are described.
Conclusions
This case collection expands the genetic and clinical spectrum of VPS16‐related disease, prompting movement disorder specialists to suspect mutations of this gene not only in patients with isolated dystonia.
Keywords: choreoathetosis, freezing, GPi‐DBS, HOPSANDs, myoclonus, pharyngeal spasm
Dominant and recessive VPS16 pathogenic variants have been associated with inherited dystonia. 1 , 2 , 3 Initially, a homozygous mutation was found to co‐segregate with juvenile‐onset progressive generalized dystonia in a consanguineous Chinese family. 2 Subsequently, heterozygous VPS16 deleterious variants were identified in patients affected by autosomal dominant dystonia with incomplete penetrance. 1 , 4 , 5 , 6 , 7 , 8 , 9 , 10
Most VPS16 patients display early‐onset isolated dystonia with prominent oromandibular, bulbar, cervical and upper limb involvement, followed by slowly progressive generalization, typically retaining the ability to walk in adulthood 1 (Table S1).
We report six patients carrying heterozygous pathogenic VPS16 variants, five of which are novel. All patients displayed various hyperkinetic features associated with dystonia Table 1.
TABLE 1.
Clinical and genetic features.
| Patient | Age at onset | Localization at onset | Other localizations | Peculiar features | VPS16 variant (heterozygous) |
|---|---|---|---|---|---|
| A.II.1 | 6 | Writer's cramp | Upper limbs, Oromandibular region | Choreoathetosis | c.2181G > A, p.Trp727* |
| B.II.6 | 52 | Blepharospasm | Pharynx, neck | Inspiratory dyspnea with stridor | c.480delT, p.Asp161Thrfs*50 |
| C.II.1 | 14 | Dysphonia | Generalized dystonia | Myoclonus and freezing | c.1939C > T, p.Arg647* |
| D.II.1 | 4 | Oral region | Upper/lower limbs | Dyskinesias | c.1389C > G, p.Tyr463* |
| E | 12 | Oromandibular | Upper/lower limbs | Myoclonus | c.2170_2171delAA, p.Lys724Glufs*44 |
| F | 12 | Oromandibular | Upper limbs, neck | Jerky dystonic tremor | c.2140C > T, p.Gln714* |
Note: Black filling denotes affected individuals.
Case Series
Case A.II.1
Patient A.II.1 came to medical attention at the age of 76 years. He was born full term and had normal psychomotor development. During primary school, he reported difficulty in writing with the right hand, associated with mild involuntary writhing movements of the right upper limb. In the sixth decade these movements worsened, speech became progressively dysarthric and severely disturbed by intrusive repetitive tongue protrusions. In the last few years, the involuntary movements spread to the left upper limb. Neurological examination showed the presence of severe speech‐induced oromandibular dystonia, involuntary choreoathetoid movements affecting the distal upper limbs (right>left), dystonic posture of the left upper limb, repetitive protrusions of the tongue, writer's cramp, and increased blinking frequency (Video 1). Brain MRI was normal.
Video 1.
Patient A.II.1: Involuntary choreiform movements affecting the distal upper limbs (right>left), dystonic posture of the left upper limb, asymmetrical posture of the shoulders, severe speech‐induced oromandibular dystonia with dysarthric speech associated with repetitive involuntary protrusions of the tongue, and increased blinking frequency. Walking was autonomous but slightly wide‐based with no imbalance.
His mother complained of involuntary late‐onset tongue protrusions, one younger sister presented choreodystonic movements, and one younger brother was affected by “slowness of movements,” diagnosed as dystonia‐parkinsonism.
SCA17, DRPLA and Huntington's disease were ruled out. A novel likely pathogenic VPS16 variant was found (NM_022575: c.2181G > A, p.Trp727*) through WES (Data S1). The relatives of the patient denied consent to genetic analysis (Fig. 1).
Figure 1.
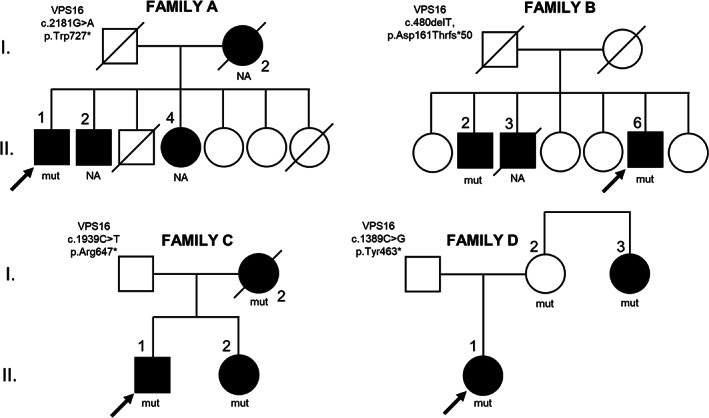
Pedigree of the families (A–D), and mutational status of the available subjects.
Case B.II.6
Patient B.II.6 is a 57‐year‐old man with unremarkable past medical history. He developed progressive blepharospasm at the age of 52, treated with botulinum toxin injections and then with bilateral blepharoplasty with significant benefit. Brain MRI was normal. At the age of 56, dystonia spread to bulbar structures causing dysphonia, dysphagia, and extremely frequent episodes of inspiratory stridor and dyspnea. His speech was often interrupted by respiratory difficulties. Videolaryngoscopy showed constrictive spasms of the oropharynx and the hypopharynx during spontaneous breathing (Video 2). Thoracic and cervical CT scans were normal. Examination at age 57 showed increased blinking frequency, mild cervical dystonia associated with dysphonia and frequent episodes of inspiratory stridor co‐occurring with bilateral blepharospasm.
Video 2.
Patient B.II.6: The patient underwent flexible videolaryngoscopy by a 4 mm endoscope introduced through a nasal fossa. While he was spontaneously breathing dramatic constrictive spasms involving the oropharynx and the hypopharynx were observed, which sub‐completely obstructed the airway and sporadically impeded the vision of the larynx. During endoscopic observation he was asked to answer questions, repeat numbers, and phonate a sustained /i/: his vocal cords were moving normally during these tasks and, while he was phonating a significant reduction of the constricting pharyngeal spasms was seen. At the end of phonation, a severe recurrence of pharyngeal constricting spasms can be seen, causing respiratory distress.
Two patient's siblings were affected by adult‐onset progressive cervical and upper limbs dystonia (B.II.2) and presented cervical dystonia since infancy (B.II.3) (Fig. 1).
WES analysis in B.II.6 and B.II.2 revealed in both a novel heterozygous likely pathogenic VPS16 variant (c.480delT, p.Asp161Thrfs*50). B.II.3 was not available for testing.
Case C.II.1
Patient C.II.1 is a 57‐year‐old man with normal psychomotor development. He presented at the age of 14 years with dysphonia and right upper limb dystonia associated with myoclonic movements. At the age of 18 dystonia spread to the cervical region with superimposed “no‐no” head dystonic tremor. In the following decades dystonia progressively generalized involving axial muscles and lower limbs. Clonazepam, pregabalin and botulinum toxin had only partial benefit. Recurrent episodes of freezing of gait appeared in the fifth decade, with partial response to levodopa. Brain MRI was normal. Cognitive and psychiatric problems were excluded. At age 49, the patient underwent bilateral Globus Pallidus Internus Deep Brain Stimulation (GPi‐DBS) with significant improvement of cervical and upper limbs dystonia but only minor effect on axial muscles and on the jerky dystonic tremor (Video 3).
Video 3.
Patient C.II.1: Segment 1 (before DBS): generalized tremulous dystonia with jerky features affecting more prominently the neck (left torticollis), right upper limb and left lower limb (striatal toe); voice tremor; writer's cramp with writing tremor; segment 2 and 3 (4 and 10 years after pallidal DBS): improvement of cervical dystonia and lower limb dystonia; persistence of dystonic head and upper limb tremor, dystonic posturing of the left upper limb when outstretched and writer's cramp/writing tremor.
The proband's mother (C.I.2) presented at the age of 40 with axial and right upper limb dystonia. In the following years dystonia spread to the cervical region (retrocollis) associated with dysphonia and dysphagia. In the seventh decade, she progressively developed gait impairment, lower limb hypokinesia, postural instability, falls and slowness of hand movements. These signs responded poorly to levodopa. She underwent GPi‐DBS at 68 years of age with improvement of retrocollis. She died at 79 years of age from aspiration pneumonia. The proband's sister (C.II.2) was affected by dysphonia, dystonic dysarthria and writer's cramp with onset during adolescence and progressive spreading with predominant cranio‐cervical and upper limbs involvement. A brief trial with levodopa up to 200 mg proved to be ineffective on these symptoms. She underwent GPi‐DBS with great benefit (Fig. 1).
WES detected a known pathogenic VPS16 variant (c.1939C > T, p.Arg647*) in the proband and affected family members.
Case D.II.1
Patient D.II.1 is a 7‐year‐old girl who was born full term to healthy unrelated parents. Spontaneous vaginal delivery was complicated with suspected birth asphyxia (APGAR score 5/8) but the neonatal and infantile periods were unremarkable except for two febrile seizures at 14 and 22 months. Interictal EEGs were unremarkable. Motor developmental milestones were normally reached, except for a moderate delay in speech development. At the age of 3 years the child manifested mouth twitches that worsened under psychological stress. She also began to present twisting movements of both hands and the right leg, often lasting for only a few seconds. These symptoms were stereotyped and had a non‐progressive course. On examination at age 7, she manifested facial dyskinesias and hand stereotypies (Video 4).
Video 4.
Patient D.II.1: tic‐like movements, hand stereotypies and choreic movements of both hands.
Generalized dystonia was reported in the 38‐year‐old patient's maternal aunt (D.I.3), who developed isolated dystonia at the age of 6 followed by additional dyskinetic movements (Fig. 1). Brain MRI was normal in the proband and aunt.
WES detected a novel VPS16 variant (c.1389C > G, p.Tyr463*) classified as pathogenic. Sanger sequencing identified the same variant in the patient's healthy mother and her affected aunt. The explanation of the healthy status of the mother is probably reduced penetrance, which is a well‐established phenomenon for VPS16‐related dystonia.
Case E
Patient E is 33‐year‐old woman without family history of neurological disorders and normal development. She presented at the age of 12 with dystonic dysarthria, dysphonia, oromandibular and tongue dystonia. She progressively developed dystonia of right upper limb with myoclonic jerks, orthostatic axial jerks, lower limb dystonia with superimposed dystonic tremor, and dystonic movements of the head with jerky features. She displayed some benefit with anticholinergic treatment. Neuropsychological assessment was normal. At the age of 23 she underwent GPi‐DBS with clear improvement of upper limb dystonia, dystonic tremor and dystonic dysarthria and dysphonia but with scarce efficacy on myoclonus (Video 5).
Video 5.
Patient E: Segment 1 (before DBS): severe generalized tremulous dystonia with prominent laryngeal, oromandibular and appendicular involvement (right>left), writer's cramp; segment 2 (5 years after pallidal DBS): Mild improvement of generalized tremulous dystonia and speech. Presence of orthostatic axial jerks.
WES revealed a novel likely pathogenic VPS16 variant c.2170_2171delAA (p.Lys724Glufs*44).
Case F
Patient F is 47‐year‐old man with no family history and normal psychomotor development. Dystonic dysarthria, oromandibular and tongue dystonia appeared at the age of 12. During adolescence, he developed dystonic jerky tremor of upper limbs with writer's cramp. At 20 years of age cervical dystonia and dysphagia became evident. Cervical and lower limbs dystonia progressively worsened. Tetrabenazine, clonazepam, and gabapentin were attempted with only partial benefit on dystonic tremor. No cognitive and behavioral deficits were detected. Bilateral GPi‐DBS at 43 years of age lead to an improvement of dystonic dysarthria and cranio‐cervical dystonia and a slight benefit on dystonic upper limb tremor (Video 6).
Video 6.
Patient F: Segment 1 (before DBS): severe speech‐induced oromandibular dystonia, right upper limb jerky dystonia and writer's cramp; mild right torticollis; segment 2 (4 years after pallidal DBS): persistence of mild right torticollis, right upper limb dystonic tremor; improvement of oromandibular dystonia.
A novel likely pathogenic VPS16 variant (c.2140C > T, p.Gln714*) was found through WES.
Discussion
This case series of VPS16‐related dystonic patients expands the phenomenology associated with this form by also including dyspnea due to pharyngeal spasms, myoclonus, choreic movements, and gait freezing.
VPS16 mutation carriers seem to almost invariably present dystonia as part of their clinical phenotype and evolution. Interestingly, five out of six patients presented with childhood–adolescence onset dystonia. This suggests that VPS16 pathogenic variants may be a relatively frequent cause of early onset dystonia with the cervical region as a common anatomical area affected at disease onset. Isolated cervical, laryngeal and oromandibular dystonia is otherwise uncommon in early ages of life and is mostly linked to KMT2B and THAP1 mutations or it can be the manifesting symptom of some metabolic diseases and NBIA disorders. Conversely, oromandibular involvement at the disease onset appears to be rare in the newly described AOPEP‐related dystonia. Similar to other genetic types of early‐onset dystonia, spreading of this movement disorder occurred in most patients, leading to generalization in some. Bulbar signs developed in four patients including dysphagia. This may be a red flag for VPS16 mutations, in that bulbar muscles are generally spared even in the most severe cases of other genetically determined childhood‐onset dystonia, with oral feeding being generally preserved during disease course.
In our series, dystonic tremor sometimes of a jerky appearance and myoclonus were observed over disease course in most patients as the most disabling feature, reminding of ANO3‐patients. In line with our observations, two patients carrying VPS16 mutations were previously reported to display myoclonus. 9 , 10 These peculiar phenomenologies are unlikely determined solely by the type of pathogenic variant in VPS16, since isolated dystonic phenotypes were observed in some affected relatives of these patients.
The favorable response to GPi‐DBS in four patients support a possible beneficial role of this approach in treating VPS16‐related dystonia, as observed in other cases. 1 , 8 Notably, in the cases reported here, GPi‐DBS was more effective in treating dystonic features than other hyperkinetic manifestations such as myoclonus and jerky tremor.
In conclusion, we widen the spectrum of movement disorders in VPS16‐related disease and report five novel likely pathogenic variants. The phenotypic heterogeneity appeared to be significant, even within single families. Large cohorts will help in further defining the phenotypic spectrum and prognosis, including actual improvement after DBS, of VPS16‐related disorders.
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution. (2) Manuscript Preparation: A. Writing of the first draft, B. Review and Critique.
E.M.: 1A, 1B, 1C, 2A
L.A.: 1C, 2B
G.P.: 1C, 2B
G.B.o.: 1B, 2B
G.B.r: 1C
R.C.: 1C, 2B
G.C.: 1C, 2B
P.M.: 1B, 2B
H.P.: 2B
K.V.G.: 1C, 2A
G.S.: 1C, 2B
A.E.: 1C, 2B
C.R.: 1C, 2B
C.P.: 1C, 2B
G.Z.: 1C, 2B
R.E.l.: 2B
R.E.r.: 2A, 2B
M.C.: 2A, 2B
B.G.: 2B
M.Z.: 1C, 2B
L.R.: 1B, 1C, 2B
A.D.F.: 1A, 1B, 2A, 2B.
Disclosures
Ethical Compliance Statement: Written informed consent for publication of clinical details and video recording were obtained from all involved subjects. The Ethics Committee of the IRCCS Foundation Ca′ Granda Ospedale Maggiore Policlinico (Milan, Italy) approved the study.
Funding Sources and Conflict of Interest: This study was partially funded by the Italian Ministry of Health (Ricerca Corrente). ADF and MZ acknowledge grant support by the EJP RD (EJP RD Joint Transnational Call 2022) and “Fondazione Regionale per la Ricerca Biomedica” awarded to the project PreDYT (PREdictive biomarkers in DYsTonia, grant agreement number: 825575). The authors declare that there are no conflicts of interest relevant to this work.
Financial Disclosures for the Previous 12 Months: MZ receives research support from the German Research Foundation (DFG 458949627; ZE 1213/2–1). MZ acknowledges grant support by the German Federal Ministry of Education and Research (BMBF, Bonn, Germany), awarded to the project PreDYT (01GM2302), by the Federal Ministry of Education and Research (BMBF) and the Free State of Bavaria under the Excellence Strategy of the Federal Government and the Länder, as well as by the Technical University of Munich—Institute for Advanced Study. HP acknowledges grant support from the German Federal Ministry of Education and Research (BMBF, Bonn, Germany) awarded to the German Network for Mitochondrial Disorders (mitoNET, 01GM1906A). RE receives royalties from Springer and has received honoraria for speaking from the International Parkinson's Disease and Movement Disorders Society.
Supporting information
Data S1. Genetic methods and American College of Medical Genetics and Genomics (ACMG) classification of variants.
Table S1. Clinical and genetic features of all the patients carrying pathogenic VPS16 variants reported so far.
References
- 1. Steel D, Zech M, Zhao C, et al. Loss‐of‐function variants in HOPS complex genes VPS16 and VPS41 cause early onset dystonia associated with lysosomal abnormalities. Ann Neurol 2020;88:867–877. [DOI] [PubMed] [Google Scholar]
- 2. Cai X, Chen X, Wu S, et al. Homozygous mutation of VPS16 gene is responsible for an autosomal recessive adolescent‐onset primary dystonia. Sci Rep 2016;6:25834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Monfrini E, Zech M, Steel D, Kurian MA, Winkelmann J, di Fonzo A. HOPS‐associated neurological disorders (HOPSANDs): linking endolysosomal dysfunction to the pathogenesis of dystonia. Brain a journal of neurology 2021;144:2610–2615. [DOI] [PubMed] [Google Scholar]
- 4. Pott H, Brüggemann N, Reese R, et al. Truncating VPS16 mutations are rare in early onset dystonia. Ann Neurol 2021;89:625–626. [DOI] [PubMed] [Google Scholar]
- 5. Ostrozovicova M, Jech R, Steel D, et al. A recurrent VPS16 p.Arg187* nonsense variant in early‐onset generalized dystonia. Movement disorders official journal of the Movement Disorder Society 2021;36:1984–1985. [DOI] [PubMed] [Google Scholar]
- 6. Li X‐Y, Wang L, Guo Y, Wan XH. Mutations in the VPS16 gene in 56 early‐onset dystonia patients. Mov Disord 2021;36:780–781. [DOI] [PubMed] [Google Scholar]
- 7. Li L‐X, Jiang L‐T, Liu Y, et al. Mutation screening of VPS16 gene in patients with isolated dystonia. Parkinsonism Relat Disord 2021;83:63–65. [DOI] [PubMed] [Google Scholar]
- 8. Petry‐Schmelzer JN, Park J, Haack TB, Visser‐Vandewalle V, Barbe MT, Wunderlich G. Long‐term benefit of pallidal deep brain stimulation in a patient with VPS16‐associated dystonia. Neurological research and practice 2022;4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gu X, Lin J, Hou Y, Zhang L, Shang H. De novo missense mutation of VPS16 in a Chinese patient with generalized dystonia with myoclonus. Movement disorders clinical practice 2022;9:551–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Park J, Reilaender A, Petry‐Schmelzer JN, et al. Transcript‐specific loss‐of‐function variants in VPS16 are enriched in patients with dystonia. Neurology Genetics 2022;8:e644. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Genetic methods and American College of Medical Genetics and Genomics (ACMG) classification of variants.
Table S1. Clinical and genetic features of all the patients carrying pathogenic VPS16 variants reported so far.