

Abstract
Optically active functional noncentrosymmetric architectures might be achieved through the combination of molecules with inscribed optical responses and species of dedicated tectonic character. Herein, we present the new series of noncentrosymmetric cocrystal salt solvates (PPh4)3[M(CN)6](L)n·msolv (M = Cr(III), Fe(III), Co(III); L = polyresorcinol coformers, multiple hydrogen bond donors: 3,3′,5,5′-tetrahydroxy-1,19-biphenyl, DiR, n = 2, or 5′-(3,5-dihydroxyphenyl)-3,3″,5,5″-tetrahydroxy-1,19:3′,1″-terphenyl, TriRB, n = 1) denoted as MDiR and MTriRB, respectively. The hydrogen-bonded subnetworks {[M(CN)6]3–;Ln}∞ of dmp, neb, or dia topology are formed through structural matching between building blocks within supramolecular cis-bis(chelate)-like {[M(CN)6]3–;(H2L)2(HL)2} or tris(chelate)-like {[M(CN)6]3–;(H2L)3} fragments. The quantum-chemical analysis demonstrates the mixed electrostatic and covalent character of these interactions, with their strength clearly enhanced due to the negative charge of the hydrogen bond acceptor metal complex. The corresponding interaction energy is also dependent on the geometry of the contact and size matching of its components, rotational degree of freedom and extent of the π-electron system of the coformer, and overall fit to the molecular surroundings. Symmetry of the crystal lattices is correlated with the local symmetry of coformers and {complex;(coformer)n} hydrogen-bonded motifs characterized by the absence of the inversion center and mirror plane. All compounds reveal second-harmonic generation activity and photoluminescence diversified by individual UV–vis spectral characteristics of the components, and interesting low-frequency Raman scattering spectra within the subterahertz spectroscopic domain. Vibrational (infrared/Raman), UV–vis electronic absorption (experimental and calculated), and 57Fe Mössbauer spectra together with electrospray ionization mass spectrometry (ESI-MS) data are provided for the complete description of our systems.
Short abstract
The series of non-centrosymmetric cocrystal salt solvates reveal hydrogen-bonded architectures {[M(CN)6]3–;Ln}∞ involving supramolecular cis-bis(chelate) like {[M(CN)6]3–;(H2L)2(HL)2} or tris(chelate) like {[M(CN)6]3–;(H2L)3} motifs, L standing for linear bis-resorcinol DiR or triangular tris-resorcinol TriRB multiple hydrogen bond donors. All networks show second harmonic generation (SHG) and photoluminescence responses conditioned by the individual optical UV-Vis absorption cut-off thresholds.
Introduction
Optical properties of matter based on nonlinear response and/or photoluminescence have attracted strong interest in fields such as bioimaging,1−5 anticancer therapy,6 or multifunctional molecular materials for photonic application, e.g. toward optical modulation of light7 and for luminescent thermometry.8,9 The phenomenon known as second-harmonic generation (SHG) has been extensively researched and utilized due to its ability to convert two incident photons into a single emitted photon with a doubled frequency. This process relies on the interaction between matter and the incident beam, specifically, its polarization and orientation. The advantage of SHG lies in its narrow operating band, which allows for the precise control and manipulation of the generated signal.10,11 The SHG function in the solid state requires a noncentrosymmetric space group and proper light absorption cutoff, which stimulated extended studies on the relevant inorganic,12,13 organic,14−17 as well as hybrid inorganic–organic18−24 phases. In particular, the two latter composition strategies have recently been employed within the cocrystallization approach25,26 considering the tunable intrinsic features of organic counterparts, e.g. hyperpolarizability, and tectonic character that allows for the desired supramolecular organization through noncovalent interactions.14,16,17,27 For example, hydrogen-bonded synthons are exploited in directional and cooperative extension toward supramolecular architectures,27−31 especially if one combines multisite complementary H-bond donor and acceptor or/and slightly modifies the already established contacts via judicious molecular replacement.14,29 The effective self-assembly process is usually supported by other ubiquitous weak interactions (π···π, ion···π, C–H···π, etc.). The potential of such a strategy was illustrated by the acquisition of materials that fulfill the technological demands for SHG performance competitive with the traditional ones (see above) as well as by efficient enantiospecific molecular recognition toward enantiopure crystal growth.15,32−35 Moreover, a hybrid approach involving 3d and 4f metal ion coordination complexes and their salts allows for the introduction of other functions, such as anion binding,4,23 specific luminescence for ratiometric or thermometric performance,8,9 single-ion magnetic properties for ultrahigh density data recording, storage, and processing,8,9 or switchable structural and dielectric loss properties owing to the phase transitions inscribed in the structure of molecular fragments and their positioning in the crystal, as commonly observed in molecular hybrid perovskites.36−39 In parallel, phonon properties of molecular materials have been studied by terahertz (THz) spectroscopy9,40−42 in the context of the optimization of their luminescent response (“management” of nonradiative vibrational loss)43,44 and slow magnetic relaxation (“management” of the effective energy barrier)45 or the generation of the THz signal through optical stimulation.41
In our research, we focus on the development of new noncovalent synthons for the construction of molecular architectures offering tunable optical properties. Over the past few years, we have been exploring the realm of cocrystal salt solvates that involve polycyanidometallate [M(CN)x]n– and organic coformers with the objective to design and generate modular patterns of noncovalent interactions within these structures. In this context, a family of structurally related charge-transfer (CT) systems comprising 1,4,5,8,9,12-hexaazatriphenylene-hexacarbonitrile (HAT(CN)6) and tetracyanopyrazine (TCP) π-acids was investigated both in the solid state and solution. These assemblies exhibited anion-π interactions, the nature and strength of which were influenced by the size and shape of the contact components.46−50 Notably, our studies introduced the first instances of binary core–shell crystals based on anion-π interactions.50 Then, following the works of Desiraju and Paul on multicomponent topological CT-colored resorcinol (1,3-dihydroxybenzene) based systems29 and the pioneering works of Oshio and co-workers on phloroglucinol-conditioned (H3PG, 1,3,5-trihydroxybenzene) spin-crossover cyanido-bridged square Co2Fe2 complexes,51 we explored further possible schemes of noncovalent interactions between multiple hydrogen bond donor H3PG (Scheme 1) and mononuclear52 and polynuclear53 cyanido-complexes. As a result, we have discovered the unique supramolecular cis-bis(chelate) {[M(CN)6]3–;(H3PG)4} (M = Cr(III), Fe(III), Co(III)) motifs within the MH3PG architectures involving the following: (i) two double cyclic hydrogen bond synthons M(−CN···HO−)2Ar, {[M(CN)6]3–;H2PGH}, between cis-oriented cyanido ligands of [M(CN)6]3– and the resorcinol-like face of H3PG, and (ii) two single hydrogen bonds M-CN···HO-Ar, {[M(CN)6]3–;HPGH2}, involving the remaining two cyanide ligands (Figure 1).52 Spectroscopic and computational descriptions revealed notable strength of the underlying interactions. While the local symmetry of the {[M(CN)6]3–;(H3PG)4} motif might be approached with the chiral C2 point group, interesting from the standpoint of the noncentrosymmetric and enantiopure resolution, regrettably, the centrosymmetric C2/c space group was observed for these crystals. Thus, we have expanded the boundaries for the synthesis of noncentrosymmetric architectures by introducing cocrystal salt solvates of (PPh4)3[M(CN)6](L)n·msolv (M = Cr(III), Fe(III), Co(III); L = coformers (Scheme 1): 3,3′,5,5′-tetrahydroxy-1,19-biphenyl, DiR,54n = 2, or 5′-(3,5-dihydroxyphenyl)-3,3″,5,5″-tetrahydroxy-1,19:3′,1″-terphenyl, TriRB,54n = 1) hereafter denoted as CrDiR, FeDiR, CoDiR, CrTriRB, FeTriRB, and CoTriRB, with the lattice symmetry dictated by the effective symmetry of coformers and the {complex;coformer} arrangement guided by structurally matched hydrogen bond synthons. The presented systems were thoroughly characterized via both experimental and computational (density functional theory, DFT) studies. These compounds are SHG active according to the nonzero second-order nonlinear optical (NLO) susceptibility tensor (χijk). Additionally, they exhibit diverse photoluminescence properties influenced by the distinct UV–vis spectral characteristics of the individual components. Finally, they display intriguing low-frequency (LF) Raman scattering spectra within the sub-THz spectroscopic range, which provides further insights into their structural and vibrational properties.
Scheme 1. Molecular Structure of Hydrogen Bond Donor Coformers.

DiR and TriRB were not exploited previously in the syntheses of cocrystals.
Figure 1.
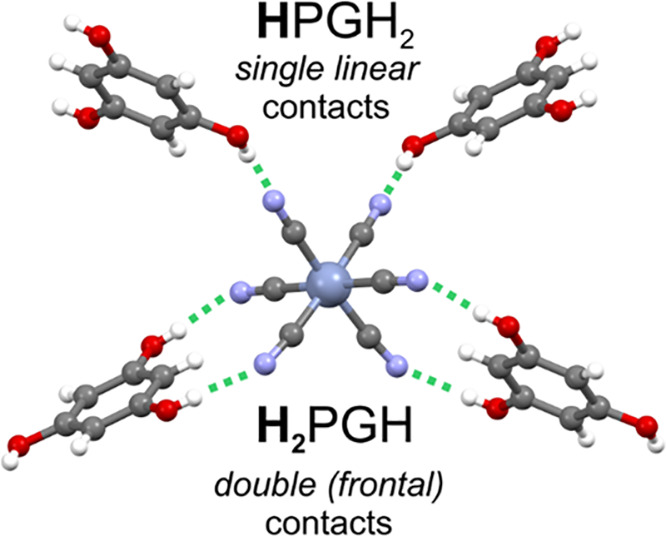
Supramolecular cis-bis(chelate) hydrogen-bonded motifs in MH3PG(52) comprising two double cyclic ring-type synthons {[M(CN)6]3–;H2PGH}, each formed between the cis-oriented cyanido ligands of [M(CN)6]3– and resorcinol-like face of H3PG, and two single linear synthons {[M(CN)6]3–;HPGH2}. The above cyclic synthons represent the frontal mode of hydrogen bond donation; for the depiction of all cyclic H-bond synthons appearing in the whole family, see Figures 2 and 3.
Results and Discussion
Structural Studies
All MDiR compounds, comprising the coformer DiR, crystallize in the orthorhombic system with a noncentrosymmetric space group of Pna21. This results in the formation of a series of isomorphous analogs. On the other hand, the MTriRB networks, involving the coformer TriRB, display noticeable diversity as far as the space groups are concerned. Specifically, CrTriRB and CoTriRB crystallize in the monoclinic system, with CrTriRB having a noncentrosymmetric Cc space group and CoTriRB having a P21 Sohncke space group. In contrast, FeTriRB crystallizes in the orthorhombic system, with a noncentrosymmetric P21212 Sohncke space group. Nevertheless, the local structural arrangement is similar across the whole MTriRB series. For detailed crystal data and structure refinement parameters, see the Supporting Information (SI) and Tables S1 and S2. The uniformity of the powder samples and the identity of the crystals examined with single-crystal X-ray diffraction (SC XRD) experiments were confirmed by room-temperature (RT) powder X-ray diffraction (PXRD) measurements (Figure S1). The symmetrically independent parts are presented in Figures S2 and S3 in the SI. All crystal structures consist of PPh4+ cations, [M(CN)6]3– anions, polyresorcinol DiR or TriRB coformer molecules (denoted also as L), and crystallization solvent MeCN and/or MeOH molecules.
Detailed information on the most important bond lengths and angles is presented in Tables S3 and S4 in the SI. As shown in Figures 2 and 3, MDiR and MTriRB uniformly feature the 3D hydrogen-bonded {[M(CN)6]3–;Ln}∞subnetworks exploiting the LO–H···NM–C≡N contacts, which coexist with the multiple phenyl embraces (MPE) based subnetwork composed of PPh4+ cations assisted by solvent molecules (Figure S4). The observed N···O and N···H distances and O–H···N angles allow classification of them as medium-strength hydrogen bonding interactions (Table 1, Tables S5, S6, and S7).55 The lone pairs of O atoms additionally contribute to the stabilization of the network acting as the hydrogen bond acceptors, mainly from C–H groups (MDiR and MTriRB) and from some of the O–H groups (MDiR), Tables S8 and S9.
Figure 2.

Crystal structure of MDiR: (a)
and (b) two projections
of the supramolecular hydrogen-bonded mixed cis-bis(chelate)/tris(chelate)
{[M(CN)6]3–;(HAHBDiR)2(HA2DiR)(HADiR)} fragment
highlighting the side double cyclic 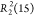 synthon and composed contact
including one single linear D and two frontal
double
synthon and composed contact
including one single linear D and two frontal
double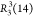 and
and 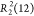 synthons (for metric parameters,
see Table S5) together with the pictorial
illustration
of the canonical cis-chelated [ML3] coordination
complex; (c) the side mode of coformer DiR with two
neighboring [M(CN)6]3– anions; (d) the frontal mode of coformer DiR with two neighboring [M(CN)6]3– anions; (e) projection of the hydrogen-bonded
{[M(CN)6]3–;(HAHBDiR)2 (H2DiR)(HDiR)} layer along the [010] crystallographic direction. Legend:
gray-blue – Cr, Fe, or Co; gray – C; blue – N;
red – O; white – H; PPh4+ cations
in (e) – purple. MeCN and MeOH solvent molecules were omitted
for clarity.
synthons (for metric parameters,
see Table S5) together with the pictorial
illustration
of the canonical cis-chelated [ML3] coordination
complex; (c) the side mode of coformer DiR with two
neighboring [M(CN)6]3– anions; (d) the frontal mode of coformer DiR with two neighboring [M(CN)6]3– anions; (e) projection of the hydrogen-bonded
{[M(CN)6]3–;(HAHBDiR)2 (H2DiR)(HDiR)} layer along the [010] crystallographic direction. Legend:
gray-blue – Cr, Fe, or Co; gray – C; blue – N;
red – O; white – H; PPh4+ cations
in (e) – purple. MeCN and MeOH solvent molecules were omitted
for clarity.
Figure 3.

Crystal structure of MTriRB: (a)
and (b) two projections
of the supramolecular hydrogen-bonded cis-bis(chelate)
{[M(CN)6]3–;(HBHCTriRB)2(HTiRB)2} fragment highlighting
the side double cyclic 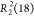 and single linear D synthons
(for metric parameters, see Tables S6 and S7) together with the pictorial illustration of the canonical cis-chelated [ML2A2] coordination
complex; (c) the coformer TriRB with four neighboring [M(CN)6]3– anions; (d) projection of the hydrogen-bonded
{[M(CN)6]3–;(HBHCTriRB)2(HTriRB)2} layer along the [010]
crystallographic direction. Legend: gray-blue – Cr, Fe, or
Co; gray – C; blue – N; red – O; white –
H; PPh4+ cations in (d) – purple. MeCN
and MeOH solvent molecules were omitted for clarity.
and single linear D synthons
(for metric parameters, see Tables S6 and S7) together with the pictorial illustration of the canonical cis-chelated [ML2A2] coordination
complex; (c) the coformer TriRB with four neighboring [M(CN)6]3– anions; (d) projection of the hydrogen-bonded
{[M(CN)6]3–;(HBHCTriRB)2(HTriRB)2} layer along the [010]
crystallographic direction. Legend: gray-blue – Cr, Fe, or
Co; gray – C; blue – N; red – O; white –
H; PPh4+ cations in (d) – purple. MeCN
and MeOH solvent molecules were omitted for clarity.
Table 1. Average Distances and Angles for Hydrogen Bond Contacts in the {[M(CN)6]3–;Ln} Synthons of MDiR and MTriRBa,b.
| Contact | D···A/Å | H···A/Å | D–H···A/° | D···A/Å | H···A/Å | D–H···A/° | D···A/Å | H···A/Å | D–H···A/° |
|---|---|---|---|---|---|---|---|---|---|
| CrDiR | FeDiR | CoDiR | |||||||
| Side | 2.72 | 1.89 | 169.1 | 2.75 | 1.93 | 166.1 | 2.75 | 1.93 | 170.3 |
| Frontal | 2.82 | 1.99 | 172.0 | 2.79 | 1.95 | 175.7 | 2.81 | 1.97 | 175.4 |
| CrTriRB | FeTriRB | CoTriRB | |||||||
| Side | 2.77 | 1.95 | 169.2 | 2.80 | 1.96 | 174.9 | 2.70 | 1.85 | 173.5 |
| Single linear | 2.69 | 1.87 | 166.2 | 2.71 | 1.87 | 174.4 | 2.72 | 1.89 | 169.0 |
D – hydrogen bond donor L = DiR or TriRB, A – hydrogen bond acceptor [M(C)N6]3–.
Compare with Figures 2 and 3 and Tables S5, S6, and S7 in the SI.
Within the {[M(CN)6]3–;DiR2}∞ subnetwork of MDiR, each
[M(CN)6]3– anion is surrounded by four
DiR molecules through seven hydrogen bond contacts (Figure 2a,b). Among them, two DiR molecules
are involved in the formation of double cyclic {[M(CN)6]3–;(HAHBDiR)} synthons of the 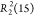 pattern56 to
establish the {[M(CN)6]3–;(HAHBDiR)2} fragments (Figure 2a,b; see the motifs outside the dashed gray
frames). Such contacts engage two O–H groups of different rings
of one DiR molecule and two cis-oriented cyanido
ligands, hereafter referred to as side synthons.
Two other DiR molecules form a composed synthon {[M(CN)6]3–;(HA2DiR)(HADiR)} through engaging the remaining pair of cis-oriented cyanido-ligands and one of the cyanido ligands involved
in the {[M(CN)6]3–;(HAHBDiR)}
interaction. Within this motif, both DiR molecules exploit two groups
of the same ring to realize double frontal proton
donation. Two cyclic synthons coexist in this fragment, featuring
the frontal
pattern56 to
establish the {[M(CN)6]3–;(HAHBDiR)2} fragments (Figure 2a,b; see the motifs outside the dashed gray
frames). Such contacts engage two O–H groups of different rings
of one DiR molecule and two cis-oriented cyanido
ligands, hereafter referred to as side synthons.
Two other DiR molecules form a composed synthon {[M(CN)6]3–;(HA2DiR)(HADiR)} through engaging the remaining pair of cis-oriented cyanido-ligands and one of the cyanido ligands involved
in the {[M(CN)6]3–;(HAHBDiR)}
interaction. Within this motif, both DiR molecules exploit two groups
of the same ring to realize double frontal proton
donation. Two cyclic synthons coexist in this fragment, featuring
the frontal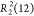 {[M(CN)6]3–;HA2DiR} pattern and frontal
{[M(CN)6]3–;HA2DiR} pattern and frontal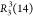 pattern, with the latter one involving
an additional O–H···O hydrogen bond between
two DiR molecules that accompanies the simple linear D pattern {[M(CN)6]3–;HADiR} (Figure 2a,b; see the motifs within the dashed gray frames).
Such an arrangement is the signature of strong competition between
the resorcine-like fragments of different DiR molecules in the formation
of the frontal cyclic
pattern, with the latter one involving
an additional O–H···O hydrogen bond between
two DiR molecules that accompanies the simple linear D pattern {[M(CN)6]3–;HADiR} (Figure 2a,b; see the motifs within the dashed gray frames).
Such an arrangement is the signature of strong competition between
the resorcine-like fragments of different DiR molecules in the formation
of the frontal cyclic 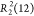 synthons with [M(CN)6]3–. Accordingly, two types of DiR linkers were
found to interconnect
M(CN)6]3– anions, exploiting exclusively
either the side mode or the frontal mode of hydrogen bond donation (Figure 2c,d). The resulting five-component {[M(CN)6]3–;(HAHBDiR)2(HA2DiR)(HADiR)} fragment provides a unique
low-symmetry supramolecular aggregate representing a hybrid of noncovalently
bonded tris(chelate) and cis-bis(chelate) motifs,
by analogy to the canonical six-coordinative [ML12L2] and cis-[ML12A2] complexes, respectively.
synthons with [M(CN)6]3–. Accordingly, two types of DiR linkers were
found to interconnect
M(CN)6]3– anions, exploiting exclusively
either the side mode or the frontal mode of hydrogen bond donation (Figure 2c,d). The resulting five-component {[M(CN)6]3–;(HAHBDiR)2(HA2DiR)(HADiR)} fragment provides a unique
low-symmetry supramolecular aggregate representing a hybrid of noncovalently
bonded tris(chelate) and cis-bis(chelate) motifs,
by analogy to the canonical six-coordinative [ML12L2] and cis-[ML12A2] complexes, respectively.
Within the {[M(CN)6]3–;TriRB}∞ subnetworks
of MTriRB, each [M(CN)6]3– anion is surrounded by four TriRB molecules
through six hydrogen bond contacts (Figure 3a,b). Two TriRB molecules are involved in
the formation of double cyclic ring-type {[M(CN)6]3–;(HBHCTriRB)} synthons of the 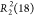 pattern to give the {[M(CN)6]3–;(HBHCTriRB)2}
fragment
(Figure 3a,b). These
synthons exploit exclusively the side mode of hydrogen
bonding interactions engaging two O–H groups of different rings
of one TriRB molecule and two pairs of cis-oriented
cyanido ligands. Two other TriRB molecules are connected with the
remaining pair of cis-oriented cyanido groups via
the single contacts of the linear D pattern, to produce
two {[M(CN)6]3–;(HTriRB)} synthons. Accordingly, each TriRB linker interconnects four
[M(CN)6]3– anions, two of them through
the double cyclic ring-type
pattern to give the {[M(CN)6]3–;(HBHCTriRB)2}
fragment
(Figure 3a,b). These
synthons exploit exclusively the side mode of hydrogen
bonding interactions engaging two O–H groups of different rings
of one TriRB molecule and two pairs of cis-oriented
cyanido ligands. Two other TriRB molecules are connected with the
remaining pair of cis-oriented cyanido groups via
the single contacts of the linear D pattern, to produce
two {[M(CN)6]3–;(HTriRB)} synthons. Accordingly, each TriRB linker interconnects four
[M(CN)6]3– anions, two of them through
the double cyclic ring-type 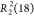 synthons and two via the single
linear D synthons, acting effectively as the 4-fold
node (Figure 3c,d).
The resulting
five-component {[M(CN)6]3–;(HBHCTriRB)2(HTriRB)2}
fragment realizes a unique low-symmetry supramolecular bis(chelate)
aggregate, by analogy to the six-coordinative [ML12A2] complexes.
synthons and two via the single
linear D synthons, acting effectively as the 4-fold
node (Figure 3c,d).
The resulting
five-component {[M(CN)6]3–;(HBHCTriRB)2(HTriRB)2}
fragment realizes a unique low-symmetry supramolecular bis(chelate)
aggregate, by analogy to the six-coordinative [ML12A2] complexes.
A strong preference for the formation of the presented above hydrogen-bonded synthons in the gas phase was confirmed by the ESI-MS spectra exhibiting the representative progressive peak-sets attributable to the {(PPh4)2[Fe(CN)6]}−, {(PPh4)2[Fe(CN)6]L}−, and {(PPh4)2[Fe(CN)6]L2}− aggregates in the negative ionization mode and to the {(PPh4)4[Fe(CN)6]}+ and {(PPh4)4[Fe(CN)6]L}+ aggregates in the positive ionization mode (Figures S5 and S6), which was properly reproduced by EnviPat software.57 The relative stability of the selected aggregates (in terms of DFT-computed interaction energy values) is discussed in the following.
The presented hydrogen-bonded subnetworks were achieved thanks to structural matching between the pairs of cis-oriented cyanido ligands in the metal complex acting as Brønsted bases and various pairs of O–H groups in polyresorcines, supported by an appropriate adaptive twist of their rings due to the natural degree of intramolecular rotation freedom. The occurrence of noncentrosymmetric or even enantiopure architectures might be related to the local symmetry of supramolecular bis(chelate) or tris(chelate) motifs and the local symmetry of coformers, all lacking both the inversion center and the mirror plane. Considering the intramolecular twist expressed by the interplanar angles between the rings A and B of DiR and between the central A ring and external B, C, and D rings of TriRB (Table S10 and Figures S7 and S8), the D2 point group might be assigned for DiR, whereas the C2 or C1 point group might be accessible in the case of TriRB (disregarding the positions of the phenolic protons and the resulting orientations of the O–H bonds with respect to the ring). It is important to note that the native crystals of DiR·2H2O and TriRB·2Me2CO·H2O grow in the centrosymmetric space groups P-1 and P21/c, respectively; for the exact conformations within these crystals, see Table S10.54 One should also consider the role of the acentric PPh4+ cation. Screening of CSD database showed that out of 4523 crystal structures containing XPh4+ cations (with Rint not exceeding 10), 543 (12%) structures were noncentrosymmetric, the percentage being 2-fold or even 3-fold smaller compared to other structures involving the selected tetraalkylammonium or trialkylmethylammonium cations (see the SI). Such statistical results indicate that XPh4+ cations cannot provide a simple key to achieve a noncentrosymmetric solution. It is also plausible that a strong tendency of PPh4+ to form MPE interactions might impose additional limiting conditions on the local symmetry and space group that might be achieved. Interestingly, a considerable number of 251 structures containing XPh4+ (46.2% of the noncentrosymmetric structures found) belong to Sohncke space groups, whereas 193 structures crystallize in the space groups P21 (87, 16.0% of 543), Cc (55, 10.2%) Pna21 (39, 7.18%), and P21212 (12, 2.21%) achieved in our studies (for details see the SI). This suggests that the results obtained within our series tend somehow to follow the trends of noncentrosymmetric space groups observed in the database within the regime of the search. To conclude, the occurrence of noncentrosymmetric space groups in our series is the result of the concentration of low-symmetry species lacking inversion centers or improper axes, with a possible decisive role of coformers and motifs they form with [M(CN)6]3– complexes. We believe that exploration of the crystal structures and properties of similar cocrystal salts involving other organic cations (both having and lacking inversion centers) might shed more light on the above complex problem.
The hydrogen-bonded subnetworks were then described by topological analysis using TOPOS Pro software.58 The underlying building blocks were simplified to single points in space: [M(CN)6]3– complexes were represented by metal ion sites, DiR molecules were represented by the centroids of the C–C bond between the rings, whereas TriRB molecules were represented by the centroids of their central ring (compare Figures 2 and 3). In line with the previous description, [M(CN)6]3– and TriRB were treated as 4-connected nodes, and DiR was consistently considered the linker, disregarding the qualitative differences of particular intermolecular hydrogen-bonded connections. The hydrogen-bonded MDiR architecture revealed the dmp topology (Figure S9), whereas among the hydrogen-bonded MTriRB architectures two separate topologies are distinguished: CrTriRB (Cc space group) revealed rather the rare neb topology, while FeTriRB (P21212 space group) and CoTriRB (P21 space group) showed the frequently encountered dia topology (Figure S10). Consistently, CrTriRB includes 66 topological motifs, which differ from the 64 motifs noted for FeTriRB and CoTriRB (Figure S10c).59
DFT Study on Hydrogen Bonding Interactions
The extended transition state-natural orbitals for chemical valence (ETS-NOCV)60 charge and bonding-energy decomposition analyses performed for the closed-shell motifs [Co(CN)6]3–/DiR and [Co(CN)6]3–/TriRB (see the SI for a description of the computational details and additional comments, and also Tables S12 and S14, Figures S14–S17 and S19–S20 for the calculated results) indicate that, similarly to what was previously found for MH3PG, hydrogen bonding {[M(CN)6]3–;DiR} and {[M(CN)6]3–;TriRB} interactions in MDiR and MTriRB are dominated by the electrostatic and orbital energy components, both enhanced by the negative charge of the hydrogen bond acceptor [M(CN)6]3–. In particular, the latter contribution stems not only from the σ-CT interaction between the occupied lone pair of nitrogen and the unoccupied σ* orbital of the O–H bond but also from the strong polarization (intra-CT) of the π-electron system within the hydrogen bond donor DiR or TriRB molecules, facilitated by the ion-dipole interaction.
Table 2 shows the DFT-calculated (B3LYP+D4//TZP)
interaction energy values between the [M(CN)6]3– and DiR, TriRB, and H3PG52 (as reference) building blocks in the molecular clusters extracted
from the respective crystal structures (see also Tables S11 and S13). The interaction energies for side double synthons in MDiR (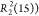 and MTriRB (
and MTriRB (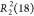 ) are between −50 and −54.5
kcal mol–1 and between −54.5 and −60
kcal mol–1, respectively. Notably smaller stabilization
is observed for the frontal double interactions of
the
) are between −50 and −54.5
kcal mol–1 and between −54.5 and −60
kcal mol–1, respectively. Notably smaller stabilization
is observed for the frontal double interactions of
the 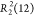 pattern for MDiR and MH3PG, represented
by
the interaction energy values ranging from ca. −46
to −49 kcal mol–1 and from −44.5 to
−48 kcal mol–1, respectively. A distinct
trend in the absolute values of these energies can be thus established: MTriRB (side double – the strongest
interaction) > MDiR (side double)
> MDiR (frontal double) ≥ MH3PG (frontal
double – the weakest interaction), which might be
related to the
size of π-electron system and degree of intramolecular rotation
freedom of L. The frontal double synthons are rather
rigid as the interacting −OH groups are attached to the same
ring; this is reflected, for example, in the diversified metric parameters
of the single LO–H···NM–C≡N component in MH3PG congeners and the relevant energy of interactions.52 The flexibility of L increases in the order H3PG (rigid) < DiR < TriRB, which results in the increasing adaptability
of the pair of phenolic groups attached to different rings to the
steric demands of rigid cis-oriented pairs of cyanido
ligands in [M(CN)6]3–. In the same order,
an increase in the extent of the π-electron system across the
molecule can be noted, which facilitates the possibility of the aforementioned
intra-CT interactions within L leading to overall stronger orbital
interactions and finally stronger total interactions between L and
[M(CN)6]3–. Moreover, in the MTriRB and MH3PG molecular
clusters, topologically identical single linear D contacts can be noted, characterized by interaction energies ranging
from −27.5 to −31.5 kcal mol–1 and
from −22 to −24.5 kcal mol–1, respectively,
again reflecting the potentially larger adjustment freedom of TriRB
in the overall scheme of intermolecular contacts and its larger size
of the π-electron system. Finally, the interaction energy values
for the whole molecular clusters {[M(CN)6]3–;L4} (all in kcal mol–1) vary from −163
to −171.5 for MDiR, from −148.5 to −158.5
for MTriRB, and from −124.5 to −133.5 for MH3PG. Accordingly, in
this case, we observe a different increasing stabilization trend of MH3PG < MTriRB < MDiR, resulting from the coformer structural and
electronic adaptability order H3PG < DiR < TriRB
discussed above and also from the presence of an additional LO–H···NM–C≡N hydrogen
bond in MDiR (showing in total seven hydrogen bond contacts
vs. six in MTriRB) that naturally produces an excess
of interaction energy compared to MTriRB. The variations
in the absolute interaction energy values of all motifs in question
observed across the series of compounds with DiR and TriRB coformers
correlate with the increasing ionic radius of the metal and [M(CN)6]3– anion size according to a general trend
of Cr(III) < Fe(III) < Co(III). However, the energetic stabilization
does not change in a perfectly linear manner, which might be attributed
to the fine collective adjustment of all intermolecular contacts.
pattern for MDiR and MH3PG, represented
by
the interaction energy values ranging from ca. −46
to −49 kcal mol–1 and from −44.5 to
−48 kcal mol–1, respectively. A distinct
trend in the absolute values of these energies can be thus established: MTriRB (side double – the strongest
interaction) > MDiR (side double)
> MDiR (frontal double) ≥ MH3PG (frontal
double – the weakest interaction), which might be
related to the
size of π-electron system and degree of intramolecular rotation
freedom of L. The frontal double synthons are rather
rigid as the interacting −OH groups are attached to the same
ring; this is reflected, for example, in the diversified metric parameters
of the single LO–H···NM–C≡N component in MH3PG congeners and the relevant energy of interactions.52 The flexibility of L increases in the order H3PG (rigid) < DiR < TriRB, which results in the increasing adaptability
of the pair of phenolic groups attached to different rings to the
steric demands of rigid cis-oriented pairs of cyanido
ligands in [M(CN)6]3–. In the same order,
an increase in the extent of the π-electron system across the
molecule can be noted, which facilitates the possibility of the aforementioned
intra-CT interactions within L leading to overall stronger orbital
interactions and finally stronger total interactions between L and
[M(CN)6]3–. Moreover, in the MTriRB and MH3PG molecular
clusters, topologically identical single linear D contacts can be noted, characterized by interaction energies ranging
from −27.5 to −31.5 kcal mol–1 and
from −22 to −24.5 kcal mol–1, respectively,
again reflecting the potentially larger adjustment freedom of TriRB
in the overall scheme of intermolecular contacts and its larger size
of the π-electron system. Finally, the interaction energy values
for the whole molecular clusters {[M(CN)6]3–;L4} (all in kcal mol–1) vary from −163
to −171.5 for MDiR, from −148.5 to −158.5
for MTriRB, and from −124.5 to −133.5 for MH3PG. Accordingly, in
this case, we observe a different increasing stabilization trend of MH3PG < MTriRB < MDiR, resulting from the coformer structural and
electronic adaptability order H3PG < DiR < TriRB
discussed above and also from the presence of an additional LO–H···NM–C≡N hydrogen
bond in MDiR (showing in total seven hydrogen bond contacts
vs. six in MTriRB) that naturally produces an excess
of interaction energy compared to MTriRB. The variations
in the absolute interaction energy values of all motifs in question
observed across the series of compounds with DiR and TriRB coformers
correlate with the increasing ionic radius of the metal and [M(CN)6]3– anion size according to a general trend
of Cr(III) < Fe(III) < Co(III). However, the energetic stabilization
does not change in a perfectly linear manner, which might be attributed
to the fine collective adjustment of all intermolecular contacts.
Table 2. DFT-Computed (B3LYP+D4//TZP) Interaction Energy Values (in kcal mol–1) between Hexacyanometallate Anion [M(CN)6]3– (M = Cr, Fe, Co) and (A) Polyresorcinol DiR Molecule(s), (B) Polyresorcinol TriRB Molecule(s), and (C) Phloroglucinol H3PG Molecule(s) in the Molecular Clusters Extracted from the Respective Crystal Structures of MDiR, MTriRB, and MH3PG(52),d.
| A | |||
|---|---|---|---|
| [Cr(CN)6]3– | [Fe(CN)6]3– | [Co(CN)6]3– | |
| (HAHBDiR)2(HA2DiR)(HADiR) | –163.34 | –168.43 | –171.23 |
| (HAHBDiR) side 2HB | –51.07, –50.23a | –53.64, –50.77a | –54.50, –52.13a |
| HA2DiR frontal 2HB | –45.97 | –47.94 | –49.06 |
| B | ||||
|---|---|---|---|---|
| [Cr(CN)6]3– | [Fe(CN)6]3– | [Co01(CN)6]3– | [Co02(CN)6]3– | |
| (HBHCTriRB)2(HTriRB)2 | –148.82 | –156.65 | –158.27 | –157.17 |
| (HBHCTriRB) side 2HB | –54.54, –54.90a | –58.86, –55.72a | –60.00, –57.14a | –58.63, –57.84a |
| (HTriRB) frontal 1HB | –27.79, –29.46a | –b | –29.87, –30.42a | –31.32, –28.92a |
| Cc | |||
|---|---|---|---|
| [Cr(CN)6]3– | [Fe(CN)6]3– | [Co(CN)6]3– | |
| (H2PGH)2(HPGH2)2 | –124.87 | –133.38 | –130.88 |
| H2PGH 2HB | –44.65 | –47.98 | –46.40 |
| HPGH21HB | –22.30 | –24.29 | –23.99 |
Two numbers correspond to two (slightly different) motifs of the same type found in the crystal structure.
Obtained results were considered unreliable due to significant spin-contamination in the wave function of the molecular cluster.
Taken from ref (52).
For cluster visualization, see Figures S11–S13 and ref (52). 2HB/1HB stands for the double/single hydrogen bonding interaction.
IR, Raman, Mössbauer, and UV–Vis Spectra
IR and Raman spectra contain bands corresponding to molecular components and intermolecular synthons indicated by SC XRD analysis (Figures S21–23). The shape and spectral position of some absorption peaks are notably modified with respect to the reference solids, which is attributed to the enhancement of noncovalent interactions within the charge-assisted hydrogen-bond network. This holds for the bathochromic shift of ca. 200 cm–1 in the 3600–2500 cm–1 range of ν(O–H) vibrations of DiR and TriRB (IR spectra), the hypsochromic shift of ca. 20 cm–1 in the 2200–2000 cm–1 range of ν(C≡N) vibrations of [M(CN)6]3– (IR and Raman spectra), and some shifts for the specific skeletal coformers vibrations (IR and Raman spectra).52,61,62
LF-Raman scattering spectra for all the presented materials exhibit numerous symmetrical LF-Raman peaks (Figures 4 and S24) with a few peaks in the sub-THz region: 25 cm–1 (0.75 THz) for FeDiR, 31 cm–1 (0.93 THz) for CoDiR, 22 cm–1 (0.66 THz) for CrDiR, 18 cm–1 (0.54 THz) for CrTriRB as well as FeTriRB, and 24 cm–1 (0.72 THz) for CoTriRB. A noteworthy observation is that the lowest Raman peak for the Co(III) based compounds is shifted to higher energy by 6 cm–1 compared to the other analogs. The low-frequency Raman peaks are associated with weak van der Waals and hydrogen bonding networks. Therefore, the observed differences in the Raman peak between the Co(III) compounds and the Fe(III) or Cr(III) analogs can be attributed to the stronger hydrogen bonding present in the former, as supported by the results of the DFT calculations (Table 2). A similar Raman shift toward higher energy was observed for [YbIII(TPPO)3(NCX)3] (X = S and Se) compounds but related rather to the substitution of the heavier Se atom with the lighter S one.42
Figure 4.

Representative low-frequency Raman scattering spectra for FeDiR and CoTriRB.
57Fe Mössbauer parameters are perfectly in line with the structural data (Figures S25) and previous findings for the MH3PG series.52
UV–vis spectra in the solid state in the form of the Kubelka–Munk function show the spectral features of all involved components and indicate diverse absorption threshold points depending on the [M(CN)6]3– used, in line with the results presented previously for the MH3PG networks (Figure S26).52 Interestingly, unlike the case of [Cr(CN)6]3– and [Co(CN)6]3– congeners, the spectra of all [Fe(CN)6]3– containing phases systematically show some additional spectral features fairly distinguished within the wavelength ranges above ligand-to-metal charge-transfer (LMCT) σ(CN–) → π(t2g) transition bands (maxima between 400 and 430 nm), spread up to ca. 750 nm for FeDiR, ca. 700 nm for FeTriRB, ca. 700 nm for FeH3PG,52 and only ca. 580 nm for K3[Fe(CN)6], and not present for PPh4[Fe(CN)6]·7H2O salt (Figure 5). The TD-DFT (PBE//TZVP) UV–vis calculations and the subsequent MO-pair analysis of the computed excitations correctly reproduced the LMCT assignment of the absorption centered around 410 nm (see LMCT1 in Figure 5b) and more importantly indicated that the additional lowest-energy absorption for FeDiR (Figure 5b), FeTriRB, and FeH3PG might be attributed to the intermolecular optical outer-sphere charge-transfer (OSCT) [Fe(CN)6]3– → coformer transitions through the O–H···Ncomplex hydrogen bonding orbital pathway (for details, see the section on UV–vis electronic absorption spectra in the SI, Figures S27–S31). In particular, two different FeDiR models, FeDiR-side and FeDiR-frontal, representing two structurally and energetically different hydrogen bonding interactions (vide supra), demonstrated differences in the basal discrete OSCT absorption position and in the intensity ratio ILMCT1/IOSCT (Figures S29 and S30), which is in qualitative agreement with the measured spectra. In the case of the pristine [Fe(CN)6]3– salts, the feature attributed to OSCT was not reproduced in the calculations, which remains in line with the lack of the proper orbital pathway. However, one important issue, namely, the absorption designated as LMCT2 appearing systematically for all the examined models in this study, requires a comment. In the relevant spectral range just above 500 nm, a very weak absorption might be observed experimentally for the [Fe(CN)6]3– anion in the aqueous solution and in the solid state, to which the spin-forbidden ligand-field (LF) character was assigned by using the ligand-field parameter approach63 and, very recently, by combining CASSCF+NEVPT2 calculations with resonant inelastic X-ray scattering (RIXS).64 While the expected spin-forbidden LF transitions could not be directly modeled using the adopted computational protocol, their contribution might be indirectly reflected in the observed LMCT2 transitions (Figures S28–S31), considering a strong mixing of LF transitions with LMCT transitions found in the case of [Fe(CN)6]3− 64 as well as in the case of other Fe(III) complexes.65 Thus, while the computational data presented herein (due to the methodological limitations and the specific character of the adopted molecular models) give a rather tentative description of UV–vis electronic spectra for FeL compounds in the solid state, it is clear that they confirm a strong impact of hydrogen bonding interactions on the valence electronic structure of FeL. Finally, it is worth highlighting that the described optical absorption cutoff thresholds (ca. 450–470 nm for the Cr(III), ca. 700 nm for the Fe(III), and ca. 375 nm for the Co(III) DiR and TriRB compounds) vividly shape the quality of the 520 nm → 1040 nm second-harmonic generation and luminescent properties of the tested samples (see below).
Figure 5.

(a) Experimental UV–vis electronic absorption spectra for FeDiR, FeTriRB, and FeH3PG along with spectra for the reference salts and coformers in the solid state, in the Kubelka–Munk (K-M) form. (b) Simulated (TD-DFT PBE//TZVP) UV–vis electronic absorption spectra of the FeDiR-side and FeDiR-frontal models, representative of the fundamental hydrogen-bonded motifs observed in the crystal structures of FeDiR, and of the [Fe(CN)6]3– anion as a reference. The absorption assigned to LMCT transitions within the [Fe(CN)6]3– anion (schematically highlighted by the pink areas) is observed in all the considered cases, whereas the additional lowest-energy OSCT [Fe(CN)6]3– → DiR transitions (the orange area) are shown for both models of FeDiR (for details, see the SI, Figures S27–S31).
SHG Studies
Considering the noncentrosymmetric space groups found for CrDiR, FeDiR, CoDiR (Pna21), CrTriRB (Cc), FeTriRB (P21212), and CoTriRB (P21) (Figure S1, Tables S1 and S2), their SHG response was examined for the powdered samples (Figures 6, S32, and S33). The room-temperature SHG studies were conducted in the reflectance mode using an in-house SHG setup42 and aimed at the observation of the 520 nm SH light (maximum of the SH light intensity vs wavelength plot) upon the 1040 nm fundamental light illumination in the reflection mode. Determined SH vs fundamental light intensity plots were fitted with the quadratic function (y = A × x2) and matched to the potassium dihydrogen phosphate (KDP) standard.66 Consequently, the SH susceptibilities (χSH) of 6.0 × 10–12 (0.5% KDP) for CrDiR, 7.2 × 10–12 (0.6% KDP) for FeDiR, 2.2 × 10–11 (1.8% KDP) for CoDiR, 6.2 × 10–11 (5.2% KDP) for CrTriRB, 3.2 × 10–11 (2.7% KDP) for FeTriRB, and 1.4 × 10–10 (11.7% KDP) for CoTriRB were determined.
Figure 6.

Second-harmonic signals for CoTriRB plotted against the wavelength to confirm the chromaticity aberration of the SH signal (a). Average SHG signals of CoTriRB (b); red solid lines correspond to the results of fitting using a quadratic function (y = Ax2). Relation between tensor elements of the SH susceptibility corresponding to space group P21 and SH polarization (c).
It is worth noting that for both DiR- and TriRB-based series of samples, a tendency to increase the SHG signal value for the compounds containing Co(III) can be observed with respect to the other metal ions, which might be due to the optical transparency of CoDiR and CoTriRB in the visible region. Moreover, the values are comparable with previous reports for polycrystalline samples of cyanido-bridged assemblies (Table S15),67−76 and they are an outcome of nonzero elements of the second-order NLO susceptibility tensor (χijk) for the P21 space group (symmetry class: 2 with polar tensor: B3) (Figure 7), for the Cc space group (symmetry class: m with polar tensor: C3) (Figure S33), for the P21212 space group (symmetry class: 222 with polar tensor: D3) (Figure S33), and for the Pna21 space group (symmetry class: mm2 with polar tensor: E3) (Figure S32). The DiR-containing materials have lower SHG efficiency than the TriRB-based cocrystals, possibly due to smaller electron polarization, which directly affects the SHG signal.
Figure 7.

Photoluminescence spectra of CoDiR and CoTriRB. Emission spectra for the corresponding excitation wavelengths of 340 nm for CoDiR (a) and 325 nm for CoTriRB (c). Excitation spectra followed the emission wavelength of 450 nm for CoDiR (b) and CoTriRB (d).
Photoluminescent Properties
Both DiR and TriRB ligands exhibit intense blue luminescence with a maximum at around 400 nm upon excitation with light of ca. 325 nm (Figures S34 and S35). Furthermore, pristine DiR and TriRB showed respectively a 5- and 1.5-fold increase in the excitation and emission intensities upon cooling to 77 K (the boiling point of liquid nitrogen), accompanied by a slight fluorescence color shift from dark blue to bright blue. Spectral analysis for CrDiR and CrTriRB (Figures S36 and S37), FeDiR and FeTriRB (Figures S38 and S39), and CoDiR and CoTriRB (Figures 7, S40, and S41) revealed that the compounds based on Cr(III) and Fe(III) emit relatively weak visible light, predominantly originating from their corresponding organic coformers. On the other hand, the compounds based on Co(III) display an intense cyan luminescence due to the synergistic combination of red visible emission from Co(III) ions and bluish emission from the organic ligands. The corresponding CIE1931 uniform chromaticity scale diagrams are shown in Figure S42. It is worth emphasizing that the emission intensities for all the compounds increased upon cooling from room to cryogenic temperatures, often attributable to the suppression of nonradiative vibrational loss. Interestingly, for the CrDiR and CrTriRB samples, there are additional emission bands with two distinctive maxima at ca. 810 and 825 nm from the 2Eg → 4A2g emission centered at the Cr(III) ion.52
Looking at the diverse features in the radiative emission of all of these compounds, we also performed quantum yield (QY) measurements for the powder samples at room temperature (Table S16). The previous findings were substantiated, confirming that pristine organic ligands exhibit very low emission quantum yields of less than 1%. The QY values for CrDiR and CrTriRB were determined to be 8.9% and 2.4%, respectively, when they were excited with 375 nm light. Similarly, the QY values for CoDiR and CoTriRB were found to be 1.7% and 1.3%, respectively, under 325 nm light irradiation. Significantly, a higher QY for the Cr(III) systems is due to the presence of near-infrared emission around 810 and 825 nm.
Conclusion and Perspectives
Realizing the cocrystallization approach, we combined linear bis-resorcinol DiR or triangular tris-resorcinol TriRB multiple hydrogen bond donors with [M(CN)6]3– (M = Cr, Fe, Co) hydrogen bond acceptors provided in the form of the PPh4+ salts. As expected, the resulting crystal phases showed the extended 3D hydrogen-bonded subnetworks {[M(CN)6]3–;Ln}∞ accompanied by the subnetwork of PPh4+ stabilized by MPE interactions assisted by solvent molecules. Within the {[M(CN)6]3–;Ln}∞ subnetworks, we recognized a range of interesting hydrogen-bonded motifs. The most elegant ones are “intuitive” (e.g., expected based on the results of our previous studies involving phloroglucinol H3PG coformer)52 supramolecular cis-bis(chelate)-like {[M(CN)6]3–;(H2L)2(HL)2} or tris(chelate)-like {[M(CN)6]3–;(H2L)3} fragments. In the current work, they are composed of the cyclic, either frontal or side {[M(CN)6]3–;(H2L)}, synthons, involving phenolic groups attached to the same resorcinol-like ring of an L molecule or the different rings of one L, respectively. The image is completed with single {[M(CN)6]3–;(HL)} synthons as well as more complex aggregates involving more than two molecules. The quantum-chemical analysis demonstrates the mixed electrostatic and covalent character of the underlying hydrogen bonding interactions, the strength of which is clearly enhanced due to the negative charge of the hydrogen bond acceptor metal complex. The DFT-computed energy of interactions between [M(CN)6]3– and L per representative {[M(CN)6]3–;(H2L)} motifs ranges from −46 to −60 kcal mol–1 and depends on the geometry of the contact and size matching of its components, rotational degree of freedom and extent of π-electron system of the coformer, and finally, the overall fit to the molecular surroundings. The symmetry of the crystal lattices of the obtained compounds is correlated with the effective local symmetry of coformers and {complex;(coformer)n} hydrogen-bonded motifs characterized by the absence of the inversion center and mirror plane, which is reflected by their D2, C2, or C1 point groups. It is important to note that the obtained acentric structural resolution remains in contrast with the centrosymmetric space groups observed for the native solvate crystals of DiR and TriRB. As our studies are the first to show the use of these molecules as building blocks, in the future, it would be interesting to examine the structural self-assembly in the solid state involving other molecular or ionic counterparts bearing prerequisites toward molecular functionality.
The SHG activity of our compounds is comparable to that of other polycyanidometallate-based molecular solids. Although the SHG performance is weaker than that of the KDP standard, the presented systems pave the way for a new strategy toward achieving noncentrosymmetric molecular architectures. Furthermore, as some of our crystals possess enantiopure Sohncke space groups, it would be interesting to examine crystallization of the enantiopure phases and circularly polarized photoluminescent response according to this still dynamically developing field in natural science. The interesting LF-Raman scattering spectral characteristics within the sub-THz spectroscopic domain might be further considered in the fabrication of the sub-THz response by optical stimulation. Finally, the obtained phases might be used as modular precursors toward the assembly of advanced multicomponent constructs, such as solid solutions, crystal-of-crystal arrays, and core–shell crystals, to test their optical performance. Also, the new multisite anion receptors might be designed and synthesized based on the arrangements achieved in this study. The work is in progress in our groups along the above lines.
Materials and Methods
A description of synthetic procedures,54 X-ray diffraction analysis,77−79 structural data presentation,80 physicochemical techniques, and computational methods and protocols60,81−97 used in this study can be found in the SI.
Acknowledgments
We gratefully acknowledge the main financial support from the National Science Centre (Poland) research project UMO-2019/35/B/ST5/01481 (to R.P.). Measurements were carried out with equipment funded by the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (contract POIG.02.01.00-12-023/08). We thank the Polish high-performance computing infrastructure PLGrid (HPC Centers: ACK Cyfronet AGH) for providing computer facilities and support within computational grants nos. PLG/2021/015125 and PLG/2022/015911. These studies were cofinanced by a JSPS Grant-in-Aid for Scientific Research (A) (Grant Number 20H00369), the CNRS-University of Tokyo “Excellence Science” Joint Research Program, and DYNACOM International Research Laboratory (CNRS). We acknowledge the Cryogenic Research Center, The University of Tokyo, the Center for Nano Lithography & Analysis, The University of Tokyo supported by MEXT, and the MEXT Quantum Leap Flagship Program (Grant Number JPMXS0118068681) for the support. O.S. is thankful to JSPS KAKENHI (Grant Number 21K14582). G.L. is grateful to JSPS KAKENHI (Grant Number 23KJ0736). T.M.M. would like to thank the programme Excellence Initiative – Research University for funding the research group of Crystal Engineering and Advanced Solid-State Characterisation.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.3c03153.
Detailed description of the used materials, synthetic procedures, physical techniques, crystal structure determination, structural, computational, and physicochemical data presentation, and additional figures and tables (PDF)
Author Contributions
K.J.: investigation—syntheses, measurements and analysis of PXRD, ESI-MS, TGA, IR spectroscopy, and UV–vis electronic absorption spectroscopy data; structural data analysis, description; data visualization; data curation; writing—original draft preparation, writing—review; J.K.: investigations—SC XRD structural measurements; crystal structure solution and refinement; D.T.: investigation—DFT calculations; data analysis and visualization; M.S.-H.: conceptualization and investigation—DFT calculations; data analysis and visualization; writing—original draft fragments preparation, writing—review; K.K.: investigation—low-frequency (LF) and RT Raman scattering spectroscopies, SHG, photoluminescence measurements and data visualization; UV–vis electronic absorption calculations; writing—original draft preparation; writing—review; G.L.: investigation—photoluminescence, SHG measurements; O.S.: investigation—low-frequency (LF) and RT Raman scattering spectroscopies, SHG, photoluminescence measurements and data visualization; UV–vis electronic absorption calculations; writing—original draft preparation; writing-review; T.M.M.: SC XRD structural measurements; crystal structure solution and refinement; writing—review; K.D.-K.: investigation—measurements of 57Fe Mössbauer spectra; data visualization; writing—review; S.O.: funding acquisition; project administration; conceptualization; supervision; writing—review; corresponding author; R.P.: funding acquisition; project administration; conceptualization; supervision; writing—original draft preparation; writing—review; corresponding author. All authors have read and agreed to the published version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Yang Y.; Wang Q.; Li G.; Guo W.; Yang Z.; Liu H.; Deng X. Cysteine-Derived Chiral Carbon Quantum Dots: A Fibrinolytic Activity Regulator for Plasmin to Target the Human Islet Amyloid Polypeptide for Type 2 Diabetes Mellitus. ACS Appl. Mater. Interfaces 2023, 15 (2), 2617–2629. 10.1021/acsami.2c17975. [DOI] [PubMed] [Google Scholar]
- Chen C.; Ibrahim Z.; Marchand M. F.; Piolot T.; Kamboj S.; Carreiras F.; Yamada A.; Schanne-Klein M.-C.; Chen Y.; Lambert A.; Aimé C. Three-Dimensional Collagen Topology Shapes Cell Morphology, beyond Stiffness. ACS Biomater. Sci. Eng. 2022, 8 (12), 5284–5294. 10.1021/acsbiomaterials.2c00879. [DOI] [PubMed] [Google Scholar]
- Choi J.-H.; Fremy G.; Charnay T.; Fayad N.; Pécaut J.; Erbek S.; Hildebrandt N.; Martel-Frachet V.; Grichine A.; Sénèque O. Luminescent Peptide/Lanthanide(III) Complex Conjugates with Push-Pull Antennas: Application to One- and Two-Photon Microscopy Imaging. Inorg. Chem. 2022, 61 (50), 20674–20689. 10.1021/acs.inorgchem.2c03646. [DOI] [PubMed] [Google Scholar]
- Valdes-García J.; Zamora-Moreno J.; Pinzón-Vanegas C.; Viviano-Posadas A. O.; Martínez-Otero D.; Barroso-Flores J.; Ortiz-Lopez B.; Ortiz-Navarrete V. F.; Dorazco-González A. Selective Luminescent Chemosensing of Chloride Based on a Cyclometalated Platinum(II) Complex in Water: Crystal Structures, Spectroscopic Studies, Extraction, and Bioimaging. Inorg. Chem. 2023, 62 (17), 6629–6641. 10.1021/acs.inorgchem.2c04558. [DOI] [PubMed] [Google Scholar]
- Monteiro J. H. S. K. Recent Advances in Luminescence Imaging of Biological Systems Using Lanthanide(III) Luminescent Complexes. Molecules 2020, 25 (9), 2089. 10.3390/molecules25092089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D. L.; He H. Z.; Leung K. H.; Chan D. S. H.; Leung C. H. Bioactive Luminescent Transition-Metal Complexes for Biomedical Applications. Angew. Chem., Int. Ed. 2013, 52 (30), 7666–7682. 10.1002/anie.201208414. [DOI] [PubMed] [Google Scholar]
- Liu J.-M.Optical Modulation. In Principles of Photonics; Cambridge University Press: 2016; pp 297–361. [Google Scholar]
- Kumar K.; Stefanczyk O.; Chorazy S.; Nakabayashi K.; Ohkoshi S. Ratiometric and Colorimetric Optical Thermometers Using Emissive Dimeric and Trimeric {[Au(SCN)2]−}n Moieties Generated in d-f Heterometallic Assemblies. Angew. Chem., Int. Ed. 2022, 61 (20), e202201265. 10.1002/anie.202201265. [DOI] [PubMed] [Google Scholar]
- Kumar K.; Stefanczyk O.; Chorazy S.; Nakabayashi K.; Ohkoshi S. Ratiometric Raman and Luminescent Thermometers Constructed from Dysprosium Thiocyanidometallate Molecular Magnets. Adv. Opt. Mater. 2022, 10 (22), 2201675. 10.1002/adom.202201675. [DOI] [Google Scholar]
- Quimby R. S.Nonlinear Optics. In Photonics and Lasers, An Introduction; John Wiley & Sons, Inc.: Hoboken, NJ, 2006; pp 93–158. [Google Scholar]
- Boyd R. W.Nonlinear Opt., 4th ed.; Elsevier, Academic Press: 2020. [Google Scholar]
- Bonnin M. A.; Bayarjargal L.; Wolf S.; Milman V.; Winkler B.; Feldmann C. GaSeCl5O: A Molecular Compound with Very Strong SHG Effect. Inorg. Chem. 2021, 60 (20), 15653–15658. 10.1021/acs.inorgchem.1c02315. [DOI] [PubMed] [Google Scholar]
- Qu L.; Bai L.; Jin C.; Liu Q.; Wu W.; Gao B.; Li J.; Cai W.; Ren M.; Xu J. Giant Second Harmonic Generation from Membrane Metasurfaces. Nano Lett. 2022, 22 (23), 9652–9657. 10.1021/acs.nanolett.2c03811. [DOI] [PubMed] [Google Scholar]
- Gryl M.; Seidler T.; Wojnarska J.; Stadnicka K.; Matulková I.; Němec I.; Němec P. Co Crystals of 2-Amino-5 Nitropyridine Barbital with Extreme Birefringence and Large Second Harmonic Generation Effect. Chem.—Eur. J. 2018, 24 (35), 8727–8731. 10.1002/chem.201802057. [DOI] [PubMed] [Google Scholar]
- Harfouche L. C.; Couvrat N.; Sanselme M.; Brandel C.; Cartigny Y.; Petit S.; Coquerel G. Discovery of New Proxyphylline-Based Chiral Cocrystals: Solid State Landscape and Dehydration Mechanism. Cryst. Growth Des. 2020, 20 (6), 3842–3850. 10.1021/acs.cgd.0c00149. [DOI] [Google Scholar]
- Bryndal I.; Drozd M.; Lis T.; Zaręba J. K.; Ratajczak H. Structural Diversity of Hydrogen-Bonded Complexes Comprising Phenol-Based and Pyridine-Based Components: NLO Properties and Crystallographic and Spectroscopic Studies. CrystEngComm 2020, 22 (27), 4552–4565. 10.1039/D0CE00606H. [DOI] [Google Scholar]
- Wojnarska J.; Gryl M.; Seidler T.; Rydz A.; Oszajca M.; Stadnicka K. M.; Marzec M.; Matulková I.; Němec I.; Němec P. Crystal Structure and (Non)Linear Optical Properties of a Cyanuric Acid Isoniazid < 1/1>; Co-Crystal: Shortcomings of Phase Matching Determination from Powdered Samples. Cryst. Growth Des. 2019, 19 (12), 6831–6836. 10.1021/acs.cgd.9b01023. [DOI] [Google Scholar]
- Ulaganathan R. K.; Roy P. K.; Mhatre S. M.; Murugesan R. C.; Chen W. L.; Lai M. H.; Subramanian A.; Lin C. Y.; Chang Y. M.; Canulescu S.; Rozhin A.; Liang C. Te; Sankar R. High-Performance Photodetector and Angular-Dependent Random Lasing from Long-Chain Organic Diammonium Sandwiched 2D Hybrid Perovskite Non-Linear Optical Single Crystal. Adv. Funct. Mater. 2023, 33, 2214078. 10.1002/adfm.202214078. [DOI] [Google Scholar]
- Cassingham M. A.; Goh Y. G.; McClure E. T.; Hodgkins T. L.; Zhang W.; Liang M.; Dawlaty J. M.; Djurovich P. I.; Haiges R.; Halasyamani P. S.; Savory C. N.; Thompson M. E.; Melot B. C. Polarizable Anionic Sublattices Can Screen Molecular Dipoles in Noncentrosymmetric Inorganic-Organic Hybrids. ACS Appl. Mater. Interfaces 2023, 15 (14), 18006–18011. 10.1021/acsami.2c20648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayakanth T.; Sahoo S.; Kothavade P.; Bhan Sharma V.; Kabra D.; Zaręba J. K.; Shanmuganathan K.; Boomishankar R. A Ferroelectric Aminophosphonium Cyanoferrate with a Large Electrostrictive Coefficient as a Piezoelectric Nanogenerator. Angew. Chem., Int. Ed. 2023, 62 (3), e202214984. 10.1002/anie.202214984. [DOI] [PubMed] [Google Scholar]
- Mączka M.; Gągor A.; Zaręba J. K.; Trzebiatowska M.; Stefańska D.; Kucharska E.; Hanuza J.; Pałka N.; Czerwińska E.; Sieradzki A. Benzyltrimethylammonium Cadmium Dicyanamide with Polar Order in Multiple Phases and Prospects for Linear and Nonlinear Optical Temperature Sensing. Dalton Trans. 2021, 50 (30), 10580–10592. 10.1039/D1DT01675J. [DOI] [PubMed] [Google Scholar]
- Zeng Y.-L.; Ai Y.; Tang S.-Y.; Song X.-J.; Chen X.-G.; Tang Y.-Y.; Zhang Z.-X.; You Y.-M.; Xiong R.-G.; Zhang H.-Y. Axial-Chiral BINOL Multiferroic Crystals with Coexistence of Ferroelectricity and Ferroelasticity. J. Am. Chem. Soc. 2022, 144 (42), 19559–19566. 10.1021/jacs.2c08667. [DOI] [PubMed] [Google Scholar]
- Cametti M.; Ilander L.; Valkonen A.; Nieger M.; Nissinen M.; Nauha E.; Rissanen K. Non-Centrosymmetric Tetrameric Assemblies of Tetramethylammonium Halides with Uranyl Salophen Complexes in the Solid State. Inorg. Chem. 2010, 49 (24), 11473–11484. 10.1021/ic101547r. [DOI] [PubMed] [Google Scholar]
- Ohkoshi S.; Takano S.; Imoto K.; Yoshikiyo M.; Namai A.; Tokoro H. 90-Degree Optical Switching of Output Second-Harmonic Light in Chiral Photomagnet. Nat. Photonics 2014, 8, 65–71. 10.1038/nphoton.2013.310. [DOI] [Google Scholar]
- Gryl M.; Kozieł M.; Stadnicka K. M. A Proposal for Coherent Nomenclature of Multicomponent Crystals. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2019, 75 (1), 53–58. 10.1107/S2052520618015858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe E.; Meekes H.; Vlieg E.; ter Horst J. H.; de Gelder R. Solvates, Salts, and Cocrystals: A Proposal for a Feasible Classification System. Cryst. Growth Des. 2016, 16 (6), 3237–3243. 10.1021/acs.cgd.6b00200. [DOI] [Google Scholar]
- Dechambenoit P.; Ferlay S.; Hosseini M. W.; Pleneix J.-M.; Kyritsakas N. Molecular Tectonics: Control of Packing of Hybrid 1-D and 2-D H-Bonded Molecular Networks Formed between Bisamidinium Dication and Cyanometallate Anions. New. J. Chem. 2006, 30, 1403–1410. 10.1039/b606265m. [DOI] [Google Scholar]
- Lin R.-B.; He Y.; Li P.; Wang H.; Zhou W.; Chen B. Multifunctional Porous Hydrogen-Bonded Organic Framework Materials. Chem. Soc. Rev. 2019, 48 (5), 1362–1389. 10.1039/C8CS00155C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul M.; Desiraju G. R. From a Binary to a Quaternary Cocrystal: An Unusual Supramolecular Synthon. Angew. Chem., Int. Ed. 2019, 58 (35), 12027–12031. 10.1002/anie.201904339. [DOI] [PubMed] [Google Scholar]
- Trevisan L.; Bond A. D.; Hunter C. A. Quantitative Measurement of Cooperativity in H-Bonded Networks. J. Am. Chem. Soc. 2022, 144 (42), 19499–19507. 10.1021/jacs.2c08120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Chang G.; Zheng F.; Chen L.; Yang Q.; Ren Q.; Bao Z. Hybrid Hydrogen-Bonded Organic Frameworks: Structures and Functional Applications. Chem.—Eur. J. 2023, 29 (14), e202202655. 10.1002/chem.202202655. [DOI] [PubMed] [Google Scholar]
- Zhou F.; Shemchuk O.; Charpentier M. D.; Matheys C.; Collard L.; ter Horst J. H.; Leyssens T. Simultaneous Chiral Resolution of Two Racemic Compounds by Preferential Cocrystallization. Angew. Chem., Int. Ed. 2021, 60 (37), 20264–20268. 10.1002/anie.202107804. [DOI] [PubMed] [Google Scholar]
- Springuel G.; Robeyns K.; Norberg B.; Wouters J.; Leyssens T. Cocrystal Formation between Chiral Compounds: How Cocrystals Differ from Salts. Cryst. Growth Des 2014, 14 (8), 3996–4004. 10.1021/cg500588t. [DOI] [Google Scholar]
- Czapik A.; Jelecki M.; Kwit M. Chiral Cocrystal Solid Solutions, Molecular Complexes, and Salts of N-Triphenylacetyl-L-Tyrosine and Diamines. Int. J. Mol. Sci. 2019, 20 (20), 5004. 10.3390/ijms20205004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; de Groen M.; Kramer H. J. M.; de Gelder R.; Tinnemans P.; Meekes H.; ter Horst J. H. Screening Approach for Identifying Cocrystal Types and Resolution Opportunities in Complex Chiral Multicomponent Systems. Cryst. Growth Des. 2021, 21 (1), 112–124. 10.1021/acs.cgd.0c00890. [DOI] [Google Scholar]
- Zhang W.; Ye H.-Y.; Graf R.; Spiess H. W.; Yao Y.-F.; Zhu R.-Q.; Xiong R.-G. Tunable and Switchable Dielectric Constant in an Amphidynamic Crystal. J. Am. Chem. Soc. 2013, 135 (14), 5230–5233. 10.1021/ja3110335. [DOI] [PubMed] [Google Scholar]
- Qian K.; Shao F.; Yan Z.; Pang J.; Chen X.; Yang C. A Perovskite-Type Cage Compound as a Temperature-Triggered Dielectric Switchable Material. CrystEngComm 2016, 18 (40), 7671–7674. 10.1039/C6CE01421F. [DOI] [Google Scholar]
- Rok M.; Moskwa M.; Działowa M.; Bieńko A.; Rajnák C.; Boča R.; Bator G. Multifunctional Materials Based on the Double-Perovskite Organic-Inorganic Hybrid (CH3NH3)2[KCr(CN)6] Showing Switchable Dielectric, Magnetic, and Semiconducting Behaviour. Dalton Trans. 2019, 48 (44), 16650–16660. 10.1039/C9DT03553B. [DOI] [PubMed] [Google Scholar]
- Rok M.; Bator G.; Medycki W.; Zamponi M.; Balčiu̅nas S.; Šimėnas M.; Banys J. Reorientational Dynamics of Organic Cations in Perovskite-like Coordination Polymers. Dalton Trans. 2018, 47 (48), 17329–17341. 10.1039/C8DT03372B. [DOI] [PubMed] [Google Scholar]
- Parrott E. P. J.; Zeitler J. A.; Friščić T.; Pepper M.; Jones W.; Day G. M.; Gladden L. F. Testing the Sensitivity of Terahertz Spectroscopy to Changes in Molecular and Supramolecular Structure: A Study of Structurally Similar Cocrystals. Cryst. Growth Des. 2009, 9 (3), 1452–1460. 10.1021/cg8008893. [DOI] [Google Scholar]
- Esaulkov M. N.; Fokina M. I.; Zulina N. A.; Timofeeva T. V.; Shkurinov A. P.; Denisyuk I. Yu. Aminopyridines and 4-Nitrophenol Cocrystals for Terahertz Application. Opt. Laser. Technol. 2018, 108, 450–455. 10.1016/j.optlastec.2018.07.033. [DOI] [Google Scholar]
- Kumar K.; Stefanczyk O.; Nakabayashi K.; Imoto K.; Oki Y.; Ohkoshi S. Detection of Sub-Terahertz Raman Response and Nonlinear Optical Effects for Luminescent Yb(III) Complexes. Adv. Opt. Mater. 2022, 10 (2), 2101721. 10.1002/adom.202101721. [DOI] [Google Scholar]
- Luo H.; Guo S.; Zhang Y.; Bu K.; Lin H.; Wang Y.; Yin Y.; Zhang D.; Jin S.; Zhang W.; Yang W.; Ma B.; Lü X. Regulating Exciton-Phonon Coupling to Achieve a Near-Unity Photoluminescence Quantum Yield in One Dimensional Hybrid Metal Halides. Adv. Sci. 2021, 8 (14), 2100786. 10.1002/advs.202100786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T.; Zhou C.; Feng X.; Dong N.; Chen H.; Chen X.; Zhang L.; Lin J.; Wang J. Regulation of the Luminescence Mechanism of Two-Dimensional Tin Halide Perovskites. Nat. Commun. 2022, 13 (1), 60. 10.1038/s41467-021-27663-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escalera-Moreno L.; Baldoví J. J.; Gaita-Ariño A.; Coronado E. Spin States, Vibrations and Spin Relaxation in Molecular Nanomagnets and Spin Qubits: A Critical Perspective. Chem. Sci. 2018, 9 (13), 3265–3275. 10.1039/C7SC05464E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobylarczyk J.; Pinkowicz D.; Srebro-Hooper M.; Hooper J.; Podgajny R. Anion-π Recognition between [M(CN)6]3- Complexes and HAT(CN)6: Structural Matching and Electronic Charge Density Modification. Dalton Trans. 2017, 46 (11), 3482–3491. 10.1039/C7DT00293A. [DOI] [PubMed] [Google Scholar]
- Kobylarczyk J.; Pinkowicz D.; Srebro-Hooper M.; Hooper J.; Podgajny R. Anion-π Architectures of HAT(CN)6 and 5d Polycyanidometalates: [W(CN)8]3−, [Re(CN)7]3−, and [Pt(CN)6]2−. Cryst. Growth Des. 2019, 19 (2), 1215–1225. 10.1021/acs.cgd.8b01653. [DOI] [Google Scholar]
- Kuzniak E.; Pinkowicz D.; Hooper J.; Srebro Hooper M.; Hetmańczyk Ł.; Podgajny R. Molecular Deformation, Charge Flow, and Spongelike Behavior in Anion-π {[M(CN)4]2- ;[HAT(CN)6]}∞ (M = Ni, Pd, Pt) Supramolecular Stacks. Chem.—Eur. J. 2018, 24 (61), 16302–16314. 10.1002/chem.201802933. [DOI] [PubMed] [Google Scholar]
- Kuzniak E.; Hooper J.; Srebro-Hooper M.; Kobylarczyk J.; Dziurka M.; Musielak B.; Pinkowicz D.; Raya J.; Ferlay S.; Podgajny R. A Concerted Evolution of Supramolecular Interactions in a {cation; Metal Complex; π-Acid; Solvent} Anion-π System. Inorg. Chem. Front. 2020, 7 (9), 1851–1863. 10.1039/D0QI00101E. [DOI] [Google Scholar]
- Kuzniak-Glanowska E.; Glosz D.; Niedzielski G.; Kobylarczyk J.; Srebro-Hooper M.; Hooper J. G. M.; Podgajny R. Binding of Anionic Pt(II) Complexes in a Dedicated Organic Matrix: Towards New Binary Crystalline Composites. Dalton Transactions 2021, 50 (1), 170–185. 10.1039/D0DT03535A. [DOI] [PubMed] [Google Scholar]
- Sekine Y.; Nihei M.; Oshio H. Dimensionally Controlled Assembly of an External Stimuli Responsive [Co2Fe2] Complex into Supramolecular Hydrogen Bonded Networks. Chem.—Eur. J. 2017, 23 (22), 5193–5197. 10.1002/chem.201605817. [DOI] [PubMed] [Google Scholar]
- Jędrzejowska K.; Kobylarczyk J.; Glosz D.; Kuzniak-Glanowska E.; Tabor D.; Srebro-Hooper M.; Zakrzewski J. J.; Dziedzic-Kocurek K.; Muzioł T. M.; Podgajny R. Supramolecular cis-“Bis(Chelation)” of [M(CN)6]3- (M = CrIII, FeIII, CoIII) by Phloroglucinol (H3PG). Molecules 2022, 27 (13), 4111. 10.3390/molecules27134111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobylarczyk J.; Pakulski P.; Potępa I.; Podgajny R. Manipulation of the Cyanido-Bridged Fe2W2 Rhombus in the Crystalline State: Co-Crystallization, Desolvation and Thermal Treatment. Polyhedron 2022, 224, 116028. 10.1016/j.poly.2022.116028. [DOI] [Google Scholar]
- Chaumont C.; Mobian P.; Kyritsakas N.; Henry M. Synthesis, Topology and Energy Analysis of Crystalline Resorcinol-Based Oligophenylene Molecules with Various Symmetries. CrystEngComm 2013, 15 (34), 6845–6862. 10.1039/c3ce40761f. [DOI] [Google Scholar]
- Steiner T. The Hydrogen Bond in the Solid State. Angew. Chem., Int. Ed. 2002, 41 (1), 48–76. . [DOI] [PubMed] [Google Scholar]
- Bernstein J.; Davis R. E.; Shimoni L.; Chang N. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem., Int. Ed. 1995, 34 (15), 1555–1573. 10.1002/anie.199515551. [DOI] [Google Scholar]
- Loos M.; Gerber C.; Corona F.; Hollender J.; Singer H. Accelerated Isotope Fine Structure Calculation Using Pruned Transition Trees. Anal. Chem. 2015, 87 (11), 5738–5744. 10.1021/acs.analchem.5b00941. [DOI] [PubMed] [Google Scholar]
- Blatov V. A.; Shevchenko A. P.; Proserpio D. M. Applied Topological Analysis of Crystal Structures with the Program Package ToposPro. Cryst. Growth Des. 2014, 14 (7), 3576–3586. 10.1021/cg500498k. [DOI] [Google Scholar]
- O’Keeffe M.; Brese N. E. Uninodal 4-Connected 3D Nets. I. Nets without 3- or 4-Rings. Acta Crystallogr. A 1992, 48 (5), 663–669. 10.1107/S0108767392001260. [DOI] [Google Scholar]
- Mitoraj M. P.; Michalak A.; Ziegler T. A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theory. Comput. 2009, 5 (4), 962–975. 10.1021/ct800503d. [DOI] [PubMed] [Google Scholar]
- Erdogdu Y.; Başköse Ü. C.; Sağlam S. Conformational, Structural, Electronic, and Vibrational Investigations on 5-Methyl-4-(2-Thiazolylazo)Resorcinol by FT-IR, FT-Raman, NMR, and DFT. Chem. Pap. 2019, 73 (8), 1879–1891. 10.1007/s11696-019-00739-4. [DOI] [Google Scholar]
- Drużbicki K.; Mikuli E.; Pałka N.; Zalewski S.; Ossowska-Chruściel M. D. Polymorphism of Resorcinol Explored by Complementary Vibrational Spectroscopy (FT-RS, THz-TDS, INS) and First-Principles Solid-State Computations (Plane-Wave DFT). J. Phys. Chem. B 2015, 119 (4), 1681–1695. 10.1021/jp507241j. [DOI] [PubMed] [Google Scholar]
- Naiman C. S. Interpretation of the Absorption Spectra of K3Fe(CN)6. J. Chem. Phys. 1961, 35 (1), 323–328. 10.1063/1.1731909. [DOI] [Google Scholar]
- Hahn A. W.; Van Kuiken B. E.; Chilkuri V. G.; Levin N.; Bill E.; Weyhermüller T.; Nicolaou A.; Miyawaki J.; Harada Y.; DeBeer S. Probing the Valence Electronic Structure of Low-Spin Ferrous and Ferric Complexes Using 2p3d Resonant Inelastic X-Ray Scattering (RIXS). Inorg. Chem. 2018, 57 (15), 9515–9530. 10.1021/acs.inorgchem.8b01550. [DOI] [PubMed] [Google Scholar]
- Steube J.; Kruse A.; Bokareva O. S.; Reuter T.; Demeshko S.; Schoch R.; Argüello Cordero M. A.; Krishna A.; Hohloch S.; Meyer F.; Heinze K.; Kühn O.; Lochbrunner S.; Bauer M. Janus-Type Emission from a Cyclometalated Iron(III) Complex. Nat. Chem. 2023, 15 (4), 468–474. 10.1038/s41557-023-01137-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemla D. S.; Zyss J.. Nonlinear Optical Properties of Organic Molecules and Crystals; Academic Press: London, 1987. [Google Scholar]
- Komine M.; Imoto K.; Namai A.; Yoshikiyo M.; Ohkoshi S. Photoswitchable Nonlinear-Optical Crystal Based on a Dysprosium-Iron Nitrosyl Metal Assembly. Inorg. Chem. 2021, 60 (4), 2097–2104. 10.1021/acs.inorgchem.0c03493. [DOI] [PubMed] [Google Scholar]
- Hozumi T.; Nuida T.; Hashimoto K.; Ohkoshi S. Crystal Structure and Nonlinear Optical Effect of a Pyroelectric Crystal Composed of a Cyano-Bridged Cu-Mo Assembly. Cryst. Growth Des. 2006, 6 (8), 1736–1737. 10.1021/cg060093h. [DOI] [Google Scholar]
- Komine M.; Chorazy S.; Imoto K.; Nakabayashi K.; Ohkoshi S. SHG-Active LnIII -[MoI(CN)5(NO)]3- (Ln = Gd, Eu) Magnetic Coordination Chains: A New Route towards Non-Centrosymmetric Molecule-Based Magnets. CrystEngComm 2017, 19 (1), 18–22. 10.1039/C6CE02214F. [DOI] [Google Scholar]
- Kawabata S.; Nakabayashi K.; Imoto K.; Klimke S.; Renz F.; Ohkoshi S. Second Harmonic Generation on Chiral Cyanido-Bridged FeII-NbIV Spin-Crossover Complexes. Dalton Trans. 2021, 50 (24), 8524–8532. 10.1039/D1DT01324F. [DOI] [PubMed] [Google Scholar]
- Kumar K.; Stefanczyk O.; Chilton N. F.; Nakabayashi K.; Imoto K.; Winpenny R. E. P.; Ohkoshi S. Magnetic Properties and Second Harmonic Generation of Noncentrosymmetric Cyanido-Bridged Ln(III)-W(V) Assemblies. Inorg. Chem. 2021, 60 (16), 12009–12019. 10.1021/acs.inorgchem.1c01113. [DOI] [PubMed] [Google Scholar]
- Ohkoshi S.; Saito S.; Matsuda T.; Nuida T.; Tokoro H. Continuous Change of Second-Order Nonlinear Optical Activity in a Cyano-Bridged Coordination Polymer. J. Phys. Chem. C 2008, 112 (34), 13095–13098. 10.1021/jp8021216. [DOI] [Google Scholar]
- Kumar K.; Stefanczyk O.; Nakabayashi K.; Imoto K.; Ohkoshi S. Studies of Er(III)-W(V) Compounds Showing Nonlinear Optical Activity and Single-Molecule Magnetic Properties. CrystEngComm 2019, 21 (39), 5882–5889. 10.1039/C9CE00822E. [DOI] [Google Scholar]
- Kosaka W.; Nuida T.; Hashimoto K.; Ohkoshi S. Crystal Structure, Magnetic Properties, and Second Harmonic Generation of a Three-Dimensional Pyroelectric Cyano-Bridged Mn-Mo Complex. Bull. Chem. Soc. Jpn. 2007, 80 (5), 960–962. 10.1246/bcsj.80.960. [DOI] [Google Scholar]
- Tsunobuchi Y.; Kosaka W.; Nuida T.; Ohkoshi S. Magnetization-Induced Second Harmonic Generation in a Three-Dimensional Manganese Octacyanoniobate-Based Pyroelectric Ferrimagnet. CrystEngComm 2009, 11 (10), 2051. 10.1039/b906778g. [DOI] [Google Scholar]
- Jankowski R.; Zakrzewski J. J.; Zychowicz M.; Wang J.; Oki Y.; Ohkoshi S.; Chorazy S.; Sieklucka B. SHG-Active NIR-Emissive Molecular Nanomagnets Generated in Layered Neodymium(III)-Octacyanidometallate(IV) Frameworks. J. Mater. Chem. C 2021, 9 (33), 10705–10717. 10.1039/D1TC00825K. [DOI] [Google Scholar]
- Sheldrick G. M. A Short History of SHELX. Acta Crystallogr. A 2008, 64 (1), 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71 (1), 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2 : A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42 (2), 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- Macrae C. F.; Sovago I.; Cottrell S. J.; Galek P. T. A.; McCabe P.; Pidcock E.; Platings M.; Shields G. P.; Stevens J. S.; Towler M.; Wood P. A. Mercury 4.0 : From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53 (1), 226–235. 10.1107/S1600576719014092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Velde G.; Bickelhaupt F. M.; Baerends E. J.; Fonseca Guerra C.; van Gisbergen S. J. A.; Snijders J. G.; Ziegler T. Chemistry with ADF. J. Comput. Chem. 2001, 22 (9), 931–967. 10.1002/jcc.1056. [DOI] [Google Scholar]
- ADF 2019.304; SCM, Theoretical Chemistry, Vrije Universiteit: Amsterdam, The Netherlands. http://www.scm.com (accessed 2023-12-09).
- Caldeweyher E.; Ehlert S.; Hansen A.; Neugebauer H.; Spicher S.; Bannwarth C.; Grimme S. A Generally Applicable Atomic-Charge Dependent London Dispersion Correction. J. Chem. Phys. 2019, 150, 154122. 10.1063/1.5090222. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98 (7), 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37 (2), 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98 (45), 11623–11627. 10.1021/j100096a001. [DOI] [Google Scholar]
- Becke A. D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38 (6), 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- van Lenthe E.; Baerends E. J.; Snijders J. G. Relativistic Regular Two-Component Hamiltonians. J. Chem. Phys. 1993, 99 (6), 4597–4610. 10.1063/1.466059. [DOI] [Google Scholar]
- van Lenthe E.; Baerends E. J.; Snijders J. G. Relativistic Total Energy Using Regular Approximations. J. Chem. Phys. 1994, 101 (11), 9783–9792. 10.1063/1.467943. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132 (15), 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32 (7), 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chemi. Chem. Phys. 2005, 7 (18), 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Mitoraj M.; Michalak A. Natural Orbitals for Chemical Valence as Descriptors of Chemical Bonding in Transition Metal Complexes. J. Mol. Model. 2007, 13 (2), 347–355. 10.1007/s00894-006-0149-4. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Ernzerhof M.; Burke K. Rationale for Mixing Exact Exchange with Density Functional Approximations. J. Chem. Phys. 1996, 105 (22), 9982–9985. 10.1063/1.472933. [DOI] [Google Scholar]
- Schäfer A.; Huber C.; Ahlrichs R. Fully Optimized Contracted Gaussian Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr. J. Chem. Phys. 1994, 100 (8), 5829–5835. 10.1063/1.467146. [DOI] [Google Scholar]
- Dennington R.; Keith T. A.; Millam J. M.. GaussView, Version 6.1.1; Semichem Inc.: Shawnee Mission, KS, 2016.
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian16, Revision C.01; 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.