

Abstract
The intracellular pathway of Janus kinase/signal transducer and activator of transcription (JAK/STAT) and modification of nucleosome histone marks regulate the expression of proinflammatory mediators, playing an essential role in carcinogenesis, antiviral immunity and the interaction of host proteins with Herpesviral particles. The pathway has also been suggested to play a vital role in the clinical course of the acute infection caused by severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2; known as coronavirus infection-2019), a novel human coronavirus initially identified in the central Chinese city Wuhan towards the end of 2019, which evolved into a pandemic affecting nearly two million people worldwide. The infection mainly manifests as fever, cough, myalgia and pulmonary involvement, while it also attacks multiple viscera, such as the liver. The pathogenesis is characterized by a cytokine storm, with an overproduction of proinflammatory mediators. Innate and adaptive host immunity against the viral pathogen is exerted by various effectors and is regulated by different signaling pathways notably the JAK/STAT. The elucidation of the underlying mechanism of the regulation of mediating factors expressed in the viral infection would assist diagnosis and antiviral targeting therapy, which will help overcome the infection caused by SARS-CoV-2.
Keywords: JAK/STAT, carcinoma, herpesviruses, EBV, KSHV, SARS-CoV-2
1. Introduction
The mammalian signal transducer and activator of transcription (STAT) family of proteins comprises seven members, namely STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B and STAT6, which function to initiate the transcription of genes, hence playing a role in advance of all cytokine-driven signaling present in the extracellular environment (1). The coding genes for STAT family proteins are mapped in three chromosomal clusters: Chromosomes 2, 12 and 17 (2); the molecular size ranges from 748 amino acids for STAT4 to 851 amino acids for STAT2 (https://www.uniprot.org/uniprotkb?query), as summarized in Table I.
Table I.
Chromosomal mapping and length in amino acid residue numbers of STATs.
| Member of STAT family | Chromosomal localization of coding genes | Molecular length (amino acids) |
|---|---|---|
| STAT1 | Chromosome 2 | 750 |
| STAT4 | Chromosome 2 | 748 |
| STAT2 | Chromosome 12 | 851 |
| STAT6 | Chromosome 12 | 847 |
| STAT3 | Chromosome 17 | 770 |
| STAT5A | Chromosome 17 | 794 |
| STAT5B | Chromosome 17 | 787 |
The coding genes for STAT family members are mapped in three chromosomal clusters as indicated. Each subgroup shows similar range of full length with amino acid numbers. STAT, signal transducer and activator of transcription.
The molecules share the conserved domains, exerting the activities of transducing intracellular signaling and initiating transcription, as suggested by their names. From amino (N)-terminus to carboxy (C)-terminus, the domains on the linear structure include, the NH2-terminal domain of STAT provides protein-protein interaction sites and is required for the interactions of dimer-dimer to form tetramers or oligomers, a coiled-coil domain, a DNA-binding domain, and the Src homology 2 (SH2) domain. The SH2 domain is required for the recruitment of STATs to phosphorylated receptors. The tyrosine residues modified by phosphorylation are Tyr 701 for STAT1, Tyr 690 for STAT2, Tyr 705 for STAT3, Tyr 693 for STAT4, Tyr 694 for STAT5 and Tyr 641 for STAT6, required for SH-phosphotyrosine interaction. The transactivation-domain on C-terminus is essential for co-factor interactions (3). The scheme of linear cluster of motifs in STAT proteins is depicted in Table II. The molecules of STATs are recruited by activated receptors of different cytokines in forms of homo- and heterodimers (4).
Table II.
Motif composition of STAT family members.
| Motif | |||||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| STAT | Protein interact domain | All-alpha domain | Tetramerization domain of TRPM | DNA binding domain | Linker | SH2 domain | TAZ2 binding domain |
| STAT1 | 2-119 | 144-308 | 266-286 | 322-458 | 481-558 | 578-622 | 715-738 |
| STAT2 | 2-122 | 146-308 | - | 321-456 | 482-557 | 578-648 | 783-838 |
| STAT3 | 2-119 | 145-313 | - | 326-464 | 488-565 | 584-651 | - |
| STAT4 | 2-119 | 144-308 | - | 321-454 | 478-554 | 573-627 | - |
| STAT5a | 3-123 | 146-324 | - | 336-469 | 492-574 | 595-669 | - |
| STAT5b | 2-123 | 146-324 | - | 336-469 | 492-574 | 595-668 | - |
| STAT6 | 2-113 | 182-242 | - | 277-413 | 436-518 | 538-616 | 655-847a |
Proteins of STAT family share most of the motifs except that only STAT1 possesses tetramerization domain of TRPM1, possessed by the TRP superfamily; the domain mapped in carboxyl terminus, TAZ2 binding domain is alternatively called TAD; it is absent in STAT 3, 4, 5a and 5b.
On the C-terminus, STAT6 possesses a C-terminal region rather than TAD. Based on KEGG entries hsa:6772-hsa:6778 (STAT1-STAT6). STAT, signal transducer and activator of transcription; TRPM, transient receptor potential melastatin; SH2, Src homology 2; TAD, transactivation domain.
STATs are transcription factors that latently exist in the cytoplasm; they are activated upon phosphorylation by Janus kinase (JAK). JAK is a family of intracellular tyrosine kinases, comprising JAK1-3 and tyrosine kinase 2 (TYK2) that non-covalently interact with the domain on membrane receptor or receptors of growth factor extending to the intracellular portion; they are triggered to activate when different cytokines bind their receptors. JAKs catalyze phosphorylation of tyrosine residue to transduce signals of a wide range of surface receptors on membrane. Upon phosphorylation, individual STAT molecules are dimerized and translocate to the nucleus, initiating transcription (Fig. 1). The important role of STAT in the regulation of immune response and other biological events has been revealed in gene-targeting studies (5,6).
Figure 1.
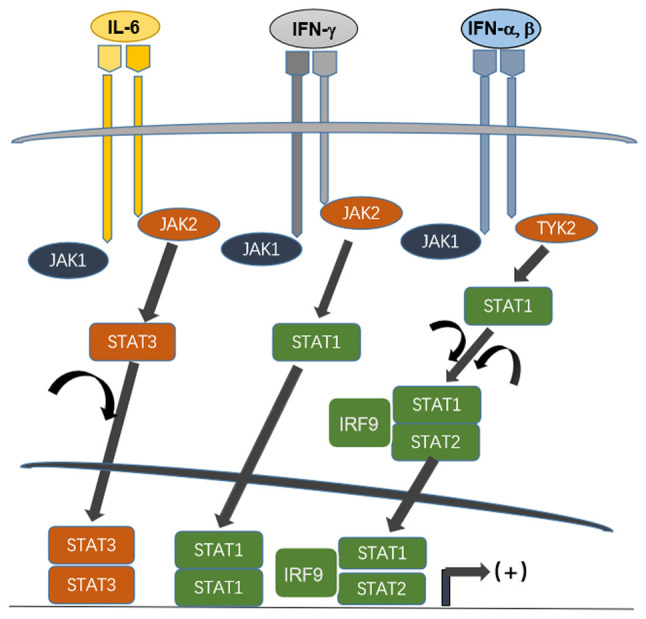
Scheme of action pattern of gene expression regulated by JAK/STAT pathway. The cellular responses elicited by three representing cytokines, IL-6, IFN-gamma and IFN alpha/beta are illustrated. The receptors associated JAKs are recruited and activated on receptor ligation, then different STATs are phosphorylated and homo- or heterodimerized. The dimerized STATs translocate to nuclei, binding regulatory elements upstream of coding portion of genes and initiate their transcription (11). JAK, Janus kinase; STAT, signal transducer and activator of transcription; IRF, interferon regulatory factor.
Three major mechanisms have been revealed to negatively regulate STAT signaling: i) The JAK and STAT dephosphorylation catalyzed by various tyrosine protein phosphatases, such T-cell specific 45 (7–9); ii) JAK inactivation by suppressor of cytokine signaling (SOCS) family proteins; and iii) interaction with protein inhibitor of activated STAT (PIAS), which has been identified as a negative regulator of STAT signaling. Previous studies have shown that PIAS proteins act on small ubiquitin, as modifier small ubiquitin-like modifier E3 ligase. In addition, biochemical studies have suggested that the list of proteins either positively or negatively regulated by the PIAS family through multiple mechanisms has rapidly expanded to >60, with the majority being transcription factors (10,11).
Upon phosphorylation, STATs are homo- or heterodimerized, translocate to the nucleus and coordinate with other coactivators of transcription or transcription factors, contributing to an increased initiation of transcription (Fig. 1). In cultured cells and experimental animals, the ligand-dependent activation of STATs has been observed as a transient process, lasting for minutes or hours. Molecules of STATs, and STAT1, 3 and 5 in particular, are persistently tyrosine-phosphorylated or tyrosine-activated. A number of experiments have revealed the importance of STAT activation for the control of growth. Data obtained strongly suggest its role in controlling cell cycle progression and initiation and occurrence of apoptosis. The JAK/STAT signaling pathway controls processes at the cellular level that is of essence in homeostasis. Alterations of this axis contribute to the progression of cancer, inflammatory and autoimmune diseases (11,12).
Evidence suggests the role of STAT family proteins in inducing and maintaining a pro-carcinogenic inflammatory microenvironment, for the initiation of malignant transformation and during cancer progression (13). The importance of inflammation in tumor initiation and malignant progression has been primarily subject to investigation. Inflammatory conditions can initiate or promote oncogenic transformation, genetic and epigenetic changes in malignant cells can also generate an inflammatory microenvironment that further supports tumor progression. In addition to tumor-promoting role of inflammation, the importance of immune responses and inflammatory mediators have been underscored in suppressing tumorigenesis and tumor growth (14–16). STAT3 and, to some extent, STAT5 and STAT6 are involved in inhibiting antitumor immunity. In view of the effector molecules of cell cycle and apoptosis regulated by JAK-STAT, it suggests that the pathway also contributes to carcinogenesis through proliferation promotion and programmed cell death potentiation (13).
Modulation of the antiviral immunity mediated by JAK/STAT pathway contributes to the signaling of interferons, notably type I. The JAK-STAT pathway is also implicated in the pathogenesis by two tumorigenic human herpesviruses, Epstein-Barr virus (EBV) and Kaposi sarcoma-associated herpesvirus (KSHV).
The gamma group of human herpesviruses contains two lymphotropic members, EBV and KSHV. In their genome, genes are located next to the terminal repeat region code for membrane proteins to transduce cellular signals. The coding products termed terminal membrane proteins (TMPs) interact with the cellular proteins involved in the activities of non-receptor protein tyrosine kinases and tumor necrosis factor receptor-associated factors. It has been demonstrated that persistent STAT activation may be implicated in EBV-driven tumorigenesis in immunocompetent individuals (17), as observed in nasopharyngeal carcinoma, tightly associated with EBV infection and endemic among certain regions in the world, including southern China and Southeast Asia.
When the cytokine receptors are activated, JAKs catalyze phosphorylation of tyrosine residue to transduce signals of a wide range of surface receptors on membrane. As aforementioned, the activity of STATs is modulated by a group of inhibitor PIAS. PIAS1 and PIAS3 block the effects of STAT1 and STAT3 respectively; it has been reported that the entry of lytic cycle is prevented if STAT3 is downregulated by PIAS3. The high expression of STAT3 has been revealed to facilitate the entry of lytic cycle of EBV (Fig. 2A) (18).
Figure 2.

Effect of STATs pathway interacting with herpesviruses EBV and KSHV during lytic cycle and interferon signaling. (A) High level of STAT3 activated by IL-6 on binding with its cognate receptor regulates EBV entry to lytic cycle; it is inhibited by the protein PIAS3. (B) The JAK/STAT pathway downstream of type I interferon counteracted by herpesviruses EBV and KSHV encoded products; IFNAR binds IFN-alpha and beta, then recruited JAK1 and TYK2, the latter is inhibited by a EBV latent protein, LMP1, while the heterotrimer STAT1-STAT2-IRF9 formed after activation of JAK1/TYK2 is inhibited by EBV kinase BGLF4 and by KSHV encoded vIRF2. STAT, signal transducer and activator of transcription; EBV, Epstein Barr virus; KSHV, Kaposi sarcoma-associated herpesvirus; PIAS3, protein inhibitor of activated STAT3; JAK, Janus kinase; IFNAR, interferon alpha/beta receptor; TYK, tyrosine kinase; vIRF, viral interferon regulatory factor.
An EBV encoded serine/threonine-protein kinase BGLF4 has been shown to enhance the production of extracellular viral particles during EBV lytic replication (19,20). BGLF4 effectively suppresses the activities of the polyinosinic:polycytidylic acid poly (I:C)-stimulated IFN-beta promoter and responsive element of interferon regulatory factor (IRF) 3. Moreover, BGLF4 represses expression of endogenous IFN-beta mRNA stimulated by the poly (I:C) and the phosphorylation of STAT1 at Tyr701 (21). The genetic coding product, viral IRF2 of KSHV/HHV-8 inhibits signaling of interferon as reported (22). The knowledge of Herpesviral interfering of JAK/STAT is summarized in Fig. 2B with the employment of the pathway mapping tool from the Kyoto Encyclopedia of Genes and Genomes database (https://www.genome.jp/entry/K11220; map05167 KSHV infection; map05169 EBV infection).
Several non-receptor tyrosine kinases, such as SH2 and the protooncogene homologous to retroviral ABL transforming gene, forming a malignancy-related cytogenetic aberration known as the Ph chromosome (19) have been found to phosphorylate STATs. It has been revealed that sorafenib, a multikinase inhibitor, inhibits cell proliferation and triggers apoptosis at much lower concentrations in cells in which the fused protein BCR/ABL is expressed (23). In Ph+ leukemia cells, apoptosis is induced by sorafenib as evidenced by that caspase-3 have been revealed and drops of mitochondrial membrane potential have been specifically identified in cells harboring BCR/ABL.
JAK/STAT pathway analysis has revealed that elements of proximal signaling such as IFN receptor 1, JAK1 and TYK2, are disrupted by SARS-CoV-2, leading to the inhibition of IFN-induced STAT phosphorylation. The mechanisms underlying STAT inhibition have been explained to uncover an immune evasion strategy against SARS-CoV-2 and the pathway involved could be targeted by anti-coronavirus therapy (24).
Coronavirus disease of 2019 (COVID-19) caused a severe pandemic in the next three years. The clinical entity of COVID-19 has been intensely studied (25), and it has been found that the infection of lower respiratory tract and multiple internal organs is characterized by cytokine release syndrome (CRS) with an increased production of interleukins (ILs), such as IL-6, IL-2, IL-7 and IL-10. Some of the cytokines involved in the pathogenesis use a distinct intracellular JAK-mediated signaling pathway. The inhibition of JAK, therefore, presents a marked therapeutic potential for CRS, which is known as a common cause of adverse clinical outcomes for COVID-19 (26,27).
A unique panel of cell culture models profiling proteomic responses to SARS-CoV-2 infection has been established and described using lung, liver, intestine, kidney, heart and brain cells. The system enables the identification of proteins and pathways in the cells that are widely targeted by SARS-CoV-2. Among the pathways traced, the most notable was the JAK-STAT signaling cascade, the key component in the interferon (IFN) response pathway. The inhibition of STAT phosphorylation combined with its nuclear translocation has been demonstrated in cells infected with SARS-CoV-2 (28–31).
JAK/STAT pathway analysis has revealed that elements of proximal signaling such as IFN receptor 1, JAK1 and TYK2, are disrupted by SARS-CoV-2, leading to the inhibition of IFN-induced STAT phosphorylation.
The JAK/STAT pathway has been implicated in carcinogenesis and viral pathogenesis, notably in pulmonary involvement from COVID-19. Progress in this field will be reviewed in the present study.
2. Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway is implicated in carcinogenesis and malignancy progression
Malignancy is achieved by the functional alteration of two types of cancer-related genes, that is, oncogenes and tumor suppressor genes (TSGs). They acquire oncogenic potential in two ways; gain-of-function for oncogenes and loss-of-function for TSGs.
Clinically, patients with colorectal carcinoma or other cancers get a prolonged and improved antitumor immunity from immunotherapy when STAT1 is expressed in the nucleus (32). When JAK1 is dissociated from the receptor of IFN-γ, the phosphorylation of STAT1 inhibition follows; concomitantly, STAT1 is translocated to the nucleus to promote tumor growth through the inhibition of apoptosis (33).
Small RNAs, such as microRNAs, which bind to the untranslated region of mRNA, may directly or indirectly impact the expression of STAT1. It has been revealed that STAT1 overexpression significantly repressed the expression of miR-181a (34).
STAT1 is phosphorylated at the Tyr 701 residue by activated JAK. Upon phosphorylation, STAT1 is then homodimerized and translocates to the nucleus, where it binds to specific gamma activated sequences (GAS), to stimulate the expression of IFN-stimulated genes (ISGs) upon the initiation of transcription (35–37). In some types of cells, STAT1 is also phosphorylated at residue Ser727, resulting in an enhancement of its transcriptional activity (38). During the induction of transcription factor IFN regulatory factor 1 (IRF1) expression is triggered by IFN-γ, IRF1 interacts with STAT1 and recruits the STAT1-IRF1 complex to the elements of GAS (32,33,39,40). Herpesvirus and coronavirus infections induce IFN-γ, which regulates the activation of the JAK/STAT pathway. It has been demonstrated that, in ovarian cancer (OC) cells, IFN-γ-induced PD-L1 expression is dependent on JAK1, STAT1 and IRF1 signaling. Furthermore, IFN-γ increases acetylation of the human PD-L1 promoter in OC and other cancer cells, and STAT1, Ser727-p-STAT1 and IRF1 are recruited to the promoter.
In OC, the elevated PD-L1 expression level is associated with poor prognosis (31). It has been further demonstrated that the levels of Tyr 701-p, STAT1 and Ser 727-p-STAT1 were increased in OC tissues (41). In OC cells, IFN-γ was found to increase STAT1, Tyr 701-p-STAT1 and Ser727-STAT1 levels when the expression of PD-L1 was increased (42).
STAT3 and other members of the STAT family are known to be oncogenic, and the JAK-STAT3 pathway is known for its potential to promote tumor cell proliferation, survival, invasion, angiogenesis and immune suppression. Previously, it has been revealed that the signaling of JAK/STAT3 contributes to inflammation-mediated carcinogenesis, maintenance of cancer stem cell (CSCs) phenotypes and of pre-metastatic niches (35,43–49). CSCs are a cancer cell subpopulation that manifests a phenotype of stem cells; they have the ability of sustaining self-renewal, resembling normal stem cells.
Upon hetero- or homodimerization, the STAT molecules bind fragments with regulatory function upstream to coding portion in genomic sequence and initiate the transcription of target genes. JAK2 activation by trans- or autophosphorylation induces the cascade of activating downstream molecules, including STATs. Experimental findings have shown that both extrinsic (environmental) and intrinsic pathways link cancer. The intrinsic pathways originate from genetic and epigenetic events in the tumor cells. STATs, notably STAT3, are crucial for both extrinsic and intrinsic pathways of inflammation (50,51). Due to its oncogenic potential, which promotes tumor cell proliferation and survival, STAT3 drives the transition from chronic inflammation to malignancy (52). In a variety of hematological malignancies, STAT5 and STAT6 are persistently activated (52–55). In cases of chronic myelogenous leukemia, STAT5 has been demonstrated to be persistently activated in malignant cells in the presence of the classic chromosomal translocation, BCR/ABL (56).
The phosphorylated form of STAT5 enhances the transcription of regulatory factors in apoptosis, such as Bcl-X, and cell cycle progression, such as cyclin D1, D2, 7E, p21Cip and other inhibitors of cyclin dependent kinase (CDK) (57,58). Mutations within the pseudokinase domain of JAK2, from valine to phenylalanine at residue 617 (JAK2 V617F) is a major activating mutation for JAK2 signaling (59,60). The mutation pattern is illustrated in Fig. 2. JAK2 V617F is a frequent mutation in myelo-proliferation disorder with absence of BCR/ABL juxtaposition caused by the cytogenetic aberration of t (9:22) observed in the Ph chromosome, in which some kinase activity is affected (61).
JAK2 engages the pathway to activate multiple STAT family members in addition to STAT3. Hyperproliferation, which is induced by JAK2 V617F involves the activation of STAT3 together with the downstream factors it targets (62). It has been reported that the inhibition of cyclin D2 transcription and enhancement of p27kip1 account for the growth arrest caused by the inhibition of JAK2 V617F (63). The p27kip1 downregulation caused by the expression of JAK2 V617F has been found to be associated with the STAT5-induced expression of Skp2, suggesting that p27kip1 degradation could result from the overexpression of the ubiquitin ligase Skp2 directed at p27kip1 (62). p27kip1 has hence been identified as a substrate of JAK2 (64).
Mutational activation frequently contributes to malignant proliferation through events such as histone marker modification. In fact, STAT3 is classified as oncogenic (14). Its engagement directing to hallmarks of cancer, such as the proliferation, apoptosis evasion and angiogenesis of tumor cells, has been reported (65). The activities control the expression of pro-tumorigenic genes, such as cyclins D1 and D2, c-Myc, MCL1 apoptosis regulator, survivin/baculoviral IAP repeat containing 5 (BIRC5), B-cell lymphoma-extra large, hepatocyte growth factor, hypoxia induced factor-α (HIF-1α and vascular endothelial growth factor A (VEGF) (66–70).
Alterations in STAT3 expression affects the host proliferation, as well as the drug resistance of cancer cells. When treated with erlotinib, p-STAT3 levels are increased in epidermal growth factor receptor (EGFR)-mutated non-small cell lung cancer (NSCLC) cell lines and drug resistance is induced. Conversely, the RNA interference-mediated knockdown of STAT3 enhances the sensitivity of cells to erlotinib (71). Of note, the findings could be extended to NSCLC, whose tumorigenesis is driven by KRAS mutation. When treated with mitogen-activated protein kinase inhibitor, NSCLC lines harboring a KRAS mutation exhibit an increased level of p-STAT3, and drug resistance is induced (72).
NSCLC lines carrying EGFR and KRAS mutations secrete cytokines, such as IL-6, which contribute to the underlying mechanisms of STAT3 activation in NSCLC-related gp130/JAK signaling. Autocrine JAK signaling-dependent STAT3 activation is followed by the engagement of gp130, thereby promoting tumor cell survival (71–73). In this context, in vivo and in vitro tumor cell growth is decreased by knocking down IL-6 or JAK1/2 (67,71,73).
Among the signaling pathways of inflammation associated with tumor development, STAT3 phosphorylation plays a key role (74). STAT3, an oncogenic transcription factor is usually constitutively activated in prostate and several other human cancers, such as breast cancer (75–77). Due to the important role it plays in cell survival and proliferation (33), STAT3 has been designated as a major target of anticancer therapy. In hepatocellular carcinoma cells, STAT3 also exerts anti-apoptotic and pro-proliferation effects (78). Indeed, in obesity (79) and fatty liver-associated inflammation (80), the IL-6/STAT3 activation is regarded as a ‘bona fide’ tumor promoter.
The central role of STAT3 is to promote and maintain the stem cell phenotype. In this population, STAT3 inhibits tumor progression. In stem cells, it has been noted that histone (H) methylation at lysine residues catalyzed by enhancer of zester homolog 2 (EZH2) regulates STAT3 (47). As the catalytic subunit of polycomb repressive complex 2, EZH2 has been known to exhibit oncoprotein-like activity to bi- or trimethylate a repressive mark on H3, lysine 27 of H3, to repress the transcription of the target gene (81). Negative cell cycle regulators, such as CDK inhibitor 1A or p21Waf1/Cip1 are downregulated. Repressive effects on these molecules lead to the promotion of malignancy through its activity of lysine transferase on histones; the target genes that are implicated in cell proliferation, apoptosis and regulation of cell cycle progression include p15 INK4b-ARF, p16 INK4A, tumor necrosis factor related apoptosis inducing ligand and Kruppel-like factor 2 (82).
The overexpression of EZH2 disrupts cell proliferation, apoptosis, migration and invasion. High EZH2 level has also been found to be associated with an unfavorable clinical outcome in cancers (83–86). It has been revealed that EZH2 promotes malignancy in NSCLC and EZH2 overexpression and is associated with poor prognosis in patients with NSCLC. Data have suggested that EZH2-siRNA increases the expression of p15INK4B p21Waf1, and p27Kip1 in NSCLC lines. Univariate analysis revealed that NSCLC patients with a high EZH2 expression have a significantly inferior overall, cancer-specific and disease-free survival. It has been demonstrated that overexpressed EZH2 is essential for NSCLC progression, and the levels of EZH2 may serve as a prognostic predicator of the same cancer (87).
EZH2 also methylates the non-histone protein STAT3 and is involved in the acetylation of STAT3 which is catalyzed by acetyltransferase p300, thus playing a role in the regulation of the formation of a transcription complex bound to the target gene promoters.
The authors have previously demonstrated that a zinc finger motif containing protein ZMYND10 encoded by a frequently lost TSG found in a variety of human tumors induces the trimethylation of a histone repressive mark (H3K9) and downregulates cyclins that promote cell cycle entry (88). Forkhead box A1 (FOXA1) is a transcription factor which plays a role in the translation of the epigenetic signatures into enhancer-driven transcriptional program characterized by cell type specificity; FOXA1 is differentially recruited to chromatin fragments and downregulated in several cancers. The positive association between FOXA1 and CDKN2A/p16 INK4a, a negative regulator of the cell cycle that can be observed in prostate and breast cancers weakly expressing EZH2, epigenetically represses CDKN2A. It has been analyzed and revealed in prostate and breast cancer cells that high expression of FOXA1 antagonizes the EZH2-mediated repression of CDKN2A, as further depletion of FOXA1 reverts the effect of CDKN2A de-silencing caused by the inhibition of EZH2. Concomitantly, the depletion of EZH2 suppresses the cancer cell cycle progression, while the presence of FOXA1 and CDKN2A optimizes this regulation (89). The modulation of proliferation-related molecules by tumor suppressors through histone and non-histone protein modification remains to be elucidated.
3. High level of STAT3 contributes to refractory state to lytic cycle switch in Epstein-Barr virus (EBV) and Kaposi sarcoma-associated herpesvirus (KSHV)
Upon viral infection, IFNs are secreted and the JAK/STAT pathway is activated, which leads to the promotion of the transcription of ISGs to defend against viral infection (18). Type I IFNs bind to the IFN-activated receptor IFNAR1, leading to JAK activation. The activated JAK in turn phosphorylates STAT1 and STAT2, enabling them to form heterodimers to bind with IRF9, forming the complex of ISG factor 3. The complex is further linked to IFN-stimulated response elements and induces the transcription of ISGs (90).
Herpesviruses are characterized by the adoption of two distinctive patterns of infection in their life cycle: Latent and lytic infection. To date, two gamma-herpesviruses among eight known human herpesviruses, the lymphotropic human herpesvirus 4 (HHV-4) or EBV and HHV-8 or KSHV are classified as tumorigenic viruses. These viruses may harbor in the body of the host throughout their life. During their latency phase, the viruses express limited genome products, without rupture of the host cells. The mechanisms underlying the switch from a latent to a lytic cycle in EBV and KSHV have been studied in B cell lines cultured in vitro.
In EBV, the activation of the lytic cycle is controlled by two virally encoded transcription factors with Zip motif; Z EB replication activator (ZEBRA) and replication and transcription activator (Rta). The viral bZIP transcription factor ZEBRA/Zta encoded by BamHI Z left frame 1 (BZLF1) regulates this cycle by binding to two classes of ZEBRA response elements. ZEBRA is a homodimeric protein related to the activating protein 1 (AP-1) family of bZIP transcription factors (91). It regulates the EBV infection cycle by contributing both in establishing viral latency and triggering lytic replication. During pre-latency, ZEBRA is transiently expressed at this time course, the EBV genome is hypomethylated and it is critical for promoting the proliferation of quiescent naive and memory B cells. During latency, when the EBV genome is methylated, the expression of ZEBRA activates a second viral transcription factor, which functions along with ZEBRA to trigger lytic replication (92,93).
The lytic cycle activator of KSHV is the open reading frame 50 encoded Rta homolog (93). Following the switch to a lytic cycle, they express all of the genomic products, ending with the lysis of the host cells and establishing infection when entering new cells (94). The lytic cycle has been linked to the oncogenic potential of the viruses.
The molecular mechanisms underlying the latent-to-lytic cycle switch with the ultimate aim of utilizing anti-viral therapy by increasing the number of cancer cells latently infected with the virus being converted to the lytic phase and thereby becoming sensitive to the antiviral agents. In fact, oncolytic therapy faces a major challenge due to the high cells fractionation rates in populations of latently infected cells, which are resistant to agents inducing the lytic cycle (95).
The lytic cycle of EBV is characterized by the expression of a cascade of regulated genes; these include intermediate-early, early and late genes. Strategies for inducing lytic infection of EBV in tumor cells are investigated as a potential therapy against EBV-related tumors; the latent-to-lytic cycle switch of EBV in infected B and epithelial cells is regulated by a panel of cellular and viral proteins and the manner in which lytic viral reactivation is harnessed may be utilized in the therapeutic approach. When the viral lytic cycle is triggered, a number of EBV-encoded proteins are expressed, including ZTA/ZEBRA, EB1, BZLF or Zta and Rta, encoded by immediate-early genes that stimulate the expression of early and late genes of EBV. The early genes code for proteins required for the replication of EBV DNA, whereas the late genes code for viral structural proteins to package infectious viral particles. In addition to BZLF1-encoded transcription factors, BZLF1 transcription activator (Zta) and Rta, the expression of lytic EBV genes is modulated by cellular proteins (96,97). RNA transcribed from BZLF1 is highly upregulated in lytic cell populations compared with refractory or untreated cells. This result has been confirmed as an effective indicator for separating refractory and lytic EBV-harboring cells.
It has been observed that a high level of STATs maintains the latent infection state of EBV. An inhibitor of activated STAT1, PIAS1, is a factor capable of restricting EBV by inhibiting transcription factors of viral and cellular origins. It has been demonstrated that PIAS1 inhibits the IRF-8 mediated activation of lytic genes through their molecular interaction (98). In the cell populations that are refractory to the induction of the lytic cycle, the preferential upregulation of STAT3 and Fos proto-oncogene, AP-1 transcription factor subunit has been observed and the expression of both factors is increased in folds in comparison with untreated cells (98).
In EBV harboring HH516-16 cells, the regulation of the lytic cycle has been investigated. When latently infected with EBV, cells highly express STAT3 protein, predominantly in its unphosphorylated form. When exposed to sodium butyrate (NaB), an agent that induces the lytic cycle and a prototype of inhibitor of histone deacetylase, entry to the lytic cycle is triggered. HH516-16 cells are also treated with IFN-γ to determine the possible phosphorylation at residue Y705 induced by the treatment, or if the pathway is detectable (88). Since the STAT3 transcript is increased primarily in refractory cells, it was examined whether the increased STAT3 protein levels correspondingly existed in the population of refractory cells. An increase in STAT3 protein in these populations compared with the untreated cells in a time-dependent manner was observed following NaB treatment. In the subpopulation of lytic cells, STAT3 protein was not significantly upregulated.
It has been observed in KSHV/HHV-8 that periodic switching from the latent to the lytic cycle contributes to an orderly expression of a large panel of viral genes to produce infectious virions (18). Clinico-epidemiological studies have revealed that the activation of the KSHV lytic cycle critically contributed to the pathogenesis of KS, pleural effusion lymphoma and multicentric Castleman disease (99–104). The lytic activation of KSHV/HHV-8 is also correlated with the progression and prognosis of the diseases.
4. Severe atypical respiratory syndrome-coronavirus-2 (SARS-CoV-2) viral genomic coding products stimulate transcription factors to synthesize pro-inflammatory mediators
COVID-19, the disease caused by SARS-CoV-2, affects the lining epithelia, leading to severe respiratory disease in humans. It also infects other viscera, including the liver and kidneys, by engaging with multiple intracellular signaling pathways, leading to the production of mediators of inflammatory response, and hence tissue damage. It was a remarkable feature of pathogenesis when widespread thrombosis with microangiopathy in the blood vessels of the lungs was identified during the clinical course of COVID-19 (105).
SARS-CoV-2 has a genome containing a single stranded RNA measuring 30 kb. Two open reading frames (ORFs), ORF 1a and 1b synthesize 27 non-structural proteins upon translation. ORFs 3, 6, 7a, 7b, 8, 9a, 9c, 10 and 14 code for spike (S), envelope (E) and nucleocapsid (N) proteins, as well as several accessory proteins. The alignment of the coding region is demonstrated in Fig. 3. The accessory proteins encoded by the aforementioned ORFs play an important role in the pathobiology attributed by the virus (106). These proteins assist the virus to establish infection to the susceptible host cells and hijack the cellular machinery of the host for particle assembly, amplification and pathogenesis of the virus. During the infection, the S protein binds to the receptor of angiotensin-converting enzyme 2 on the host cells (107); subsequently the cellular transmembrane protease serine 2 (TMPRSS2) cleaves the viral S protein, resulting in two subunits, S1 and S2. The two fragments fuse with viral and cellular membranes and initiate virus internalization (107) (Fig. 4).
Figure 3.

Linear clustering of JAK2 motifs and role of abnormal JAK/STAT signaling in carcinogenesis. (A) The JAK2 molecule contains motifs of FERM required for lower Michaelis constant (Km; the concentration of saturated substrate in case of the half of maximum catalytic speed of an enzyme being reached) of JAK2 (V617F) towards substrates, SH2, pseudo-PTK and the functional PTK. The pseudo-PTK domain starting with amino acid residue 604 is identical in human and mouse. The mutation on 617 position of amino acid sequence is responsible for hemopoietic disorders described. (B) The impact of aberrant JAK/STAT pathway in carcinogenesis. High expression of STAT1 downstream of IFN-g signaling induces PD-L1 expression, enabling the tumor cells to evade host antitumor immunity. Mutant JAK2 with the lesion of V617F alters STAT3 signaling and fused protein BCR/ABL expressed from the translocated gene on Ph1 chromosome induces abnormal STAT5 signaling to enhance transcription of cyclins D1, D2, E and anti-apoptotic regulator Bcl_xL, to promote cancer initiation. JAK, Janus kinase; STAT, signal transducer and activator of transcription; FERM, 4.1/ezrin/radixin/moesin; SH2, Src homolog 2; PTK, protein tyrosine kinase; PD-L1, programmed cell death ligand 1; Ph, Philadelphia; Bcl xL, B-cell lymphoma-extra large.
Figure 4.

Schematic pattern of genomic composition of SARS-CoV-2. (A) The arrangement of exons within SARS-CoV-2 genome is indicated and referred to the text. (B) The genomic products of SARS-CoV-2 target to several components of the axis of JAK/STAT pathway. SARS-CoV-2, severe acute respiratory syndrome coronavirus type 2; JAK, Janus kinase; STAT, signal transducer and activator of transcription; IFNAR, interferon alpha/beta receptor; IRF, interferon regulatory factor; ORF, open reading frame.
Previously, the viral peptides binding with major histocompatibility complex-1 (MHC-1) molecules to downregulate antiviral immune response have been described in human immunodeficiency virus type 1 and KSHV (108,109). A similar interaction between SARS-CoV-2 through ORF8 protein and human proteins has been identified, considered to be essential for the SARS-CoV-2-mediated immune evasion by MHC-1 suppression (110).
The coding products of the SARS-CoV-2 genome have been investigated in relation to their pathogenesis of host internal organs through engagement of intracellular pathways to trigger production of pro-inflammatory mediators. NF-κB proteins are a set of transcription factors that regulate inflammation and cell death. The transcription factors comprise several subunits, such as NF-κB 3/p65, together with negative regulator I-κB, which dimerize with the subunits to repress their activities. Modification on specific amino residues, such as phosphorylation, leads to the NF-κB activation. The inhibition of the activity of these transcription factors is removed on their degradation through phosphorylation and then ubiquitination on their amino (N) terminus. HK-2 cells derived from the proximal tubular epithelium are susceptible to SARS-CoV-2 infection. The effect of ORF3A expression on the effective activation of the NF-κB signaling pathway by increasing the phosphorylation of subunit p65 has been studied; it has been revealed that the SARS-CoV-2-encoded protein stimulates phosphorylation on positions Ser 536 and Ser 276 of the p65 protein. ORF3A also increases the amount of cleavage of caspase-3 (111), suggesting its role in triggering apoptosis, as identified in other cell lines. It has been observed that the mRNA levels of the downstream targets of TNF-α and IL-6 are increased in HK-2 cells by ORF3A. TNF-α is a cytokine that activates the NF-κB pathway and causes tubular cell injury, confirming the modulation of NF-κB by ORF3A. STAT3 phosphorylation on residue 705 leads to functional activation of STAT3 induced by the cytokines TNF-α and IL-6, the expression of STAT3 is increased by ORF3A expression. It has also been suggested that TRIM59 expression is increased by SARS-CoV-2 infection in HK-2 cells. It has been noted that SARS-CoV-2 has a similar antagonistic activity to that of IFN (112). It has therefore been hypothesized that the pathophysiology of SARS-CoV-2 infection is related to the effects of the virus on IFN and JAK/STAT signaling. It has been proposed that COVID-19 is a disease stemming from the dysregulation of STATs induced by SARS-CoV-2, leading to a catastrophic cascade of internal organ failure.
After the entry of susceptible cells, SARS-CoV-2 expresses the proteins non-structural protein 1 and ORF6 to inhibit the activity of STAT1 (113). While the activity of STAT1 is restricted, STAT3 becomes the predominant form in signaling and triggers downstream pathways. SOCS1 and SOCS3, are factors involved in the negative feedback of type I IFN signaling. They are induced by both STAT1 and STAT3 and inhibit the activity of JAKs (114). The binding of STAT1 and STAT3 to DNA is prevented by PIAS1 and PIAS3, respectively (115). When STAT3 is aberrantly activated and uncoupled from SOCS regulation, the role of PIAS3 becomes crucial in regulating the activity of STAT3. Protein tyrosine phosphorylases exert regulatory activities on activated JAKs and STATs (116), but their role in the control of viral infection requires further elucidation. In lungs infected with SARS-CoV-2, EGFR levels are induced by acute lung injury or reduced activity of STAT1; STAT3 is activated by ligation or viral protein binding of EGFR (117). PIAS3 normally inhibits STAT3 activity; however, during the viral infection, PAI-1 is produced (118) and an escalating cascade in the PAI-1/STAT3 axis is in turn established.
Immunomodulation and corticosteroids for intervention against cytokine production have been suggested as a means to limiting or minimizing the hyperactive response of inflammation (119). Immunoregulatory antagonists of IL-1 or IL-6 and of JAK inhibitors have so far been examined (120). Clinical trials on antagonists and antibodies targeting several cytokines at initial stages have produced promising results for the treatment of COVID-19, which is characterized by abnormalities in the expression of cytokines. The inhibition of JAK has been proven to have the potential to suppress inflammatory response, and endocytosis has been shown to mediate the surface viral receptor during the pathogenesis of COVID-19 (121). Ruxolitinib, an inhibitor of JAK1 and JAK2 is an orally administered drug approved for the treatment of myelofibrosis and polycythemia vera in Europe (122). Several newly developed agents have been tested to determine their efficacy in treating COVID-19, including Baricitinib (123), Clazakizumab, Siluximab and Anakinra (124).
A recent study described a COVID-19 patient suffering from systemic sclerosis (SSc) with pulmonary fibrosis (125). It has been observed that the respiratory function had rapidly improved 10 days after she was administered Ruxolitinib. The reduction in pulmonary fibrosis was compared before and after the diagnosis of COVID-19. JAK/STAT signaling has been revealed to be involved in pathogenesis and fibrosis modulation in SSc patients (125). The administration of ruxolitinib is therefore recommended as a new therapeutic strategy for patients with COVID-19 and lung fibrosis (122). To date, numerous clinical trials are ongoing to evaluate the effectiveness of biopharmaceutical drugs such as blockers of IL-1, inhibitors of IL-6 and JAKs in anti-COVID-19 therapy. The rationale behind pathogenesis and state-of-the-art therapeutic approach for blocking hyperinflammation have been described (126).
The therapeutic efficacy of Vitamin (Vit) D in COVID-19 has been tested. Ongoing studies indicate the antiviral effect of Vit D supplements in the protection against SARS-CoV-2 infection, as well as its ability to reduce the risk of other viral and bacterial infections, including influenza (127–129).
Vit D is a lipid soluble vitamin that maintains calcium levels in the body. Vit D binds to the cognate receptor, Vit D receptor (VDR) to form a complex and in turn binds to the regions of a promoter on a genomic DNA sequence to modulate target gene expression (130). In addition, Vit D binds to VDR through a non-genomic activity, activates several intracellular signaling pathways and directly regulates the transcription of multiple genes, including immune response-related genes. The ability of Vit D in vitro, in vivo and in patients with severe COVID-19 to enhance host IFN-a/b signaling have been reported (131). Higher levels of JAK/STAT signaling pathway activity have been observed with significantly higher antiviral ISGs at both the gene and protein levels; the revealed regulatory role of Vit D on IFN type I suggests the importance of maintaining a normal level of Vit D to prevent and possibly treat SARS-CoV-2 infection. Additional mechanistic studies are needed to fully elucidate the antiviral activity of Vit D against COVID-19. The recent laboratory findings support the promising use of Vit D as a therapeutic agent for COVID-19.
5. Conclusions
As a kinase molecule non-covalently associated with the intracellular portion of membrane integral surface receptor of ILs and other cytokines, JAK is activated upon ligation of the receptors to activate a downstream cascade, leading with STATs. The pathways involved are implicated in carcinogenesis, viral infection and host antiviral defense against the infection of human herpesviruses and SARS-CoV-2. The elucidation of the JAK/STAT pathway would shed light into viral pathogenesis and precise targeting therapy against cancers, herpesviruses and coronaviruses.
Acknowledgements
The authors would like to thank Mr. Jiaqing Luo and Ms. Yuan Zhang, undergraduates of biotechnology, Guangdong Medical University, China for helpful suggestions on the content of the manuscript.
Glossary
Abbreviations
- JAK
Janus kinase
- STAT
signal transducer and activator of transcription
- EBV
Epstein-Barr virus
- KSHV
Kaposi sarcoma-associated herpesvirus
- ISG
IFN-stimulated gene
- COVID-19
coronavirus infectious disease 2019
- SARS-CoV-2
severe atypical respiratory syndrome-coronavirus-2
Funding Statement
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
WL, YZ and SJS wrote draft of the manuscript. SJS and PT assisted with figures preparation, searched for references and revised the manuscript. XZ, GLH, BZ and ZH conceived ideas of the study and corrected the drafts. Data authentication is not applicable. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Malemud CJ. The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther Adv Musculoskelet Dis. 2018;10:117–127. doi: 10.1177/1759720X18776224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ihle JN. The STAT family in cytokine signaling. Curr Opin Cell Biol. 2001;13:211–217. doi: 10.1016/S0955-0674(00)00199-X. [DOI] [PubMed] [Google Scholar]
- 3.Levy DE, Darnell JE., Jr Stats: Transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 4.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 5.Simoncic PD, Lee-Loy A, Barber DL, Tremblay ML, McGlade CJ. The T cell protein tyrosine phosphatase is a negative regulator of Janus family kinases 1 and 3. Curr Biol. 2002;12:446–453. doi: 10.1016/S0960-9822(02)00697-8. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Gao Z, Jiang F, Yan H, Yang B, He Q, Luo P, Xu Z, Yang X. JAK-STAT signaling as an ARDS therapeutic target: Status and future trends. Biochem Pharmacol. 2023;208:115382. doi: 10.1016/j.bcp.2022.115382. [DOI] [PubMed] [Google Scholar]
- 7.Chen CW, Chang YH, Tsi CJ, Lin WW. Inhibition of IFN-gamma-mediated inducible nitric oxide synthase induction by the peroxisome proliferator-activated receptor gamma agonist, 15-deoxy-delta 12,14-prostaglandin J2, involves inhibition of the upstream Janus kinase/STAT1 signaling pathway. J Immunol. 2003;171:979–988. doi: 10.4049/jimmunol.171.2.979. [DOI] [PubMed] [Google Scholar]
- 8.Wiede F, Shields BJ, Chew SH, Kyparissoudis K, van Vliet C, Galic S, Tremblay ML, Russell SM, Godfrey DI, Tiganis T. T cell protein tyrosine phosphatase attenuates T cell signaling to maintain tolerance in mice. J Clin Invest. 2011;121:4758–4774. doi: 10.1172/JCI59492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, Zhu W, Tremblay M, David M, Shuai K. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol. 2002;22:5662–5668. doi: 10.1128/MCB.22.16.5662-5668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol. 2003;3:900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- 11.Shuai K. Modulation of STAT signaling by STAT-interacting proteins. Oncogene. 2000;19:2638–2644. doi: 10.1038/sj.onc.1203522. [DOI] [PubMed] [Google Scholar]
- 12.Yan Z, Gibson SA, Buckley JA, Qin H, Benveniste EN. Role of the JAK/STAT signaling pathway in regulation of innate immunity in neuroinflammatory diseases. Clin Immunol. 2018;189:4–13. doi: 10.1016/j.clim.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450:903–907. doi: 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- 15.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 16.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6:836–848. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- 17.Chen H, Lee JM, Zong Y, Borowitz M, Ng MH, Ambinder RF, Hayward SD. Linkage between STAT regulation and Epstein-Barr virus gene expression in tumors. J Virol. 2001;75:2929–2937. doi: 10.1128/JVI.75.6.2929-2937.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang K, Lv DW, Li R. Cell receptor activation and chemical induction trigger caspase-mediated cleavage of PIAS1 to facilitate epstein-barr virus reactivation. Cell Rep. 2017;21:3445–3457. doi: 10.1016/j.celrep.2017.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang JT, Doong SL, Teng SC, Lee CP, Tsai CH, Chen MR. Epstein-Barr virus BGLF4 kinase suppresses the interferon regulatory factor 3 signaling pathway. J Virol. 2009;83:1856–1869. doi: 10.1128/JVI.01099-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Guo W, Long C, Zhou H, Wang H, Sun X. The split Renilla luciferase complementation assay is useful for identifying the interaction of Epstein-Barr virus protein kinase BGLF4 and a heat shock protein Hsp90. Acta Virol. 2016;60:62–70. doi: 10.4149/av_2016_01_62. [DOI] [PubMed] [Google Scholar]
- 21.Li R, Wang L, Liao G, Guzzo CM, Matunis MJ, Zhu H, Hayward SD. SUMO binding by the Epstein-Barr virus protein kinase BGLF4 is crucial for BGLF4 function. J Virol. 2012;86:5412–5421. doi: 10.1128/JVI.00314-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuld S, Cunningham C, Klucher K, Davison AJ, Blackbourn DJ. Inhibition of interferon signaling by the Kaposi's sarcoma-associated herpesvirus full-length viral interferon regulatory factor 2 protein. J Virol. 2006;80:3092–3097. doi: 10.1128/JVI.80.6.3092-3097.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aurer I, Butturini A, Gale RP. BCR-ABL rearrangements in children with Philadelphia chromosome-positive chronic myelogenous leukemia. Blood. 1991;78:2407–2410. doi: 10.1182/blood.V78.9.2407.bloodjournal7892407. [DOI] [PubMed] [Google Scholar]
- 24.Dan S, Naito M, Tsuruo T. Selective induction of apoptosis in Philadelphia chromosome-positive chronic myelogenous leukemia cells by an inhibitor of BCR-ABL tyrosine kinase, CGP 57148. Cell Death Differ. 1998;5:710–715. doi: 10.1038/sj.cdd.4400400. [DOI] [PubMed] [Google Scholar]
- 25.Miller G, El-Guindy A, Countryman J, Ye J, Gradoville L. Lytic cycle switches of oncogenic human gammaherpesviruses. Adv Cancer Res. 2007;97:81–109. doi: 10.1016/S0065-230X(06)97004-3. [DOI] [PubMed] [Google Scholar]
- 26.Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, Qiu Y, Wang J, Liu Y, Wei Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet. 2020;395:507–513. doi: 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo W, Li YX, Jiang LJ, Chen Q, Wang T, Ye DW. Targeting JAK-STAT signaling to control cytokine release Syndrome in COVID-19. Trends Pharmacol Sci. 2020;41:531–543. doi: 10.1016/j.tips.2020.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia H, Cao Z, Xie X, Zhang X, Chen JY, Wang H, Menachery VD, Rajsbaum R, Shi PY. Evasion of type I interferon by SARS-CoV-2. Cell Rep. 2020;33:108234. doi: 10.1016/j.celrep.2020.108234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuen CK, Lam JY, Wong WM, Mak LF, Wan X, Chu H, Cai JP, Jin DY, To KK, Chan JF, et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg Microbes Infect. 2020;9:1418–1428. doi: 10.1080/22221751.2020.1780953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miorin L, Kehrer T, Sanchez-Aparicio MT, Zhang K, Cohen P, Patel RS, Cupic A, Makio T, Mei M, Moreno E, et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc Natl Acad Sci USA. 2020;117:28344–28354. doi: 10.1073/pnas.2016650117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen DY, Khan N, Close BJ, Goel RK, Blum B, Tavares AH, Kenney D, Conway HL, Ewoldt JK, Chitalia VC, et al. SARS-CoV-2 disrupts proximal elements in the JAK-STAT pathway. J Virol. 2021;95:e0086221. doi: 10.1128/JVI.00862-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Montero P, Milara J, Roger I, Cortijo J. Role of JAK/STAT in interstitial lung diseases; molecular and cellular mechanisms. Int J Mol Sci. 2021;22:6211. doi: 10.3390/ijms22126211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simpson JA, Al-Attar A, Watson NF, Scholefield JH, Ilyas M, Durrant LG. Intratumoral T cell infiltration, MHC class I and STAT1 as biomarkers of good prognosis in colorectal cancer. Gut. 2010;59:926–933. doi: 10.1136/gut.2009.194472. [DOI] [PubMed] [Google Scholar]
- 34.Jia H, Song L, Cong Q, Wang J, Xu H, Chu Y, Li Q, Zhang Y, Zou X, Zhang C, et al. The LIM protein AJUBA promotes colorectal cancer cell survival through suppression of JAK1/STAT1/IFIT2 network. Oncogene. 2017;36:2655–2666. doi: 10.1038/onc.2016.418. [DOI] [PubMed] [Google Scholar]
- 35.Zhang X, Li X, Tan F, Yu N, Pei H. STAT1 Inhibits MiR-181a expression to suppress colorectal cancer cell proliferation through PTEN/Akt. J Cell Biochem. 2017;118:3435–3443. doi: 10.1002/jcb.26000. [DOI] [PubMed] [Google Scholar]
- 36.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: An overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 37.Stark GR, Darnell JE., Jr The JAK-STAT pathway at twenty. Immunity. 2012;36:503–514. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Varinou L, Ramsauer K, Karaghiosoff M, Kolbe T, Pfeffer K, Müller M, Decker T. Phosphorylation of the STAT1 transactivation domain is required for full-fledged IFN-gamma-dependent innate immunity. Immunity. 2003;19:793–802. doi: 10.1016/S1074-7613(03)00322-4. [DOI] [PubMed] [Google Scholar]
- 39.Garda-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19:1189–1201. doi: 10.1016/j.celrep.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ivashkiv LB. IFNγ: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. 2018;18:545–558. doi: 10.1038/s41577-018-0029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abiko K, Mandai M, Hamanishi J, Yoshioka Y, Matsumura N, Baba T, Yamaguchi K, Murakami R, Yamamoto A, Kharma B, et al. PD-L1 on tumor cells is induced in asdtes and promotes peritoneal dissemination of ovarian cancer through CTL dysfunction, din. Cancer Res. 2013;19:1363–1374. doi: 10.1158/1078-0432.CCR-12-2199. [DOI] [PubMed] [Google Scholar]
- 42.Tian X, Guan W, Zhang L, Sun W, Zhou D, Lin Q, Ren W, Nadeem L, Xu G. Physical interaction of STAT1 isoforms with TGF-β receptors leads to functional crosstalk between two signaling pathways in epithelial ovarian cancer. J Exp Clin Cancer Res. 2018;37:103. doi: 10.1186/s13046-018-0773-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Padmanabhan S, Gaire B, Zou Y, Uddin MM, Vancurova I. IFNγ-induced PD-L1 expression in ovarian cancer cells is regulated by JAK1, STAT1 and IRF1 signaling. Cell Signal. 2022;97:110400. doi: 10.1016/j.cellsig.2022.110400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 45.Priceman SJ, Kujawski M, Shen S, Cherryholmes GA, Lee H, Zhang C, Kruper L, Mortimer J, Jove R, Riggs AD, Yu H. Regulation of adipose tissue T cell subsets by Stat3 is crucial for diet-induced obesity and insulin resistance. Proc Natl Acad Sci USA. 2013;110:13079–13084. doi: 10.1073/pnas.1311557110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deng J, Liu Y, Lee H, Herrmann A, Zhang W, Zhang C, Shen S, Priceman SJ, Kujawski M, Pal SK, et al. S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell. 2012;21:642–654. doi: 10.1016/j.ccr.2012.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2012;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ, Choudhury SA, Maruyama R, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+CD24-stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121:2723–2735. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schroeder A, Herrmann A, Cherryholmes G, Kowolik C, Buettner R, Pal S, Yu H, Müller-Newen G, Jove R. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res. 2014;74:1227–1237. doi: 10.1158/0008-5472.CAN-13-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, Nebelsiek T, Lundgren-May T, Canli O, Schwitalla S, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 52.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, Karin M. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu H, Jove R. The STATs of cancer-new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 54.Lin TS, Mahajan S, Frank DA. STAT signaling in the pathogenesis and treatment of leukemias. Oncogene. 2000;19:2496–2504. doi: 10.1038/sj.onc.1203486. [DOI] [PubMed] [Google Scholar]
- 55.Battle TE, Frank DA. The role of STATs in apoptosis. Curr Mol Med. 2002;2:381–392. doi: 10.2174/1566524023362456. [DOI] [PubMed] [Google Scholar]
- 56.Bruns HA, Kaplan MH. The role of constitutively active Stat6 in leukemia and lymphoma. Crit Rev Oncol Hematol. 2006;57:245–253. doi: 10.1016/j.critrevonc.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 57.Sorger H, Dey S, Vieyra-Garcia PA, Pölöske D, Teufelberger AR, de Araujo ED, Sedighi A, Graf R, Spiegl B, Lazzeri I, et al. Blocking STAT3/5 through direct or upstream kinase targeting in leukemic cutaneous T-cell lymphoma. EMBO Mol Med. 2022;14:e15200. doi: 10.15252/emmm.202115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao R, Xing S, Li Z, Fu X, Li Q, Krantz SB, Zhao ZJ. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem. 2005;280:22788–22792. doi: 10.1074/jbc.C500138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao L, Ma Y, Seemann J, Huang LJ. A regulating role of the JAK2 FERM domain in hyperactivation of JAK2(V617F) Biochem J. 2010;426:91–98. doi: 10.1042/BJ20090615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 61.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 62.Walz C, Crowley BJ, Hudon HE, Gramlich JL, Neuberg DS, Podar K, Griffin JD, Sattler M. Activated Jak2 with the V617F point mutation promotes G1/S phase transition. J Biol Chem. 2006;281:18177–18183. doi: 10.1074/jbc.M600064200. [DOI] [PubMed] [Google Scholar]
- 63.Wernig G, Gonneville JR, Crowley BJ, Rodrigues MS, Reddy MM, Hudon HE, Walz C, Reiter A, Podar K, Royer Y, et al. The Jak2V617F oncogene associated with myeloproliferative diseases requires a functional FERM domain for transformation and for expression of the Myc and Pim protooncogenes. Blood. 2008;111:3751–3759. doi: 10.1182/blood-2007-07-102186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Furuhata A, Kimura A, Shide K, Shimoda K, Murakami M, Ito H, Gao S, Yoshida K, Tagawa Y, Hagiwara K, et al. p27 deregulation by Skp2 overexpression induced by the JAK2V617 mutation. Biochem Biophys Res Commun. 2009;383:411–416. doi: 10.1016/j.bbrc.2009.04.015. [DOI] [PubMed] [Google Scholar]
- 65.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 66.Jäkel H, Weinl C, Hengst L. Phosphorylation of p27Kip1 by JAK2 directly links cytokine receptor signaling to cell cycle control. Oncogene. 2011;30:3502–3512. doi: 10.1038/onc.2011.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mohrherr J, Uras IZ, Moll HP, Casanova E. STAT3: Versatile functions in non-small cell lung cancer. Cancers (Basel) 2020;12:1107. doi: 10.3390/cancers12051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bromberg J. Stat proteins and oncogenesis. J Clin Investig. 2002;109:1139–1142. doi: 10.1172/JCI0215617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huynh J, Etemadi N, Hollande F, Ernst M, Buchert M. The JAK/STAT3 axis: A comprehensive drug target for solid malignancies. Semin Cancer Biol. 2017;45:13–22. doi: 10.1016/j.semcancer.2017.06.001. [DOI] [PubMed] [Google Scholar]
- 70.Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, Settleman J. Drug resistance via feedback activation of Stat3 oncogene-addicted cancer cells. Cancer Cell. 2014;26:207–221. doi: 10.1016/j.ccr.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 71.Gao SP, Mark KG, Leslie K, Pao W, Motoi N, Gerald WL, Travis WD, Bornmann W, Veach D, Clarkson B, Bromberg JF. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Investig. 2007;117:3846–3856. doi: 10.1172/JCI31871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhu Z, Aref AR, Cohoon TJ, Barbie TU, Imamura Y, Yang S, Moody SE, Shen RR, Schinzel AC, Thai TC, et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014;4:452–465. doi: 10.1158/2159-8290.CD-13-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu D, Huang Y, Zeng J, Chen B, Huang N, Guo N, Liu L, Xu H, Mo X, Li W. Down-regulation of JAK1 by RNA interference inhibits growth of the lung cancer cell line A549 and interferes with the PI3K/mTOR pathway. J Cancer Res Clin Oncol. 2011;137:1629–1640. doi: 10.1007/s00432-011-1037-6. [DOI] [PubMed] [Google Scholar]
- 74.Xu Y, Jin J, Xu J, Shao YW, Fan Y. JAK2 variations and functions in lung adenocarcinoma. Tumour Biol. 2017;39:1010428317711140. doi: 10.1177/1010428317711140. [DOI] [PubMed] [Google Scholar]
- 75.Lee JH, Kim C, Baek SH, Ko JH, Lee SG, Yang WM, Um JY, Sethi G, Ahn KS. Capsazepine inhibits JAK/STAT3 signaling, tumor growth, and cell survival in prostate cancer. Oncotarget. 2017;8:17700–17711. doi: 10.18632/oncotarget.10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee JH, Kim JE, Kim BG, Han HH, Kang S, Cho NH. STAT3-induced WDR1 overexpression promotes breast cancer cell migration. Cell Signal. 2016;28:1753–1760. doi: 10.1016/j.cellsig.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 77.Subramaniam A, Shanmugam MK, Ong TH, Li F, Perumal E, Chen L, Vali S, Abbasi T, Kapoor S, Ahn KS, et al. Emodin inhibits growth and induces apoptosis in an orthotopic hepatocellular carcinoma model by blocking activation of STAT3. Br J Pharmacol. 2013;170:807–821. doi: 10.1111/bph.12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Paul A, Das S, Das J, Samadder A, Bishayee K, Sadhukhan R, Khuda-Bukhsh AR. Diarylheptanoid-myricanone isolated from ethanolic extract of Myrica cerifera shows anticancer effects on HeLa and PC3 cell lines: Signalling pathway and drug-DNA interaction. J Integr Med. 2013;11:405–415. doi: 10.3736/jintegrmed2013057. [DOI] [PubMed] [Google Scholar]
- 79.He G, Karin M. NF-kappaB and STAT3-key players in liver inflammation and cancer. Cell Res. 2011;21:159–168. doi: 10.1038/cr.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu Z, Chen T, Lu X, Xie H, Zhou L, Zheng S. Overexpression of variant PNPLA3 gene at I148M position causes malignant transformation of hepatocytes via IL-6-JAK2/STAT3 pathway in low dose free fatty acid exposure: A laboratory investigation in vitro and in vivo. Am J Transl Res. 2016;8:1319–1338. [PMC free article] [PubMed] [Google Scholar]
- 81.Miller AM, Wang H, Bertola A, Park O, Horiguchi N, Ki SH, Yin S, Lafdil F, Gao B. Inflammation-associated interleukin-6/signal transducer and activator of transcription 3 activation ameliorates alcoholic and nonalcoholic fatty liver diseases in interleukin-10-deficient mice. Hepatol. 2011;54:846–856. doi: 10.1002/hep.24517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, Oh YT, Kim H, Rheey J, Nakano I, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013;23:839–852. doi: 10.1016/j.ccr.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 84.Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22:128–134. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cebria F, Kobayashi C, Umesono Y, Nakazawa M, Mineta K, Ikeo K, Gojobori T, Itoh M, Taira M, Sánchez Alvarado A, Agata K. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:620–624. doi: 10.1038/nature01042. [DOI] [PubMed] [Google Scholar]
- 87.Cao W, Ribeiro Rde O, Liu D, Saintigny P, Xia R, Xue Y, Lin R, Mao L, Ren H. EZH2 promotes malignant behaviors via cell cycle dysregulation and its mRNA level associates with prognosis of patient with non-small cell lung cancer. PLoS One. 2012;7:e52984. doi: 10.1371/journal.pone.0052984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wu LJ, Zhang X, Wang J, Kong X, Zheng BY, Yu H. HeZ: ZMYND10 downregulates cyclins B1 and D1 to arrest cell cycle by trimethylating lysine 9 on histone 3. Life Res. 2021;4:17–24. doi: 10.53388/life2021-0727-132. [DOI] [Google Scholar]
- 89.Zhang Y, Tong T. FOXA1 antagonizes EZH2-mediated CDKN2A repression in carcinogenesis. Biochem Biophys Res Commun. 2014;453:172–178. doi: 10.1016/j.bbrc.2014.09.092. [DOI] [PubMed] [Google Scholar]
- 90.Ganem D. KSHV infection and the pathogenesis of Kaposi's sarcoma. Annu Rev Pathol. 2006;1:273–296. doi: 10.1146/annurev.pathol.1.110304.100133. [DOI] [PubMed] [Google Scholar]
- 91.Farrell PJ, Rowe DT, Rooney CM, Kouzarides T. Epstein-Barr virus BZLF1 trans-activator specifically binds to a consensus AP-1 site and is related to c-fos. EMBO J. 1989;8:127–132. doi: 10.1002/j.1460-2075.1989.tb03356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Feederle R, Kost M, Baumann M, Janz A, Drouet E, Hammerschmidt W, Delecluse HJ. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 2000;19:3080–3089. doi: 10.1093/emboj/19.12.3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kenney SC, Mertz JE. Regulation of the latent-lytic switch in Epstein-Barr virus. Semin Cancer Biol. 2014;26:60–68. doi: 10.1016/j.semcancer.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang Y, Ma R, Wang Y, Sun W, Yang Z, Han M, Han T, Wu XA, Liu R. Viruses run: the evasion mechanisms of the antiviral innate immunity by Hantavirus. Front Microbiol. 2021;12:759198. doi: 10.3389/fmicb.2021.759198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mesev EV, LeDesma RA, Ploss A. Decoding type I and III interferon signaling during viral infection. Nat. Microbiol. 2019;4:914–924. doi: 10.1038/s41564-019-0421-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Boneschi V, Brambilla L, Berti E, Ferrucci S, Corbellino M, Parravicini C, Fossati S. Human herpesvirus 8 DNA in the skin and blood of patients with Mediterranean Kaposi's sarcoma: Clinical correlations. Dermatology. 2001;203:19–23. doi: 10.1159/000051697. [DOI] [PubMed] [Google Scholar]
- 97.Campbell TB, Borok M, Gwanzura L, MaWhinney S, White IE, Ndemera B, Gudza I, Fitzpatrick L, Schooley RT. Relationship of human herpesvirus 8 peripheral blood virus load and Kaposi's sarcoma clinical stage. AIDS. 2000;14:2109–2116. doi: 10.1097/00002030-200009290-00006. [DOI] [PubMed] [Google Scholar]
- 98.Murray PG, Young LS. The Role of the Epstein-Barr virus in human disease. Front Biosci. 2002;7:d519–d540. doi: 10.2741/murray. [DOI] [PubMed] [Google Scholar]
- 99.Chen J, Ueda K, Sakakibara S, Okuno T, Parravicini C, Corbellino M, Yamanishi K. Activation of latent Kaposi's sarcoma-associated herpesvirus by demethylation of the promoter of the lytic transactivator. Proc Natl Acad Sci USA. 2001;98:4119–4124. doi: 10.1073/pnas.051004198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fardet L, Blum L, Kerob D, Agbalika F, Galicier L, Dupuy A, Lafaurie M, Meignin V, Morel P, Lebbé C. Human herpesvirus 8-associated hemophagocytic lymphohistiocytosis in human immunodeficiency virus-infected patients. Clin Infect Dis. 2003;37:285–291. doi: 10.1086/375224. [DOI] [PubMed] [Google Scholar]
- 101.Grandadam M, Dupin N, Calvez V, Gorin I, Blum L, Kernbaum S, Sicard D, Buisson Y, Agut H, Escande JP, Huraux JM. Exacerbations of clinical symptoms in human immunodeficiency virus type 1-infected patients with multicentric Castleman's disease are associated with a high increase in Kaposi's sarcoma herpesvirus DNA load in peripheral blood mononuclear cells. J Infect Dis. 1997;175:1198–1201. doi: 10.1086/593567. [DOI] [PubMed] [Google Scholar]
- 102.Oksenhendler E, Carcelain G, Aoki Y, Boulanger E, Maillard A, Clauvel JP, Agbalika F. High levels of human herpesvirus 8 viral load, human interleukin-6, interleukin-10, and C reactive protein correlate with exacerbation of multicentric Castleman disease in HIV-infected patients. Blood. 2000;96:2069–2073. doi: 10.1182/blood.V96.6.2069. [DOI] [PubMed] [Google Scholar]
- 103.Robles R, Lugo D, Gee L, Jacobson MA. Effect of antiviral drugs used to treat cytomegalovirus end-organ disease on subsequent course of previously diagnosed Kaposi's sarcoma in patients with AIDS. J Acquir Immune Defic Syndr Hum Retrovirol. 1999;20:34–38. doi: 10.1097/00042560-199901010-00005. [DOI] [PubMed] [Google Scholar]
- 104.King CA, Li X, Barbachano-Guerrero A, Bhaduri-McIntosh S. STAT3 regulates lytic activation of Kaposi's sarcoma-associated herpesvirus. J Virol. 2015;89:11347–11355. doi: 10.1128/JVI.02008-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mousavizadeh L, Ghasemi S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J Microbiol Immunol Infect. 2021;54:159–163. doi: 10.1016/j.jmii.2020.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Robson F, Khan KS, Le TK, Paris C, Demirbag S, Barfuss P, Rocchi P, Ng WL. Coronavirus RNA proofreading: molecular basis and therapeutic targeting. Mol Cell. 2020;79:710–727. doi: 10.1016/j.molcel.2020.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chou JM, Tsai JL, Hung JN, Chen IH, Chen ST, Tsai MH. The ORF8 protein of SARS-CoV-2 modulates the spike protein and its implications in viral transmission. Front Microbiol. 2022;13:883597. doi: 10.3389/fmicb.2022.883597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kim D, Lee JY, Yang JS, Kim JW, Kim VN, Chang H. The architecture of SARS-CoV-2 transcriptome. Cell. 2020;181:914–921. e10. doi: 10.1016/j.cell.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu DX, Fung TS, Chong KK, Shukla A, Hilgenfeld R. Accessory proteins of SARS-CoV and other coronaviruses. Antiviral Res. 2014;109:97–109. doi: 10.1016/j.antiviral.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280. e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Valcarcel A, Bensussen A, Álvarez-Buylla ER, Díaz J. Structural analysis of SARS-CoV-2 ORF8 protein: Pathogenic and therapeutic implications. Front Genet. 2021;12:693227. doi: 10.3389/fgene.2021.693227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Leifert JA, Holler PD, Harkins S, Kranz DM, Whitton JL. The cationic region from HIV tat enhances the cell-surface expression of epitope/MHC class I complexes. Gene Ther. 2003;10:2067–2073. doi: 10.1038/sj.gt.3302115. [DOI] [PubMed] [Google Scholar]
- 113.Haque M, Ueda K, Nakano K, Hirata Y, Parravicini C, Corbellino M, Yamanishi K. Major histocompatibility complex class I molecules are down-regulated at the cell surface by the K5 protein encoded by Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8. J Gen Virol. 2001;82:1175–1180. doi: 10.1099/0022-1317-82-5-1175. [DOI] [PubMed] [Google Scholar]
- 114.Selvaraj C, Dinesh DC, Pedone EM, Alothaim AS, Vijayakumar R, Rudhra O, Singh SK. SARS-CoV-2 ORF8 dimerization and binding mode analysis with class I MHC: computational approaches to identify COVID-19 inhibitors. Brief Funct Genomics. 2023;22:227–240. doi: 10.1093/bfgp/elac046. [DOI] [PubMed] [Google Scholar]
- 115.Cai H, Chen Y, Feng Y, Asadi M, Kaufman L, Lee K, Kehrer T, Miorin L, Garcia-Sastre A, Gusella GL, et al. SARS-CoV-2 viral protein ORF3A injures renal tubules by interacting with TRIM59 to induce STAT3 activation. Mol Ther. 2023;31:774–787. doi: 10.1016/j.ymthe.2022.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, Vanstapel A, Werlein C, Stark H, Tzankov A, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020;383:120–128. doi: 10.1056/NEJMoa2015432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers DAMPJ, Kant KM, Kaptein FHJ, van Paassen J, Stals MAM, Huisman MV, Endeman H. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145–147. doi: 10.1016/j.thromres.2020.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Matsuyama T, Kubli SP, Yoshinaga SK, Pfeffer K, Mak TW. An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 2020;27:3209–3225. doi: 10.1038/s41418-020-00633-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Martineau AR, Jolliffe DA, Hooper RL, Greenberg L, Aloia JF, Bergman P, Dubnov-Raz G, Esposito S, Ganmaa D, Ginde AA, et al. Vitamin D supplementation to prevent acute respiratory tract infections: Systematic review and meta-analysis of individual participant data. BMJ. 2017;356:i6583. doi: 10.1136/bmj.i6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, HLH Across Speciality Collaboration UK: COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033–1034. doi: 10.1016/S0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jamilloux Y, Henry T, Belot A, Viel S, Fauter M, El Jammal T, Walzer T, Francois B, Seve P. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmun Rev. 2020;19:102567. doi: 10.1016/j.autrev.2020.102567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Richardson P, Griffin I, Tucker C, Smith D, Oechsle O, Phelan A, Rawling M, Savory E, Stebbing J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet. 2020;395:e30–e31. doi: 10.1016/S0140-6736(20)30304-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vannucchi AM, Mortara A, D'Alessio A, Morelli M, Tedeschi A, Festuccia MB, Monforte AD, Capochiani E, Selleri C, Simonetti F, et al. JAK Inhibition with Ruxolitinib in Patients with COVID-19 and severe pneumonia: multicenter clinical experience from a compassionate use program in Italy. J Clin Med. 2021;10:3752. doi: 10.3390/jcm10163752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dobosh B, Zandi K, Giraldo DM, Goh SL, Musall K, Aldeco M, LeCher J, Giacalone VD, Yang J, Eddins DJ, et al. Baricitinib attenuates the proinflammatory phase of COVID-19 driven by lung-infiltrating monocytes. Cell Rep. 2022;39:110945. doi: 10.1016/j.celrep.2022.110945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ucciferri C, Auricchio A, Marinari S, Vecchiet J, Falasca K. COVID-19 in a patient with SISTEMIC sclerosis: The role of ruxolitinib. Eur J Inflammation. 2021;19:1–4. doi: 10.1177/20587392211036813. [DOI] [Google Scholar]
- 126.Ucciferri C, Vecchiet J, Falasca K. Role of monoclonal antibody drugs in the treatment of COVID-19. World J Clin Cases. 2020;8:4280–4285. doi: 10.12998/wjcc.v8.i19.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hashemi R, Morshedi M, Asghari Jafarabadi M, Altafi D, Saeed Hosseini-Asl S, Rafie-Arefhosseini S. Anti-inflammatory effects of dietary vitamin D. Neurol Genet. 2018;4:e278. doi: 10.1212/NXG.0000000000000278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hashemi R, Hosseini-Asl SS, Arefhosseini SR, Morshedi M. The impact of vitamin D3 intake on inflammatory markers in multiple sclerosis patients and their first-degree relatives. PLoS One. 2020;15:e0231145. doi: 10.1371/journal.pone.0231145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Grant WB, Lahore H, McDonnell SL, Baggerly CA, French CB, Aliano JL, Bhattoa HP. Evidence that Vitamin D Supplementation Could Reduce Risk of Influenza and COVID-19 Infections and Deaths. Nutrients. 2020;12:988. doi: 10.3390/nu12040988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hii CS, Ferrante A. The Non-Genomic Actions of Vitamin D. Nutrients. 2016;8:135. doi: 10.3390/nu8030135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hafezi S, Saheb Sharif-Askari F, Saheb Sharif-Askari N, Ali Hussain Alsayed H, Alsafar H, Al Anouti F, Hamid Q, Halwani R. Vitamin D enhances type I IFN signaling in COVID-19 patients. Sci Rep. 2022;12:17778. doi: 10.1038/s41598-022-22307-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.