

Abstract
Alpha-1-antitrypsin (AAT), a serine protease inhibitor, is the third most abundant protein in plasma. While the best-known function of AAT is irreversible inhibition of elastase, AAT is an acute-phase reactant and is increasingly recognized to have a panoply of other functions including as an anti-inflammatory mediator and a host-protective molecule against various pathogens. While a canonical receptor for AAT has not been identified, AAT can be internalized into the cytoplasm and is known to affect gene regulation. Since AAT has anti-inflammatory properties, we examined whether AAT binds the cytoplasmic glucocorticoid receptor (GR) in human macrophages. We report the novel finding that AAT binds to GR using several approaches, including co-immunoprecipitation, mass spectrometry, and microscale thermophoresis. We also performed in silico molecular modeling and found that binding between AAT and GR has a plausible stereochemical basis. The significance of this interaction in macrophages is evinced by AAT inhibition of lipopolysaccharide-induced nuclear factor-kappa B activation and interleukin-8 production as well as AAT induction of angiopoietin-like-4 protein are, in part, dependent on GR. Furthermore, this AAT–GR interaction contributes to a host-protective role against mycobacteria in macrophages. In summary, this study identifies a new mechanism for the gene regulation, anti-inflammatory, and host-defense properties of AAT.
INTRODUCTION
Severe alpha-1-antitrypsin (AAT) deficiency most commonly occurs in individuals who possess the protease inhibitor (Pi) ZZ genotype, resulting in a substantially increased risk of precocious emphysema. Other disorders associated with PiZZ include bronchiectasis, panniculitis, cirrhosis, and anti-neutrophil cytoplasmic antibody-mediated vasculitis (1–3). Additionally, AAT has potent anti-inflammatory properties and is host-protective against HIV, influenza, Pseudomonas aeruginosa, and non-tuberculous mycobacteria (4–12); however, the molecular basis of these AAT-associated activities is not well understood.
AAT resides in the cytoplasm of monocytes and macrophages and may be produced by them (13). Alternatively, elastase-bound AAT may enter cells from the extracellular milieu through endocytosis by low-density lipoprotein receptor-related protein-1 (LRP1) or through clathrin-coated vesicles and caveolae-mediated endocytosis (14–16). The scavenger receptor B type I has also been proposed as a mechanism for endocytosis of AAT (17). In the cytoplasm, an anti-inflammatory effect of AAT occurs via binding to IκBα (nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha), the basal inhibitor of nuclear factor-kappa B (NFκB). Such binding by AAT to IκBα prevents IκBα phosphorylation, ubiquitination, and targeted proteasome degradation (12). Through these actions, AAT inhibits NFκB activation. Additional proposed anti-inflammatory functions of AAT include binding of interleukin-8 (IL-8) (18) and inhibiting the ability of ADAM-17 (A Disintegrin And Metalloprotease 17) to cleave membrane-bound tumor necrosis factor (TNF) to soluble TNF (19). Via its function as a serine protease inhibitor (serpin), AAT also irreversibly binds and inhibits thrombin, plasmin, and caspase-3, with the lattermost interaction preventing the apoptosis of endothelial cells (14, 20, 21).
Independent of its serpin activity, the mechanism(s) underlying anti-inflammatory and disease-mitigating properties of AAT remains poorly understood. AAT regulates transcription and is found in the nucleus (22, 23); however, its ability to translocate into the nucleus is unknown. In contrast, the pleiotropic actions of the glucocorticoid (GC)-glucocorticoid receptor (GR) complex have been extensively studied (24). GR regulates pro- and anti-inflammatory genes by binding to transcriptional activators, co-activators, repressors, and co-repressors (24). In the absence of binding to its ligand, GR resides in the cytoplasm bound to the chaperones Hsp90 and Hsp70. Upon binding to its canonical ligand, cortisol, the chaperone molecules dissociate from GR, and the GC–GR complex enters the nucleus to inhibit or induce transcription. GR signaling in response to GC leads to anti-inflammatory effects and immune suppression. A known connection between AAT and GR is that GC activation of GR increases AAT production at the transcriptional level (25, 26).
Given that both AAT and GCs have anti-inflammatory properties and reside in the cytoplasmic compartment, we investigated the possibility that AAT interacts directly with GR. We found that AAT binds GR and that this molecular interaction has anti-inflammatory and anti-mycobacterial functions in macrophages. Furthermore, this AAT–GR interaction lends itself to manipulation within the context of the anti-inflammatory, immunosuppressive, and lesser well-known immune-enhancing activities of the GR. Potential prospects include the development of small peptides that serve as functional surrogates for AAT to bind GR but with more favorable benefit to risk ratio than GC. This notion is plausible given that the many synthetic GCs are known to have differences in potency and severity of adverse effects.
MATERIALS AND METHODS
Materials
The human monocytic cell line (THP-1) and Mycobacterium tuberculosis H37Rv were obtained from the ATCC (Manassas, Virginia). M. intracellulare NJH9141 is a clinical strain isolated at National Jewish Health. Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Lawrenceville, GA) and heat-inactivated at 56°C for 30 minutes. Recombinant human GR protein (#A15663), GR-specific rabbit polyclonal antibody purified by antigen affinity chromatography (PA1–511A) with robust GR specificity in applications such as GR chromatin IP (27), AAT-specific rabbit polyclonal antibody purified by ammonium sulfate precipitation and dialysis (PA5–26439), AAT-specific mouse monoclonal antibody purified by recombinant fragment of human AAT (clone 2B12), and the human angiopoietin-like 4 protein (ANGPTL4) ELISA Kit, goat anti-mouse IgG (H+L) Alexa Fluor™ Plus 488, Lipofectamine RNAi-MAX Transfection reagent (Cat#13778075), and CyQUANT™ MTT Cell Viability Assay Kit (#V13154) were purchased from Invitrogen-ThermoFisher Scientific (Carlsbad, CA). TLR2-specific polyclonal antibody (rabbit) (#12276), tubulin polyclonal antibody (#2144), lamin B polyclonal antibody (#12586), and β-actin polyclonal antibody (rabbit) were purchased from Cell Signaling Technology (Danvers, MA). Human IL-8/CXCL8 Quantikine ELISA kit and non-immune human IgG control were purchased from R&D Systems (Minneapolis, MN). Phorbol myristate acetate (PMA), lipopolysaccharide (#L-2880), cortisol (#C-106), and dexamethasone (#1756) were purchased from Sigma (St. Louis, MO). The TransAM® NFκB p65 kit was purchased from Active Motif (Carlsbad, CA). An aliquot of the glucocorticoid receptor (NR3C1) human shRNA (shRNA-GR) Lentiviral Particle was a kind gift from Miles Pufall, Ph.D. (University of Iowa Carver College of Medicine). Protein A Sepharose™ 4 Fast Flow sepharose beads were purchased from GE Healthcare Life Sciences (Pittsburgh, PA). AAT (Glassia®) was acquired from Kamada Ltd., Israel. Cy3 goat anti-rabbit IgG (H+L) A10520 was purchased from Life Technologies Inc. GRa-siRNA (sc-35505) was purchased from Santa Cruz Biotechnologies (Dallas, TX).
Macrophage culture
Human THP-1 cells were cultured in RPMI-1640 medium containing 2 mM L-glutamine (Gibco; Grand Island, NY), 10% FBS, penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37°C and 5% CO2. THP-1 cells were differentiated into macrophages following incubation with 15 ng/mL PMA for 24 hours. Peripheral blood mononuclear cells (PBMC) from a healthy adult donor (known to possess the PiMM genotype for AAT) were isolated from blood collected in CPT™ tubes after informed consent based on an approved Institutional Review Board protocol (HS-2651) that abides by the Declaration of Helsinki principles. Differentiation of human monocytes to monocyte-derived macrophages (MDM) was performed in the presence of 20 ng/mL monocyte-colony stimulating factor and cultured in 10% autologous plasma as previously described (4).
Fluorescent immunocytochemistry
Differentiated human THP-1 macrophages and MDMs, were seeded onto 4-well chamber slides (Lab-Tek; ThermoFisher Scientific Inc.) at a density of 0.5–1 ×105 cells/well. Cells were fixed with 4% paraformaldehyde for 30 minutes. Fixed cells were permeabilized for 20 minutes using 0.3% Triton X-100 in PBS. Non-specific binding of antibodies was blocked by incubating the cells for 1–1.5 hours with 3% BSA dissolved in 1X PBST. Cells were incubated with polyclonal anti-GR (1:300 dilution) and monoclonal anti-AAT (1:300 dilution) antibodies overnight at 4°C, washed thrice, and incubated with anti-rabbit Cy3 and anti-mouse Alexa Fluor™ Plus 488 secondary antibodies, respectively (both 1:1000 dilution), at room temperature for 1–2 hours. The slides were sealed with ProLong Gold™ Antifade Mountant with DAPI (ThermoFisher Scientific). The THP-1 macrophages were imaged using a SP5 confocal microscope (Leica Microsystems) with a magnification of 63X and a 1.4 numerical aperture (NA) oil immersion objective. The fluorescent labels DAPI, Cy3, and Alexa Fluor™ Plus 488 were excited using 359 nm, 552 nm, and 495 nm laser excitation, respectively. The MDM were analyzed at 40X magnification using a fluorescent microscope (Carl Zeiss Axiovert 200M) equipped with DAPI and fluorescent filters.
Immunoprecipitation (IP) and immunoblot analyses
Immunoprecipitation (IP) was performed according to a previously published protocol (28). Briefly, the cells were lysed with 50 mM Tris–HCl, pH 7.4, containing 0.5% (v/v) Nonidet P-40, 1 mM EDTA, 150 mM NaCl, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 2 mM Na3VO4, and 1 mM PMSF. The Bradford protein assay (Bio-Rad) was used to quantify the protein concentrations of the whole cell lysates. Then 300 μg of protein from each preparation was incubated with 3 μg of anti-GR polyclonal antibodies, anti-AAT polyclonal antibodies, or control non-immune IgG at 4°C overnight with gentle rotary mixing. The protein-antibody complexes were isolated by incubation with 20 μL protein A-sepharose beads with gentle mixing for 2 hours at 4°C. The beads were washed three times with wash buffer (10 mM Tris buffer, pH 7.4 with protease inhibitor cocktail).
Each immunoprecipitated (IP’d) pellet was resuspended in 30 μL 1X Laemmli/DTT buffer and heated at 95°C for 5 minutes for immunoblot analysis. The IP’d preparations and whole-cell lysates were fractionated using 12% SDS-PAGE and transferred onto PVDF membranes using the iBlot 2 dry blotting System from Invitrogen (ThermoFisher Scientific). For immunodetection of AAT, the membranes were blocked in PBST buffer containing 5% fat-free milk powder (blocking buffer) for one hour, then incubated overnight at 4°C with anti-AAT polyclonal antibody, anti-AAT monoclonal antibody, or anti-β-actin antibody (each at a dilution of 1:1000 (v/v)), followed by detection with HRP-conjugated anti-rabbit IgG (1:2000 dilution) or HRP-conjugated anti-mouse IgG (1:2000). To detect GR, polyclonal anti-GR polyclonal (1:1000) or anti-β-actin (1:1000) were diluted in PBST buffer with 5% BSA overnight at 4°C with light shaking, followed by detection using HRP-conjugated anti-rabbit IgG at 1:2000 to detect GR or 1:5000 to detect β-actin-antibody binding in blocking buffer at room temperature for 1–2 hours. Immunoblotting for lamin and tubulin was performed with polyclonal anti-lamin (1:3000 dilution) and polyclonal anti-tubulin (1:3000), respectively, diluted in blocking buffer overnight at 4°C, and then incubated at room temperature for one hour with anti-rabbit-HRP conjugated antibodies at 1:2000 dilution with blocking buffer. The bands were visualized using the SuperSignal West Femto Maximum Sensitivity Substrate System (ThermoFisher Scientific). TLR2 immunoblotting was performed with anti-TLR2-specific polyclonal antibody (1:1000) and β-actin specific polyclonal antibody (1:3000).
Mass spectrometry
IP was performed as described above. The bound immune complexes were eluted by heating in 1X Laemmli/DTT loading buffer, separated with 12% SDS-PAGE, and the gel stained with Coomassie. The gel pieces were de-stained in 200 μL of 25 mM ammonium bicarbonate in 50% v/v acetonitrile for 15 minutes and washed with 200 μL of 50% (v/v) acetonitrile. Disulfide bonds in proteins were reduced by incubation in 10 mM DTT at 60°C for 30 minutes and cysteine residues were alkylated with 20 mM iodoacetamide in the dark at room temperature for 45 minutes. Gel pieces were subsequently washed with 100 μL of distilled water followed by addition of 100 mL of acetonitrile and dried on SpeedVac (Savant ThermoFisher). Then 100 ng of trypsin was added to each sample and allowed to rehydrate the gel plugs at 4°C for 45 minutes and then incubated at 37°C overnight. The tryptic mixtures were acidified with formic acid up to a final concentration of 1%. Peptides were extracted twice from the gel plugs using 1% formic acid in 50% acetonitrile. The collected extractions were pooled with the initial digestion supernatant and dried on SpeedVac. Samples were desalted on Thermo Scientific Pierce C18 Tip.
Samples were analyzed on a LTQ Orbitrap Velos mass spectrometer (ThermoFisher Scientific) coupled to an Eksigent nanoLC-2D system through a nanoelectrospray LC −MS interface. An autosampler injected 8 μL of sample into a 10 μL loop. To desalt the sample, the material was flushed out of the loop, loaded onto a trapping column (ZORBAX 300SB-C18, dimensions 5×0.3 mm×5 μm), and washed with 0.1% formic acid at a flow rate of 5 μL/minute for 5 minutes. The analytical column was then switched on-line at 0.6 μL/minute over an in-house made 100 μm i.d. × 200 mm fused silica capillary packed with 4 μm 80 Å Synergi Hydro C18 resin (Phenomex; Torrance, CA). After 10 minutes of sample loading, the flow rate was adjusted to 0.35 mL/minute, and each sample was run on a 90-minute linear gradient of 4–40% acetonitrile with 0.1% formic acid to separate the peptides. LC mobile phase solvents and sample dilutions used 0.1% formic acid in water (Buffer A) and 0.1% formic acid in acetonitrile (Buffer B) (Chromasolv LC–MS grade; Sigma-Aldrich, St. Louis, MO).
Data were acquired using the instrument supplied Xcalibur™(version 2.1) software. The mass spectrometer was operated in the positive ionmode. Each survey scan of m/z 400–2000 was followed by collisionassisted dissociation (CAD) MS/MS of the twenty most intense precursor ions. Singly charged ions were excluded from CAD selection. Doubly charged and higher ions were included. Normalized collision energies were employed using helium as the collision gas.
MS/MS spectra were extracted from raw data files and converted into mascot generic format (mgf) files using a PAVA script (UCSF, MSF, San Francisco, CA). These .mgf files were then independently searched against the human SwissProt database (13188 entries) using an in-house Mascot server (Version 2.6, Matrix Science). Mass tolerances were +/− 10 ppm for MS peaks, and +/− 0.6 Da for MS/MS fragment ions. Trypsin specificity was used allowing for one missed cleavage. Met oxidation, protein N-terminal acetylation, and peptide N-terminal pyroglutamic acid formation were allowed as variable modifications while carbamidomethyl of Cys was set as a fixed modification.
Scaffold (version 4.8, Proteome Software, Portland, OR, USA) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 95.0% probability as specified by the Peptide Prophet algorithm. Protein identifications were accepted if they could be established at greater than 99.0% probability and contained at least two identified unique peptides. All peptide sequences assigned are provided in Supplementary Table S1. The full mass spectrometry data are available via ProteomeXchange with identifier PXD030989.
Microscale thermophoresis
To measure the binding affinity between AAT and GR in a cell-free assay, microscale thermophoresis (MST) was performed using the Monolith NT.115 Pico instrument (Nanotemper Technologies, München, Germany) according to manufacturer’s instructions. Instrument performance was confirmed prior to each experiment using the Monolith NT Control Kit RED Molecular Interaction Control Kit for Monolith NT.115 Blue/Red. AAT protein was fluorescently labeled using a Protein Labeling Kit RED-NHS 2nd Generation kit (Amine Reactive) (Nanotemper Technologies) according to the manufacturer’s specifications. Recombinant human GR was serially diluted two-fold in nano-pure water two-fold from the stock concentration to create a 16-member concentration series (1.91 nM to 6.25 × 103 nM). Fluorescent-labeled 10 nM AAT, prepared in PBS buffer with 0.05% P20, was added to the different GR aliquots. Samples were then loaded into premium Monolith NT.115 capillary tubes and the tubes were loaded onto the Monolith NT.115 Pico instrument. For all the experiments, a Pico.RED detector was used at 20% laser power and medium MST power. Optimal laser and MST power settings, as well as protein stability, were confirmed by measuring a capillary tube containing only 10 nM of the fluorescent-labeled AAT diluted in PBS. Data were analyzed using the MO Affinity Analysis software suite (Nanotemper Technologies).
Molecular modeling and docking
A model of GR was constructed using the known structures of DNA-binding domain (DBD, PDB ID 5HN5) and ligand-binding domain (LBD, PDB ID 1M2Z) and AlphaFold2 (29) to construct the hinge region connecting them. Structure refinement was performed using the Rosetta Relax protocol (30). Given the intrinsic disorder of the N-terminal activation function-1 domain (NTD), it was not modeled. Structural representations were produced using Schrodinger PyMOL version 2.5.2. Structural alignments were performed using MUSTANG-MR (31). Docking simulations were performed using AlphaFold2 and ClusPro (29, 32).
Isolation of nuclear and cytoplasmic fractions
Nuclear and cytoplasmic fractions of THP-1 macrophages were isolated using the NE-PER™ Nuclear and Cytoplasmic Extraction Reagents kit (ThermoFisher Scientific) according to the manufacturer’s instructions. Briefly, THP-1 macrophages were washed with 1X PBS and re-suspended in cell extraction buffer supplemented with 1 mM PMSF and a protease inhibitor cocktail (Cell Signaling Technology (Danvers, MA). Nuclear and cytoplasmic protein fraction concentrations were quantified prior to co-IP of AAT and GR.
Stable knockdown of the glucocorticoid receptor (GR) in THP-1 cells
We employed shRNA-lentivirus technology to develop a pool of THP-1 cells stably depleted for GR. In brief, THP-1 cells were plated in 12-well tissue culture plates at a density of 1×104 cells/well and infected with GR-directed shRNA or scrambled shRNA lentiviral particles at a multiplicity-of-infection of ≥1. THP-1 cells infected with the scrambled shRNA (THP-1control) and those stably depleted (knocked down, KD) for GR (THP-1GR-KD) were selected by adding 1 μg/mL puromycin dihydrochloride to the cell culture medium.
Transient knockdown of GR in primary human monocyte-derived macrophages
PBMC were isolated from the same PiMM individual as described above differentiated to MDM. MDM (6.2 × 105) were transfected with one of two siRNA (scrambled as a control or targeting GR) using Lipofectamine® RNAiMAX Transfection reagent (ThermoFisher Scientific). After 24 hours, the cells were harvested and immunoblotted for GR.
RNA sequencing (RNAseq)
RNAseq and immunoblotting were used to confirm the depletion of GR mRNA and protein, respectively. For the former approach, we analyzed the expression of the NR3C1 (GR) gene as part of another study of total RNA sequencing using the Illumina Next Generation Sequencing service from Genomics Shared Resource at the University of Colorado Cancer Center in THP-1control and THP-1GR-KD cells. RNAseq data were analyzed using DAVID Bioinformatics Resources 6.8 Vision software.
p65 NFκB binding assay
The binding of the p65 subunit of NFκB was quantified using the TransAM™ NFκB p65 kit (Active Motif, Carlsbad, CA), as we previously reported (33).
ELISA for interleukin-8 (IL-8) and angiopoietin-like 4 (ANGPTL4)
Differentiated THP-1control and THP-1GR-KD macrophages were left untreated or treated with 1 μg/mL LPS alone or in the presence of 10 μM cortisol, 1 μM dexamethasone, or 3 or 5 mg/mL AAT at 37°C in a 5% CO2 incubator. After 6 and 24 hours, the culture supernatants were quantified for IL-8 (R&D Systems) or ANGPTL4 (ThermoFisher Scientific) expression according to the manufacturer’s instructions. MDMcontrol and MDMGR-KD were left untreated or treated with 1 μg/mL LPS alone or in the presence of either 10 μM cortisol, or 3 or 5 mg/mL AAT. The cells were incubated at 37°C in a 5% CO2 incubator for 24 hours and the supernatant assayed for IL-8.
Infection of macrophages with mycobacteria and quantitation of cell-associated mycobacteria
Infection of THP-1 macrophages with M. tuberculosis or M. intracellulare and quantitation of cell-associated mycobacteria were performed as previously described (33). In brief, differentiated THP-1 macrophages in 24-well tissue culture plates were infected with M. tuberculosis H37Rv or M. intracellulare at a multiplicity-of-infection (MOI) of 10 bacilli to 1 macrophage. For cells infected for one hour (Day 0), the supernatants were collected after one hour of infection, the cells were washed twice with a 1:1 solution of RPMI:1X PBS, and the adherent cells were lysed with 250 μL of a 0.25% SDS per well, followed by addition of 250 μL of 7H9 plating broth. Serial dilutions of cell lysates were prepared, and then 5 μL of each dilution was plated on Middlebrook 7H10 agar. For Day 2 and 4 infections, the cells were washed twice after the initial one hour of infection and replaced with RPMI medium containing 10% FBS. After two or four days of incubation, the supernatants were centrifuged to recover any non-adherent macrophages; these macrophages were lysed together with the adherent macrophages and M. tuberculosis or M. intracellulare cultured as described above.
Statistical Analysis
Replicate experiments (two for the MST experiments and three for the cellular experiments with the latter done in duplicate wells) are independent, and data are presented as means ± SEM or representative experiment. Group means were compared by repeated-measures ANOVA using Fisher’s least significant test or by two-way ANOVA with Bonferroni’s post-hoc test. Data were graphed in Prism 9® and comparisons were considered significant when p<0.05.
DATA AVAILABILITY
The mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium via the PRIDE [1] partner repository with the dataset identifier PXD030989 and 10.6019/PXD030989.
RESULTS
AAT co-localizes with GR
Fluorescent immunocytochemical staining of macrophages was performed using anti-AAT and anti-GR antibodies conjugated to different fluorochromes to determine whether AAT and GR are expressed under basal conditions. Both AAT and the GR co-localized in the cytoplasm and, to a lesser extent, within the nuclei of differentiated THP-1 macrophages by confocal microscopy (Figure 1A). MDM from an individual who possesses two normal M alleles for AAT (PiMM) also showed that AAT and GR are found intracellularly by fluorescent microscopy (Figures 1B). Similarly, AAT and GR expression in human macrophages were confirmed by immunoblotting whole-cell lysates (Figure 1C).
Figure 1. Immunofluorescence staining for glucocorticoid receptor (GR) and alpha-1-antitrypsin (AAT).
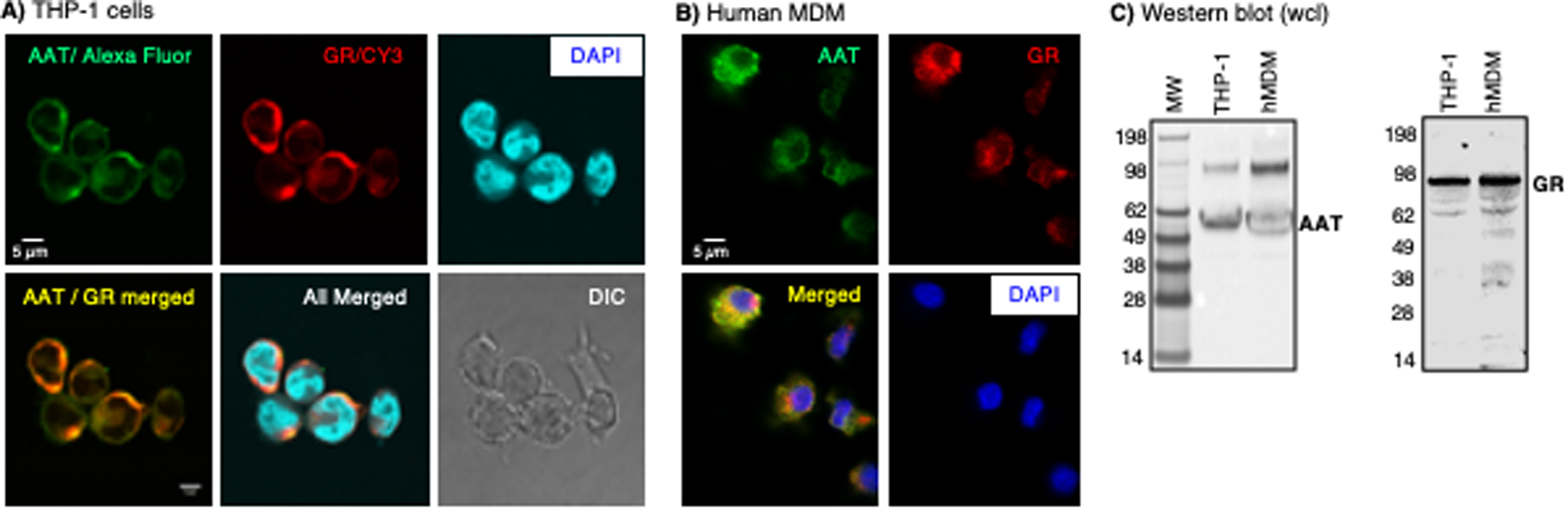
(A) Human THP-1 differentiated macrophages and (B) human monocyte-derived macrophages (MDM) were immunostained using anti-AAT and anti-GR antibodies conjugated to the fluorochromes Alexa Fluor™ Plus 488 and Cy3, respectively. THP-1 macrophages were imaged with a confocal microscope. Human monocyte-derived macrophages (MDM) were imaged with a fluorescent microscope. The scale bars represent 5 μm. (C) Immunoblotting for AAT and GR in human macrophages. Images shown are representative of three independent experiments.
AAT binds to the GR
Co-immunoprecipitation and immunoblot approach
To investigate whether AAT binds to the GR in differentiated THP-1 macrophages, whole-cell lysates were immunoprecipitated (IP’d) for GR with polyclonal non-immune IgG control or anti-GR antibodies using protein A sepharose beads. The beads were washed, centrifuged, the bound proteins extracted, separated by SDS-PAGE, and immunoblotted with an anti-AAT polyclonal antibody. In contrast to non-immune IgG, immunoprecipitation (IP) for the GR followed by immunoblot for AAT revealed a distinct band at ~52 kDa, which is consistent with the molecular mass of pharmacologic AAT (Glassia®) and AAT from whole-cell lysates (Figure 2A, left side of immunoblot). To corroborate the interaction between AAT and GR in primary human macrophages, MDMs were prepared from an individual with the PiMM AAT phenotype. Compared to the non-immune IgG control, IP of MDM lysates with anti-GR revealed the presence of AAT within the IP’d lysates (Figure 2A, right side of immunoblot). Conversely, THP-1 cell lysates were IP’d for AAT and the separated proteins were immunoblotted for GR. As shown in Figure 2B, GR was detected in the fraction IP’d for AAT but not in the fraction IP’d with non-immune IgG.
Figure 2. Alpha-1-antitrypsin (AAT) co-immunoprecipitates with glucocorticoid receptor (GR).
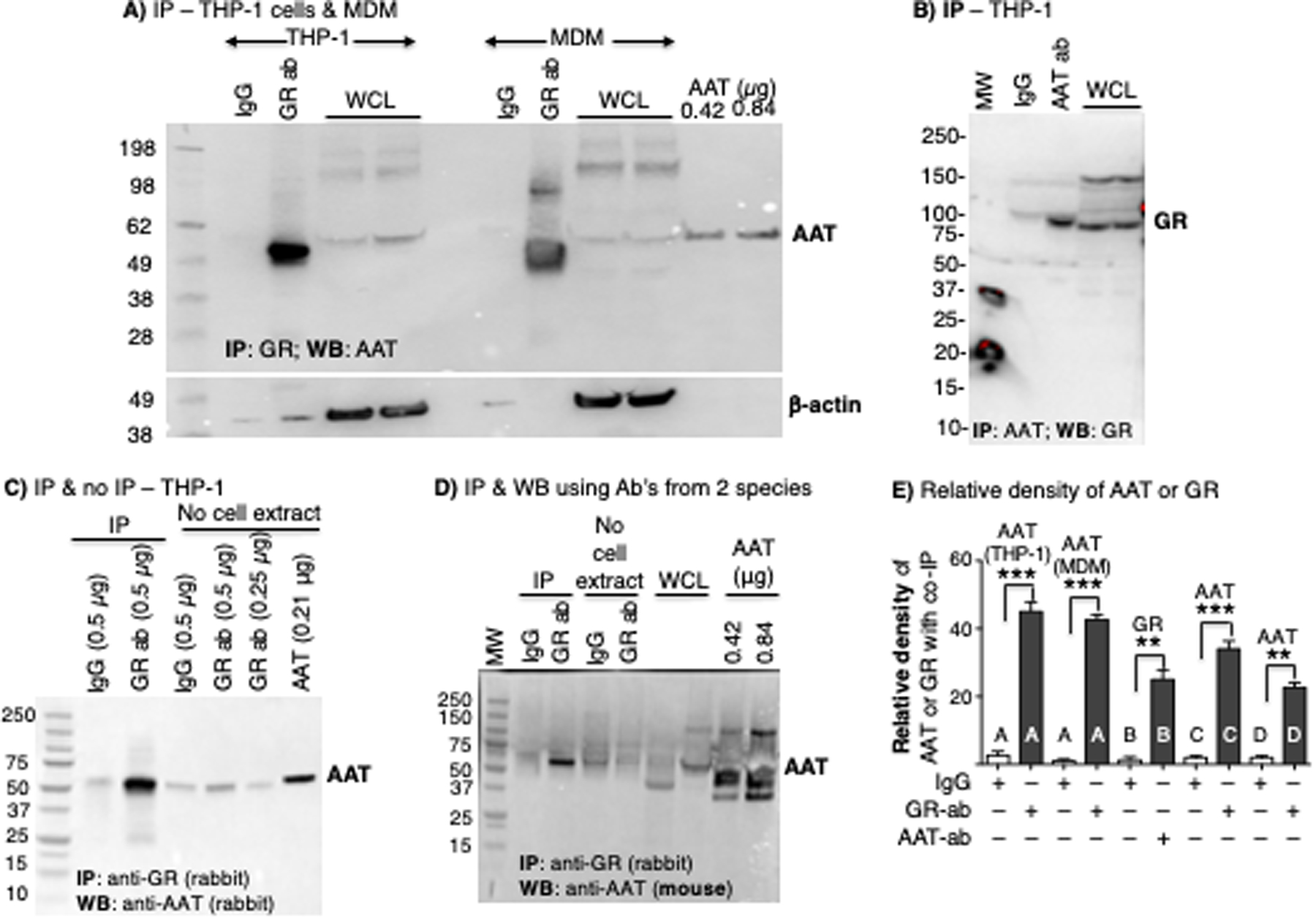
(A) THP-1 macrophage and human monocyte-derived macrophage (MDM) lysates were immunoprecipitated (IP’d) with anti-GR polyclonal antibody, followed by immunoblotting of the IP’d fraction to detect AAT and β-actin. (B) THP-1 macrophage lysates were IP’d with anti-AAT polyclonal antibody, followed by immunoblotting of the IP’d fraction for GR. (C) Comparing the relative amount of IgG and anti-GR antibody alone (“No cell extract”) used in the immunoprecipitation (IP) experiments, with THP-1 macrophage lysates IP’d with non-immune IgG or anti-GR, followed by immunoblotting for AAT. (D) THP-1 macrophage lysates were IP’d with anti-GR polyclonal antibodies followed by immunoblotting of the IP’d fraction with a mouse anti-AAT monoclonal antibody. (E) Densitometry of the co-IP protein using non-immune IgG, anti-GR antibody, or anti-AAT antibody for immunoblots represented in Figures A-D (letters A to D shown in the graph correspond to the IP’d bands in Figures A-D). The proteins listed above each bar is the protein detected in the immunoblot. Images in A-D are representative of at least three independent experiments. Data shown in E are the mean ± SD of three independent experiments. AAT=alpha-1-antitrypsin; ab=antibody; GR=glucocorticoid receptor; MDM=monocyte-derived macrophages; WB=western blot; WCL=whole cell lysates. Lanes labeled “No cell extract” contains only IgG or anti-GR antibody alone with no IP performed.
We further confirmed the integrity of the AAT–GR binding interaction by excluding an experimental artifact resulting from the molecular weight similarity between heavy chain immunoglobulins and AAT. We employed two approaches. In the first approach, the THP-1 macrophage lysates were IP’d as previously indicated with the anti-GR polyclonal antibody. In addition, 0.5 μg of anti-GR or non-immune IgG antibodies – the amount used for IP in each condition – were loaded onto separate wells of the SDS-PAGE gel. While the migration of IgG heavy chain is similar to that of AAT, the intensities of the relevant bands in the lanes that contained only either non-immune IgG or anti-GR (with no IP of cell extract) are significantly less than the IP’d band, indicating that the prominent band at 52 kDa following IP for GR mostly represents AAT and not the heavy chain of IgG (Figure 2C). In the second approach, we conducted IP with rabbit anti-GR polyclonal antibody as before but used a mouse anti-AAT monoclonal antibody for immunoblotting. This approach excluded detection of the anti-GR antibody (or the non-immune IgG) used for the IP in the immunoblot. This strategy still demonstrated a prominent band at 52 kDa (representing co-IP’d AAT) following IP with anti-GR and immunoblotting for AAT but not with the control IgG (Figure 2D). To semi-quantify the co-IP’d proteins in these experiments, we performed densitometry of the relevant bands detected in the immunoblots following IP; the antibodies used in the IP experiments are labeled on the x-axis and the protein probed by the immunoblot is labeled on top of each of the bars in Figure 2E. In summary, the data provide evidence that AAT binds the GR to form a complex in THP-1 macrophages and MDM.
Co-IP and mass spectrometry approach
We adopted a co-IP and mass spectrometry approach to further validate the physical interaction between AAT and the GR. THP-1 macrophage lysates were incubated with non-immune IgG or anti-GR antibody as before, and the IP’d proteins were separated with SDS-PAGE under reducing conditions. After electrophoresis, the gel was stained with Coomassie blue. Protein bands that corresponded in molecular weight to AAT following IP with non-immune IgG and anti-GR antibody were excised from the gel and prepared for mass spectrometry (Figure 3A, dashed and solid black line boxes). Since the molecular weight of AAT may vary based on its glycosylation, we also excised adjacent areas of the gel just beneath the first excised sites and performed mass spectrometry of the samples (Figure 3A, dashed and solid red line boxes).
Figure 3. Immunoprecipitation-mass spectrometry and microscale thermophoresis demonstrate AAT–GR interaction.
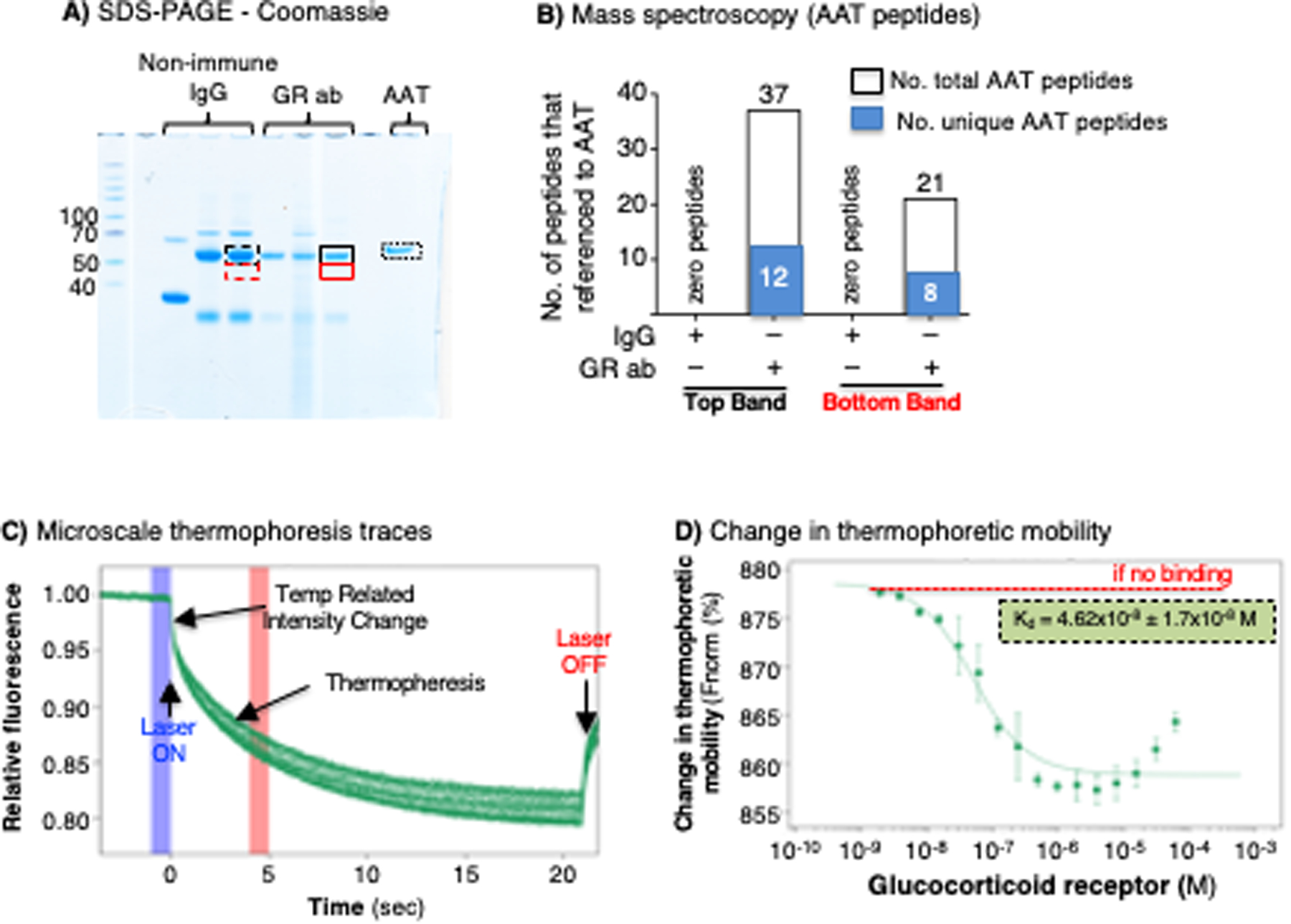
(A) THP-1 macrophages were immunoprecipitated (IP’d) with non-immune IgG or anti-GR antibody, the IP’d fractions were separated by SDS-PAGE, and the gel was stained with Coomassie blue. Stained bands in the gel that ran similarly to AAT (dash and solid black line boxes) were excised along with an area just beneath the aforementioned band (dash and solid red line boxes) and subjected to mass spectrometry. (B) Graphical and numerical representation of the number of total and unique peptides that referenced to the AAT protein in samples IP’d with control IgG (zero AAT peptides for either the top or bottom bands) or with anti-GR antibody (37 total AAT peptides with 12 unique AAT peptides in the top band, and 21 total AAT peptides with 8 unique to AAT in the bottom band). Data shown are representative of two independent experiments. (C) Microscale thermophoresis time-course tracings following the mixing of fluorescent-tagged AAT with 16 different GR concentrations and subjected to a thermogradient. (D) The presence of a “change in thermophoretic mobility” of the fluorescent-tagged AAT with varying GR concentrations demonstrates that the two molecules interact in vitro. Data shown in (D) are the mean ± SD of two independent experiments. AAT=alpha-1-antitrypsin; ab=antibody; GR=glucocorticoid receptor.
Mass spectrometry of the band corresponding to the pharmacologic control AAT (Glassia®) (Figure 3A, dotted black box, far right AAT lane) revealed 1762 total peptides and 62 unique peptides that referenced to the complete AAT protein. For the cell lysates IP’d with non-immune IgG, mass spectrometry of both the upper band and lower adjacent area (Figure 3A, dashed back and red line boxes, respectively) yielded zero peptides corresponding to AAT (Figure 3B). For the cell lysates IP’d with anti-GR, mass spectrometry of the upper band (Figure 3A, solid black line box) yielded 37 total peptides that corresponded to amino acid sequences of AAT, 12 of which are unique to the complete AAT protein (Figure 3B). Similarly, the lower adjacent area (Figure 3A, solid red line box) revealed a total of 21 peptides that referenced to AAT, 8 of which were unique peptides to AAT (Figure 3B). As anticipated, the heavy chain of IgG also migrated to a similar position as AAT in that the upper band and the adjacent lower area of the migrated lysates IP’d with non-immune IgG or anti-GR contained 540 and 1,175 peptides (or 265 and 848 peptides) that referenced to their respective IgG heavy chains. These findings further corroborated that AAT co-IP’d with GR.
Microscale Thermophoresis approach
To determine whether AAT binds to the GR in a cell-free system, we employed Microscale Thermophoresis (MST) technology. MST is a highly sensitive method that measures the mobility of a fluorescently labeled protein in the presence of a thermal gradient, a process known as thermophoresis (34). The thermophoretic mobility of a molecule is dependent on multiple factors, including charge, hydrodynamic radius, and interactions with the solvent. If the fluorescently labeled molecule binds another molecule, the thermophoretic mobility of the labeled molecule increases by a measurable amount. For this specific experiment, 10 nM of fluorescently labeled AAT was combined with 16 GR concentrations ranging from 1.91 nM to 6.25 μM. The thermophoretic mobility of AAT changed in a GR concentration-dependent manner (Figure 3C/D). We speculate that the partial reversal of the AAT–GR thermophoretic mobility at the highest GR concentrations suggests the possibility that more than one GR binding to AAT as seen biologically as well as the molecular modeling (below). The calculated Kd of 4.62×10−8 ± 1.7×10−8 M indicates a strong interaction between the AAT and GR and validates in a cell-free system that AAT and GR bind to each other.
Molecular modeling of AAT–GR interaction
To begin to understand how AAT and GR may interact stereochemically, we performed in silico molecular modeling and docking calculations using two state-of-the-art approaches: AlphaFold2 (29) and ClusPro (32). GR is organized into three major domains: an intrinsically disordered N-terminal activation function-1 domain (NTD) and two ordered domains comprised of a DNA binding domain (DBD) and a C-terminal ligand binding domain (LBD) (Figure 4A). In addition to its role in ligand recognition, the LBD also contains a ligand-dependent activation function region (AF-2) that is tightly regulated by hormone binding (dotted box). Structural modeling of the GR protein reveals that an unstructured hinge region links the LBD and DBD (Figure 4B). Docking analyses revealed several plausible modes of interaction between AAT and GR both in the cytosol and the nucleus. The top docking solution from AlphaFold2 shows AAT interacting with the LBD via its reactive center loop (RCL), in a binding mode compatible with the GR–Hsp90/p23 cochaperone complex (35) (Figure 4C). The AAT–GR complex is also compatible with the known dimeric arrangement of LBD (36) (Figure 4D). The RCL of AAT interacts hydrophobically with the ligand-dependent AF-2 region of the LBD in a similar fashion to that of the two nuclear coregulator proteins – nuclear receptor corepressor (NCoR) and transcriptional intermediary factor-2 (TIF2; Figure 4D). Superposed is a monomeric NCoR-bound LBD showing key structural differences within the AF-2 site that favor repression over activation. Once translocated to the nucleus, the DBD of GR recognizes Glucocorticoid Response Elements (GREs). The top docking result using ClusPro positions AAT at the opposite end of the LBD, again interacting via its RCL, bridging the interaction with a monomeric DBD, but showing some degree of steric overlap with the second DBD formed by D-loop-mediated dimerization (Figure 4E). Thus, we hypothesize that the model shown in Figure 4D is stereochemically more plausible than the one shown in Figure 4E.
Figure 4. Molecular modeling of alpha-1-antitrypsin (AAT)–glucocorticoid receptor (GR) complex.
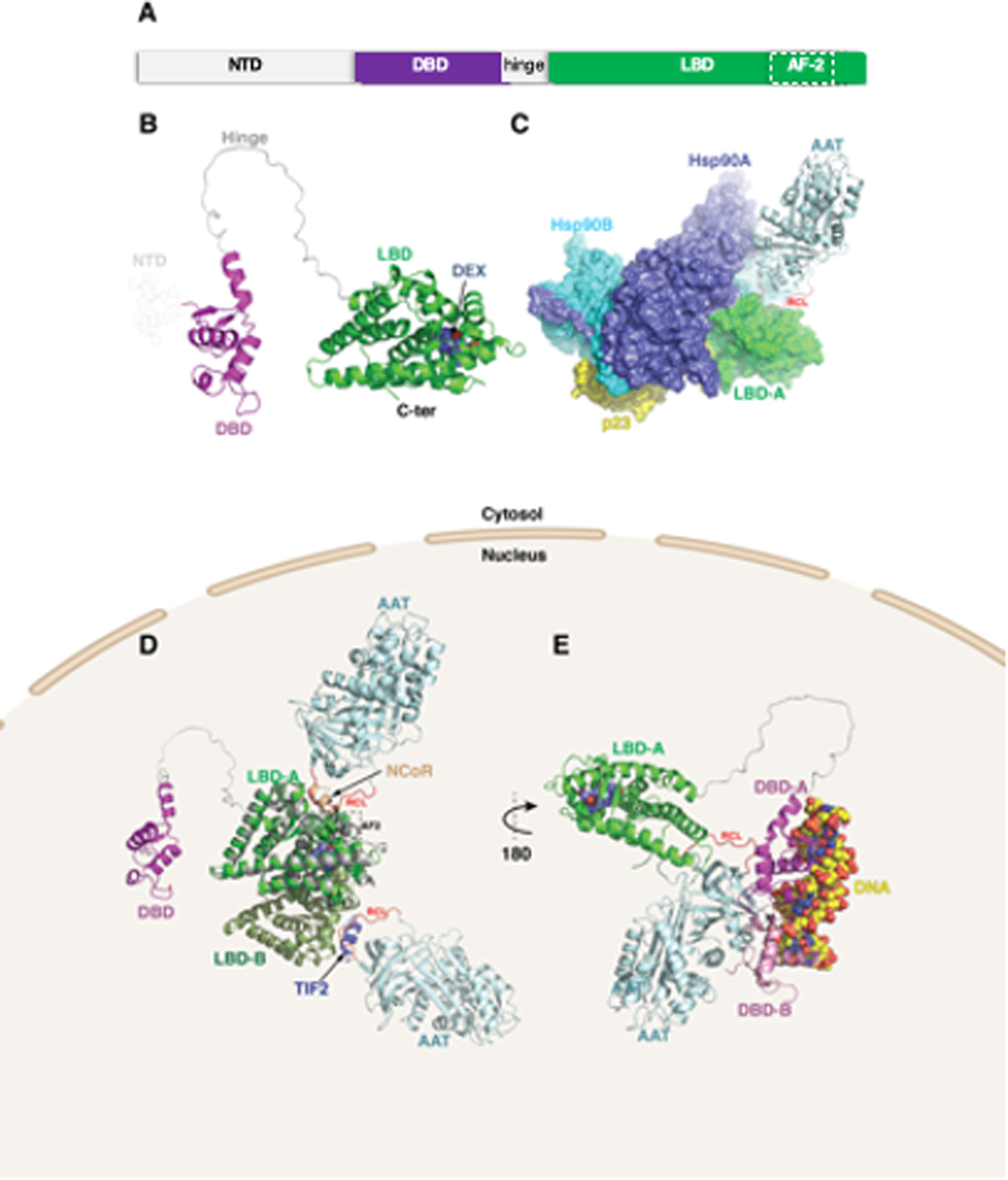
(A) GR is organized into three major domains: an intrinsically disordered N-terminal activation function-1 domain (NTD), a DNA binding domain (DBD), and a C-terminal ligand binding domain (LBD). (B) Structural modeling of the GR protein reveals the presence of the LBD (green) and DBD (magenta), linked by an unstructured hinge region (grey). The intrinsically disordered NTD was not modeled and is depicted by dotted lines. Dexamethasone bound to the LBD is shown as steel blue spheres. (C) AlphaFold2 docking simulations of LBD and AAT (PDB ID 3NE4) superimposed on the GR-Hsp90-p23 co-chaperone complex (PDB ID 7KRJ) show AAT interacting with the LBD via its RCL. (D) The same AAT–GR complex expanded to the LBD dimer (monomers labeled LBD-A and LBD-B). Superimposed are the nuclear coregulator proteins nuclear receptor corepressor (NCoR; derived from PDB ID 3H52) and transcriptional intermediary factor-2 (TIF2; derived from PDB ID 1M2Z) bound to the AF-2 site (broken rectangle) are shown as brown and slate-blue cartoons, respectively. (E) The second model using ClusPro docking shows the RCL of AAT interacting with the LBD of GR. In addition, the DBD is shown complexed with DNA (space-filling atoms), modeled using the GR DNA Binding Domain monomer – TSLP nGRE Complex (magenta; PDB ID 5HN5). Also shown (pink) is a second DBD formed by D-loop-mediated dimerization (PDB ID 5E69), having some degree of steric overlap with AAT. RCL (red)=reactive center loop.
The AAT–GR complex is found in both the cytoplasmic and nuclear fractions
Given that GR can localize to the nuclear compartment, our next step in characterizing the importance of the AAT–GR interaction was to identify whether the AAT–GR complex exists in the cytoplasm, nucleus, or both. We prepared nuclear and cytoplasmic fractions of the THP-1 macrophage lysates (Figure 5A) and performed IP for GR, followed by AAT immunoblotting. We identified the presence of the AAT–GR complex in both the nuclear and cytoplasmic fractions, suggesting that this interaction may play a role in the transcriptional function of GR (Figure 5B).
Figure 5. The alpha-1-antitrypsin (AAT)–glucocorticoid receptor (GR) complex is found in both the nucleus and cytoplasm.
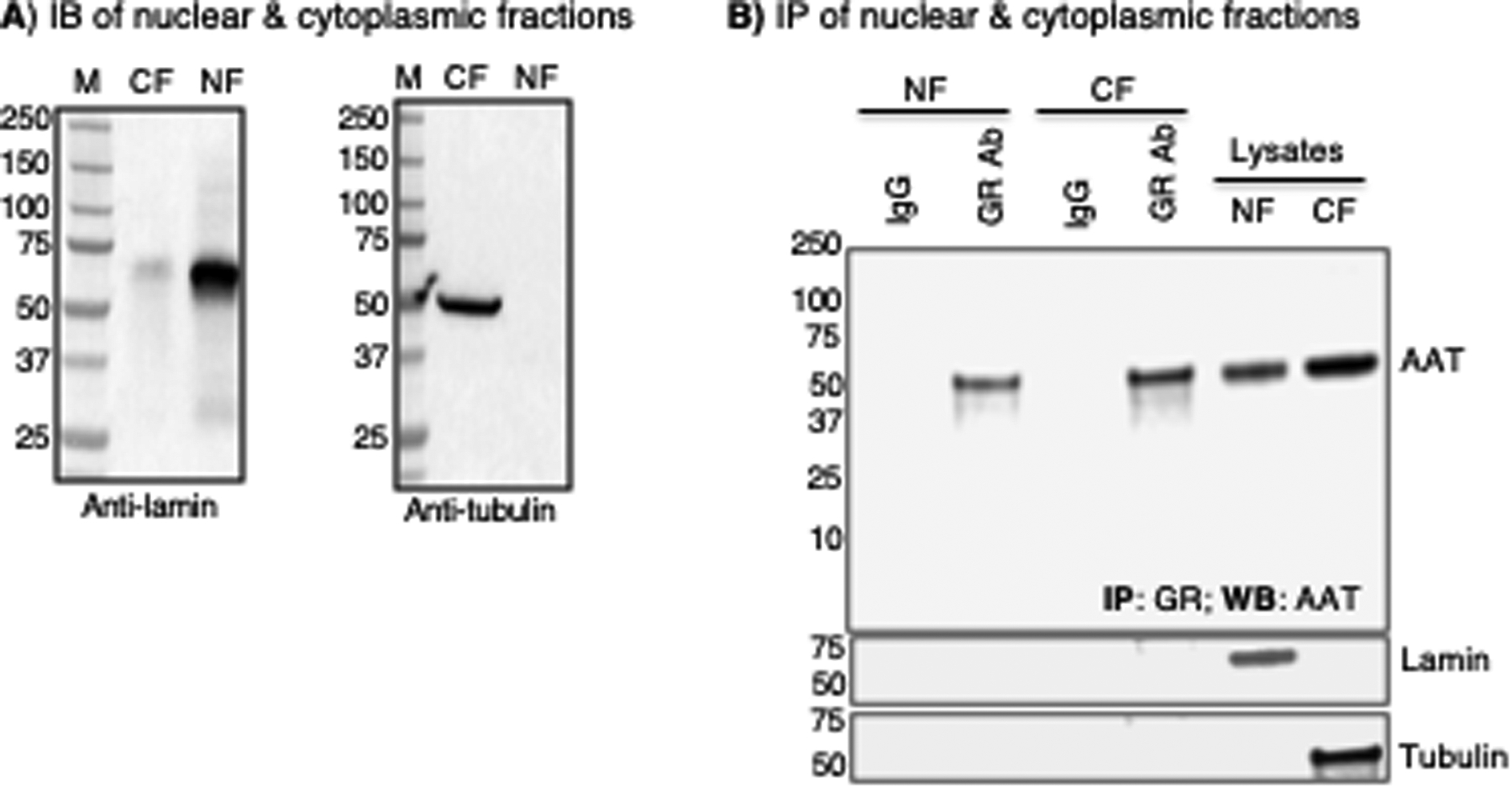
(A) Pre-immunoprecipitation immunoblot (IB) experiment to detect lamin and tubulin to confirm specific isolation of nuclear and cytoplasmic fractions, respectively. (B) Immunoprecipitation (IP) of the nuclear and cytoplasmic fractions for GR and immunoblot of immunoprecipitated lysates with an anti-AAT antibody. The same membrane was then immunoblotted for lamin and tubulin (two lower panels). All data shown are representative of three independent experiments. CF=cytoplasmic fraction, NF=nuclear fraction
Stable knockdown of GR in THP-1 macrophages
We next investigated the biological importance of the AAT–GR interaction by generating a pool of transformed THP-1 macrophages where GR was stably knocked down (KD) using a lentiviral vector that expressed a shRNA-GR construct. To confirm that GR was depleted, we immunoblotted the cell lysates of THP-1control and THP-1GR-KD for GR, demonstrating a significant down-regulation of GR in the THP-1GR-KD cells (Figure 6A, left panel). Semi-quantitation of the relative knockdown of GR by densitometry measurement of the GR immunoband revealed ~4-fold reduction of the GR protein in the THP-1GR-KD cells compared to the THP-1control cells (Figure 6A, right panel). To further validate the depletion of GR expression in THP-1GR-KD macrophages, RNA sequencing for GR mRNA in THP-1control and THP-1GR-KD cells showed that THP-1GR-KD had a 2.5-fold reduction in mRNA for GR compared to that of THP-1control cells (Figure 6B).
Figure 6. Glucocorticoid or AAT inhibition of NFκB activation, inhibition of IL-8 production, and induction of angiopoietin-like 4 is glucocorticoid receptor-dependent.
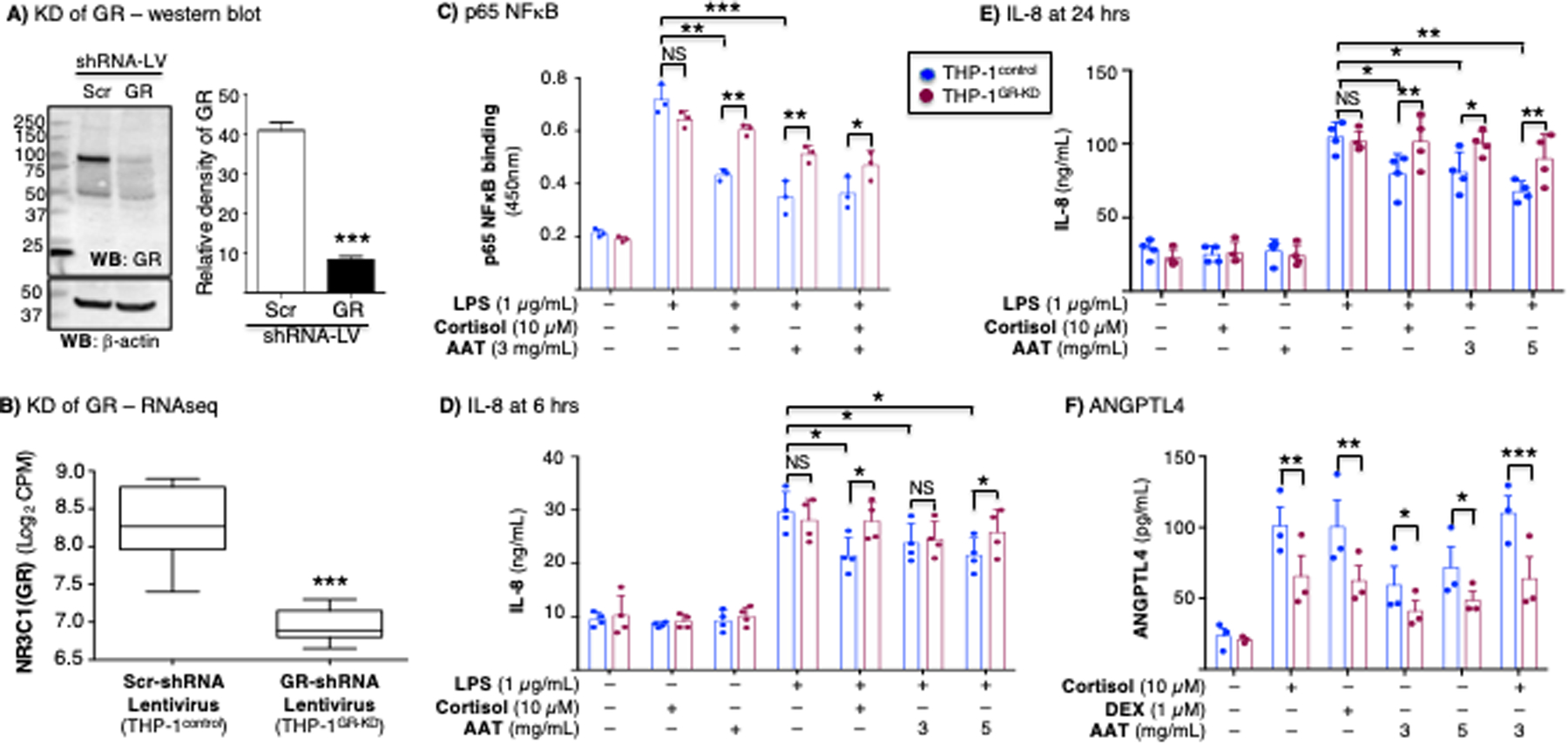
(A) Western blot of whole-cell lysates of THP-1control and THP-1GR-KD macrophages for GR and β-actin. Densitometry of the GR band on immunoblot normalized for β-actin (***p<0.001 compared to Scr-shRNA lentivirus). The immunoblot and densitometry shown are representative and the mean of three independent experiments, respectively. (B) RNA sequencing (RNAseq) for the GR gene (NR3C1) transcript of the THP-1control and THP-1GR-KD cells using shRNA-lentivirus technology (***p<0.001 compared to Scr-shRNA lentivirus). (C) THP-1control and THP-1GR-KD macrophages were left untreated or pre-treated with cortisol, AAT, or both at the indicated concentrations for 30 minutes, and after stimulation with lipopolysaccharide (LPS) for 6 hours, p65-NFκB binding assay to its consensus oligonucleotide was performed. THP-1control and THP-1GR-KD macrophages were left untreated or pre-treated with cortisol, AAT, or both at the indicated concentrations for 30 minutes, and after stimulation with LPS for (D) 6 hours or (E) 24 hours, the supernatants were assayed for IL-8 by ELISA. (F) Glucocorticoid (cortisol or dexamethasone) or AAT induction of ANGPTL4 in THP-1control and THP-1GR-KD macrophages. Experiments in (C) / (F) and (D) / (E) are the mean ± SEM of three and four independent experiments, respectively, with each experiment done in duplicates. *p<0.05, **p<0.01, ***p<0.001. Blue bars=control shRNA (THP-1control), red bars=GR shRNA (THP-1GR-KD). NS=not significant, KD=knockdown, LV=lentivirus, Scr=scrambled.
AAT–GR interaction inhibits lipopolysaccharide-induced NFκB activation and IL-8 production in THP-1 cells
Since GCs have potent anti-inflammatory function via the ability of the GC–GR complex to affect inflammatory gene transcription related to NFκB targets (24, 37), we examined the role of AAT in NFκB activation in the context of GR signaling. We pre-treated THP-1control and THP-1GR-KD macrophages with cortisol, AAT, or both for 30 minutes, then stimulated the cells with lipopolysaccharide (LPS) for 6 hours and quantified the binding of p65-NFκB to its consensus oligonucleotide. In THP-1control cells, LPS induced p65-NFκB binding was significantly inhibited by cortisol or AAT (Figure 6C, blue bars). However, although LPS-induced activation of p65-NFκB in THP-1GR-KD macrophages was comparable to THP-1control cells (Figure 6C, the second set of red and blue bars, respectively), there was less inhibition by cortisol or AAT in the THP-1GR-KD macrophages (Figure 6C, compare the second set of blue and red bars to their corresponding last three sets of bars). Furthermore, whereas the inhibitory effect of cortisol was nearly completely abrogated in the THP-1GR-KD macrophages (Figure 6C, second vs. third red bars), the inhibitory effects of AAT were not completely attenuated in the THP-1GR-KD macrophages (Figure 6C, second vs. fourth and fifth red bars), suggesting that AAT also affects NFκB signaling in a GR-independent manner.
To compare LPS-induced expression of IL-8 in the supernatants of THP-1control and THP-1GR-KD macrophage cultures, both cell types were stimulated with LPS for 6 and 24 hours alone or in the presence of cortisol (10 μM) or AAT (3 and 5 mg/mL), and the supernatants measured for IL-8 expression. The normal AAT level in plasma is 1–2 mg/mL but may increase 3- to 5-fold in states of systemic inflammation and/or infection (38, 39). There was robust induction of IL-8 by LPS alone at 6 hours (Figure 6D) and at 24 hours (Figure 6E) in both THP-1control and THP-1GR-KD macrophages, with greater amounts accumulated at 24 hours. In THP-1control macrophages, there was significant inhibition of LPS-induced IL-8 expression following incubation with cortisol or 5 mg/mL AAT, particularly at 24 hours (Figures 6D/E, blue bars). However, in THP-1GR-KD macrophages, neither cortisol nor AAT significantly inhibited LPS-induced IL-8 production (Figures 6D/E, red bars).
AAT induction of angiopoietin-like-4 protein is mediated by GR
Either AAT or GCs induce the expression and release of angiopoietin-like-4 protein (ANGPTL4) in human blood monocytes and human microvascular endothelial cells (40–42). Thus, THP-1control and THP-1GR-KD macrophages were stimulated with cortisol (10 μM), dexamethasone (1 μM), AAT (3 and 5 mg/mL), or both cortisol and AAT for 24 hours and the supernatants quantified for ANGPTL4 (43). Stimulating THP-1control macrophages with cortisol, dexamethasone, or AAT induced ANGPTL4 production, although stimulation with AAT was not as robust as the GCs (Figure 6F, blue bars). However, GC- or AAT-induced production of ANGPTL4 was significantly less in the THP-1GR-KD macrophages (Figure 6F, red bars).
AAT–GR interaction inhibits lipopolysaccharide-induced IL-8 production in MDM
To determine whether the biological effects of AAT–GR interaction is also seen in primary macrophages, transient knockdown of GR of MDM was performed using siRNA technology. Compared to MDM that were transfected with scrambled siRNA (MDMcontrol), MDM transfected with siRNA directed against GR transcript (MDMGR-KD) revealed there was a modest but significant reduction in GR protein as determined by immunoblotting (Figure 7A). In MDMcontrol, LPS induced IL-8 which was inhibited by either cortisol or AAT (Figure 7B, blue bars); this inhibition was partially but significantly abrogated in MDMGR-KD cells (Figure 7B, red bars), similar to that seen with the THP-1 cells.
Figure 7. AAT inhibition of IL-8 production is glucocorticoid receptor-dependent in MDM.
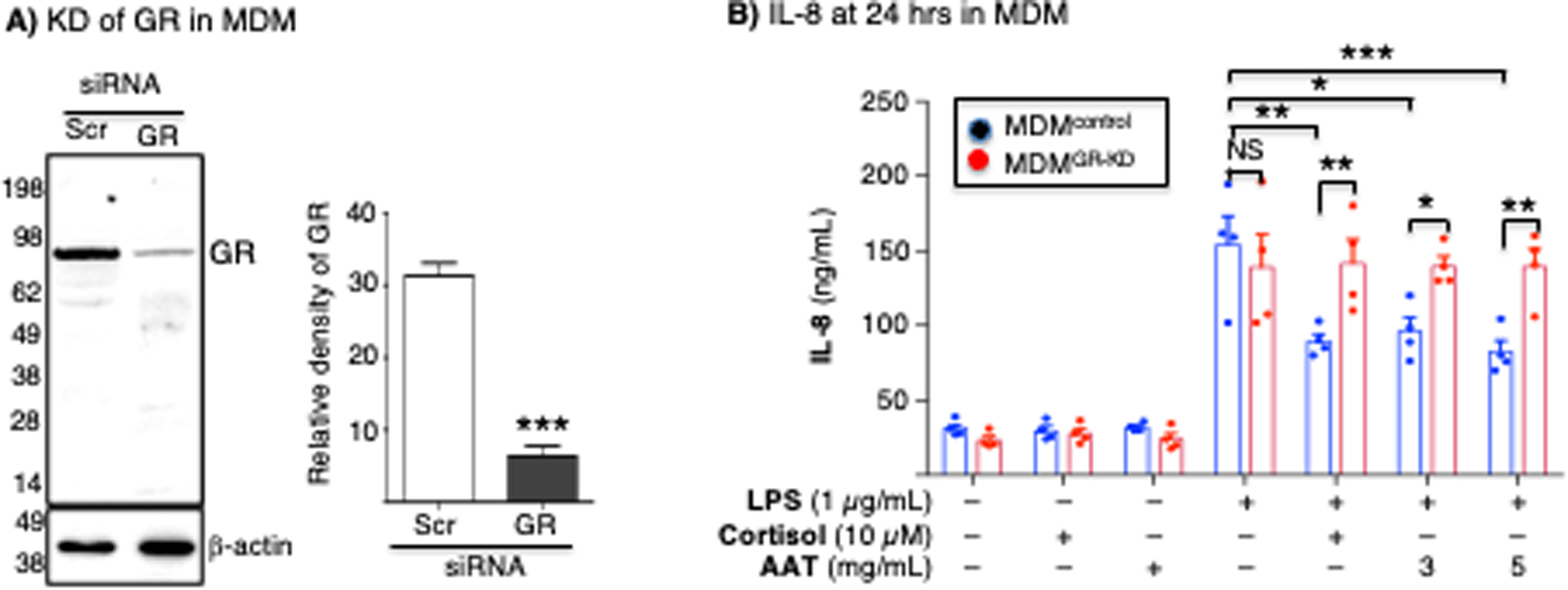
(A) MDM differentiated from the PBMC from a PiMM individual were transfected with scrambled siRNA (control) or GR siRNA. Cell lysates from MDMcontrol or MDMGR-KD were separated by SDS-PAGE and immunoblotted for GR. Densitometry of the GR band on the immunoblot (mean density of two independent experiments). (B) MDMcontrol and MDMGR-KD were left untreated or pre-treated with cortisol (10 μM) or AAT (3 mg/mL) for 30 minutes, followed by stimulation with LPS for 24 hours, and supernatant quantified for IL-8 by ELISA. The immunoblot and densitometry analysis of GR are representative and the mean of three independent experiments, respectively. ELISA for IL-8 is the mean of three independent experiments. *p<0.05, **p<0.01, ***p<0.001. Blue bars=control siRNA (MDMcontrol), red bars=GR siRNA (MDMGR-KD). NS=not significant, KD=knockdown, control=scrambled.
AAT–GR binding enhances mycobacterial control in THP-1 macrophages
The consensus among clinicians and scientists is that GCs increase susceptibility to Mycobacterium tuberculosis (44, 45) and possibly non-tuberculous mycobacteria (NTM) (46). On the other hand, AAT enhances autophagy, an effector mechanism known to kill intracellular mycobacteria (4, 47–49). Since canonical GC–GR signaling and the cellular effector function(s) induced by AAT appear to have opposing effects in controlling mycobacterial infection, we examined in macrophages the role of the AAT–GR interaction in the control of M. tuberculosis and Mycobacterium intracellulare (an NTM) infection. THP-1control and THP-1GR-KD macrophages were initially infected with M. tuberculosis H37Rv or M. intracellulare at a multiplicity-of-infection of 10 mycobacteria:1 macrophage for 1 hour, 2 days, and 4 days (without exogenous GC or AAT added), and then the mycobacteria quantified. We detected a productive M. tuberculosis and M. intracellulare infection of the THP-1control macrophages, peaking at day 2 (Figure 8A, top and bottom panels, blue bars), and which, unexpectedly, consistently increased in the THP-1GR-KD macrophages (Figure 8A, top and bottom panels, red bars). Compared to uninfected THP-1control and THP-1GR-KD macrophages, there was ~10–15% reduction in viability in both cell phenotypes (as measured by the MTT assay) after 4 days of infection with either mycobacterium (data not shown), similar to what we previously reported (50).
Figure 8. Mycobacterial infection of THP-1control and THP-1GR-KD macrophages in the absence or presence of alpha-1-antitrypsin (AAT) or glucocorticoid.
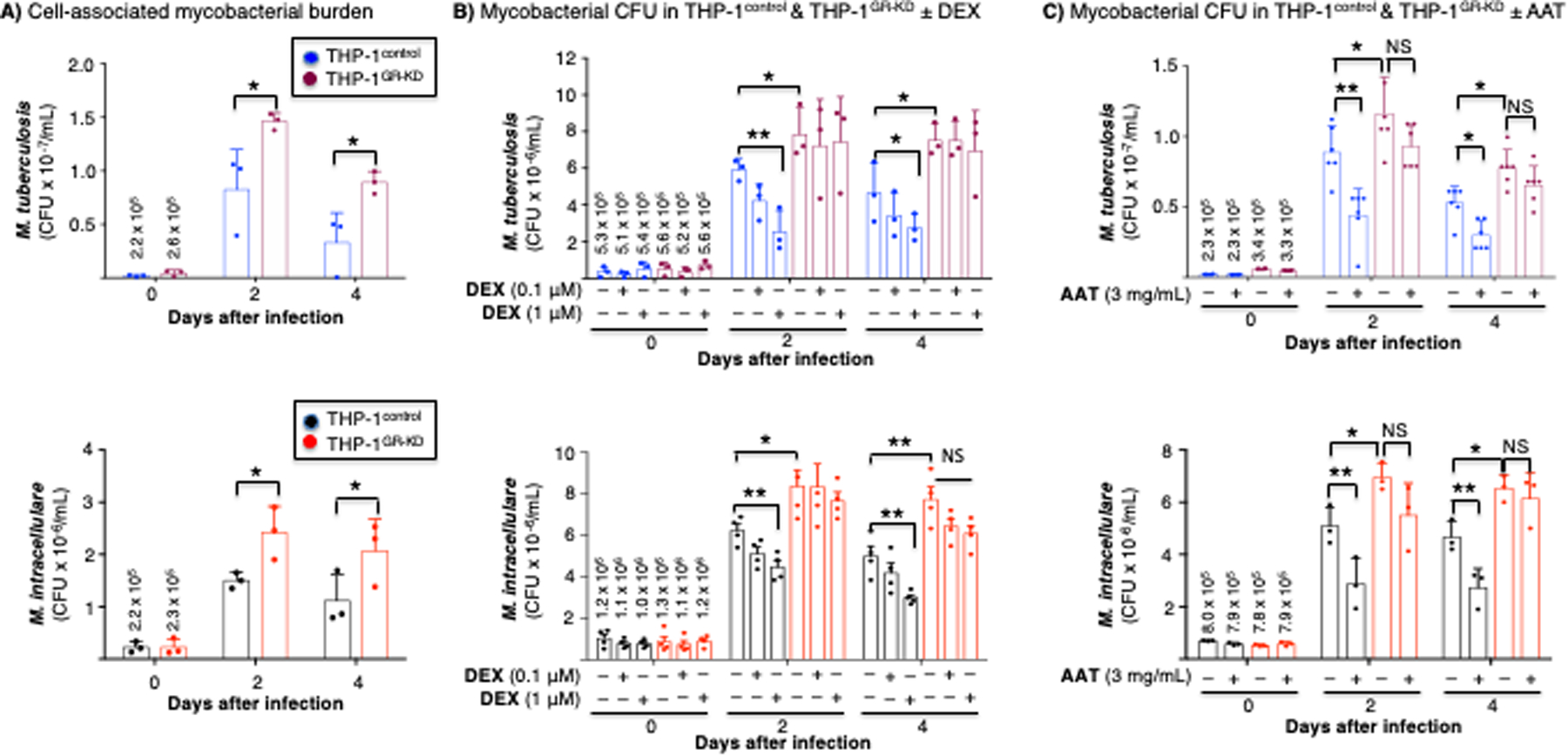
(A) THP-1control and THP-1GR-KD macrophages were infected with M. tuberculosis H37Rv or M. intracellulare for 1 hour, 2 and 4 days. The cells were washed, and intracellular mycobacteria quantified. (B) THP-1control and THP-1GR-KD macrophages were left untreated or pre-treated with dexamethasone (0.1 or 1 μM) for 60 minutes, followed by M. tuberculosis (top panel) or M. intracellulare (bottom panel) infection for 1 hour, 2 days, and 4 days. The cells were washed and intracellular mycobacteria quantified. (C) THP-1control and THP-1GR-KD macrophages were left untreated or pre-treated with 3 mg/mL AAT for 30 minutes, infected with M. tuberculosis (top panel) or M. intracellulare (bottom panel) for 1 hour, 2 and 4 days, the cells washed, and intracellular mycobacteria quantified. Experiments in (A) and (B) are the mean ± SEM of three independent experiments, and in (C), the mean ± SEM of six independent experiments, with each experiment done in duplicates. *p<0.05, **p<0.01. The numbers shown above the bars at t=0 in (B) and (C) are the mean cell-associated CFU at 1 hour after infection. Blue bars=control shRNA (THP-1control), red bars=GR shRNA (THP-1GR-KD). NS=not significant.
Since depletion of basal GR increased the burden of both mycobacterial species in THP-1 macrophages, we sought to determine the effects of GCs in the control of M. tuberculosis and M. intracellulare. THP-1control and THP-1GR-KD macrophages were pre-incubated with 0.1 and 1 μM dexamethasone for 60 minutes and then infected with M. tuberculosis or M. intracellulare. One hour, 2 days, and 4 days after infection, the infected macrophages were washed, lysed, and quantified for each of the mycobacterial species by counting CFUs on solid agar plates. In THP-1control macrophages, dexamethasone reduced the burden of both M. tuberculosis and M. intracellulare in a dose-dependent fashion (Figure 8B, top and bottom panels, blue bars). However, dexamethasone had no significant effect on the number of mycobacterial CFUs in the THP-1GR-KD macrophages (Figure 8B, top and bottom panels, red bars).
To determine the effects of AAT on M. tuberculosis and M. intracellulare infection in the context of GR, THP-1control and THP-1GR-KD macrophages were then pre-incubated with AAT (3 mg/mL) for 30 min, infected with M. tuberculosis or M. intracellulare, and mycobacteria quantified 1 hour, 2 days, and 4 days after infection. In THP-1control macrophages, pre-incubation with AAT significantly reduced the burden of both M. tuberculosis and M. intracellulare (Figure 8C, top and bottom panels, blue bars), whereas this did not occur in THP-1GR-KD macrophages (Figure 8C, top and bottom panels, red bars). While the ability of dexamethasone to enhance macrophage control of M. tuberculosis (or M. intracellulare) is not consistent with the paradigm that GC use may predispose patients to tuberculosis (TB) and NTM infections, other studies have shown that GCs can induce a host-protective phenotype in innate immune cells through increased expression of Toll-like receptor (TLR)2 and enhanced TLR4 signaling (51–54)(51–53). Thus, to determine whether GC alone can induce TLR2, a pattern recognition receptor capable of recognizing lipoproteins of mycobacteria, we immunoblotted for TLR2 in unstimulated cells or with dexamethasone stimulation in THP-1control and THP-1GR-KD macrophages. Notably, dexamethasone induced TLR2 production in the THP-1control macrophages, but its expression was markedly diminished in the THP-1GR-KD macrophages (Figure 9A/B).
Figure 9. Glucocorticoid induces expression of a pattern-recognition receptor.
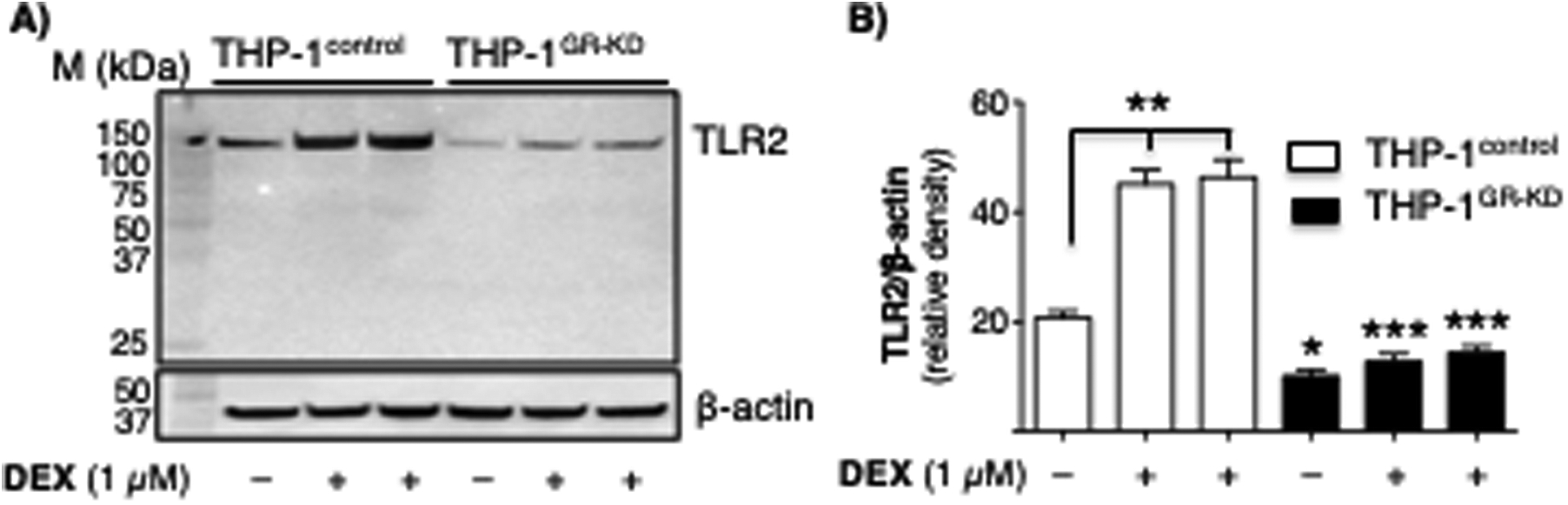
(A) THP-1control and THP-1GR-KD macrophages were left untreated or pre-treated with dexamethasone (1 μM) for 24 hours, followed by immunoblotting of the whole-cell lysates for TLR2 (top). The same membrane was stripped and immunblotted for β-actin (bottom). Data shown in representative of three independent experiments. (B) Relative densitometric measurement of TLR2 protein normalized for β-actin. Data shown are the mean density (arbitrary units) of three independent experiments. *p<0.05, **p<0.01, ***p<0.001. p values for the THP-1GR-KD cells are in comparison to their corresponding THP-1control cells.
DISCUSSION
Herein we demonstrated that AAT binds directly to the GR in THP-1 macrophages and primary human MDMs. We further showed in THP-1GR-KD macrophages that AAT–GR interaction inhibited LPS-induced NFκB activation and IL-8 production, induced ANGPTL4 expression, and reduced M. tuberculosis and M. intracellulare burden in macrophages. Similar to that seen in THP-1 cells, the ability of cortisol or AAT to inhibit LPS-induced IL-8 was abrogated in MDMGR-KD compared to MDMcontrol. We recognize that alternative pathways, such as Akt signaling, may also inhibit both LPS-induced NFκB activation and IL-8 production through GR (55). Because AAT-mediated inhibition on NFκB activation was less abrogated than the inhibition by GC in the THP-1GR-KD cells (Figure 6C), it suggests that AAT affects signaling and gene regulation in both GR-dependent and GR-independent fashion (9, 56).
Individuals with AAT deficiency have increased neutrophilic pulmonary infiltration and proclivity to neutrophil-mediated panniculitis or vasculitis due to the ability of AAT to inhibit neutrophil infiltration by two reported mechanisms: (i) the ability of the glycan moieties of AAT to bind IL-8, preventing the neutrophil chemokine from binding to its receptor (CXCR1) and (ii) the ability of AAT to inhibit ADAM17, a disintegrin capable of cleaving cell membrane-bound FcgRIIIb bound to immune complexes which then binds complement receptor 3 to induce cytoskeletal rearrangements necessary for neutrophil chemotaxis (18, 57). We now demonstrate a third mechanism where AAT promotes GR-mediated inhibition of IL-8 production via antagonizing proinflammatory NFκB activation.
Ultimately, while we have established the AAT–GR interaction and the physiologic role of this interaction in antagonizing NFκB-mediated inflammation, promoting the transcription of target genes, and mediating anti-mycobacterial function (Figure 10A), the molecular mechanism by which AAT binds GR and regulates transcription remains to be defined. While AAT is unlikely to bind to the canonical GC binding site of the LBD of GR (although molecular modeling predicts AAT binding to the AF-2 site of LBD of GR), several alternative avenues that are not necessarily mutually exclusive may explain its role in GR function, forming the basis for further investigations (Figure 10B). First, our co-IP findings of the nuclear and cytoplasmic fractions suggest GR may shuttle between the two compartments (Figure 10B-1). A possible role of AAT binding to GR may be to increase GR localization to the nuclear compartment compared to absent or lower AAT levels. This hypothesis is consistent with the acute phase reactant nature of both cortisol and AAT in states of systemic inflammation and infection. Second, exogenous or supraphysiologic AAT may act primarily in the cytoplasm to modulate the disassembly of GR from chaperone proteins and facilitate nuclear translocation and transactivation upon binding to canonical ligands (Figure 10B-2). We observed GR-dependent inhibition of NFκB activity in response to AAT but in the absence of exogenous GC. This may be due to low concentrations of GC in the cell culture conditions potentiated by AAT. A less likely possibility is that of a direct transcriptional role for AAT that facilitates translocation of GR to the nucleus where GR activates transcription in a GC-independent manner, although each is not necessarily mutually exclusive of the other. A third possibility of the cooperation between AAT and the GC–GR is that the serpin activity of AAT may exert an indirect, stabilizing effect on some transcriptional complexes that interact with GR, which may occur via its serpin or by a yet uncharacterized mechanism (Figure 10B-3).
Figure 10. Illustration of the key findings and hypotheses of the AAT–GR interactions.
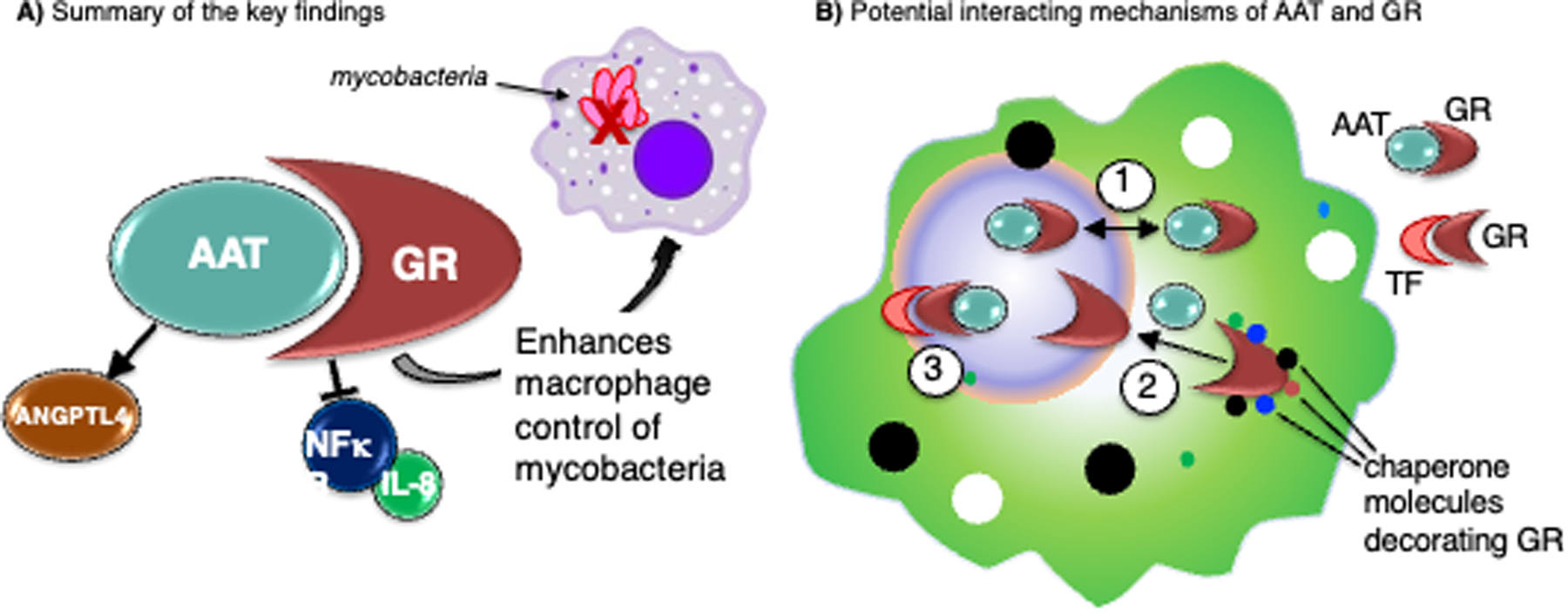
(A) Summarizing the key findings, alpha-1-antitrypsin–glucocorticoid receptor (AAT–GR) interaction inhibits nuclear factor-kappa B (NFκB) activation and interleukin-8 (IL-8) production, induces angiopoietin-like 4 (ANGPTL4), and enhances macrophage control of mycobacteria. (B) Several hypotheses of how AAT–GR interaction may affect cellular function: (1) AAT may shuttle GR between the nuclear and cytoplasmic compartments; (2) AAT may facilitate disassembly of GR–chaperone complexes; (3) AAT may stabilize transcriptional complexes of GR–transcription factor (TF, orange structure) or locally modulate the activity of proteases.
We performed molecular modeling of AAT–GR complexes to explore these possibilities further. AAT binding to the LBD of GR is compatible with cochaperone-GR interactions in the cytosol (Figure 4C), suggesting that AAT may play a role in the assembly/disassembly of the cochaperone-GR complex. Intriguingly, this complex positions the RCL of AAT interacting with the AF-2 region of the LBD of GR in a similar mode to the binding of the nuclear coregulator proteins NCoR and TIF2 to GR (Figure 4D). Although the RCL does not contain the conserved Lxxx(I/L)xxx(I/L) motif found in corepressors (58), its hydrophobic nature and flexibility are compatible with the hydrophobic AF-2 site. Conformational dynamics of the AF-2 site dictate its function as a coactivator or a corepressor site (59, 60), and this is apparent from structural comparisons of LBD-ligand complexes (Figure 4D). Coregulator molecules binding to AF-2 tune its conformational dynamics and its cooperativity with the GC ligand binding site, and thus govern downstream events; for example, the release of bound chaperones, nuclear translocation, dimerization, DNA-binding, and ultimately transcription. Therefore, it is plausible that AAT binding to the AF-2 site may similarly achieve allosteric regulation of downstream activation and repression pathways. An alternative docking solution positions AAT at the opposite end of the LBD, again interacting via its RCL and bridging the interaction with a monomeric DBD; however, this model showed some degree of steric overlap with the second DBD formed by D-loop-mediated dimerization (Figure 4E). This mode of interaction within the nucleus prompts speculation that AAT binding may favor monomer over dimer and thus the repression of pro-inflammatory genes (61). However, our finding that AAT induces ANGPTL4 expression, together with the known dimeric GR DBD that binds to the ANGPTL4 GRE (41), is more consistent with a scenario where AAT is released upon dimerization, which can then exert localized effects, be it protease-inhibition or other binding activity that may be key for gene induction. However, the flexible nature of the hinge region limits the accuracy of the modeling and therefore such interpretations must be made with caution. Nevertheless, overall, modeling highlights several experimentally-testable hypotheses on the role of AAT in GR biology.
Our findings suggest that short-term GR activation (whether by GC or AAT) in differentiated THP-1 macrophages directs protective responses against mycobacteria. On the surface, this notion is counterintuitive given the well-described pharmacologic role of GC in suppressing immunity, resulting in predisposition to tuberculosis or NTM infection (44, 45). However, it is important to emphasize that GC-induced susceptibility to TB or NTM infection occurs with GC doses that are significantly greater than physiologic output and for an “extended period” of time in vivo. Yet, we demonstrate in two lines of evidence that GR activation enhances THP-1 macrophage control of M. tuberculosis and M. intracellulare: (i) the downregulation of GR in the absence of exogenous AAT (or GC) resulting in increased M. tuberculosis and M. intracellulare burden in the differentiated mononuclear cells and (ii) the treatment of THP-1 macrophages with either AAT or GC reduced the burden of M. tuberculosis and M. intracellulare in a GR-dependent fashion. Published literature supports this seemingly paradoxical finding. Tukenmez et al (62) found that different GC formulations individually decreased M. tuberculosis burden in human MDMs. Bongiovanni and co-workers (63) reported that cortisol decreased phagocytosis of M. tuberculosis (by ~10%) with further reduction in the intracellular burden of M. tuberculosis by day 4 of culture in THP-1 macrophages. In contrast, Wang et al (64) found that dexamethasone enhanced the growth of M. bovis BCG in primary mouse and human macrophages, raising the possibility that the effects of GC on macrophage control of mycobacterial infection may be mycobacterial species- and host-specific. It cannot be overemphasized that such potential host-protective effects of GCs in pure macrophage cultures are likely to be different in vivo, where, for example, GCs in pharmacologic doses induce apoptosis in lymphocytes, subsets of which are critical in controlling mycobacterial infections. Thus, future elucidation on how GC may be exploited to maximize innate immune function while minimizing their immunosuppressive effects on lymphocytes may bring new approaches to treating mycobacterial infections.
In support of the aforementioned findings, other studies have shown that low-dose GC has immunoenhancing effects in myeloid cells (51, 53, 65–67). GCs have been shown to enhance immunity by inducing TLR2 expression, especially in the presence of TNF and IL-1β (51, 52). We also showed dexamethasone induced TLR2 expression in a GR-dependent fashion (Figure 9). TLR2 activation may also induce autophagy (68), and various polymorphisms of TLR2 have been associated with an increased risk for TB and NTM lung disease (69, 70). Zhang and Daynes (53) demonstrated that during differentiation of murine myeloid progenitors into macrophages, incubation with corticosterone increased the cellular responsiveness to LPS through increased expression of a phosphatase that inhibits PI3 kinase/Akt with subsequent increase in TLR4 signaling. Since we found that GC inhibited LPS-induction of NFκB activation and IL-8 production, possible explanations that may account for these disparities are: (i) host species differences accounting for variations in the macrophage phenotypic response to GC, (ii) differential responses to singular GC formulations, (iii) different signaling pathways that may oppose each other may be similarly affected by GC, and (iv) presence of a bimodal dose response paradigm wherein lower GC doses are immunopermissive whereas higher doses are immunosuppressive (53). Our previous findings that inhibition of NFκB activation in macrophages (by AAT or a small molecule inhibitor of IκBα) enhanced autophagy and clearance of M. intracellulare or M. tuberculosis infections would lend further credence to the finding that GC inhibition of NFκB activation augments macrophage activity against mycobacteria (4, 47).
We previously showed that AAT induces autophagosome formation and maturation in mycobacteria-infected macrophages, enhancing bacterial clearance (4). But could AAT have effects on other cell types that are beneficial in the context of TB? One possibility is that AAT antagonizes the damaging effects of neutrophils in the later stages of TB (71) by: (i) sequestering IL-8 (18), inhibiting neutrophil chemotaxis (57), and, as we have shown, inhibiting IL-8 production via binding to GR and (ii) inhibiting the formation of neutrophil extracellular traps (NETs) – which are elastase- and IL-8-induced extruded DNA decorated with citrullinated histones, elastase, and other proteins – meant to capture extracellular bacteria but may also contribute to tissue injury (72–75). AAT has also been shown to cause decreased shedding of macrophage mannose receptors (MMR), increasing their cell surface expression and reducing soluble MMR, effects which may contribute to the anti-inflammatory effects of AAT (76, 77).
A limitation of this study is that we used mainly a differentiated monocytic cell line to knock-down GR to study the biological significance of the AAT–GR interaction, albeit similar findings were found with MDMGR-KD in the context of cortisol or AAT inhibition of IL-8 production. On the other hand, the advantage of using THP-1 cells to create a pool of immortal cells stably knocked down for GR is that transient knockdown of primary macrophages would introduce more confounding variables such as non-clonality of cells, differential epigenetics among different individuals, the influence of AAT variants and glucocorticoid resistance (latter often due to genotypic differences in GR subunits), and the acute cellular stress associated with transient transfection to accomplish gene knockdown. Nevertheless, additional future studies using primary macrophages and accounting for the aforementioned variables to determine the biological significance of the AAT–GR interaction are likely to lead to additional mechanistic insights to this paradigm. Another limitation is that we used a laboratory strain of M. tuberculosis as clinical strains of varying virulence may have more robust immune evasive mechanisms. While subjects with AAT deficiency are predisposed to NTM lung disease, perhaps a third limitation is that we are unaware such individuals are also predisposed to TB, as our findings may imply. Possible reasons for this lattermost point are that AAT anomalies are uncommon in most of the TB endemic areas of the world due mostly to racial differences (i.e., clinically significant AAT variants are more common in the Caucasian population), AAT phenotype or levels are not checked in the vast majority of cases of TB, and because M. tuberculosis is relatively more virulent than NTM and can affect healthy individuals, the number of TB cases that may be associated with AAT deficiency will be a small minority of the entire TB population.
In conclusion, AAT is a highly abundant biomolecule with a well-demonstrated protective role against multiple inflammatory disorders and disease states. The AAT–GR interaction we described intersects two crucial anti-inflammatory pathways that are highly amenable to pharmacologic manipulation. In an in vitro macrophage model, we showed that AAT–GR interaction can attenuate inflammatory markers and augment macrophage control of M. tuberculosis and M. intracellulare infections. A novel implication of AAT–GR biology is an alternative or adjunct to GC therapy in treating chronic inflammation due to non-infectious or infectious causes. Indeed, comparable induction of GR responses between exogenous AAT and GC may provide novel treatment approaches, such as the development of small peptides that could serve as surrogates for AAT in order to manipulate GR, a key target for the widely prescribed GCs. These potential benefits of the AAT–GR interaction in macrophages must be tempered by the need to corroborate the findings in vivo.
Supplementary Material
Key Points.
Alpha-1-antitrypsin (AAT) binds to the glucocorticoid receptor (GR).
The AAT–GR interaction inhibits expression of inflammatory biomarkers.
The AAT–GR interaction augments macrophage control of mycobacterial infection.
Acknowledgment
We are grateful to Dr. Robert Sandhaus for helping to fund this research project. JRH is supported by NIH grants R03 AI139822 and R01 GM135421.
Footnotes
Conflict of interest
ANG holds equity in Psammiad Therapeutics. All other authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.de Serres F, and Blanco I. 2014. Role of alpha-1 antitrypsin in human health and disease. J Intern Med 276: 311–335. [DOI] [PubMed] [Google Scholar]
- 2.Parr DG, Guest PG, Reynolds JH, Dowson LJ, and Stockley RA. 2007. Prevalence and impact of bronchiectasis in alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 176: 1215–1221. [DOI] [PubMed] [Google Scholar]
- 3.Strnad P, McElvaney NG, and Lomas DA. 2020. Alpha(1)-Antitrypsin Deficiency. N Engl J Med 382: 1443–1455. [DOI] [PubMed] [Google Scholar]
- 4.Bai X, Bai A, Honda JR, Eichstaedt C, Musheyev A, Feng Z, Huitt G, Harbeck R, Kosmider B, Sandhaus RA, and Chan ED. 2019. Alpha-1-antitrypsin enhances primary human macrophage immunity against non-tuberculous mycobacteria,. Front Immunol 10: 1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bryan CL, Beard KS, Pott GB, Rahkola J, Gardner EM, Janoff EN, and Shapiro L. 2010. HIV infection is associated with reduced serum alpha-1-antitrypsin concentrations. Clin Invest Med 33: E384–E389. [DOI] [PubMed] [Google Scholar]
- 6.Chan ED, Kaminska AM, Gill W, Chmura K, Feldman NE, Bai X, Floyd CM, Fulton KE, Huitt GA, Strand MJ, Iseman MD, and Shapiro L. 2007. Alpha-1-antitrypsin (AAT) anomalies are associated with lung disease due to rapidly growing mycobacteria and AAT inhibits Mycobacterium abscessus infection of macophages. Scand J Infect Dis 39: 690–696. [DOI] [PubMed] [Google Scholar]
- 7.Harbig A, Mernberger M, Bittel L, Pleschka S, Schughart K, Steinmetzer T, Stiewe T, Nist A, and Böttcher-Friebertshäuser E. 2020. Transcriptome profiling and protease inhibition experiments identify proteases that activate H3N2 influenza A and influenza B viruses in murine airway. J Biol Chem Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pott GB, Beard KS, Bryan CL, Merrick DT, and Shapiro L. 2013. Alpha-1 antitrypsin reduces severity of pseudomonas pneumonia in mice and inhibits epithelial barrier disruption and pseudomonas invasion of respiratory epithelial cells. Front Public Health 1: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shapiro L, Pott GB, and Ralston AH. 2001. Alpha-1-antitrypsin inhibits human immunodeficiency virus type 1. FASEB J 15: 115–122. [DOI] [PubMed] [Google Scholar]
- 10.Whitney JB, Asmal M, and Geiben-Lynn R. 2011. Serpin induced antiviral activity of prostaglandin synthetase-2 against HIV-1 replication. PLoS One 6: e18589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou X, Liu Z, Zhang J, Adelsberger JW, Yang J, and Burton GF. 2016. Alpha-1-antitrypsin interacts with gp41 to block HIV-1 entry into CD4+ T lymphocytes. BMC Microbiol 16: 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou X, Shapiro L, Fellingham G, Willardson BM, and Burton GF. 2011. HIV replication in CD4+ T lymphocytes in the presence and absence of follicular dendritic cells: inhibition of replication mediated by α−1-antitrypsin through altered IκBα ubiquitination. J Immunol 186: 3148–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barbey-Morel C, Pierce JA, Campbell EJ, and Perlmutter DH. 1987. Lipopolysaccharide modulates the expression of alpha 1 proteinase inhibitor and other serine proteinase inhibitors in human monocytes and macrophages. J Exp Med 166: 1041–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serban KA, and Petrache I. 2016. Alpha-1 antitrypsin and lung cell apoptosis. Ann Am Thorac Soc 13 (Supplement 2): S146–S149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sohrab S, Petrusca DN, Lockett AD, Schweitzer KS, Rush NI, Gu Y, Kamocki K, Garrison J, and Petrache I. 2009. Mechanism of alpha-1 antitrypsin endocytosis by lung endothelium. FASEB J 23: 3149–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou X, Liu Z, Shapiro L, Yang J, and Burton GF. 2015. Low-density lipoprotein receptor-related protein 1 mediates α1-antitrypsin internalization in CD4+ T lymphocytes. J Leukoc Biol 98: 1027–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lockett AD, Petrusca DN, Justice MJ, Poirier C, Serban KA, Rush NI, Kamocka M, Predescu D, Predescu S, and Petrache I. 2015. Scavenger receptor class B, type I-mediated uptake of A1AT by pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol 309: L425–L434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergin DA, Reeves EP, Meleady P, Henry M, McElvaney OJ, Carroll TP, Condron C, Chotirmall SH, Clynes M, O’Neill SJ, and McElvaney NG. 2010. α−1 Antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8. J Clin Invest 120: 4236–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Queiroz TM, Lakkappa N, and Lazartigues E. 2020. ADAM17-Mediated Shedding of Inflammatory Cytokines in Hypertension. Front Pharmacol 11: 1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petrache I, Fijalkowska I, Medler TR, Skirball J, Cruz P, Zhen L, Petrache HI, Flotte TR, and Tuder RM. 2006. Alpha-1 antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosis. Am J Pathol 169: 1155–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gans H, and Tan BH. 1967. α1-antitrypsin, an inhibitor for thrombin and plasmin. Clinica Chimica Acta 17: 111–117. [DOI] [PubMed] [Google Scholar]
- 22.Aggarwal N, Koepke J, Matamala N, Martinez-Delgado B, Martinez MT, Golpon H, Stolk J, Janciauskiene S, and Koczulla R. 2016. Alpha-1 Antitrypsin Regulates Transcriptional Levels of Serine Proteases in Blood Mononuclear Cells. Am J Respir Crit Care Med 193: 1065–1067. [DOI] [PubMed] [Google Scholar]
- 23.Janciauskiene S, Wrenger S, Immenschuh S, Olejnicka B, Greulich T, Welte T, and Chorostowska-Wynimko J. 2018. The Multifaceted Effects of Alpha1-Antitrypsin on Neutrophil Functions. Front Pharmacol 9: 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Necela BM, and Cidlowski JA. 2004. Mechanisms of glucocorticoid receptor action in noninflammatory and inflammatory cells. Proc Am Thorac Soc 1: 239–246. [DOI] [PubMed] [Google Scholar]
- 25.Kadiyala V, Sasse SK, Altonsy MO, Berman R, Chu HW, Phang TL, and Gerber AN. 2016. Cistrome-based Cooperation between Airway Epithelial Glucocorticoid Receptor and NF-κB Orchestrates Anti-inflammatory Effects. J Biol Chem 291: 12673–12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ni K, Umair Mukhtar Mian M, Meador C, Gill A, Barwinska D, Cao D, Justice MJ, Jiang D, Schaefer N, Schweitzer KS, Chu HW, March KL, and Petrache I. 2017. Oncostatin M and TNF-α Induce Alpha-1 Antitrypsin Production in Undifferentiated Adipose Stromal Cells. Stem Cells Dev 26: 1468–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, Hager GL, and Stamatoyannopoulos JA. 2011. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet 43: 264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bai X, Silvius D, Chan ED, Escalier D, and Xu X. 2009. Identification and characterization of a novel testis-specific gene CKT2, which encodes a substrate for Protein Kinase CK2. Nucleic Acid Res 37: 2699–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, and Hassabis D. 2021. Highly accurate protein structure prediction with AlphaFold. Nature 596: 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nivón LG, Moretti R, and Baker D. 2013. A Pareto-Optimal Refinement Method for Protein Design Scaffolds. PLoS ONE 8: e59004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Konagurthu AS, Reboul CF, Schmidberger JW, Irving JA, Lesk AM, Stuckey PJ, Whisstock JC, and Buckle AM. 2010. MUSTANG-MR structural sieving server: applications in protein structural analysis and crystallography. PLoS One 5: e10048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, and Vajda S. 2017. The ClusPro web server for protein-protein docking. Nat Protocols 12: 255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bai X, Stitzel JA, Bai A, Zambrano CA, Phillips M, Marrack P, and Chan ED. 2017. Nicotine impairs macrophage control of Mycobacterium tuberculosis. Am J Respir Mol Cell Biol 57: 324–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jerabek-Willemsen M, Wienken CJ, Braun D, Baaske P, and Duhr S. 2011. Molecular interaction studies using microscale thermophoresis. Assay Drug Dev Technol 9: 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noddings CM, Wang RY, Johnson JL, and Agard DA. 2022. Structure of Hsp90-p23-GR reveals the Hsp90 client-remodelling mechanism. Nature 601: 465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, Consler TG, Parks DJ, Stewart EL, Willson TM, Lambert MH, Moore JT, Pearce KH, and Xu HE. 2002. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 110: 93–105. [DOI] [PubMed] [Google Scholar]
- 37.Van Bogaert T, De Bosscher K, and Libert C. 2010. Crosstalk between TNF and glucocorticoid receptor signaling pathways. Cytokine Growth Factor Rev 21: 275–286. [DOI] [PubMed] [Google Scholar]
- 38.Hazari YM, Bashir A, Habib M, Bashir S, Habib H, Qasim MA, Shah NN, Haq E, Teckman J, and Fazili KM. 2017. Alpha-1-antitrypsin deficiency: Genetic variations, clinical manifestations and therapeutic interventions. Mutat Res 773: 14–25. [DOI] [PubMed] [Google Scholar]
- 39.de Serres FJ, Blanco I, and Fernández-Bustillo E. 2003. Genetic epidemiology of alpha-1 antitrypsin deficiency in North America and Australia/New Zealand: Australia, Canada, New Zealand and the United States of America. Clin Genet. 64: 382–397. [DOI] [PubMed] [Google Scholar]
- 40.Frenzel E, Wrenger S, Brügger B, Salipalli S, Immenschuh S, Aggarwal N, Lichtinghagen R, Mahadeva R, Marcondes AM, Dinarello CA, Welte T, and Janciauskiene S. 2015. α1-Antitrypsin Combines with Plasma Fatty Acids and Induces Angiopoietin-like Protein 4 Expression. J Immunol 195: 3605–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koliwad SK, Kuo T, Shipp LE, Gray NE, Backhed F, So AY, Farese RV Jr., and Wang JC. 2009. Angiopoietin-like 4 (ANGPTL4, fasting-induced adipose factor) is a direct glucocorticoid receptor target and participates in glucocorticoid-regulated triglyceride metabolism. J Biol Chem 284: 25593–25601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakamoto M, Ishihara K, Watanabe T, Hirosue A, Hino S, Shinohara M, Nakayama H, and Nakao M. 2017. The Glucocorticoid Receptor Regulates the ANGPTL4 Gene in a CTCF-Mediated Chromatin Context in Human Hepatic Cells. PLoS One 12: e0169225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yin W, Romeo S, Chang S, Grishin NV, Hobbs HH, and Cohen JC. 2009. Genetic Variation in ANGPTL4 Provides Insights Into Protein Processing and Function. J Biol Chem 284: 13213–13222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jick SS, Lieberman ES, Rahman MU, and Choi HK. 2006. Glucocorticoid use, other associated factors, and the risk of tuberculosis. Arthritis Rheum 55: 19–26. [DOI] [PubMed] [Google Scholar]
- 45.Lai C-C, Lee M-TG, Lee S-H, Lee S-H, Chang S-S, and Lee C-C. 2015. Risk of incident active tuberculosis and use of corticosteroids. Int J Tuberc Lung Dis 19: 936–942. [DOI] [PubMed] [Google Scholar]
- 46.Xie Y, Xie J, Meijer AH, and Schaaf MJM. 2021. Glucocorticoid-Induced Exacerbation of Mycobacterial Infection Is Associated With a Reduced Phagocytic Capacity of Macrophages. Front Immunol 12: 618569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bai X, Feldman NE, Chmura K, Ovrutsky AR, Su W-L, Griffin L, Pyeon D, McGibney MT, Strand MJ, Numata M, Murakami S, Gaido L, Honda JR, Kinney WH, Oberley-Deegan RE, Voelker DR, Ordway DJ, and Chan ED. 2013. Inhibition of nuclear factor-kappa B activation decreases survival of Mycobacterium tuberculosis in human macrophages. PLoS One 8: e61925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deretic V, Singh S, Master S, Harris J, Roberts E, Kyei G, Davis A, de Haro S, Naylor J, Lee H-H, and Vergne I. 2006. Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cell Microbiol 8: 719–727. [DOI] [PubMed] [Google Scholar]
- 49.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, and Deretic V. 2004. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell Immunol 119: 753–766. [DOI] [PubMed] [Google Scholar]
- 50.Bai X, Kim S-H, Azam T, McGibney MT, Huang H, Dinarello CA, and Chan ED. 2010. IL-32 is a host protective cytokine against Mycobacterium tuberculosis in differentiated THP-1 human macrophages. J Immunol 184: 3830–3840. [DOI] [PubMed] [Google Scholar]
- 51.Busillo JM, and Cidlowski JA. 2013. The five Rs of glucocorticoid action during inflammation: ready, reinforce, repress, resolve, and restore. Trends Endocrinol Metab 24: 109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chinenov Y, and Rogatsky I. 2007. Glucocorticoids and the innate immune system: crosstalk with the toll-like receptor signaling network. Mol Cell Endocrinol 275: 30–42. [DOI] [PubMed] [Google Scholar]
- 53.Zhang TY, and Daynes RA. 2007. Glucocorticoid conditioning of myeloid progenitors enhances TLR4 signaling via negative regulation of the phosphatidylinositol 3-kinase-Akt pathway. J Immunol 178: 2517–2526. [DOI] [PubMed] [Google Scholar]
- 54.Bansal A, Mostafa MM, Kooi C, Sasse SK, Michi AN, Shah SV, Leigh R, Gerber AN, and Newton R. 2022. Interplay between nuclear factor-κB, p38 MAPK, and glucocorticoid receptor signaling synergistically induces functional TLR2 in lung epithelial cells. J Biol Chem 298: 101747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hattori Y, Hattori S, and Kasai K. 2003. Lipopolysaccharide activates Akt in vascular smooth muscle cells resulting in induction of inducible nitric oxide synthase through nuclear factor-kappa B activation. Eur J Pharmacol 481: 153–158. [DOI] [PubMed] [Google Scholar]
- 56.Chan ED, Pott GB, Silkoff PE, Ralston AH, Bryan CL, and Shapiro L. 2012. Alpha-1-antitrypsin inhibits nitric oxide production. J Leukoc Biol 92: 1251–1260. [DOI] [PubMed] [Google Scholar]
- 57.McCarthy C, Reeves EP, and McElvaney NG. 2016. The Role of Neutrophils in Alpha-1 Antitrypsin Deficiency. Ann Am Thorac Soc 13 Suppl 4: S297–S304. [DOI] [PubMed] [Google Scholar]
- 58.Frank F, Ortlund EA, and Liu X. 2021. Structural insights into glucocorticoid receptor function. Biochem Soc Trans 49: 2333–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Köhler C, Carlström G, Gunnarsson A, Weininger U, Tångefjord S, Ullah V, Lepistö M, Karlsson U, Papavoine T, Edman K, and Akke M. 2020. Dynamic allosteric communication pathway directing differential activation of the glucocorticoid receptor. Sci Adv 6: eabb5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schoch GA, D’Arcy B, Stihle M, Burger D, Bär D, Benz J, Thoma R, and Ruf A. 2010. Molecular switch in the glucocorticoid receptor: active and passive antagonist conformations. J Mol Biol 395: 568–577. [DOI] [PubMed] [Google Scholar]
- 61.Hudson WH, Youn C, and Ortlund EA. 2013. The structural basis of direct glucocorticoid-mediated transrepression. Nat Struct Mol Biol 20: 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tükenmez H, Edström I, Kalsum S, Braian C, Ummanni R, Fick SB, Sundin C, Lerm M, Elofsson M, and Larsson C. 2019. Corticosteroids protect infected cells against mycobacterial killing in vitro. Biochem Biophys Res Commun 511: 117–121. [DOI] [PubMed] [Google Scholar]
- 63.Bongiovanni B, Mata-Espinosa D, D’Attilio L, Leon-Contreras JC, Marquez-Velasco R, Bottasso O, Hernandez-Pando R, and Bay ML. 2015. Effect of cortisol and/or DHEA on THP1-derived macrophages infected with Mycobacterium tuberculosis. Tuberculosis (Edinb) 95: 562–569. [DOI] [PubMed] [Google Scholar]
- 64.Wang J, Wang R, Wang H, Yang X, Yang J, Xiong W, Wen Q, and Ma L. 2017. Glucocorticoids Suppress Antimicrobial Autophagy and Nitric Oxide Production and Facilitate Mycobacterial Survival in Macrophages. Sci Rep 7: 982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barber AE, Coyle SM, Marano MA, Fischer E, Calvano SE, Fong Y, Moldawer LL, and Lowry SF. 1993. Glucocorticoid therapy alters hormonal and cytokine responses to endotoxin in man. J Immunol 150: 1999–2006. [PubMed] [Google Scholar]
- 66.Fantuzzi G, Galli G, Zinetti M, Fratelli M, and Ghezzi P. 1995. The upregulating effect of dexamethasone on tumor necrosis factor production is mediated by a nitric oxide-producing cytochrome P450. Cell Immunol 160: 305–308. [DOI] [PubMed] [Google Scholar]
- 67.Smyth GP, Stapleton PP, Freeman TA, Concannon EM, Mestre JR, Duff M, Maddali S, and Daly JM. 2004. Glucocorticoid pretreatment induces cytokine overexpression and nuclear factor-kappaB activation in macrophages. J Surg Res 116: 253–261. [DOI] [PubMed] [Google Scholar]
- 68.Seto S, Tsujimura K, and Koide Y. 2012. Coronin-1a inhibits autophagosome formation around Mycobacterium tuberculosis-containing phagosomes and assists mycobacterial survival in macrophages. Cell Microbiol 14: 710–727. [DOI] [PubMed] [Google Scholar]
- 69.Bento CF, Empadinhas N, and Mendes V. 2015. Autophagy in the fight against tuberculosis. DNA Cell Biol 34: 228–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yim J-J, Kim HJ, Kwon OJ, and Koh W-J. 2008. Association between microsatellite polymorphisms in intron II of the human Toll-like receptor 2 gene and nontuberculous mycobacterial lung disease in a Korean population. Hum Immunol 69: 572–576. [DOI] [PubMed] [Google Scholar]
- 71.Lowe DM, Redford PS, Wilkinson RJ, O’Garra A, and Martineau AR. 2012. Neutrophils in tuberculosis: friend or foe? Trends Immunol 33: 14–25. [DOI] [PubMed] [Google Scholar]
- 72.de Melo MGM, Mesquita EDD, Oliveira MM, da Silva-Monteiro C, Silveira AKA, Malaquias TS, Dutra TCP, Galliez RM, Kritski AL, Silva EC, and Rede TBSG. 2018. Imbalance of NET and Alpha-1-Antitrypsin in Tuberculosis Patients Is Related With Hyper Inflammation and Severe Lung Tissue Damage. Front Immunol 9: 3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Frenzel E, Korenbaum E, Hegermann J, Ochs M, Koepke J, Koczulla AR, Welte T, Köhnlein T, and Janciauskiene S. 2012. Does augmentation with alpha1-antitrypsin affect neutrophil extracellular traps formation? Int J Biol Sci 8: 1023–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wartha F, Beiter K, Normark S, and Henriques-Normark B. 2007. Neutrophil extracellular traps: casting the NET over pathogenesis. Curr Opin Microbiol 10: 52–56. [DOI] [PubMed] [Google Scholar]
- 75.Dömer D, Walther T, Möller S, Behnen M, and Laskay T. 2021. Neutrophil Extracellular Traps Activate Proinflammatory Functions of Human Neutrophils. Front Immunol 12: 636954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Serban KA, Petrusca DN, Mikosz A, Poirier C, Lockett AD, Saint L, Justice MJ, Twigg HL 3rd., Campos MA, and Petrache I. 2017. Alpha-1 antitrypsin supplementation improves alveolar macrophages efferocytosis and phagocytosis following cigarette smoke exposure. PLoS One 12: e0176073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Embgenbroich M, van der Zande HJP, Hussaarts L, Schulte-Schrepping J, Pelgrom LR, García-Tardón N, Schlautmann L, Stoetzel I, Händler K, Lambooij JM, Zawistowska-Deniziak A, Hoving L, de Ruiter K, Wijngaarden M, Pijl H, Willems van Dijk K, Everts B, van Harmelen V, Yazdanbakhsh M, Schultze JL, Guigas B, and Burgdorf S. 2021. Soluble mannose receptor induces proinflammatory macrophage activation and metaflammation. Proc Natl Acad Sci U S A 118: e2103304118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium via the PRIDE [1] partner repository with the dataset identifier PXD030989 and 10.6019/PXD030989.